
A Rat α-Fetoprotein Binding Activity Prediction Model to Facilitate Assessment of the Endocrine Disruption Potential of Environmental Chemicals

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
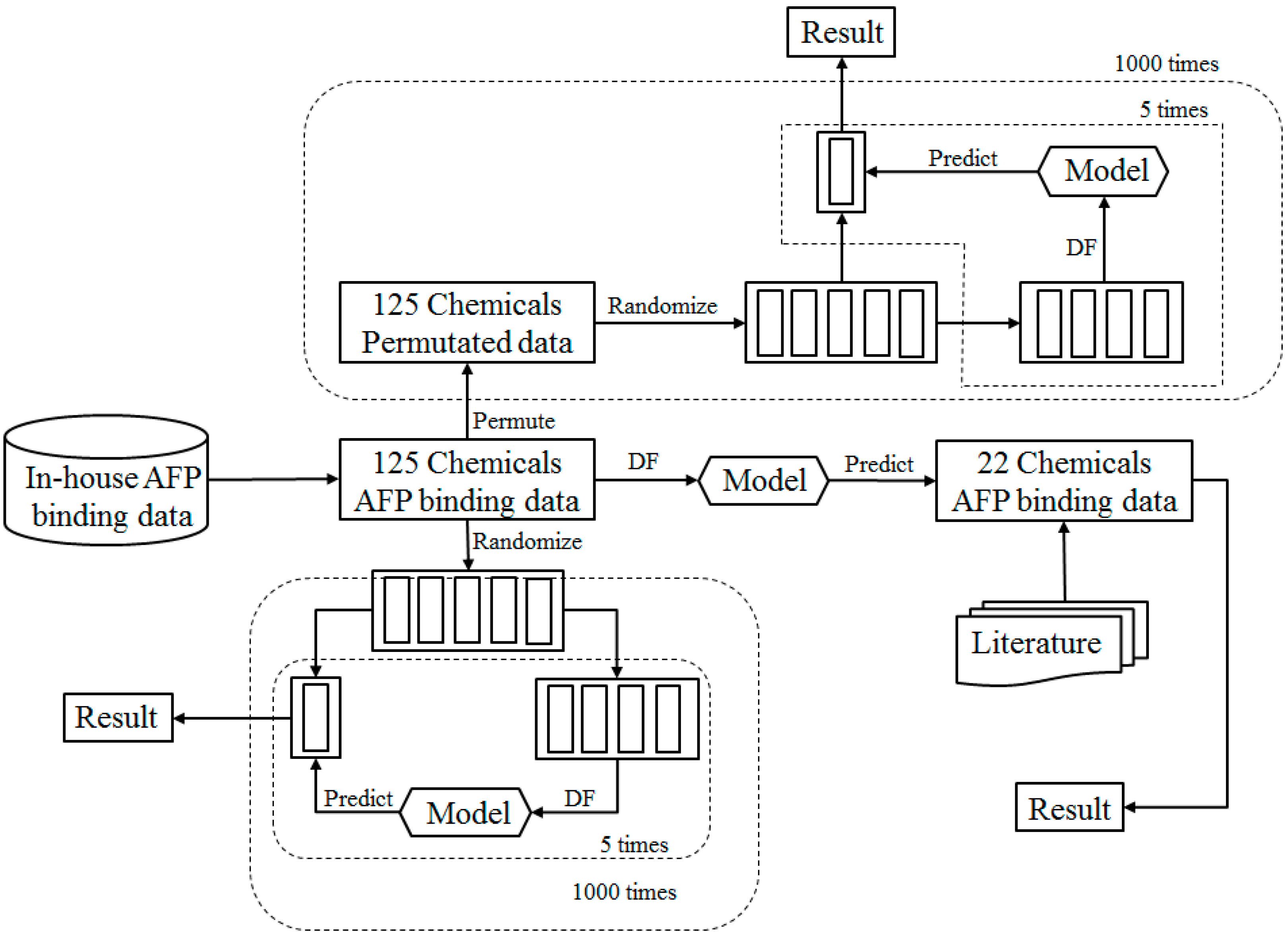
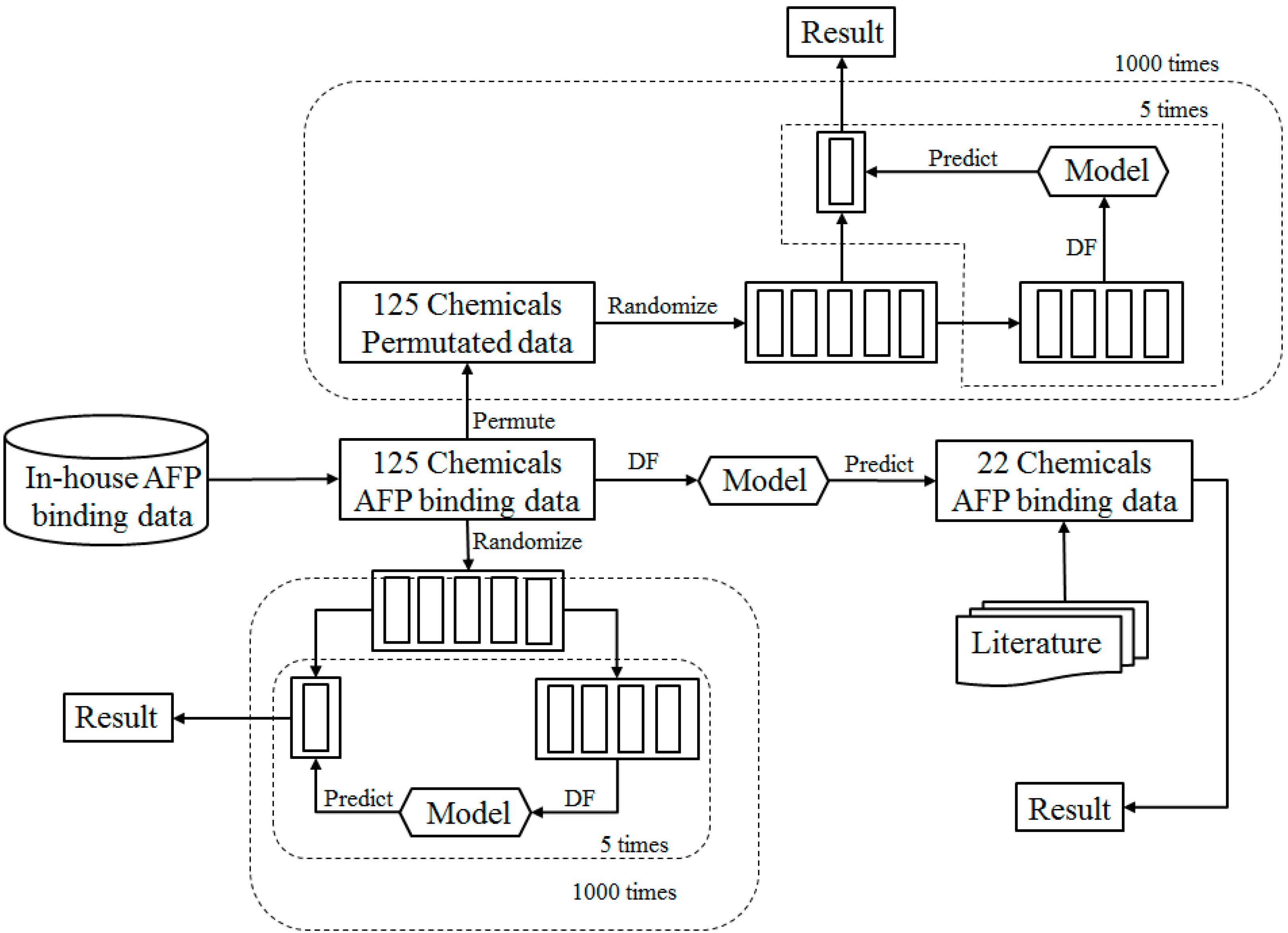
2.1. Study Design
2.2. Data Sets
2.3. Molecular Descriptors
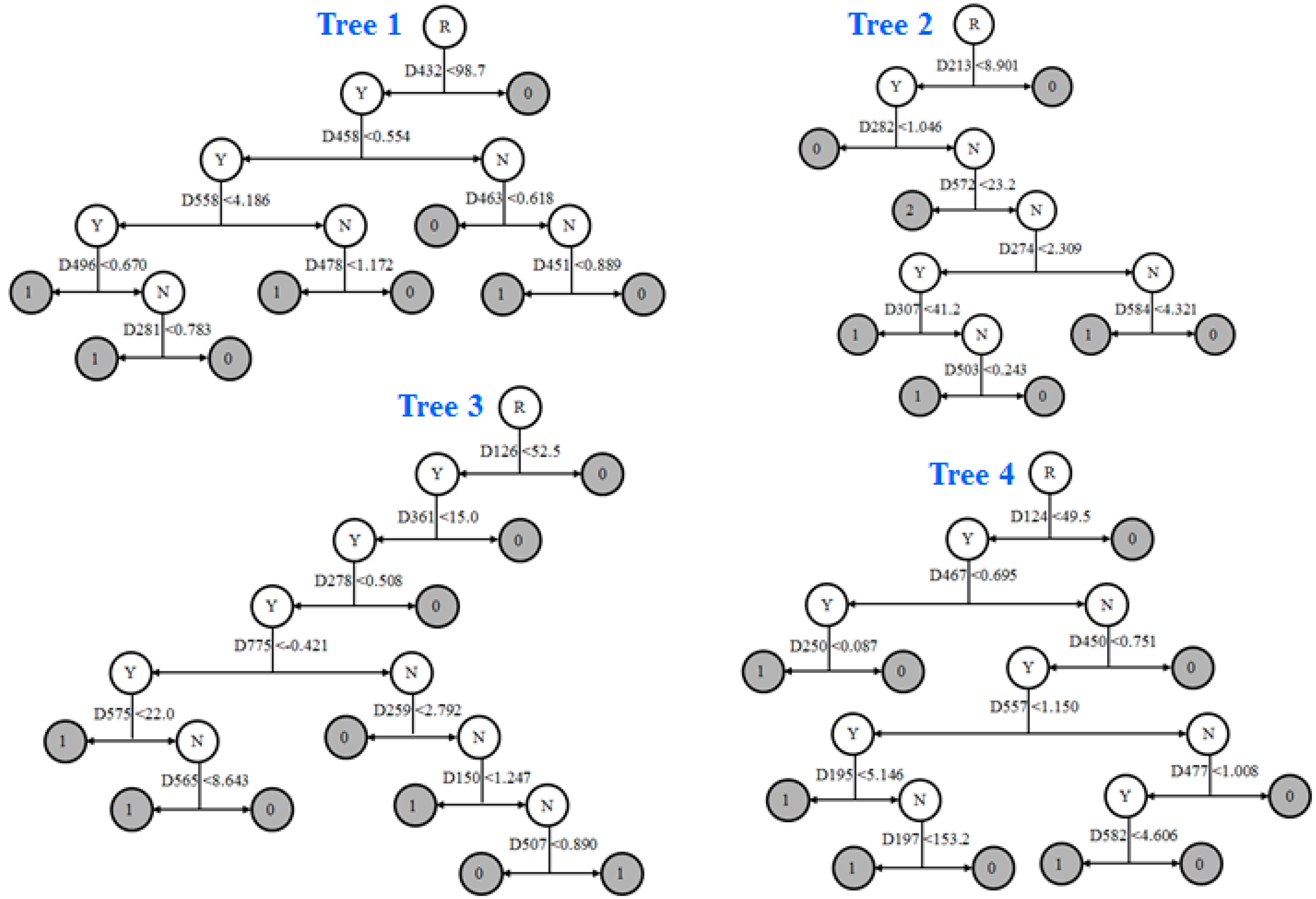
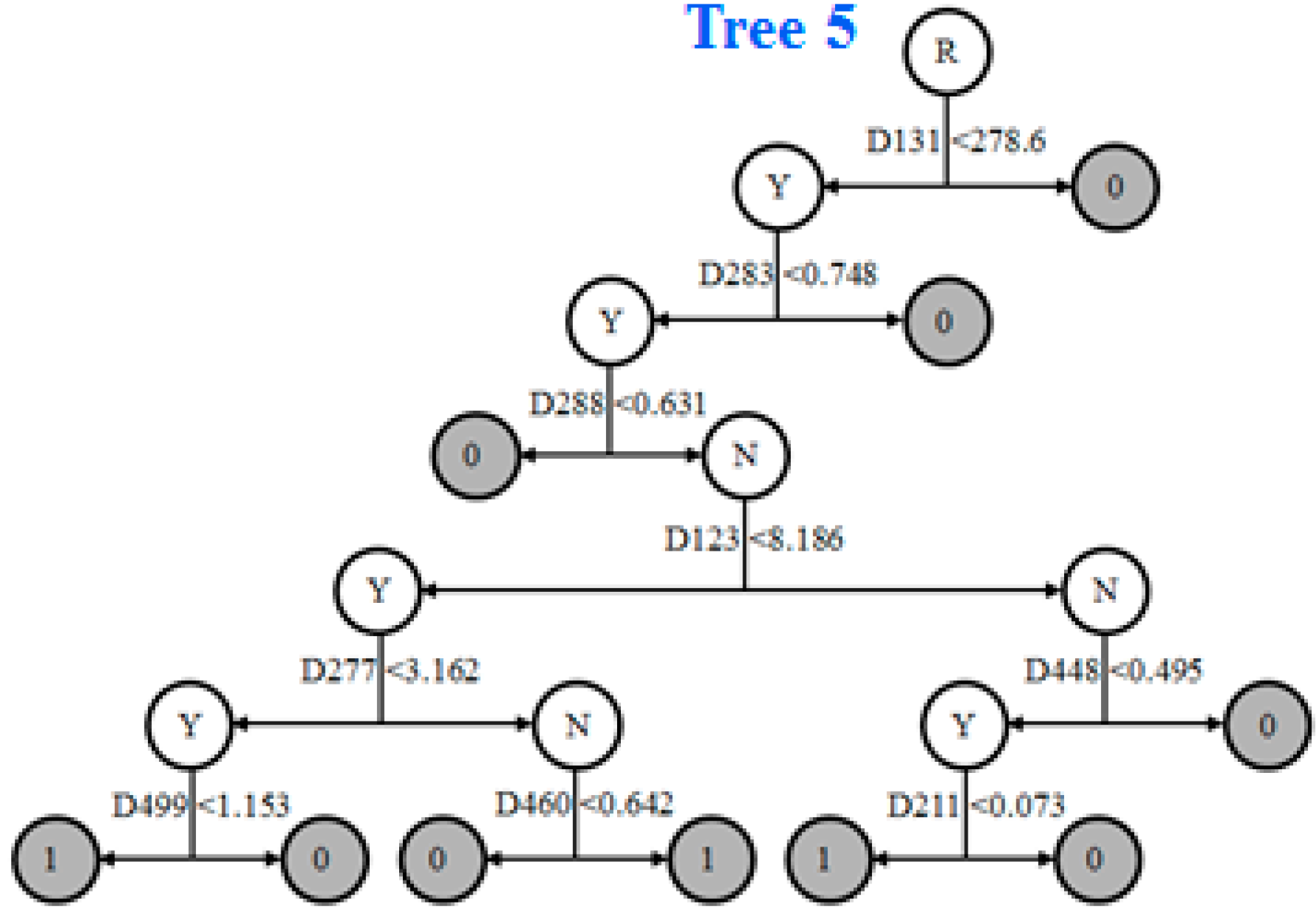
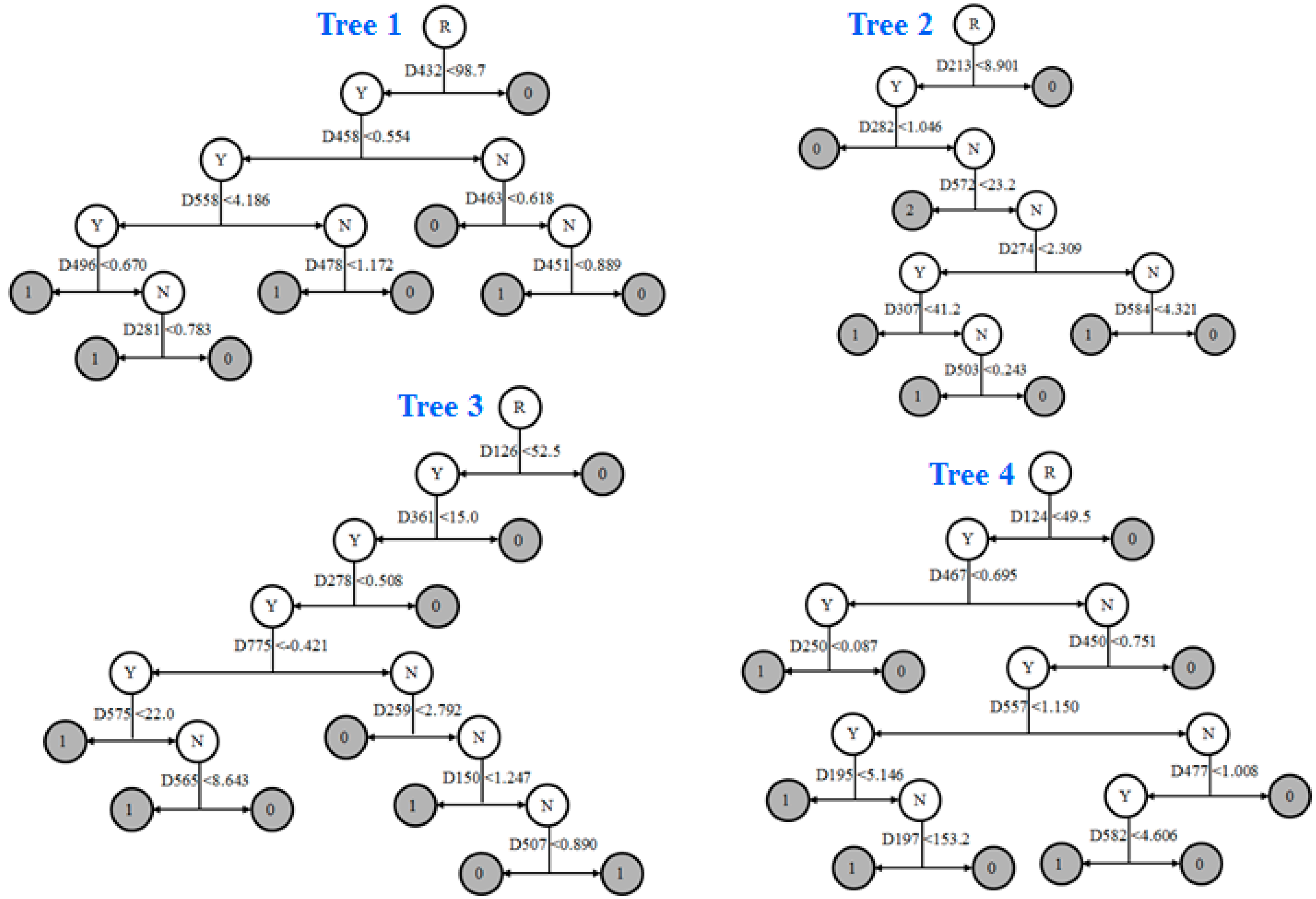
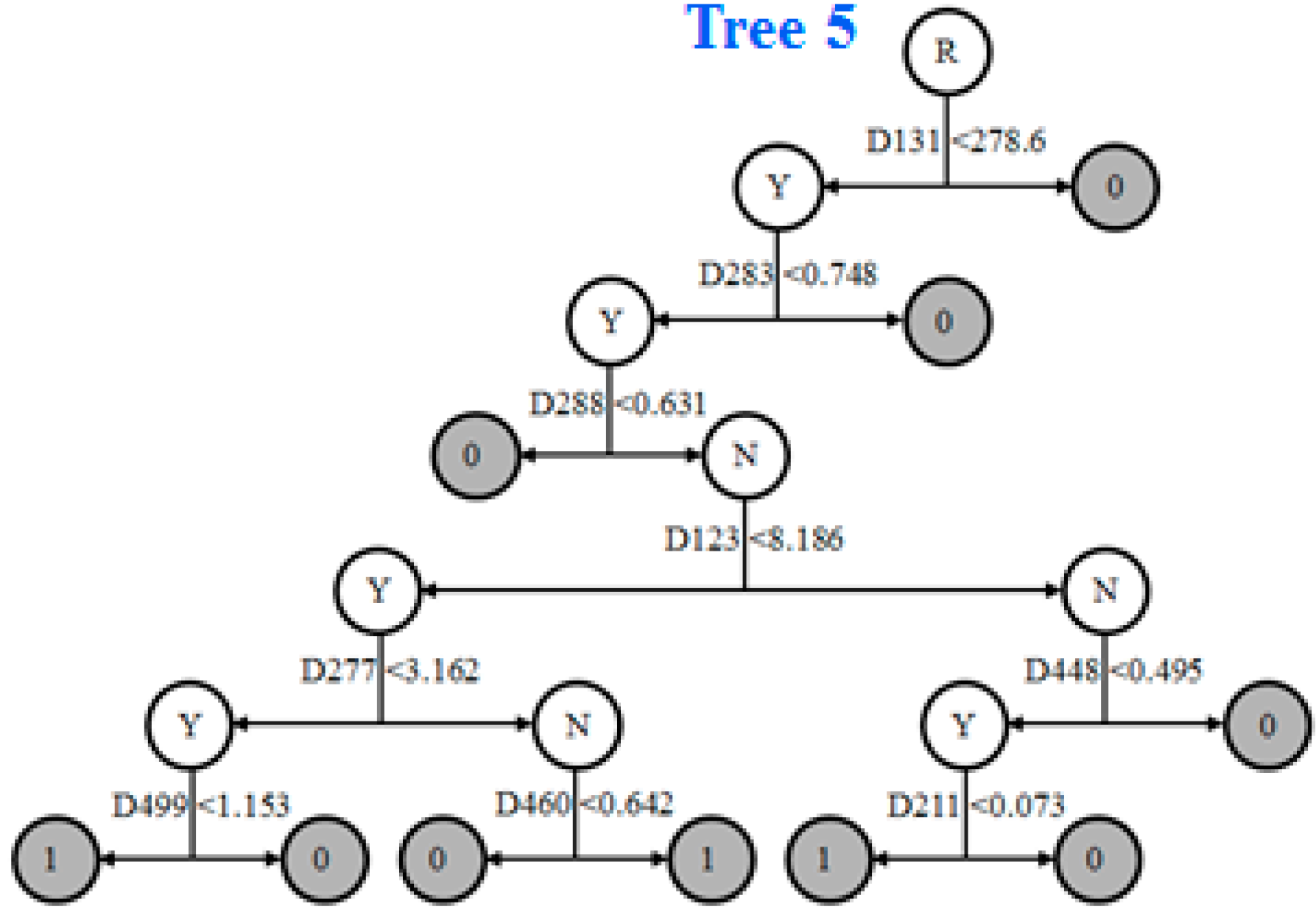
2.4. Prediction Model
2.5. Cross Validations
2.6. Permutation Tests
2.7. Prediction Confidence Analysis
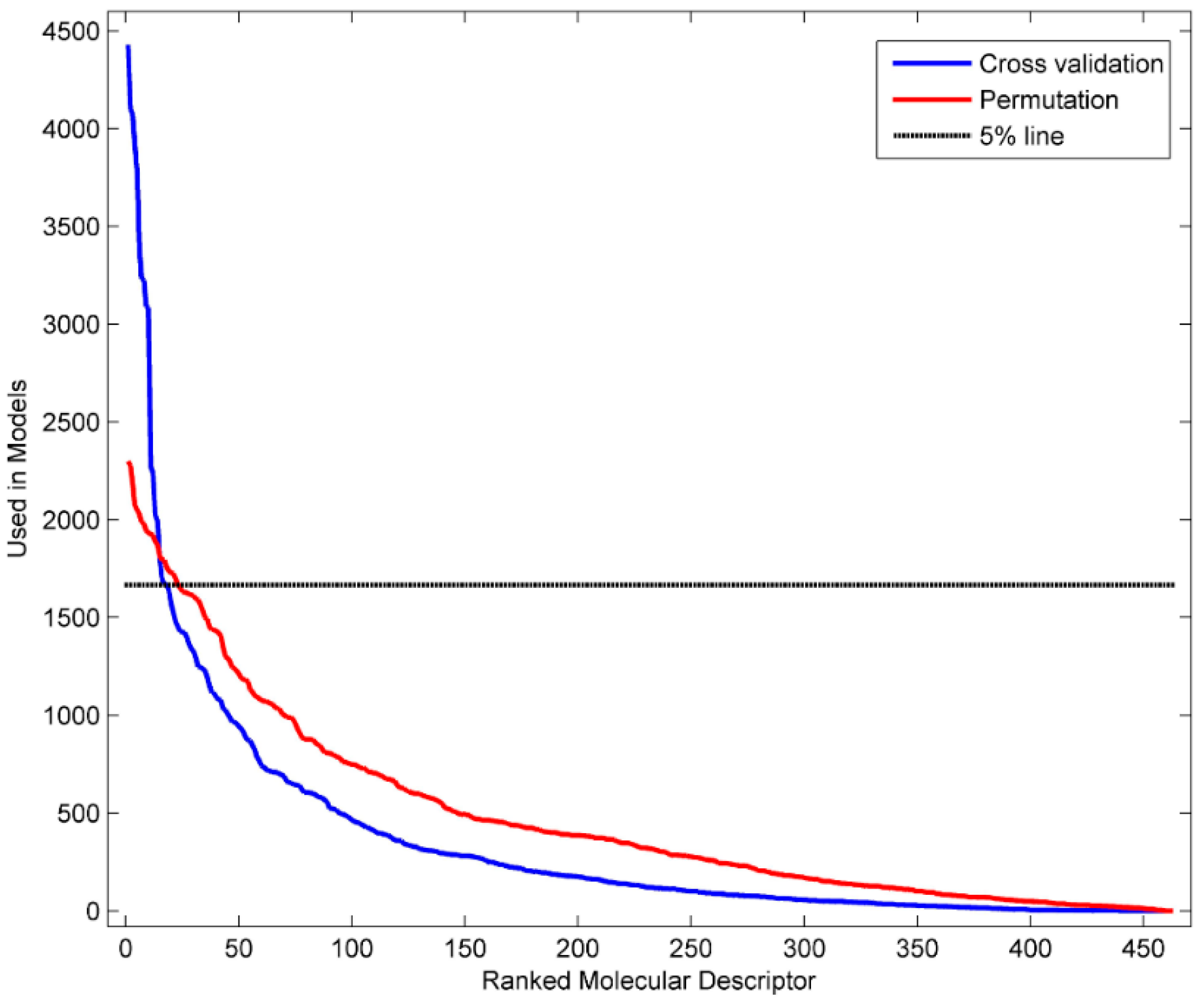
2.8. Informative Molecular Descriptors Identification
2.9. External Validation
3. Results
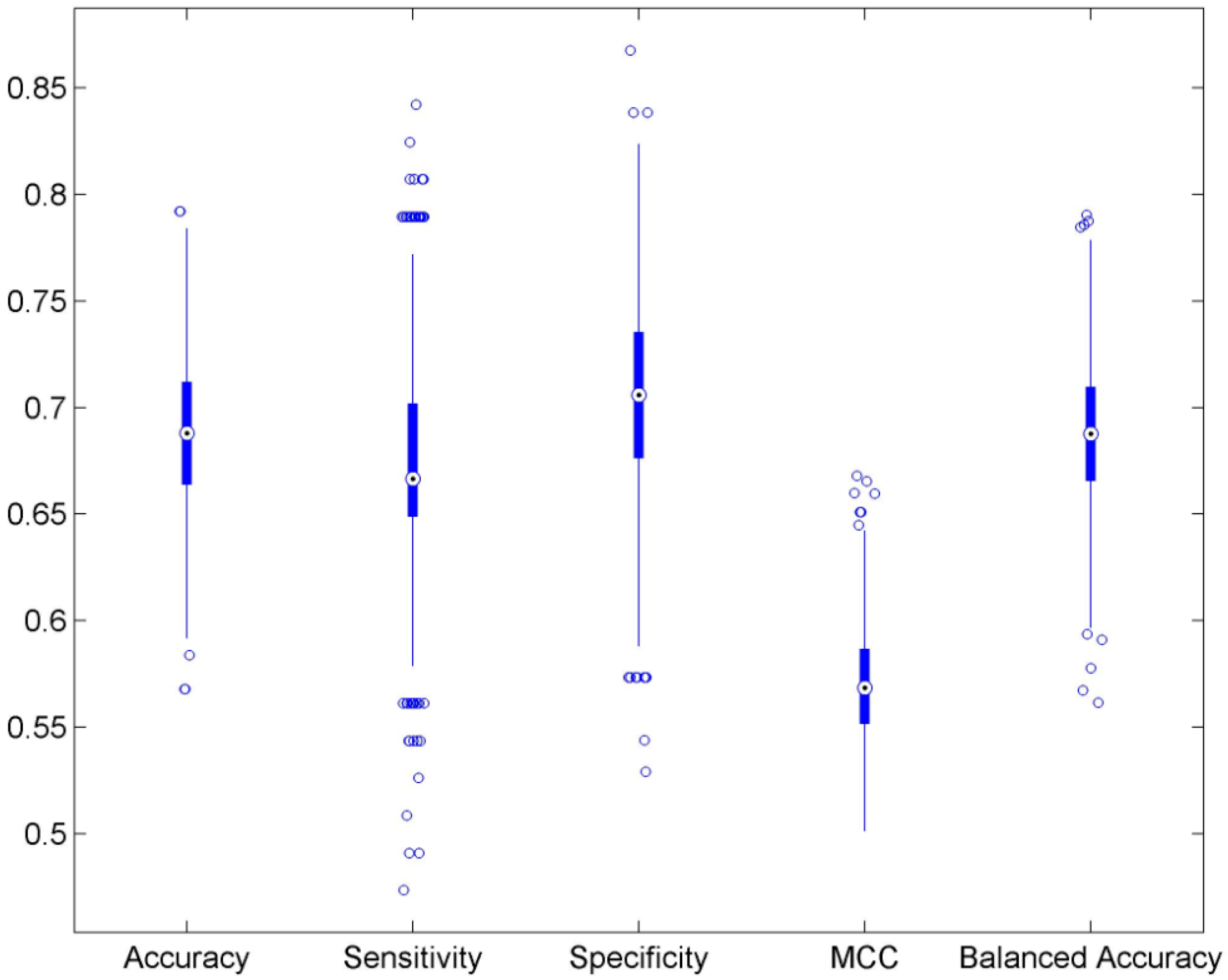
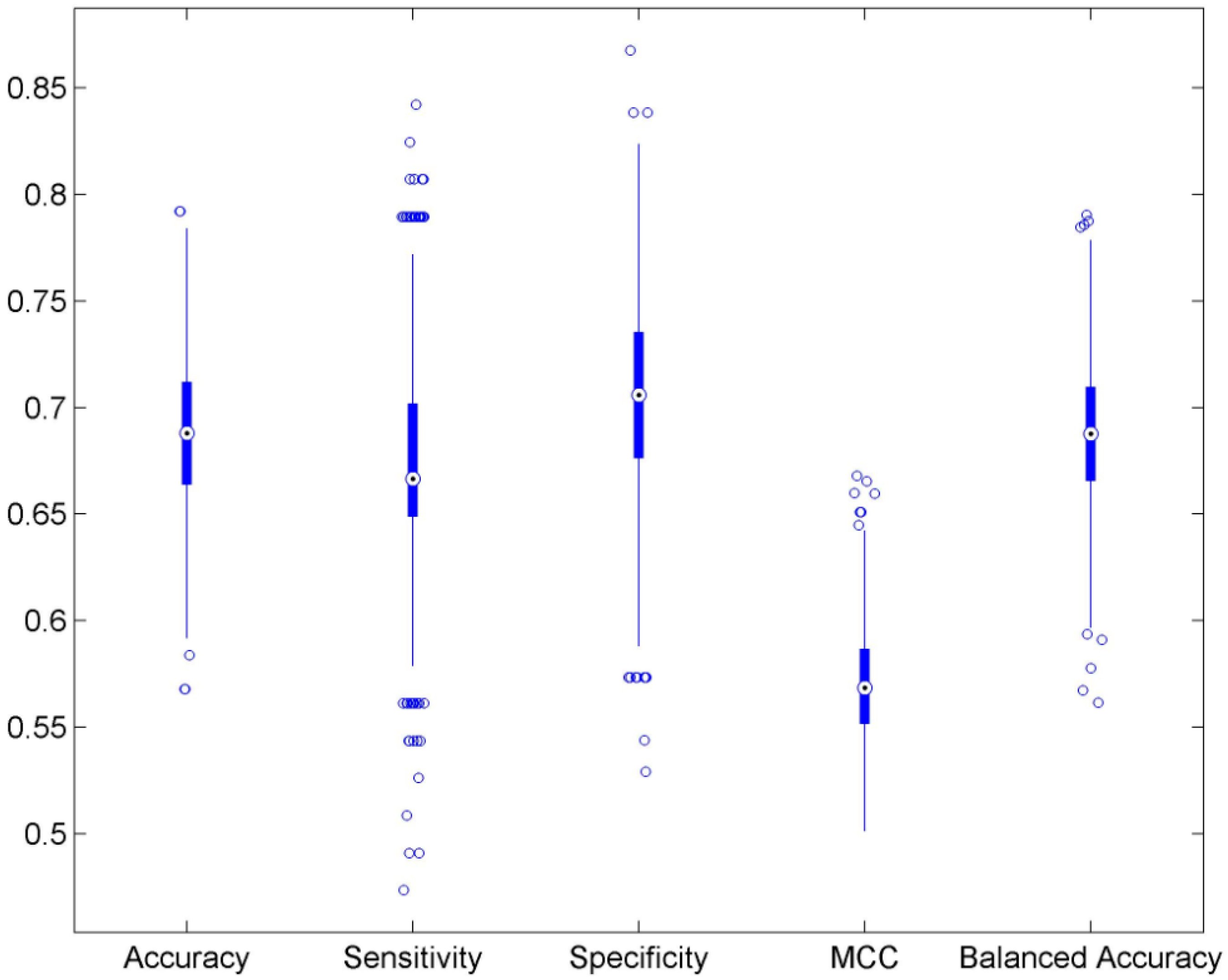
3.1. Cross Validations
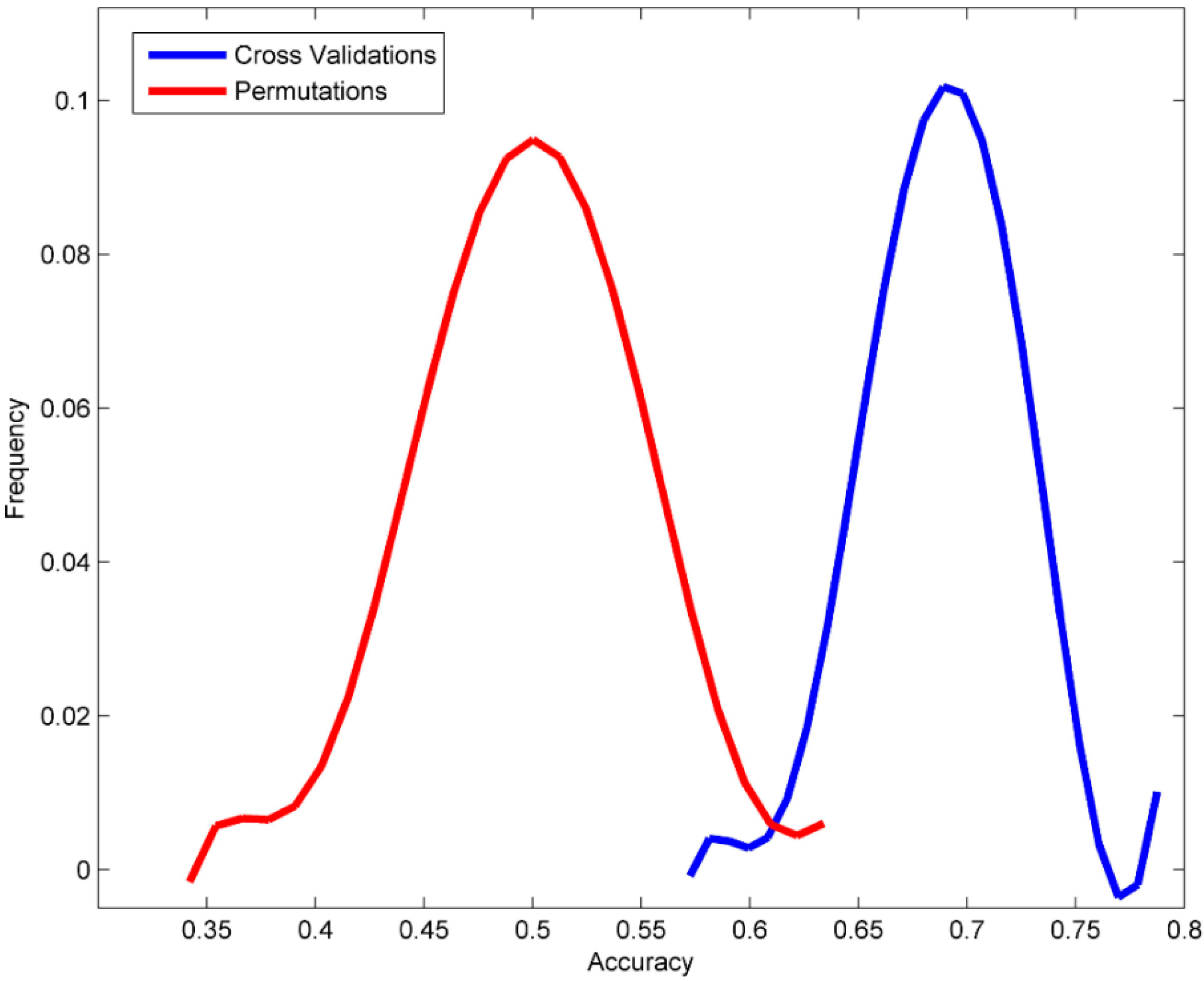
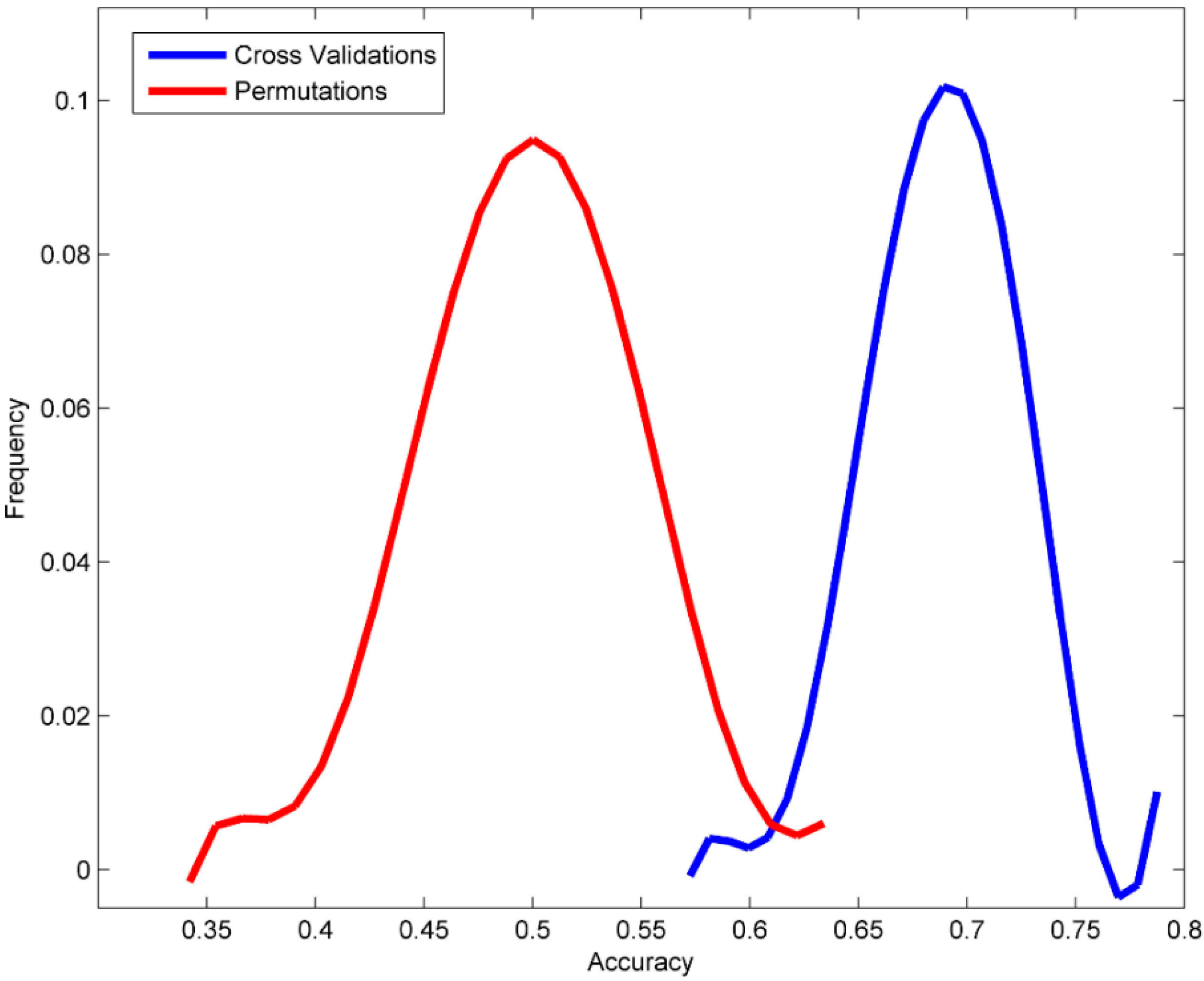
3.2. Permutation Tests
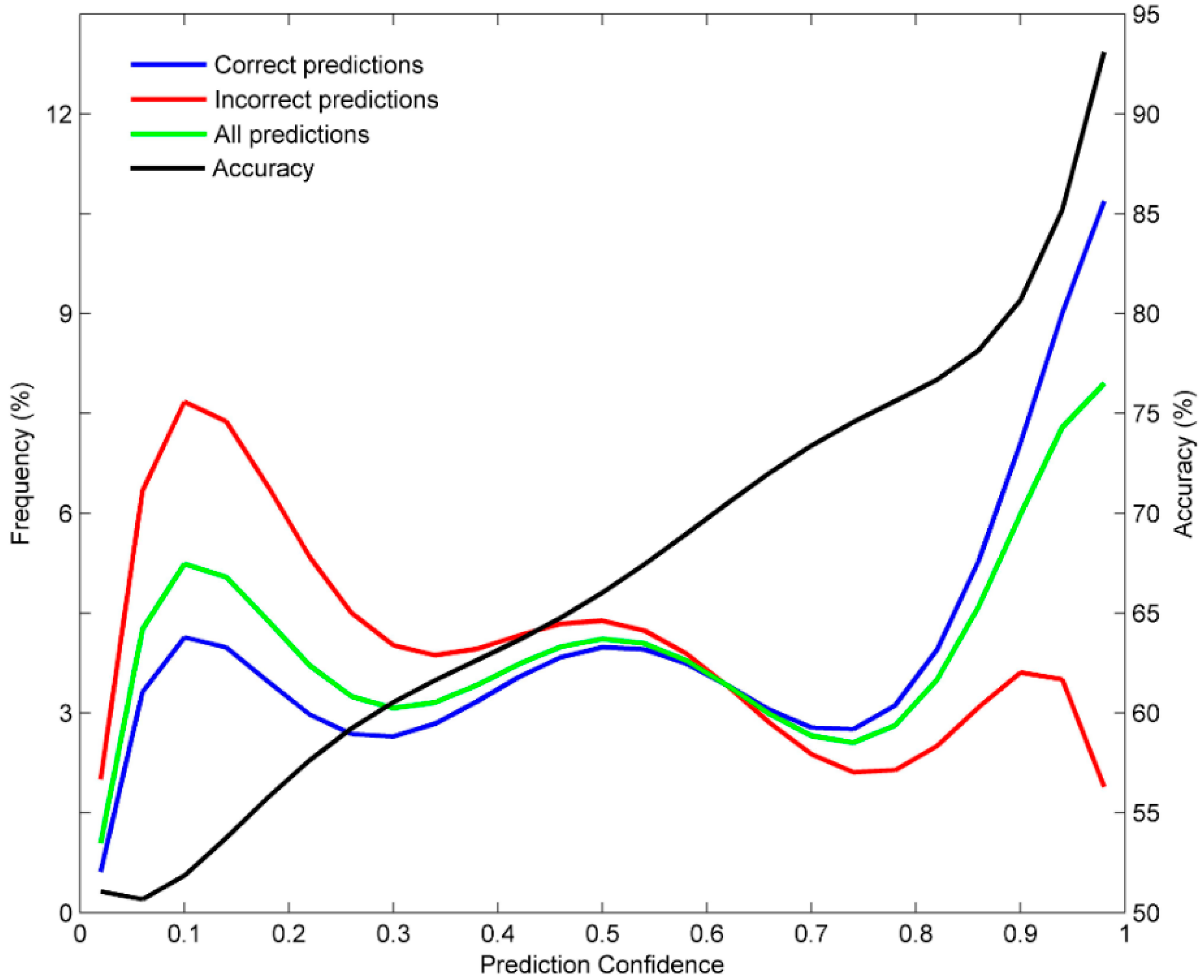
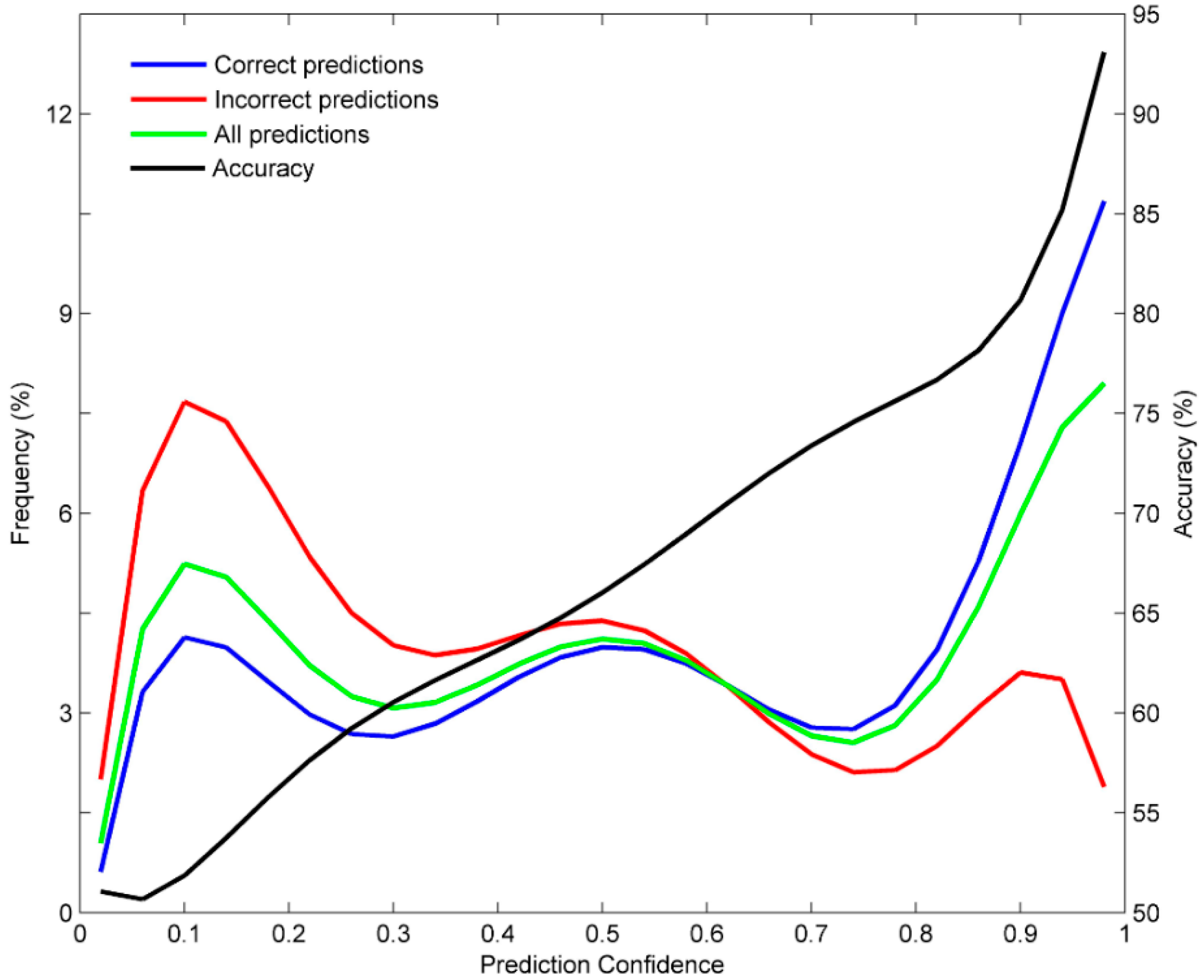
3.3. Prediction Confidence Analysis
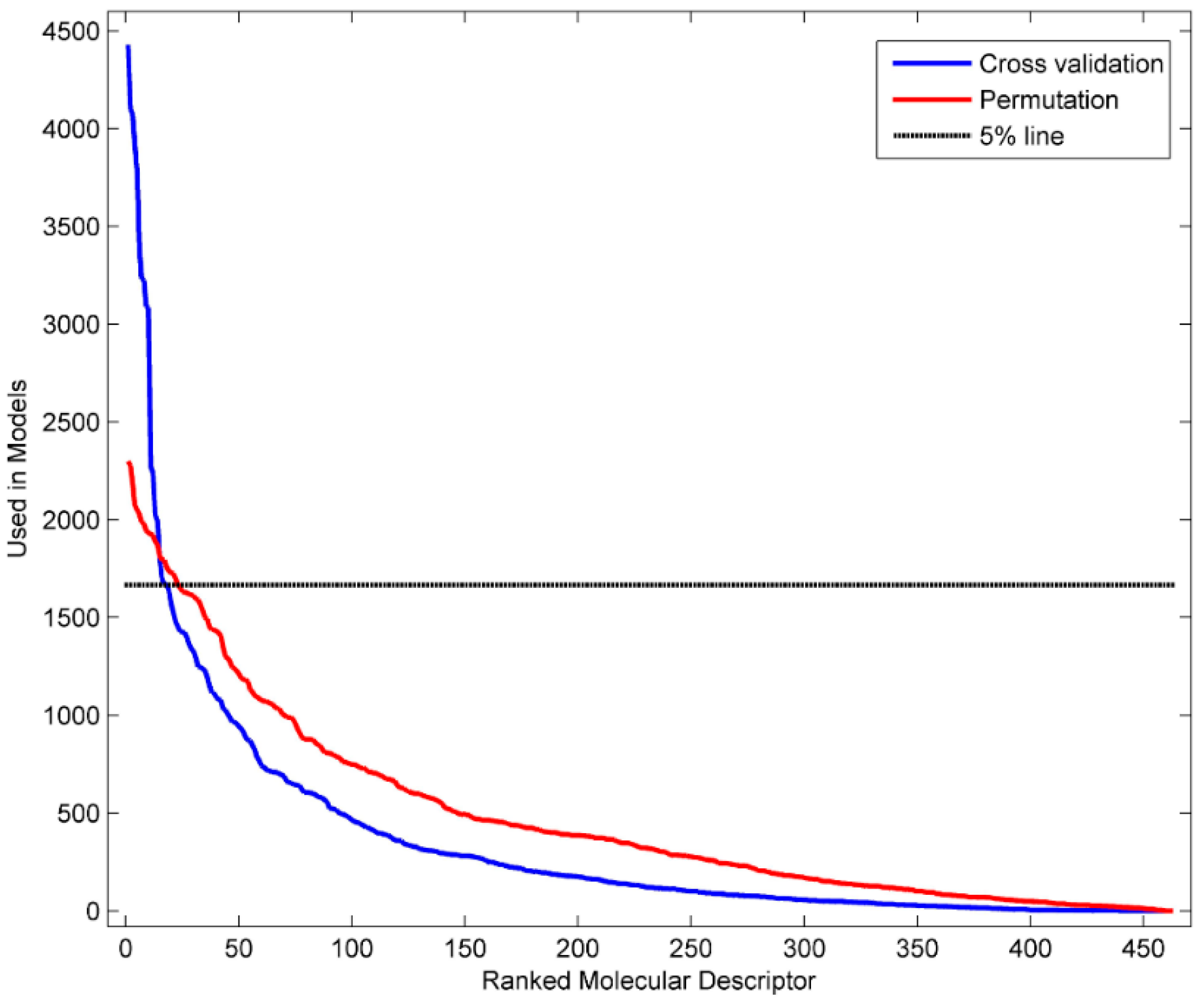
3.4. Identification of Informative Descriptors
3.5. Prediction Model and External Validation
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D | Two-Dimensional |
| 3D | Three-Dimensional |
| AFP | Alpha-Fetoprotein |
| AR | Androgen Receptor |
| EADB | Estrogenic Activity Database |
| ED | Endocrine Disruptor |
| EDKB | Endocrine Disruptors Knowledge Base |
| EDSP | Endocrine Disruptor Screening Program |
| EPA | Environmental Protection Agency |
| DF | Decision Forest |
| ER | Estrogen Receptor |
| FDA | Food and Drug Administration |
| FN | False Negative |
| FP | False Positive |
| MCC | Matthews Correlation Coefficient |
| NMR | Nuclear Magnetic Resonance |
| QSAR | Quantitative Structure-Activity Relationship |
| SDF | Structure-Data File |
| SHBG | Sex Hormone-Binding Globulin |
| STD | Standard Deviation |
| TN | True Negative |
| TP | True Positive |
References
- Dodds, E.C.; Goldberg, L.; Lawson, W.; Robinson, R. Estrogenic activity of certain synthetic compounds. Nature 1938, 141, 247–248. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Diethylstilbestrol (DES) and Cancer. Available online: http://www.cancer.gov/about-cancer/causes-prevention/risk/hormones/des-fact-sheet (accessed on 8 March 2016).
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.E.; Soto, A.M.; Woodruff, T.J.; Vom Saal, F.S. Endocrine-disrupting chemicals and public health protection: A statement of principles from the endocrine society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef] [PubMed]
- Nohynek, G.J.; Borgert, C.J.; Dietrich, D.; Rozman, K.K. Endocrine disruption: Fact or urban legend? Toxicol. Lett. 2013, 223, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Willett, C.E.; Bishop, P.L.; Sullivan, K.M. Application of an integrated testing strategy to the U.S. EPA endocrine disruptor screening program. Toxicol. Sci. 2011, 123, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Hewison, M.; Adams, J.S. Hormone response element binding proteins: Novel regulators of vitamin D and estrogen signaling. Steroids 2011, 76, 331–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anand-Ivell, R.; Ivell, R. Insulin-like factor 3 as a monitor of endocrine disruption. Reproduction 2014, 147, 87–95. [Google Scholar]
- Martinez-Arguelles, D.B.; Papadopoulos, V. Mechanisms mediating environmental chemical-induced endocrine disruption in the adrenal gland. Front. Endocrinol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Jégou, B. Paracetamol-induced endocrine disruption in human fetal testes. Nat. Rev. Endocrinol. 2015, 11, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Rieger, S.; Zhao, H.; Martin, P.; Abe, K.; Lisse, T.S. The role of nuclear hormone receptors in cutaneous wound repair. Cell Biochem. Funct. 2015, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Grabowski, T.B.; Patiño, R. Thyroid endocrine disruption and external body morphology of Zebrafish. Gen. Comp. Endocrinol. 2016, 226, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Perkins, R.; Tong, W.; Hong, H. Versatility or promiscuity: The estrogen receptors, control of ligand selectivity and an update on subtype selective ligands. Int. J. Environ. Res. Public Health. 2014, 11, 8709–8742. [Google Scholar] [CrossRef] [PubMed]
- Beekmann, K.; de Haan, L.H.; Actis-Goretta, L.; Houtman, R.; van Bladeren, P.J.; Rietjens, I.M. The effect of glucuronidation on isoflavone induced estrogen receptor (ER)α and ERβ mediated coregulator interactions. J. Steroid Biochem. Mol. Biol. 2015, 154, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Bulzomi, P.; Lecis, M.; Leone, S.; Campesi, I.; Franconi, F.; Marino, M. Endocrine disruptors differently influence estrogen receptor β and androgen receptor in male and female rat VSMC. J. Cell. Physiol. 2014, 229, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Kharlyngdoh, J.B.; Pradhan, A.; Asnake, S.; Walstad, A.; Ivarsson, P.; Olsson, P.E. Identification of a group of brominated flame retardants as novel androgen receptor antagonists and potential neuronal and endocrine disrupters. Environ. Int. 2015, 74, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xu, L.; Fang, H.; Hong, H.; Perkins, R.; Harris, S.; Bearden, E.D.; Shi, L.; Tong, W. The EDKB: An established knowledge base for endocrine disrupting chemicals. BMC Bioinform. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xu, L.; Fang, H.; Richard, A.M.; Bray, J.D.; Judson, R.S.; Zhou, G.; Colatsky, T.J.; Aungst, J.L.; Teng, C.; et al. EADB: An estrogenic activity database for assessing potential endocrine activity. Toxicol. Sci. 2013, 135, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Shu, M.; Luo, H.; Ye, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Estrogenic activity data extraction and in silico prediction show the endocrine disruption potential of bisphenol a replacement compounds. Chem. Res. Toxicol. 2015, 28, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Doughty, S.W.; Luo, H.; Ye, H.; Ge, W.; Tong, W.; Hong, H. Development and validation of decision forest model for estrogen receptor binding prediction of chemicals using large data sets. Chem. Res. Toxicol. 2015, 28, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Zhang, W.; Shu, M.; Luo, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Competitive molecular docking model for predicting estrogen receptor agonists and antagonists. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Fang, H.; Xie, Q.; Perkins, R.; Sheehan, D.M.; Tong, W. Comparative Molecular Field Analysis (CoMFA) model using a large diverse set of natural, synthetic and environmental chemicals for binding to the androgen receptor. SAR QSAR Environ. Res. 2003, 14, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tong, W.; Fang, H.; Xie, Q.; Hong, H.; Perkins, R.; Wu, J.; Tu, M.; Blair, R.M.; Branham, W.S.; et al. An integrated “4-Phase” approach for setting endocrine disruption screening priorities—Phase I and II predictions of estrogen receptor binding affinity. SAR QSAR Environ. Res. 2002, 13, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W.; Fang, H.; Shi, L.; Xie, Q.; Wu, J.; Perkins, R.; Walker, J.D.; Branham, W.; Sheehan, D.M. Prediction of estrogen receptor binding for 58,000 chemicals using an integrated system of a tree-based model with structural alerts. Environ. Health Perspect. 2002, 110, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Hong, H.; Xie, Q.; Shi, L.; Fang, H.; Perkins, R. Assessing QSAR limitations-a regulatory perspective. Curr. Comput.-Aided Drug Des. 2005, 1, 195–205. [Google Scholar] [CrossRef]
- Hornung, M.W.; Tapper, M.A.; Denny, J.S.; Kolanczyk, R.C.; Sheedy, B.R.; Hartig, P.C.; Aladjov, H.; Henry, T.R.; Schmieder, P.K. Effects-based chemical category approach for prioritization of low affinity estrogenic chemicals. SAR QSAR Environ. Res. 2014, 25, 289–323. [Google Scholar] [CrossRef] [PubMed]
- Devillers, J.; Bro, E.; Millot, F. Prediction of the endocrine disruption profile of pesticides. SAR QSAR Environ. Res. 2015, 26, 831–852. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bai, F.; Cao, H.; Li, J.; Liu, H.; Gramatica, P. A combined quantitative structure-activity relationship research of quinolinone derivatives as androgen receptor antagonists. Comb. Chem. High Throughput Screen. 2015, 18, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Niinivehmas, S.P.; Manivannan, E.; Rauhamäki, S.; Huuskonen, J.; Pentikäinen, O.T. Identification of estrogen receptor α ligands with virtual screening techniques. J. Mol. Graph. Model. 2016, 64, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Branham, W.S.; Dial, S.; Moland, C.L.; Fang, H.; Shen, J.; Perkins, R.; Sheehan, D.; Tong, W. Rat alpha-fetoprotein binding affinities of a large set of structurally diverse chemicals elucidated the relationships between structures and binding affinities. Chem. Res. Toxicol. 2012, 25, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, W.; Fang, H.; Perkins, R.; Tong, W.; Hong, H. Homology modeling, molecular docking, and molecular dynamics simulations elucidated alpha-fetoprotein binding modes. BMC Bioinform. 2013, 14, S6. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Branham, W.S.; Ng, H.W.; Moland, C.L.; Dial, S.L.; Fang, H.; Perkins, R.; Sheehan, D.; Tong, W. Human sex hormone binding globulin binding affinities of 125 structurally diverse chemicals and comparison with their binding to androgen receptor, estrogen receptor and a-fetoprotein. Toxicol. Sci. 2015, 143, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.C. Sex-hormone-binding globulin. Clin. Endocrinol. 1974, 3, 69–96. [Google Scholar] [CrossRef]
- Bergstrand, C.G.; Czar, B. Paper electrophoretic study of human fetal serum proteins with demonstration of a new protein fraction. Scand. J. Clin. Lab. Investig. 1956, 9, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Mizejewski, G.J.; MacColl, R. Alpha-fetoprotein growth inhibitory peptides: Potential leads for cancer therapeutics. Mol. Cancer Ther. 2003, 2, 1243–1255. [Google Scholar] [PubMed]
- Mizejewski, G.J. Biological role of alpha-fetoprotein in cancer: Prospects for anticancer therapy. Expert. Rev. Anticancer Ther. 2002, 2, 89–115. [Google Scholar] [CrossRef] [PubMed]
- Alava, M.A.; Sturralde, M.; Lampreave, F.; Pineiro, A. Specific uptake of alpha-fetoprotein and albumin by rat Morris 777 hepatoma cells. Tumour. Biol. 1999, 20, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Uriel, J.; Faivre-Bauman, A.; Trojan, J.; Foiret, D. Immunocytochemical demonstration of alpha-fetoprotein uptake by primary cultures of fetal hemisphere cells from mouse brain. Neurosci. Lett. 1981, 272, 171–175. [Google Scholar] [CrossRef]
- Laborda, J.; Naval, J.; Allouche, M.; Calvo, M.; Georgoulias, V.; Mishal, Z.; Uriel, J. Specific uptake of alpha-fetoprotein by malignant human lymphoid cells. Int. J. Cancer 1987, 40, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Hajeri-Germond, M.; Trojan, J.; Uriel, J.; Hauw, J.J. In vitro uptake of exogenous AFP by chicken dorsal root ganglia. Dev. Neurosci. 1983, 6, 11–15. [Google Scholar] [CrossRef]
- Uriel, J.; Trojan, J.; Dubouch, P.; Pineiro, A. Intracellular alpha-fetoprotein and albumin in the developing nervous system of the baboon. Pathol. Biol. 1982, 302, 79–83. [Google Scholar]
- Brock, D.J.; Bolton, A.E.; Monaghan, J.M. Prenatal diagnosis through maternal serum AFP measurement. Lancet 1973, 2, 293–294. [Google Scholar]
- Leek, A.E.; Ruoss, C.F.; Kitau, M.J.; Chard, T. Raised AFP in maternal serum with anenecephalic pregnancy. Lancet 1973, 2, 385–386. [Google Scholar] [CrossRef]
- Benassayag, C.; Migrot, T.M.; Haurigui, M.; Civel, C.; Hassid, J.; Carbonne, B.; Nunez, E.A.; Ferre, F. High polyunsaturated fatty acid, thromboxane A2, and alpha-fetoprotein concentrations at the human feto-maternal interface. J. Lipid Res. 1997, 38, 276–286. [Google Scholar] [PubMed]
- Gross, S.; Catillo, W.; Crane, M.; Espinosa, B.; Carter, S.; DeVeaux, R.; Salafia, C. Maternal serum AFP and HCG levels in women with human HIV. Am. J. Obstet. Gynecol. 2003, 188, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Savu, L.; Benassayag, C.; Vallette, G.; Christeff, N.; Nunez, E. Mouse alpha 1-fetoprotein and albumin. Comparison of their binding properties with estrogen and fatty acid ligands. J. Biol. Chem. 1981, 256, 9414–9418. [Google Scholar] [PubMed]
- Uriel, J.; Bouillon, D.; Aussel, C.; Dupiers, M. Alpha-fetoprotein: The major high-affinity estrogen binder in rat uterine cytosols. Proc. Natl. Acad. Sci. USA 1976, 73, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Pomper, M.G.; VanBrocklin, H.; Thieme, A.M.; Thomas, R.D.; Kiesewetter, D.O.; Carlson, K.E.; Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A. 11 beta-methoxy-, 11 beta-ethyl- and 17 alpha-ethynyl-substituted 16 alpha-fluoroestradiols: Receptor-based imaging agents with enhanced uptake efficiency and selectivity. J. Med. Chem. 1990, 33, 3143–3155. [Google Scholar] [CrossRef] [PubMed]
- VanBrocklin, H.F.; Brodack, J.W.; Mathiss, C.J.; Welch, M.J.; Katzenellenbogen, J.A.; Keenan, J.F.; Mizejewski, G.J. Binding of a 16 a-F18-fluoro-17B-estradiol to alpha-fetoprotein in Sprague-Dawley female rats affects blood levels. Nucl. Med. Biol. 1992, 17, 769–773. [Google Scholar]
- Milligan, S.R.; Khan, O.; Nash, M. Competitive binding of xenobiotic oestrogens to rat alpha-fetoprotein and to sex steroid binding proteins in human and rainbow trout (Oncorhynchus mykiss) plasma. Gen. Comp. Endocrinol. 1998, 112, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Nunez, E.A.; Bennassayag, C.; Sava, L.; Vallette, G.; Delorme, J. Oestrogen binding function of AFP. J. Steroid Biochem. 1979, 11, 237–243. [Google Scholar] [CrossRef]
- Garreau, B.; Vallette, G.; Adlercreutz, H.; Wähälä, K.; Mäkelä, T.; Benassayag, C.; Nunez, E.A. Phytoestrogens: New ligands for rat and human alpha-fetoprotein. Biochim. Biophys. Acta 1991, 1094, 339–345. [Google Scholar] [CrossRef]
- Kleinstreuer, N.C.; Ceger, P.C.; Allen, D.G.; Strickland, J.; Chang, X.; Hamm, J.T.; Casey, W.M. A Curated Database of Rodent Uterotrophic Bioactivity. Environ. Health Perspectect. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Mold2, Descriptors Generator Software. Available online: http://www.fda.gov/ScienceResearch/BioinformaticsTools/Mold2/ (accessed on 13 January 2016).
- Hong, H.; Xin, X. ESSESA: An expert system for structure elucidation from spectra analysis. 2. A novel algorithm of perception of the linear independent smallest set of smallest rings. Anal. Chim. Acta 1992, 262, 179–191. [Google Scholar] [CrossRef]
- Hong, H.; Xin, X. ESSESA: An expert system for structure elucidation from spectra analysis. 3. LNSCS for chemical knowledge representation. J. Chem. Inf. Comput. Sci. 1992, 32, 116–120. [Google Scholar] [CrossRef]
- Hong, H.; Xin, X. ESSESA: An expert system for structure elucidation from spectra analysis. 4. Canonical representation of structures. J. Chem. Inf. Comput. Sci. 1994, 34, 730–734. [Google Scholar] [CrossRef]
- Hong, H.; Xin, X. ESSESA: An expert system for structure elucidation from spectra analysis. 1. The knowledge base of infrared spectra and analysis and interpretation program. J. Chem. Inf. Comput. Sci. 1990, 30, 203–210. [Google Scholar] [CrossRef]
- Hong, H.; Xin, X. ESSESA: An expert system for structure elucidation from spectra analysis. 5. Substructure constraints from from analysis of first-order 1H-NMR spectra. J. Chem. Inf. Comput. Sci. 1994, 34, 1259–1266. [Google Scholar] [CrossRef]
- Hong, H.; Han, Y.; Xin, X.; Shi, Y. ESSESA: An expert system for structure elucidation from spectra. 6. Substructure constraints from analysis of 13C-NMR spectra. J. Chem. Inf. Comput. Sci. 1994, 35, 979–1000. [Google Scholar]
- Masui, H.; Hong, H. Spec2D: A structure elucidation system based on 1H NMR and H-H COSY spectra in organic chemistry. J. Chem. Inf. Model. 2006, 46, 775–787. [Google Scholar] [CrossRef] [PubMed]
- McPhail, B.; Tie, Y.; Hong, H.; Pearce, B.A.; Schnackenberg, L.K.; Ge, W.; Valerio, L.G.; Fuscoe, J.C.; Tong, W.; Buzatu, D.A.; et al. Modeling chemical interaction profiles: I. Spectral data-activity relationship and structure-activity relationship models for inhibitors and non-inhibitors of cytochrome P450 CYP3A4 and CYP2D6 isozymes. Molecules 2012, 17, 3283–3406. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; McPhail, B.; Hong, H.; Pearce, B.A.; Schnackenberg, L.K.; Ge, W.; Buzatu, D.A.; Wilkes, J.G.; Fuscoe, J.C.; Tong, W.; et al. Modeling chemical interaction profiles: II. Molecular docking, spectral data-activity relationship, and structure-activity relationship models for potent and weak inhibitors of cytochrome p450 cyp3A4 isozyme. Molecules 2012, 17, 3407–3460. [Google Scholar] [CrossRef] [PubMed]
- Neamati, N.; Hong, H.; Owen, J.M.; Sunder, S.; Winslow, H.E.; Christensen, J.L.; Zhao, H.; Burke, T.R., Jr.; Milne, G.W.; Pommier, Y. Salicylhydrazine-Containing Inhibitors of Hiv-1 Integrase: Implication for a Selective Chelation in the Integrase Active Site. J. Med. Chem. 1998, 41, 3202–3209. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Neamati, N.; Winslow, H.E.; Christensen, J.L.; Orr, A.; Pommier, Y.; Milne, G.W. Identification of Hiv-1 Integrase Inhibitors Based on a Four-Point Pharmacophore. Antivir. Chem. Chemother. 1998, 9, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Neamati, N.; Hong, H.; Sunder, S.; Milne, G.W.A.; Pommier, Y. Potent Inhibitors of Human Immunodeficiency Virus Type 1 Integrase: A Novel Four-Point Pharmacophore Searching of the NCI 3D Database. Mol. Pharmacol. 1997, 52, 1041–1055. [Google Scholar] [PubMed]
- Hong, H.; Neamati, N.; Wang, S.; Nicklaus, M.C.; Mazumder, A.; Zhao, H.; Burke, T.R.; Pommier, Y.; Milne, G.W.A. Discovery of Hiv-1 Integrase Inhibitors by Pharmacophore Searching. J. Med. Chem. 1997, 40, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Du, T.; Zhou, P.; Yang, L.; Mei, H.; Ng, H.W.; Zhang, W.; Shu, M.; Tong, W.; Shi, L.; et al. Molecular docking to identify associations between drugs and class I human leukocyte antigens for predicting potential idiosyncratic drug reactions. Comb. Chem. High Throughput Screen. 2015, 18, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Drake, R.R.; Neamati, N.; Hong, H.; Pilon, A.; Sunthankar, P.; Hume, S.D.; Wilne, G.W.A.; Pommier, Y. Identification of a mononucleotide binding site in human HIV-1 integrase. Proc. Natl. Accad. Sci. USA 1998, 98, 1495–1500. [Google Scholar]
- Hong, H.; Hong, Q.; Perkins, R.; Shi, L.; Fang, H.; Su, Z.; Dragan, Y.; Fuscoe, J.C.; Tong, W. The accurate prediction of protein family from amino acid sequence by measuring features of sequence fragments. J. Comput. Biol. 2009, 16, 1671–1688. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mansouri, K.; Judson, R.; Martin, M.T.; Hong, H.; Chen, M.; Xu, X.; Thomas, R.; Shah, I. Predicting hepatotoxicity using ToxCast in vitro bioactivity and chemical structure. Chem. Res. Toxicol. 2015, 28, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Ye, H.; Ng, H.W.; Shi, L.; Tong, W.; Mendrick, D.L.; Hong, H. Machine learning methods for predicting HLA-peptide binding activity. Bioinform. Biol. Insights 2015, 9, 21–29. [Google Scholar] [PubMed]
- Tong, W.; Hong, H.; Fang, H.; Xie, Q.; Perkins, R. Decision forest: Combining the predictions of multiple independent decision tree models. J. Chem. Inf. Comput. Sci. 2003, 43, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W.; Xie, Q.; Fang, H.; Perkins, R. An in silico ensemble method for lead discovery: decision forest. SAR QSAR Environ. Res. 2005, 16, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Decision Forest. Available online: http://www.fda.gov/ScienceResearch/BioinformaticsTools/DecisionForest/ (accessed on 13 January 2016).
- Xie, Q.; Ratnasinghe, L.D.; Hong, H.; Perkins, R.; Tang, Z.Z.; Hu, N.; Taylor, P.R.; Tong, W. Decision forest analysis of 61 single nucleotide polymorphisms in a case-control study of esophageal cancer; a novel method. BMC Bioinform. 2005, 6, S4. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W.; Perkins, R.; Fang, H.; Xie, Q.; Shi, L. Multiclass decision forest-a novel pattern recognition method for multiclass classification in microarray data analysis. DNA Cell Biol. 2004, 23, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Xie, Q.; Hong, H.; Shi, L.; Fang, H.; Perkins, R.; Petricoin, E.F. Using decision forest to classify prostate cancer samples on the basis of seldi-tof ms data: Assessing chance correlation and prediction confidence. Environ. Health Perspect. 2004, 112, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Guha, R. On the interpretation and interpretability of quantitative structure-activity relationship models. J. Comput. Aided Mol. Des. 2008, 22, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Nishi, S.; Shahbazzadeh, D.; Azuma, M.; Sakai, M. Estrogen binding site of rat AFP. Tumour. Biol. 1993, 14, 234–237. [Google Scholar]
- Use of High Throughput Assays and Computational Tools in the Endocrine Disruptor Screening Program. Available online: http://www.epa.gov/endocrine-disruption/use-high-throughput-assays-and-computational-tools-endocrine-disruptor (accessed on 8 March 2016).
- Mansouri, K. CERAPP: Collaborative estrogen receptor activity prediction project. Environ. Health Perspect. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nishi, S.; Matsue, H.; Yoshida, H.; Yamaoto, R.; Sakai, M. Localization of the estrogen-binding site of alpha-fetoprotein in the chimeric human-rat proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 3102–3105. [Google Scholar] [CrossRef] [PubMed]
- Herve, F.; Gentin, M.; Rajkowski, K.M.; Wong, L.T.; Hsia, C.J.; Cittanova, N. Estrogen-binding properties of rat AFP and its isoforms: Investigation of the apparent non-integrality of sites on the unfractionated protein. J. Steroid Biochem. 1990, 36, 319–324. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Cross Validations | Permutation Tests | External Validation | ||
|---|---|---|---|---|---|
| Mean | STD | Mean | STD | ||
| Accuracy | 0.689 | ±0.034 | 0.498 | ±0.049 | 0.546 |
| Sensitivity | 0.675 | ±0.054 | 0.427 | ±0.067 | 0.412 |
| Specificity | 0.700 | ±0.046 | 0.558 | ±0.061 | 1.000 |
| MCC | 0.570 | ±0.026 | 0.497 | ±0.009 | 0.371 |
| Balanced accuracy | 0.688 | ±0.034 | 0.492 | ±0.050 | 0.706 |
| ID | Models | Descriptor Definition |
|---|---|---|
| D282 | 4429 | complementary information content (neighborhood symmetry of 2-order) |
| D281 | 4099 | structural information content (neighborhood symmetry of 2-order) |
| D450 | 4075 | Geary autocorrelation-lag 4/weighted by atomic masses |
| D432 | 3916 | Broto-Moreau autocorrelation of a topological structure-lag 2/weighted by atomic Sanderson electronegativity |
| D458 | 3770 | Geary autocorrelation-lag 4/weighted by atomic van der Waals volumes |
| D361 | 3391 | ratio of multiple path counts to path counts |
| D213 | 3233 | valence connectivity index chi-1 |
| D467 | 3225 | Geary autocorrelation-lag 5/weighted by atomic Sanderson electronegativity |
| D491 | 3091 | Moran autocorrelation-lag 5/weighted by atomic van der Waals volumes |
| D259 | 3084 | mean information content on the distance degree equality |
| D496 | 2272 | Moran autocorrelation-lag 2/weighted by atomic Sanderson electronegativity |
| D478 | 2238 | Geary autocorrelation-lag 8/weighted by atomic polarizabilities |
| D463 | 2024 | Geary autocorrelation-lag 1/weighted by atomic Sanderson electronegativity |
| D246 | 1995 | Maximum of the differences between vertex distance and unipolarity |
| D473 | 1799 | Geary autocorrelation-lag 3/weighted by atomic polarizabilities |
| D595 | 1698 | highest eigenvalue n. 8 of Burden matrix/weighted by atomic polarizabilities |
| Chemical Name | Experiment | Prediction | Reference |
|---|---|---|---|
| 17-α-Ethynylestradiol | 1 | 1 | [49] |
| 11-β-Ethyloxyestradiol | 1 | 0 | [48] |
| 11-β-Methoxyestradiol | 1 | 1 | [48] |
| Compound 7b | 1 | 0 | [49] |
| 16-α-Fluoroestradiol (FES) | 1 | 1 | [48] |
| Compound 8b | 1 | 0 | [49] |
| Compound 8c | 1 | 1 | [49] |
| Compound 3 | 1 | 1 | [48] |
| Compound 1 | 1 | 0 | [48] |
| Compound 2 | 1 | 0 | [48] |
| Compound 7c | 1 | 1 | [49] |
| 11-β-Ethyl-17-α-ethynylestradiol | 1 | 0 | [49] |
| 11-β-Ethylestradiol | 1 | 0 | [49] |
| Compound 8a | 1 | 0 | [49] |
| 17-α-Ethynyl-11-β-Methoxyestradiol | 1 | 0 | [49] |
| Compound 7a | 1 | 0 | [49] |
| 4-Nonylphenoxyacetic acid (NP1EC) | 1 | 1 | [50] |
| 4-tert-Butylphenol (BP) | 0 | 0 | [50] |
| Igepal | 0 | 0 | [50] |
| 2,4’DDT | 0 | 0 | [50] |
| 2,4’-DDE | 0 | 0 | [50] |
| Kepone | 0 | 0 | [50] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, H.; Shen, J.; Ng, H.W.; Sakkiah, S.; Ye, H.; Ge, W.; Gong, P.; Xiao, W.; Tong, W. A Rat α-Fetoprotein Binding Activity Prediction Model to Facilitate Assessment of the Endocrine Disruption Potential of Environmental Chemicals. Int. J. Environ. Res. Public Health 2016, 13, 372. https://doi.org/10.3390/ijerph13040372
Hong H, Shen J, Ng HW, Sakkiah S, Ye H, Ge W, Gong P, Xiao W, Tong W. A Rat α-Fetoprotein Binding Activity Prediction Model to Facilitate Assessment of the Endocrine Disruption Potential of Environmental Chemicals. International Journal of Environmental Research and Public Health. 2016; 13(4):372. https://doi.org/10.3390/ijerph13040372
Chicago/Turabian StyleHong, Huixiao, Jie Shen, Hui Wen Ng, Sugunadevi Sakkiah, Hao Ye, Weigong Ge, Ping Gong, Wenming Xiao, and Weida Tong. 2016. "A Rat α-Fetoprotein Binding Activity Prediction Model to Facilitate Assessment of the Endocrine Disruption Potential of Environmental Chemicals" International Journal of Environmental Research and Public Health 13, no. 4: 372. https://doi.org/10.3390/ijerph13040372
APA StyleHong, H., Shen, J., Ng, H. W., Sakkiah, S., Ye, H., Ge, W., Gong, P., Xiao, W., & Tong, W. (2016). A Rat α-Fetoprotein Binding Activity Prediction Model to Facilitate Assessment of the Endocrine Disruption Potential of Environmental Chemicals. International Journal of Environmental Research and Public Health, 13(4), 372. https://doi.org/10.3390/ijerph13040372