Abstract
Both environmental factors and genetic factors are involved in the pathogenesis of autism spectrum disorders (ASDs). Epigenetics, an essential mechanism for gene regulation based on chemical modifications of DNA and histone proteins, is also involved in congenital ASDs. It was recently demonstrated that environmental factors, such as endocrine disrupting chemicals and mental stress in early life, can change epigenetic status and gene expression, and can cause ASDs. Moreover, environmentally induced epigenetic changes are not erased during gametogenesis and are transmitted to subsequent generations, leading to changes in behavior phenotypes. However, epigenetics has a reversible nature since it is based on the addition or removal of chemical residues, and thus the original epigenetic status may be restored. Indeed, several antidepressants and anticonvulsants used for mental disorders including ASDs restore the epigenetic state and gene expression. Therefore, further epigenetic understanding of ASDs is important for the development of new drugs that take advantages of epigenetic reversibility.
1. Introduction
Autism spectrum disorders (ASDs) are complex, pervasive neurodevelopmental disorders that are characterized by dysfunctions in social interactions and communications and restricted/fixated interests or repetitive behavior that manifest in early childhood [1]. ASDs include classical autism, Asperger syndrome, and pervasive developmental disorder-not otherwise Specified [2,3].
A number of environmental factors are known to be involved in the pathogenesis of ASDs, including nutritional factors and hormones [4]. Furthermore, inappropriate child rearing, such as child abuse and malnutrition by parents with psychiatric problems, can be associated with ASDs [5,6,7,8]. Viral infections with rubella and cytomegalovirus and associated immunological reactions via activation of microglia are also thought to be involved in ASDs, which has been demonstrated by pathological studies of post-mortem brains and neuroimaging studies of ASD patients [9,10,11,12,13,14], although some epidemiological studies conducted in Denmark and Taiwan did not support the hypothesis that pre- and postnatal infection and immunological reaction are involved in ASD cases with regard to herpes and influenza viral infection and Kawasaki Disease (a disorder potentially associated with corona virus) [15,16,17]. In addition, endocrine-disrupting chemicals (EDCs) are thought to be involved in the development of ASDs, including tobacco, air pollutants, solvents, metals, pesticides, and organic EDCs such as flame retardants, non-stick chemicals, phthalates, and bisphenol A (BPA) [18].
Conversely, a number of genetic factors have been identified as causes of ASDs. Mutations in genes encoding neurotransmitters such as synapsin, dopamine transporter, and neuroligin, and synapse-associated proteins such as scaffold proteins including shank and lin7B, have been identified in ASD patients [19,20,21,22,23,24]. Unexpectedly, mutations have also been identified in chromatin-remodeling factors such as histone modifying enzymes and chromodomain helicases in congenital ASDs (e.g., Kleefstra syndrome) [25,26,27]. These findings suggest that ASDs can be recognized as a “synaptic and chromatin-remodeling disorders” [25,28].
Chromatin is a genetic unit that consists of DNA and histone proteins, which are modified by enzymes for DNA methylation, histone acetylation and methylation and by chromatin-binding polycomb proteins. A recent three-dimensional resolution imaging technology provided a precise chromatin organization with epigenetic modifications [29]. Furthermore, autism susceptibility candidate 2, a nuclear protein involved in cortical neuronal migration and neuritogenesis in the developing brain [30] and whose mutations cause ASDs [31,32], forms a complex with polycomb repressive complex 1 to purge its repressive function and activates expression of neurodevelopmental genes involved in axon guidance in the developing forebrain, such as neruocan [33,34]. These results suggest that close interaction between neuronal molecules and epigenetic molecules is important for normal brain development and failure of this interaction is potentially associated with ASDs.
In this review, we introduce congenital epigenetic disorders with ASD-like phenotypes and environmental factors that affect epigenetic regulation of neuronal genes, and discuss transgenerational epigenetic inheritance and therapeutic strategies for ASDs taking advantage of use of the epigenetic reversibility.
2. Congenital Epigenetic Diseases
Rett syndrome (RTT) is a representative ASD characterized by repetitive and stereotypic hand movements, seizures, gait ataxia and autism [35] and is caused by mutations in the gene that encode methyl-CpG-binding protein 2 (MeCP2), which is associated with chromatin remodeling [36]. Since RTT is an X-linked dominant disorder, male patients are embryonic lethal and thus all patients are female.
MeCP2 interacts with the Sin3A/HDAC complex [37,38,39,40], and binds to methylated CpG in DNA to suppress a number of genes associated with synaptic function (e.g., BDNF, DLX5, ID, CRH, IGFBP3, CDKL1, PCDHB1 and PCDH7, LIN7A) in neurons and other types of brain cells [41,42,43,44,45,46,47], thereby controlling excitatory synaptic strength by regulating the number of glutamatergic synapses [48].
Induced pluripotent stem cells (iPSCs) can be used to determine how a disease develops in patients, especially inaccessible brain cells. Using iPSC technology, it is possible to generate neural cells from patients’ peripheral tissue such as skin fibroblasts. Several studies have shown that RTT iPSC-derived neurons exhibit maturation and electro-physiological defects reminiscent of those seen in RTT patients and mouse models [49,50,51], and we have shown that astrocyte-specific genes (e.g., GFAP) are aberrantly expressed in neural cells generated from iPSC lines that lack MeCP2 expression, which leads to the de-suppression of astrocyte-specific genes (Figure 1A) [52].
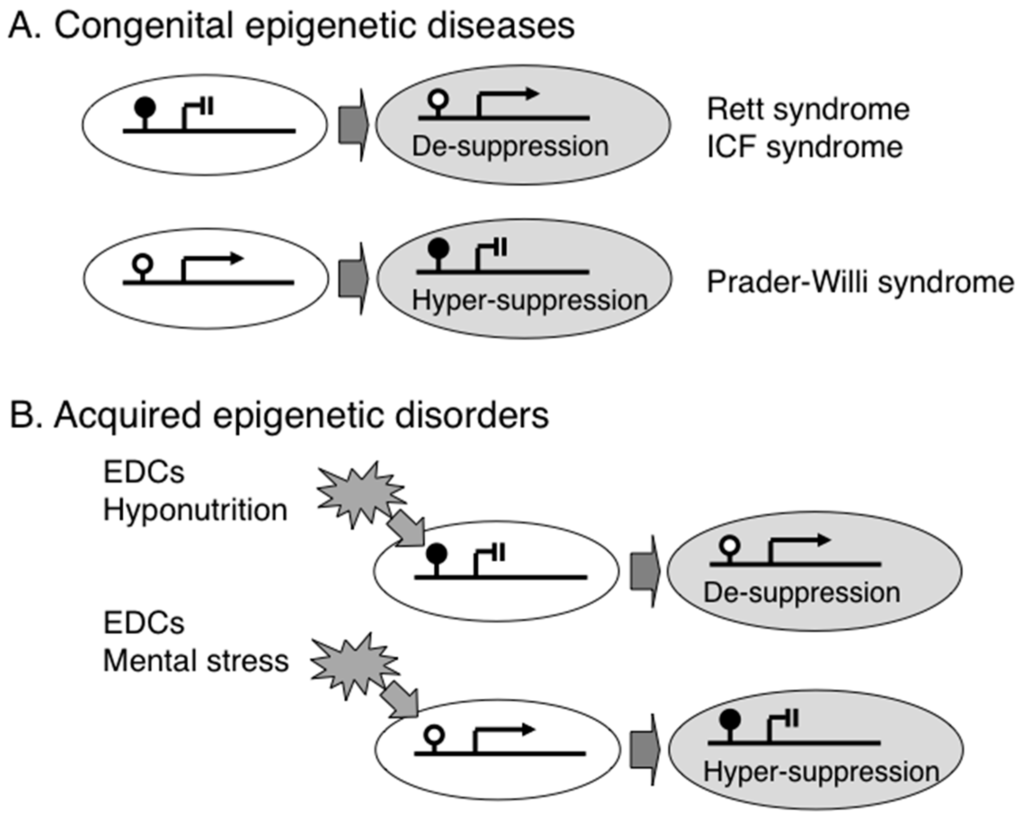
Figure 1.
Epigenetic gene regulation in congenital epigenetic diseases and acquired epigenetic disorders. (A) Deficiency of DNA-binding protein or DNA methylation causes Rett syndrome (an ASD) or ICF syndrome (an immunodeficiency disease), respectively. Congenital aberrant DNA methylation due to genomic imprinting error causes Prader-Willi syndrome; and (B) Various environmental factors such as endocrine disrupting chemicals (EDCs), hyponutrition, and mental stress are known to alter epigenetic status, resulting in aberrant gene expression.
Interestingly, not only functional deficiency of MeCP2 protein (i.e., due to mutations of MECP2) but also increased dosage of MeCP2 protein (i.e., due to duplication of MECP2) results in severe mental retardation in males [53] and cognitive impairment with learning difficulties and speech delay in females [54]. The increased dosage effect of Mecp2 on neurological function has been confirmed in a model mouse that exhibits motor coordination deficits, heightened anxiety, and impairments of learning and memory [55], and in a monkey model that exhibits a higher frequency of repetitive circular locomotion, increased stress responses, less interaction with wild-type monkeys, reduced interaction time with other transgenic monkeys, and stereotypic cognitive behaviors [56,57]. These findings indicate that the expression of MECP2 within a normal range is essential for normal brain development.
ICF syndrome is a congenital disorder named after three major features, such as Immunodeficiency, Centromere instability, and Facial anomalies [58]. Although the cause is different between RTT and ICF syndromes, the consequence is similar; both lead to the de-suppression of target genes by the failure of DNA methylation-dependent gene regulation (Figure 1A). ICF syndrome is diagnosed by specific chromosome findings with breakage of the pericentric heterochromatic regions of chromosomes 1, 9 and 16, which are normally hypermethylated but are hypomethylated due to deficiency of DNMT3B in ICF [59]. The patients show distinct low levels of immunoglobulins (e.g., IgG and IgA) and they required intravenous immunoglobulin supplementation every 2 weeks. Although a recent study has demonstrated an ICF-specific DNA hypomethylation pattern in mesenchymal stem cells differentiated from the iPSCs of ICF patients [60] and another study has shown a subset of hypomethylated genes in ICF patients [61], the precise molecular mechanism for the immune dysregulation, which is the main clinical feature in ICF, is still largely unknown. It may be necessary to analyze purified B lymphocytes from ICF patients in order to identify hypomethylated DNMT3B-driven immunological genes. Interestingly, mutations in a gene encoding another DNA methyltransferase, DNMT3A, cause intellectual disability with overgrowth [62], suggesting that DNA methyltransferases are essential for normal brain and immunological development.
Prader-Willi syndrome (PWS) is a hallmark epigenetic disease; the causative epigenetic abnormality was identified more than 20 years ago. Approximately 70% patients have a chromosomal deletion at 15q11-q13, and the remaining patients have genomic imprinting errors. In PWS patients with maternal uniparental disomy, both paternal and maternal alleles of genes within the 15q11-q13 region are hypermethylated and thus expression from both alleles is suppressed (Figure 1A) [63,64,65]. The clinical features of PWS includes neurocognitive deficits, excessive daytime sleepiness, muscle hypotonia, short stature, small hands and feet, hypergonadism, hyperphagia starting from infancy, and subsequent obesity and type 2 diabetes [66].
Angelman syndrome is characterized by severe intellectual disability, intractable epilepsy, puppet-like ataxic movement, and paroxysms of laughter. The critical region is the same as PWS (i.e., 15q11-q13), but parental-of-origin is different; either maternal deletion or paternal uniparental disomy causes Angelman syndrome, because the causative gene, ubiquitin protein ligase E3A (UBE3A), is maternally expressed [67]. Interestingly, the increased copy number (i.e., duplication or triplication) of the maternal 15q11-q13 region that leads to UBE3A overexpression causes an ASD [68]. These findings indicate that the expression of UBE3A within a normal range is essential for normal brain development.
Epigenomic studies were conducted within the regions of various neuronal genes and ASD-specific differential DNA methylation was revealed. For example, increased DNA methylation at the promoter regions subsequent reduced expression were observed within the genes of oxytocin receptor (OCTR), Engrailed-2 (EN2) and Reelin (RELN) in the postmortem brain tissues from ASD patients [69,70,71]. Increased hydroxymethylation and subsequent increased binding of MeCP2 associated with gene silencing were identified within the promoter region of glutamate decarboxylase 1 (GAD1) in the postmortem brain tissues from ASD patients [71].
Recent genome-wide DNA methylation studies using array-based Infinium BeadChip identified ASD-associated differential DNA methylation at ZFP57 associated with folate metabolism, which is a potential contributor to ASD risk, in the postmortem brain tissues [72,73,74], and at brain-derived neurotrophic factor (BDNF) in the peripheral blood tissues from ASD patients [75]. Findings through these studies potentially generate robust epigenetic biomarkers for risk, diagnosis and prognosis of ASD, which may also be used to monitor response to early interventions [76,77].
3. Acquired Epigenetic Disorders
As mentioned above, not only genetic factors (i.e., DNA mutations) but also environmental factors are involved in ASDs, and a combination of heritability (G: genetic factor such as single nucleotide polymorphism) and experience (E: environmental factor)—that is, “G, X, E”—has been the main concept for understanding common diseases, including ASDs. Recently, new G X E model has been proposed in which E dynamically changes G and causes DNA and histone chemical modifications (i.e., epigenetics), but not DNA sequence changes [78].
EDCs are compounds released from chemical, agricultural, pharmaceutical, and consumer product industries that have estrogenic activity or interfere with endogenous sex hormones. Of the many EDCs, BPA is associated with reproductive toxicity, altered growth, and immune dysregulation, and alters DNA methylation in fetal mouse brains [79]. Moreover, perinatal BPA exposure via maternal diet decreases global DNA methylation in bone marrow-derived mast cells of the offspring during adulthood [80], and it alters DNA methylation of Stat3 dose in a dose dependent manner in mouse liver [81]. High dose exposure of polybrominated diphenyl ethers (flame retardants) decreases DNA methylation at the promoter of TNFα, a proinflammatory gene, and increase TNFα protein expression in human cord blood [82]. Furthermore, the altered DNA methylation patterns in AHRR, MYO1G, CYP1A1, and CNTNAP2 caused by maternal tobacco smoking detected in cord blood was confirmed in the peripheral blood of their children at 17 years of age [83], suggesting that altered DNA methylation in the early development period can persist for a long period and it may be useful as a long-lasting signature of maternal stress or history of the offspring.
Nutrition also influences programming of an offspring’s epigenome, which includes folic acid and vitamins B2, B6 and B12 that are essential for one-carbon metabolism and are involved in DNA methylation (Figure 1B). Moreover, a calorie- or protein-restricted maternal diet decreases DNA methylation and induces the over-expression of energy storage-associated genes (e.g., PPARγ) in fetal liver to generate a “thrifty phenotype,” which promotes survival under conditions of poor nutrition before and after birth [84,85]. Once an individual is born with a thrifty phenotype in modern society with an abundance of food, the nutritional mismatch between prenatal and postnatal conditions induces metabolic and mental disorders [86,87,88,89], the concept of which is referred to as “Developmental Origins of Heath and Disease (DOHaD)” [90].
Several lines of evidence suggest that not only materials (e.g., chemicals and nutrients) but also mental stresses can alter an offspring’s epigenome (Figure 1B). For example, exposure of short-term postnatal stress by separating offspring from the mother induced hypermethylation within the promoter region of the glucocorticoid Receptor (NR3C1) gene, which encodes a hormone associated with resilience, in the hippocampal region of the offspring, leading to abnormal behavior in rats [91]. Furthermore, exposure to prenatal maternal stress also predicts a wide variety of behavioral and physical outcomes in the offspring. A recent study of women who were pregnant during a disaster (the ice storm in Quebec in 1998) revealed that DNA methylation profiles were altered in genes related to immune function in the peripheral blood of their offspring [92]. Similarly, it has been demonstrated that maternal stress during pregnancy alters DNA methylation of the imprinted genes IGF2 and GNASXL in cord blood [93], and that maternal stress also alters DNA methylation in NR3C1 and BDNF in buccal mucosa DNA samples obtained from 2 month-old infants born to mothers with depressive symptoms during pregnancy [94,95].
4. Transgenerational Epigenetic Inheritance
Environmental factors that alter a phenotype not only affect the exposed individual but also subsequent progeny for successive generations. In other words, ancestral experiences could influence subsequent generations, the concept of which is termed “transgenerational inheritance.” Furthermore, environmental factors such as EDCs and nutrition do not promote genetic mutations but instead promote epigenetic changes; the permanent programming of an altered epigenome in the germline can allow for the transmission of transgenerational epigenetic phenotypes [96]. The evidence supports the theory of Lamarckian inheritance in which an organism can pass on phenotypes that it acquired during its lifetime to its offspring. More precisely, a hypothesis has emerged that environmental stress results in epigenetic changes at some loci in the genome and these can escape the epigenetic reprogramming that normally occurs between generations [97,98].
Short-term postnatal mental stress by separating offspring from their mother alters DNA methylation not only in the brain but also in the sperm of male offspring, and then the environmentally induced epigenetic and expression alterations of Crfr2 are transmitted up to the third generation (F1 sperm and F2 brain) along with behavioral abnormalities [99]. Since this initial observation, similar findings have accumulated. For example, prenatal stress exposure induces changes in DNA methylation and miRNA expression in the placenta and brain, which leads to an increase in risk for schizophrenia, attention deficit hyperactivity disorder, ASDs, and anxiety- or depression-related disorders later in life [100]. Besides mental stress, exposure to an EDC (e.g., vinclozolin) during embryonic gonadal sex determination can alter male germ-line epigenetics, and the alteration of DNA methylation in the germ line appears to result in the transmission of transgenerational adult-onset diseases, including spermatogenic defects, prostate disease, kidney disease and cancer [101]. A recent study demonstrated that exposure to BPA in early life induces glucose intolerance and β-cell dysfunction, with hypermethylation and associated decreased expression of IGF2 in the islets of male F2 offspring; this finding suggests that BPA exposure during early life can result in generational transmission of glucose intolerance and β-cell dysfunction through the male germ line by an epigenetic mechanism [102].
However, evidence that such effects persist in the subsequent generations has been inconclusive [97,103,104]. The effects must be observed in the F3 generation to be considered transgenerational, because the in utero nature of the ancestral perturbation affects not only the somatic and germ cells of the developing F1 fetus, but also the germ cells of the F2 generation. In this context, a recent study demonstrated that treatment of pregnant mice with the EDC methoxychlor altered the methylation of all imprinted genes examined (i.e., H19, Meg3 (Gtl2), Mest (Peg1), Snrpn, and Peg3) in the F1 offspring, but these effects disappeared gradually from F1 to F3 [105]. These finding suggests that transgenerational epigenetic inheritance is not “solid” inheritance, such as genetic (DNA sequence-based) inheritance, but “soft” inheritance [106,107].
5. Conclusions
In this article, we have introduced ASDs with epigenetic abnormalities caused by genetic mutations in enzymes and proteins involved or chromosomal abnormalities such as Rett and Prader-Willi syndromes (i.e., congenital and syndromic ASDs) and ASDs with epigenetic abnormalities caused by environmental factors such as chemicals, nutrition, and mental stress (i.e., acquired and non-syndromic ASDs). Furthermore, we introduced the concept of transgenerational epigenetic inheritance in which environmental stress-induced epigenetic changes can be transmitted to the subsequent generations by escaping from erasure during epigenetic reprogramming. However, transgenerational epigenetic inheritance is not “solid” inheritance but “soft” inheritance because epigenetics is a reversible mechanism based on the addition and removal of chemical residues on DNA and histone proteins.
Taking advantage of this epigenetic reversibility, some psychotropic drugs, such as valproic acid for epilepsy and mental disorders and imipramine for depressive disorders, can restore altered histone modifications and gene expression [108,109,110]. A recent epidemiological study demonstrated that supplementation of folic acid during pregnancy, which is an important nutrient and substrate for DNA methylation, reduced the risk of ASDs in the offspring [111]. Furthermore, studies using RTT or MeCP2-duplication mouse models demonstrated that genetic supplementation of MeCP2, bone marrow transplantation, or antisense oligonucleotides after birth successfully attenuated neurological symptoms [112,113,114]. These findings support the idea that the phenotypes of ASDs caused by epigenetic dysregulation are reversible and thus treatable. Further epigenetic understanding of ASDs will offer new concepts for therapeutic strategies.
Acknowledgments
The authors would like to thank the Ministry of Education, Science, Sports and Culture (MEXT), Grants-in-Aid (KAKENHI) for Scientific Research (B) (#26293245) and Exploratory Research (#15K15388), and the Japan Agency for Medical Research and Development (AMED) for funds for the development of core technologies for innovative drug development based on IT for conducting our original research as described in this article.
Author Contributions
Takeo Kubota and Kazuki Mochizuki prepared the manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Text Revision, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- American Psychiatric Association. Proposed Revision: A 05 Autism Spectrum Disorder. DSM-5 development website. Available online: http://web-beta.archive.org/web/20121115164727/http://www.dsm5.org/ProposedRevision/Pages/proposedrevision.aspx?rid=94 (accessed on 31 January 2016).
- Sealey, L.A.; Hughes, B.W.; Sriskanda, A.N.; Guest, J.R.; Gibson, A.D.; Johnson-Williams, L.; Pace, D.G.; Bagasra, O. Environmental factors in the development of autism spectrum disorders. Environ. Int. 2016, 88, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Larsson, H.J.; Eaton, W.W.; Madsen, K.M.; Vestergaard, M.; Olesen, A.V.; Agerbo, E.; Schendel, D.; Thorsen, P.; Mortensen, P.B. Risk factors for autism: Perinatal factors, parental psychiatric history, and socioeconomic status. Am. J. Epidemiol. 2005, 161, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Jokiranta, E.; Brown, A.S.; Heinimaa, M.; Cheslack-Postava, K.; Suominen, A.; Sourander, A. Parental psychiatric disorders and autism spectrum disorders. Psychiatry Res. 2013, 207, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Lyall, K.; Rich-Edwards, J.W.; Ascherio, A.; Weisskopf, M.G. Association of maternal exposure to childhood abuse with elevated risk for autism in offspring. JAMA Psychiatry 2013, 70, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Belluscio, L.M.; Berardino, B.G.; Ferroni, N.M.; Ceruti, J.M.; Cánepa, E.T. Early protein malnutrition negatively impacts physical growth and neurological reflexes and evokes anxiety and depressive-like behaviors. Physiol. Behav. 2014, 129, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.E.; Navar-Boggan, A.M.; Omer, S.B. Congenital rubella syndrome and autism spectrum disorder prevented by rubella vaccination—United States; 2001–2010. BMC Public Health 2011, 11, 340. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Moriuchi, H.; Matsuzaki, J.; Motoyama, K.; Moriuchi, M. Retrospective diagnosis of congenital cytomegalovirus infection in children with autism spectrum disorder but no other major neurologic deficit. Brain Dev. 2015, 37, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Heilbrun, L.P.; Palmer, R.F.; Jaen, C.R.; Svoboda, M.D.; Miller, C.S.; Perkins, J. Maternal Chemical and Drug Intolerances: Potential Risk Factors for Autism and Attention Deficit Hyperactivity Disorder (ADHD). J. Am. Board Fam. Med. 2015, 28, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.I.; Kern, J.K. Evidence of microglial activation in autism and its possible role in brain under connectivity. Neuron Glia Biol. 2011, 7, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- McDougle, C.J. Toward an immune-mediated subtype of autism spectrum disorder. Brain Res. 2015, 1617, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Atladóttir, H.Ó.; Henriksen, T.B.; Schendel, D.E.; Parner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Wu, C.M.; Chang, W.P.; Kuo, C.N.; Yeter, D.; Lin, C.Y.; Pai, J.T.; Chi, Y.C.; Lin, C.H.; Wang, L.J.; Chang, W.C. Association between Kawasaki disease and autism: A population-based study in Taiwan. Int. J. Environ. Res. Public Health 2014, 11, 3705–3716. [Google Scholar] [CrossRef] [PubMed]
- Esper, F.; Shapiro, E.D.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Association between a novel human coronavirus and Kawasaki disease. J. Infect. Dis. 2005, 191, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Kalkbrenner, A.E.; Schmidt, R.J.; Penlesky, A.C. Environmental chemical exposures and autism spectrum disorders: A review of the epidemiological evidence. Curr. Probl. Pediatr. Adolesc. Health Care 2014, 44, 277–318. [Google Scholar] [CrossRef] [PubMed]
- Corradi, A.; Fadda, M.; Piton, A.; Patry, L.; Marte, A.; Rossi, P.; Cadieux-Dion, M.; Gauthier, J.; Lapointe, L.; Mottron, L.; et al. SYN2 is an autism predisposing gene: Loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 2014, 23, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, P.J.; Campbell, N.G.; Sharma, S.; Erreger, K.; Herborg Hansen, F.; Saunders, C.; Belovich, A.N.; NIH ARRA Autism Sequencing Consortium; Sahai, M.A.; Cook, E.H.; et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 2013, 18, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Hill-Yardin, E.L.; Argyropoulos, A.; Hosie, S.; Rind, G.; Anderson, P.; Hannan, A.J.; O’Brien, T.J. Reduced susceptibility to induced seizures in the Neuroligin-3(R451C) mouse model of autism. Neurosci. Lett. 2015, 589, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Matsumoto, A.; Hamada, N.; Ito, H.; Miyauchi, A.; Jimbo, E.F.; Momoi, M.Y.; Tabata, H.; Yamagata, T.; Nagata, K. Role of an adaptor protein Lin-7B in brain development: Possible involvement in autism spectrum disorders. J. Neurochem. 2015, 132, 61–69. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar]
- Balan, S.; Iwayama, Y.; Maekawa, M.; Toyota, T.; Ohnishi, T.; Toyoshima, M.; Shimamoto, C.; Esaki, K.; Yamada, K.; Iwata, Y.; et al. Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHTM2 and WIZ, in Japanese autism subjects. Mol. Autism 2014, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.A.; Pomaville, M.B.; O’Roak, B.J. Mutations and Modeling of the Chromatin Remodeler CHD8 Define an Emerging Autism Etiology. Front. Neurosci. 2015, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; FitzPatrick, D.; Tommerup, N.; Bugge, M.; Niebuhr, E.; Neumann, L.M.; Tzschach, A.; Shoichet, S.A.; Menzel, C.; Erdogan, F.; et al. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 2007, 121, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lee, P.; Stafford, J.M.; von Schimmelmann, M.; Schaefer, A.; Reinberg, D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 2014, 516, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Haliburton, G.D.; Eckalbar, W.L.; Oren, I.; Nishizaki, S.; Murphy, K.; Pollard, K.S.; Birnbaum, R.Y.; Ahituv, N. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry 2014, 4, e431. [Google Scholar] [CrossRef] [PubMed]
- GeneReview-MeCP2 Related Disorders. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1497/ (accessed on 28 February 2016).
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Ballestar, E.; Yusufzai, T.M.; Wolffe, A.P. Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 2000, 39, 7100–7106. [Google Scholar] [CrossRef] [PubMed]
- Wolffe, A.P. Histone deacetylase: A regulator of transcription. Science 1996, 272, 371–372. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsbergerv, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Bird, A. The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome. Brain Dev. 2001, 23 (Suppl. 1), S32–S37. [Google Scholar] [CrossRef]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fanv, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Peddada, S.; Yasui, D.H.; LaSalle, J.M. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet. 2006, 15, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Ide, S.; Takashima, S.; Kudo, S.; Nomura, Y.; Segawa, M.; Kubota, T.; Mori, H.; Tanaka, S.; Horie, H.; Tanabe, Y.; Goto, Y. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 2007, 66, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Carouge, D.; Host, L.; Aunis, D.; Zwiller, J.; Anglard, P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol. Dis. 2010, 38, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Hirasawa, T.; Soutome, M.; Itoh, M.; Goto, Y.; Endoh, K.; Takahashi, K.; Kudo, S.; Nakagawa, T.; Yokoi, S.; et al. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: Implication for pathogenesis of Rett syndrome. BMC Neurosci. 2011, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Farra, N.; Zhang, W.B.; Pasceri, P.; Eubanks, J.H.; Salter, M.W.; Ellisv, J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 2012, 17, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Djuric, U.; Cheung, A.Y.; Zhang, W.; Mok, R.S.; Lai, W.; Piekna, A.; Hendry, J.A.; Ross, P.J.; Pasceri, P.; Kim, D.S.; et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 2015, 76, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Andoh-Noda, T.; Akamatsu, W.; Miyake, K.; Matsumoto, T.; Yamaguchi, R.; Sanosaka, T.; Okada, Y.; Kobayashi, T.; Ohyama, M.; Nakashima, K.; et al. Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain 2015, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, LR.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Novara, F.; Simonati, A.; Sicca, F.; Battini, R.; Fiori, S.; Contaldo, A.; Criscuolo, L.; Zuffardi, O.; Ciccone, R. MECP2 duplication phenotype in symptomatic females: Report of three further cases. Mol. Cytogenet. 2014, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Na, E.S.; Nelson, E.D.; Adachi, M.; Autry, A.E.; Mahgoub, M.A.; Kavalali, E.T.; Monteggia, L.M. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 2012, 32, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, X.; Zhang, J.T.; Cai, Y.J.; Cheng, T.L.; Cheng, C.; Wang, Y.; Zhang, C.C.; Nie, Y.H.; Chen, Z.F.; et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 2016, 530, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Monkeys genetically modified to show autism symptoms. Nature 2016, 529, 449. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Kubota, T.; Furuumi, H.; Kamoda, T.; Iwasaki, N.; Tobita, N.; Fujiwara, N.; Goto, Y.; Matsui, A.; Sasaki, H.; Kajii, T. ICF syndrome in a girl with DNA hypomethylation but without detectable DNMT3B mutation. Am. J. Med. Genet. A 2004, 129A, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wu, Z.; Liu, Z.; Hu, G.; Yu, J.; Chang, K.H.; Kim, K.P.; Le, T.; Faull, K.F.; Rao, N.; et al. Selective demethylation and altered gene expression are associated with ICF syndrome in human-induced pluripotent stem cells and mesenchymal stem cells. Hum. Mol. Genet. 2014, 23, 6448–6457. [Google Scholar] [CrossRef] [PubMed]
- Simo-Riudalbas, L.; Diaz-Lagares, A.; Gatto, S.; Gagliardi, M.; Crujeiras, A.B.; Matarazzo, M.R.; Esteller, M.; Sandoval, J. Genome-Wide DNA Methylation Analysis Identifies Novel Hypomethylated Non-Pericentromeric Genes with Potential Clinical Implications in ICF Syndrome. PLoS ONE 2015, 10, e0132517. [Google Scholar]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Das, S.; Christian, S.L.; Baylin, S.B.; Herman, J.G.; Ledbetter, D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997, 16, 16–17. [Google Scholar] [PubMed]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance of Men (OMIM) #176270. Available online: http://omim.org/entry/176270 (accessed on 29 February 2016).
- Rangasamy, S.; D’Mello, S.R.; Narayanan, V. Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 2013, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Scoles, H.A.; Urraca, N.; Chadwick, S.W.; Reiter, L.T.; Lasalle, J.M. Increased copy number for methylated maternal 15q duplications leads to changes in gene and protein expression in human cortical samples. Mol. Autism 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl. Psychiatry 2013, 3, e232. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Chen, Y.; Dong, E.; Cook, E.H.; Guidotti, A.; Grayson, D.R. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 2014, 4, e349. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [PubMed]
- Castro, K.; Klein, L.D.; Baronio, D.; Gottfried, C.; Riesgo, R.; Perry, I.S. Folic acid and autism: What do we know? Nutr. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, Y.; Zhang, F.; Xu, M.; Zhang, J.; Yan, J.; Ju, W.; Brown, W.T.; Zhong, N. Hypermethylation of the enolase gene (ENO2) in autism. Eur. J. Pediatr. 2014, 173, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [PubMed]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The role of epigenetic change in autism spectrum disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Crews, D.; Gillette, R.; Miller-Crews, I.; Gore, A.C.; Skinner, M.K. Nature, nurture and epigenetics. Mol. Cell Endocrinol. 2014, 398, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Yaoi, T.; Itoh, K.; Nakamura, K.; Ogi, H.; Fujiwara, Y.; Fushiki, S. Genome-wide analysis of epigenomic alterations in fetal mouse forebrain after exposure to low doses of bisphenol A. Biochem. Biophys. Res. Commun. 2008, 376, 563–567. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Dolinoy, D.C.; Mancuso, P. Perinatal bisphenol A exposures increase production of pro-inflammatory mediators in bone marrow-derived mast cells of adult mice. J. Immunotoxicol. 2014, 11, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, C.; Bergin, I.L.; Harris, C.; Dolinoy, D.C. Stat3 is a candidate epigenetic biomarker of perinatal Bisphenol A exposure associated with murine hepatic tumors with implications for human health. Epigenetics 2015, 10, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Hong, X.; Wang, X.; Tang, W.Y. Aberrant 5’-CpG Methylation of Cord Blood TNFα Associated with Maternal Exposure to Polybrominated Diphenyl Ethers. PLoS ONE 2015, 10, e0138815. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Simpkin, A.J.; Woodward, G.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Smith, A.D.; Timpson, N.J.; Tilling, K.; et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2015, 24, 2201–2217. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPARα promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, C.A.; van Veldhoven, K.; Relton, C.; Stringhini, S.; Kyriacou, K.; Vineis, P. Biological embedding of early-life exposures and disease risk in humans: A role for DNA methylation. Eur. J. Clin. Invest. 2015, 45, 303–332. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Seng, C.Y.; Fukuoka, H.; Beedle, A.S.; Hanson, M.A. Low birthweight and subsequent obesity in Japan. Lancet 2007, 369, 1081–1082. [Google Scholar] [CrossRef]
- St Clair, D.; Xu, M.; Wang, P.; Yu, Y.; Fang, Y.; Zhang, F.; Zheng, X.; Gu, N.; Feng, G.; Sham, P.; et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA 2005, 294, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar] [PubMed]
- Silveira, P.P.; Manfro, G.G. Retrospective studies. Adv. Neurobiol. 2015, 10, 251–267. [Google Scholar] [PubMed]
- Benyshek, D.C. The “early life” origins of obesity-related health disorders: New discoveries regarding the intergenerational transmission of developmentally programmed traits in the global cardiometabolic health crisis. Am. J. Phys. Anthropol. 2013, 152 (Suppl. 57), 79–93. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lei, L.; Massart, R.; Suderman, M.J.; Machnes, Z.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS ONE 2014, 9, e107653. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, E.B.; Izzi, B.; Hompes, T.; Vansteelandt, K.; Lambrechts, D.; Freson, K.; Claes, S. DNA methylation in imprinted genes IGF2 and GNASXL is associated with prenatal maternal stress. Genes Brain Behav. 2015, 14, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.C.; Kundakovic, M.; Ramchandani, P.G.; Murphy, S.E.; Champagne, F.A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 2015, 10, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Palma-Gudiel, H.; Córdova-Palomera, A.; Eixarch, E.; Deuschle, M.; Fañanás, L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: A meta-analysis. Epigenetics 2015, 10, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, E. Disputing Lamarckian epigenetic inheritance in mammals. Genome Biol. 2015, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 2010, 68, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Skinner, M.K. Epigenetic programming of the germ line: Effects of endocrine disruptors on the development of transgenerational disease. Reprod. Biomed. Online 2008, 16, 23–25. [Google Scholar] [CrossRef]
- Mao, Z.; Xia, W.; Chang, H.; Huo, W.; Li, Y.; Xu, S. Paternal BPA exposure in early life alters IGF2 epigenetic status in sperm and induces pancreatic impairment in rat offspring. Toxicol. Lett. 2015, 238, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Dias, B.G.; Ressler, K.J. Experimental evidence needed to demonstrate inter- and trans-generational effects of ancestral experiences in mammals. Bioessays 2014, 36, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Stouder, C.; Paoloni-Giacobino, A. Specific transgenerational imprinting effects of the endocrine disruptor methoxychlor on male gametes. Reproduction 2011, 141, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Low, F.M.; Gluckman, P.D. Epigenetic epidemiology: The rebirth of soft inheritance. Ann. Nutr. Metab. 2011, 58 (Suppl. 2), 8–15. [Google Scholar] [CrossRef] [PubMed]
- Dickins, T.E.; Rahman, Q. The extended evolutionary synthesis and the role of soft inheritance in evolution. Proc. Biol. Sci. 2012, 279, 2913–2921. [Google Scholar] [PubMed]
- Jessberger, S.; Nakashima, K.; Clemenson, G.D., Jr.; Mejia, E.; Mathews, E.; Ure, K.; Ogawa, S.; Sinton, C.M.; Gage, F.H.; Hsieh, J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 2007, 27, 5967–5975. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Nghia, N.A.; Hirasawa, T.; Kasai, H.; Obata, C.; Moriishi, K.; Mochizuki, K.; Koizumi, S.; Kubota, T. Long-term imipramine treatment increases N-methyl-d-aspartate receptor activity and expression via epigenetic mechanisms. Eur. J. Pharmacol. 2015, 752, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Surén, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.; Guyenet, P.G.; Kipnis, J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 2012, 484, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Chen, H.M.; Swann, J.W.; Hao, S.; Tang, B.; Wu, Z.; Tang, J.; Wan, Y.W.; Liu, Z.; Rigo, F.; Zoghbi, H.Y. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 2015, 528, 123–126. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).