
Numerical Study of the Simultaneous Oxidation of NO and SO2 by Ozone

Abstract
:1. Introduction
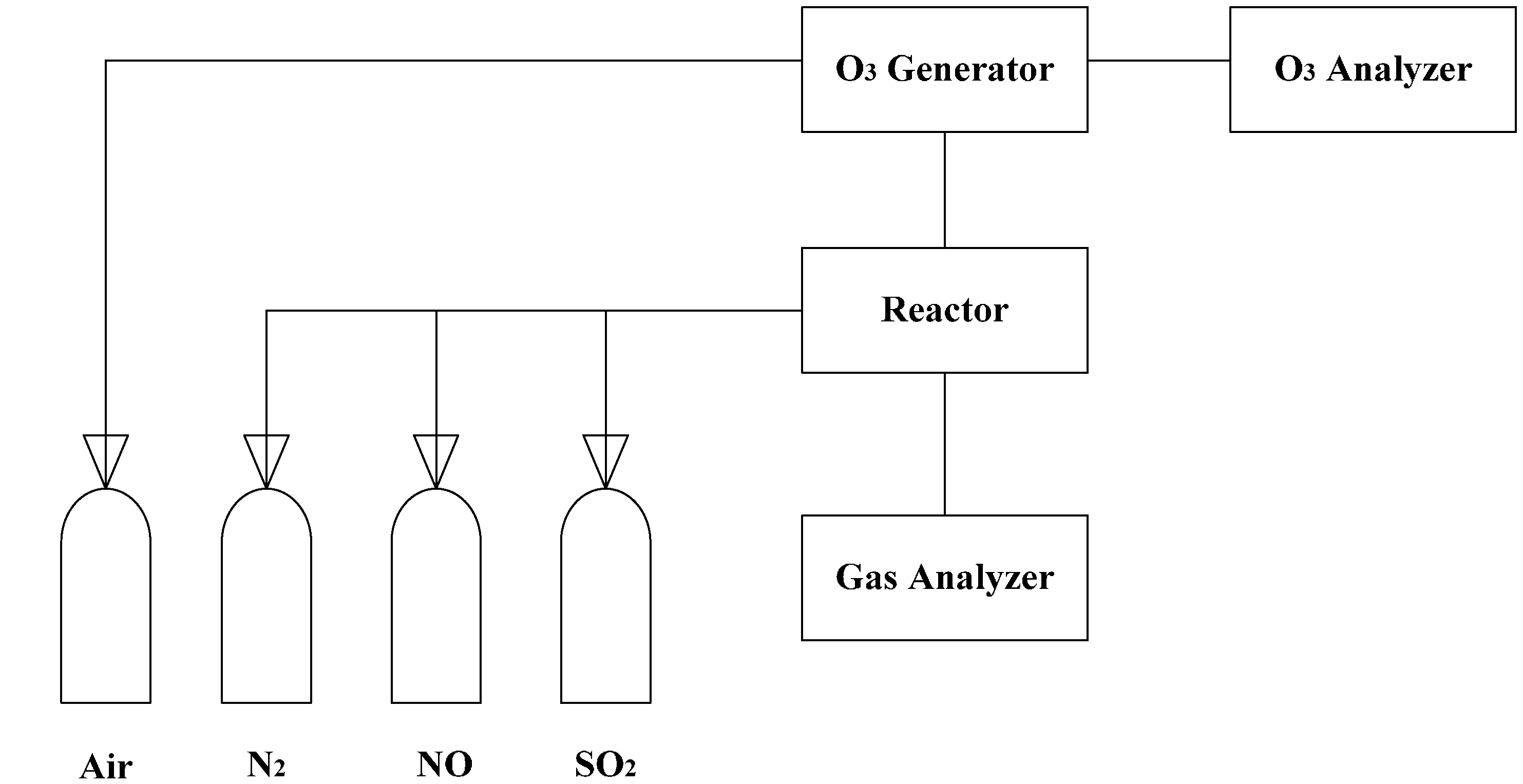
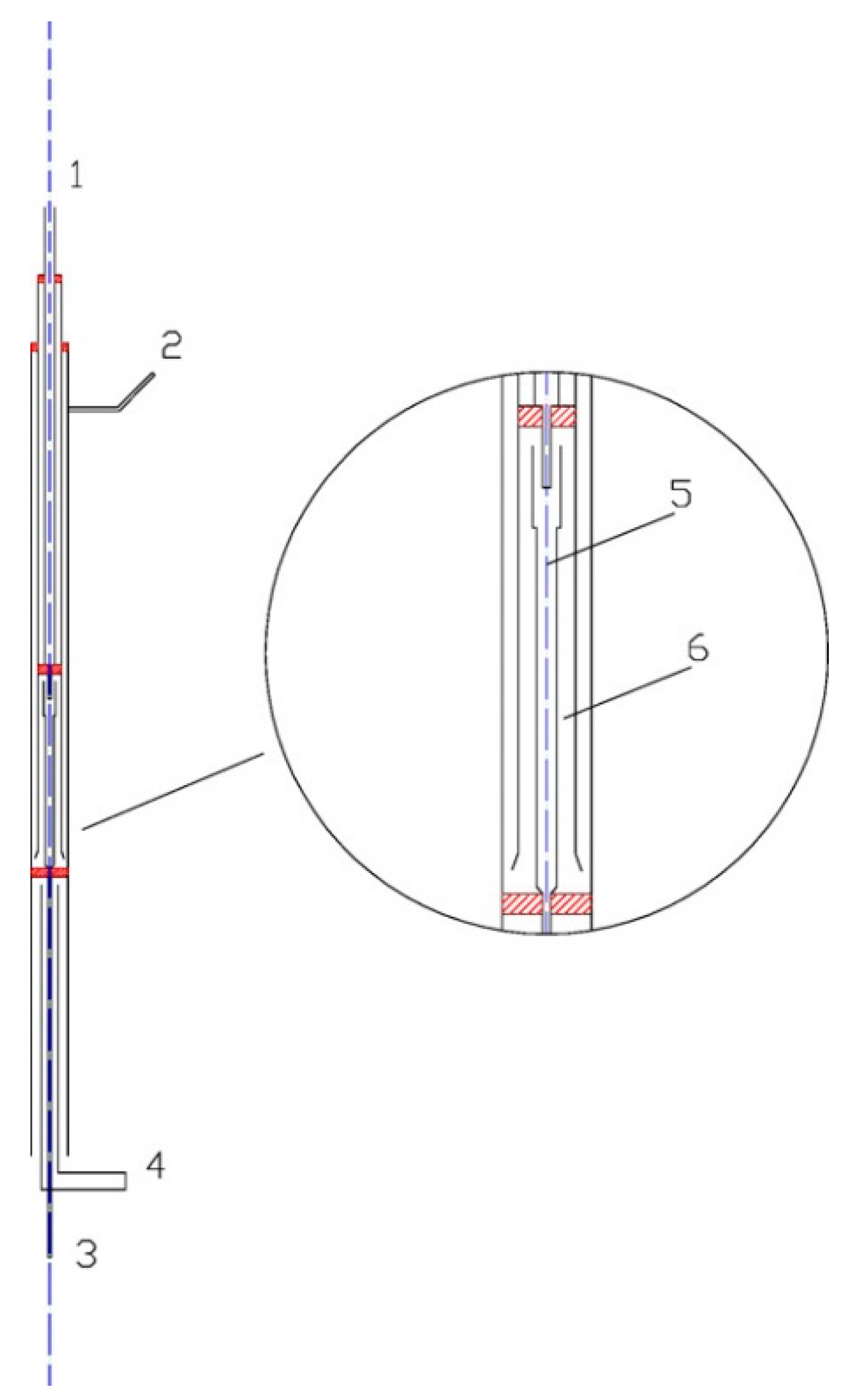
2. Experimental Setup

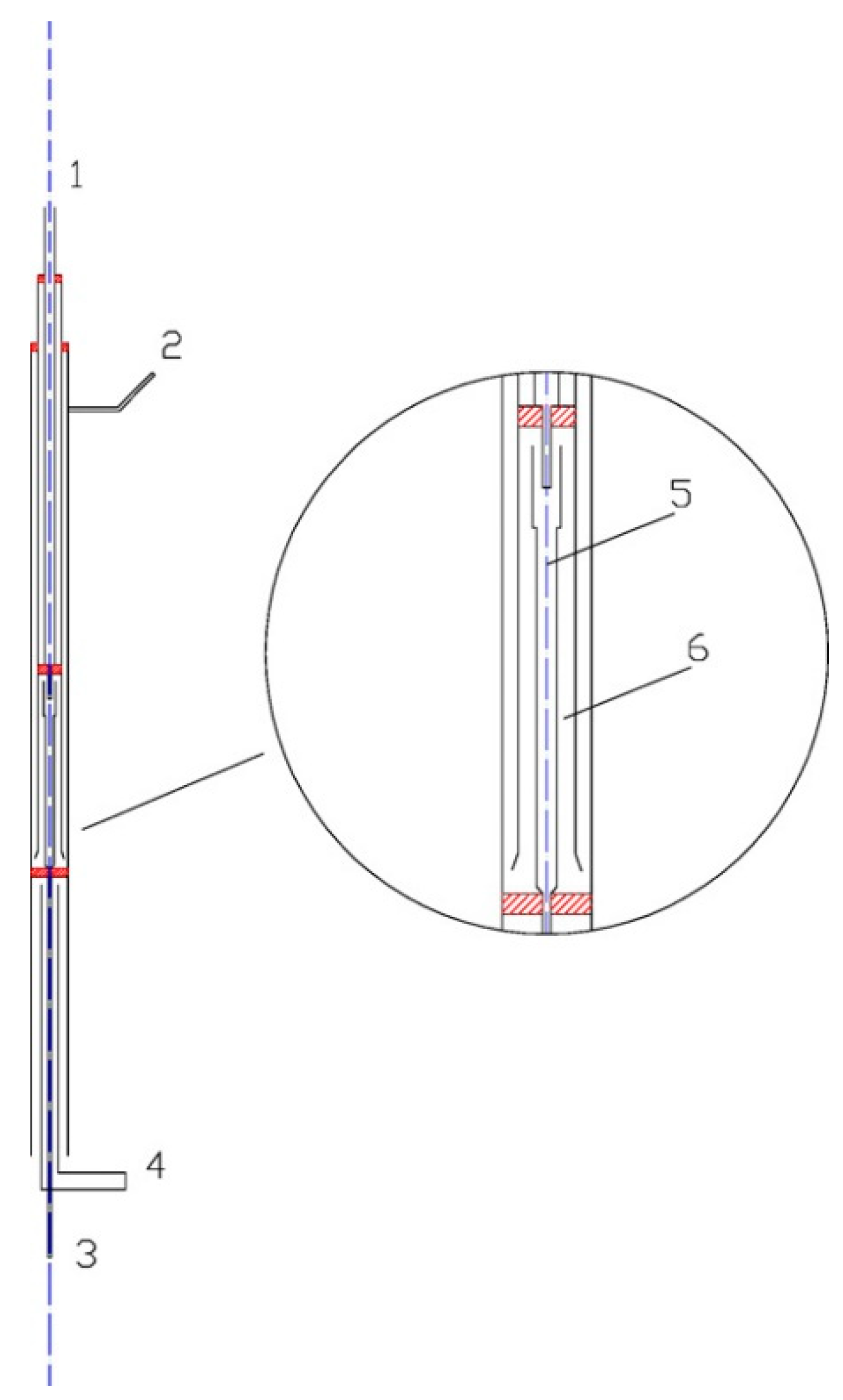
3. Kinetic Modeling and Numerical Simulation Methodology
3.1. Kinetic Mechanism Description

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactions | Model I | Model II | ||
|---|---|---|---|---|
| A (cm3/mol-s) | Ea (cal/mol) | A (cm3/mol-s) | Ea (cal/mol) | |
| O3 + NO = NO2 + O2 (R1) | 1.8E + 12 | 2722 | 8.43E + 11 | 2605 |
| O3 + NO2 = O2 + NO3 (R2) | 7.22E + 10 | 4870 | 8.43E + 10 | 4913 |
| O3 + SO2 = O2 + SO3 (R3) | 1.81E + 12 | 13,910 | 1.81E + 12 | 13,923 |
| O3 = O2 + O (R4) | 2.0E + 15 | 23,250 | 4.31E + 14 | 22,277 |
| O3 + O = O2 + O2 (R5) | 4.82E + 12 | 4093 | 4.82E + 12 | 4098 |
3.2. Simulation Strategy
4. Results and Discussion
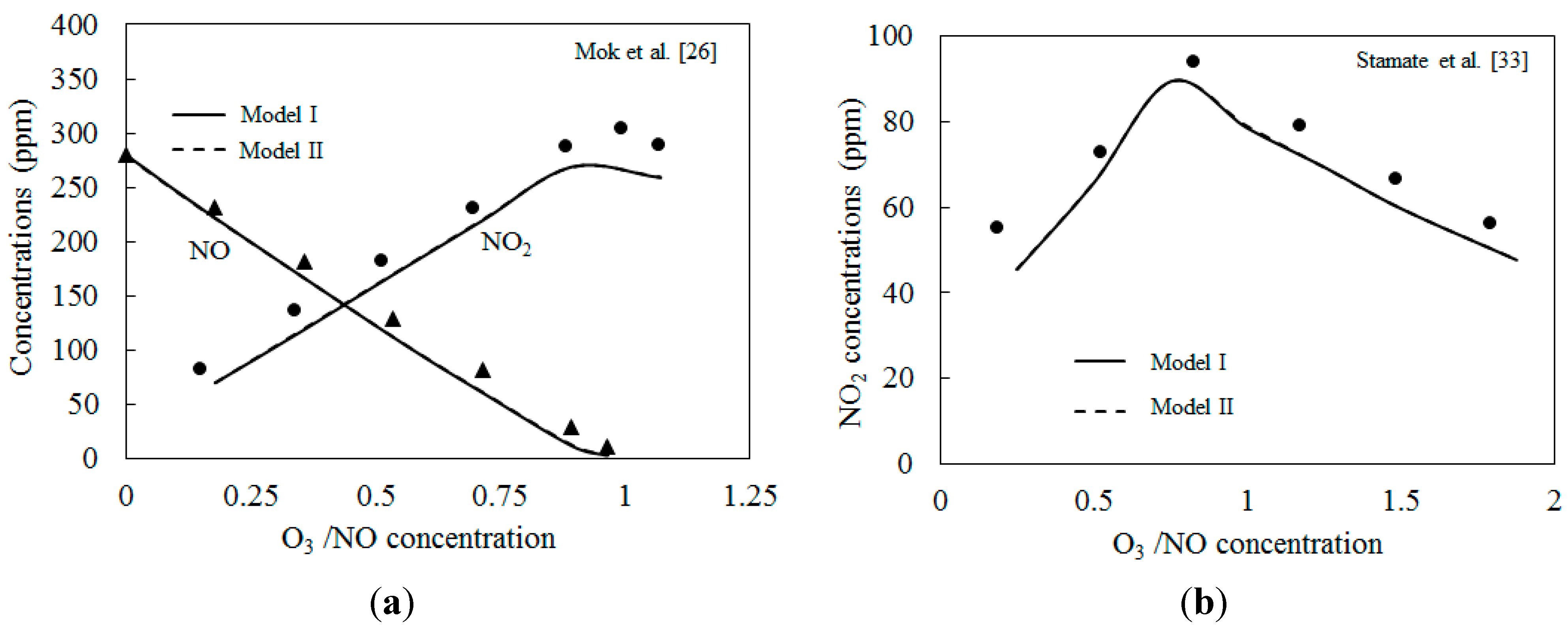
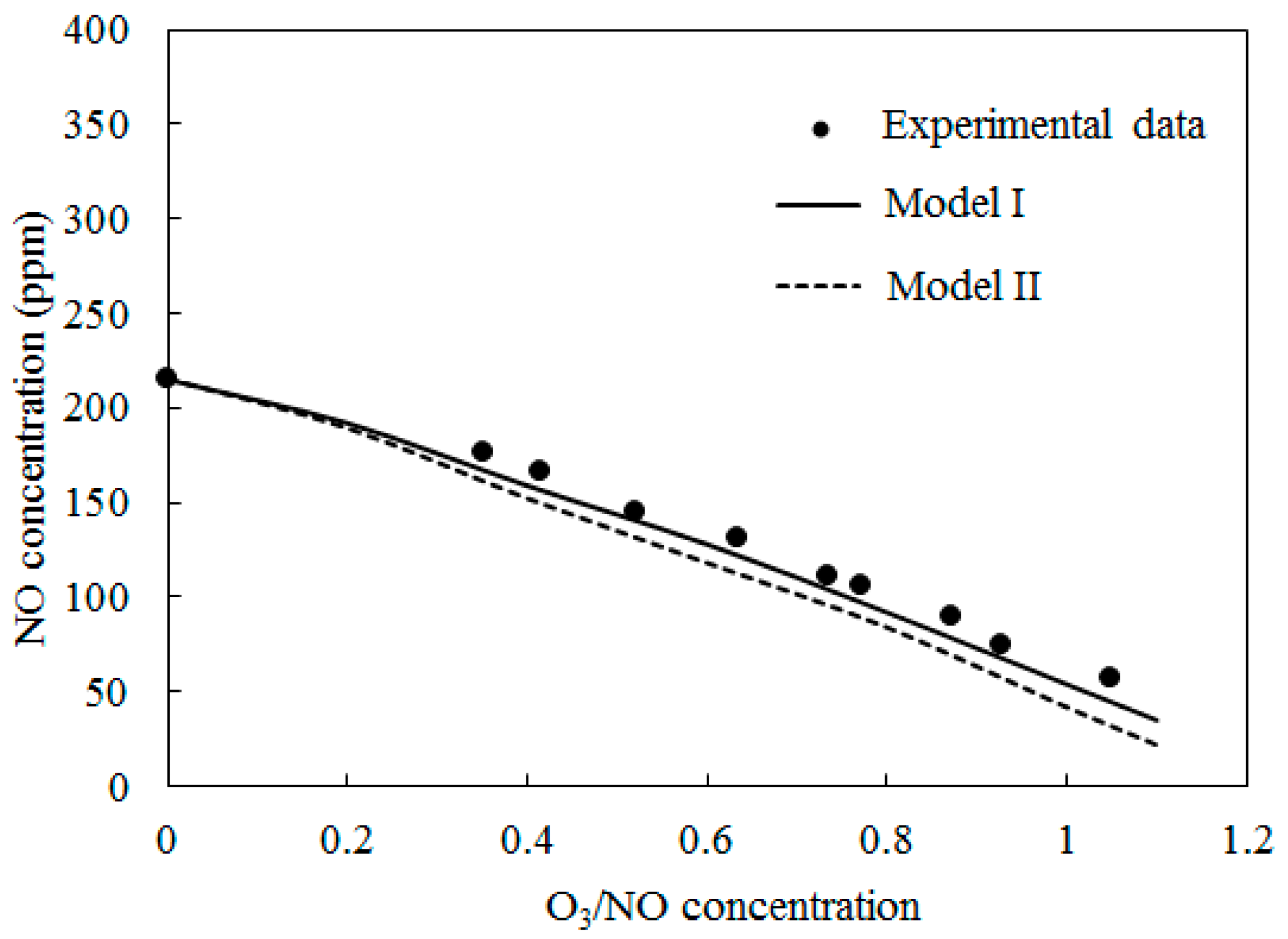
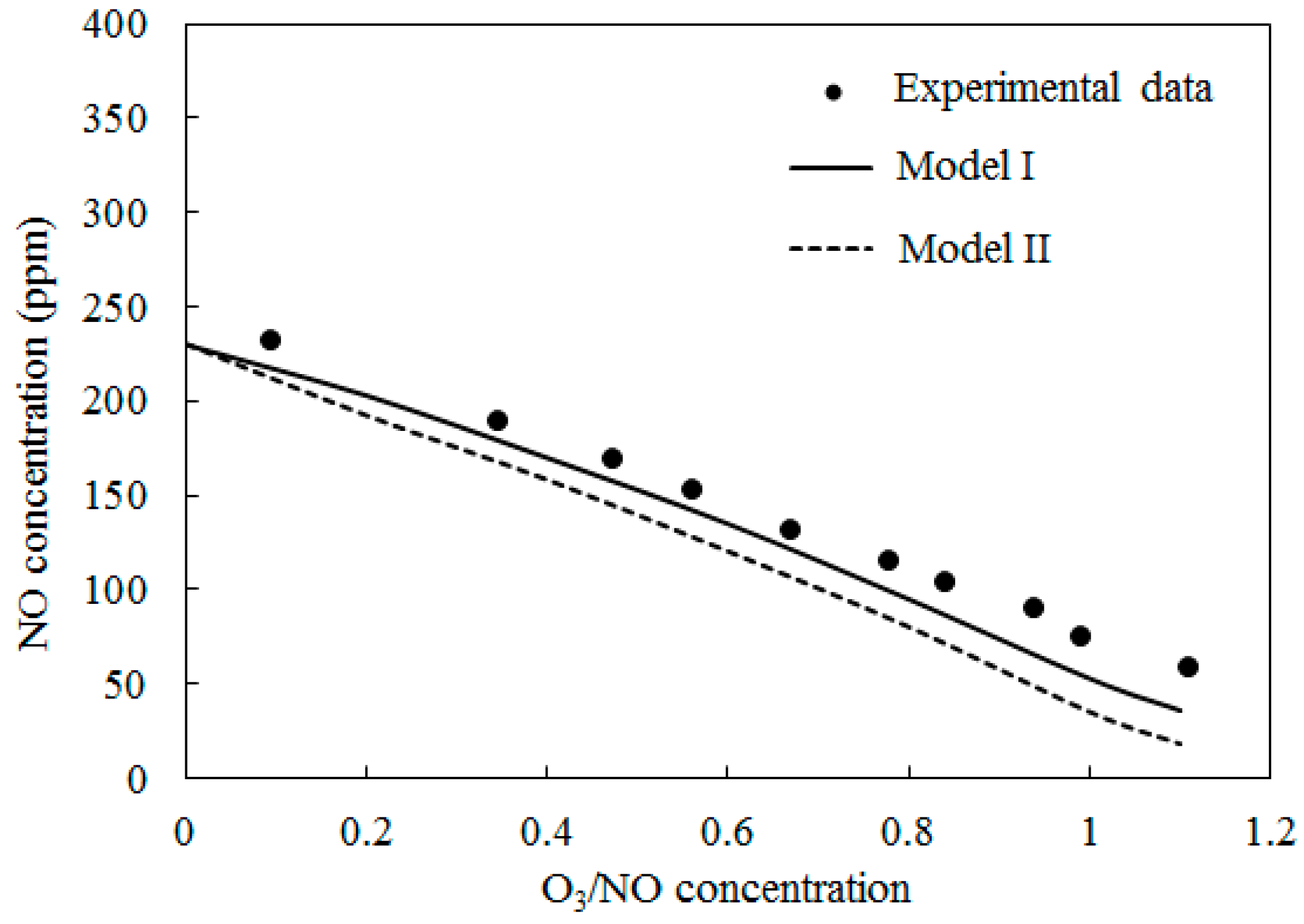
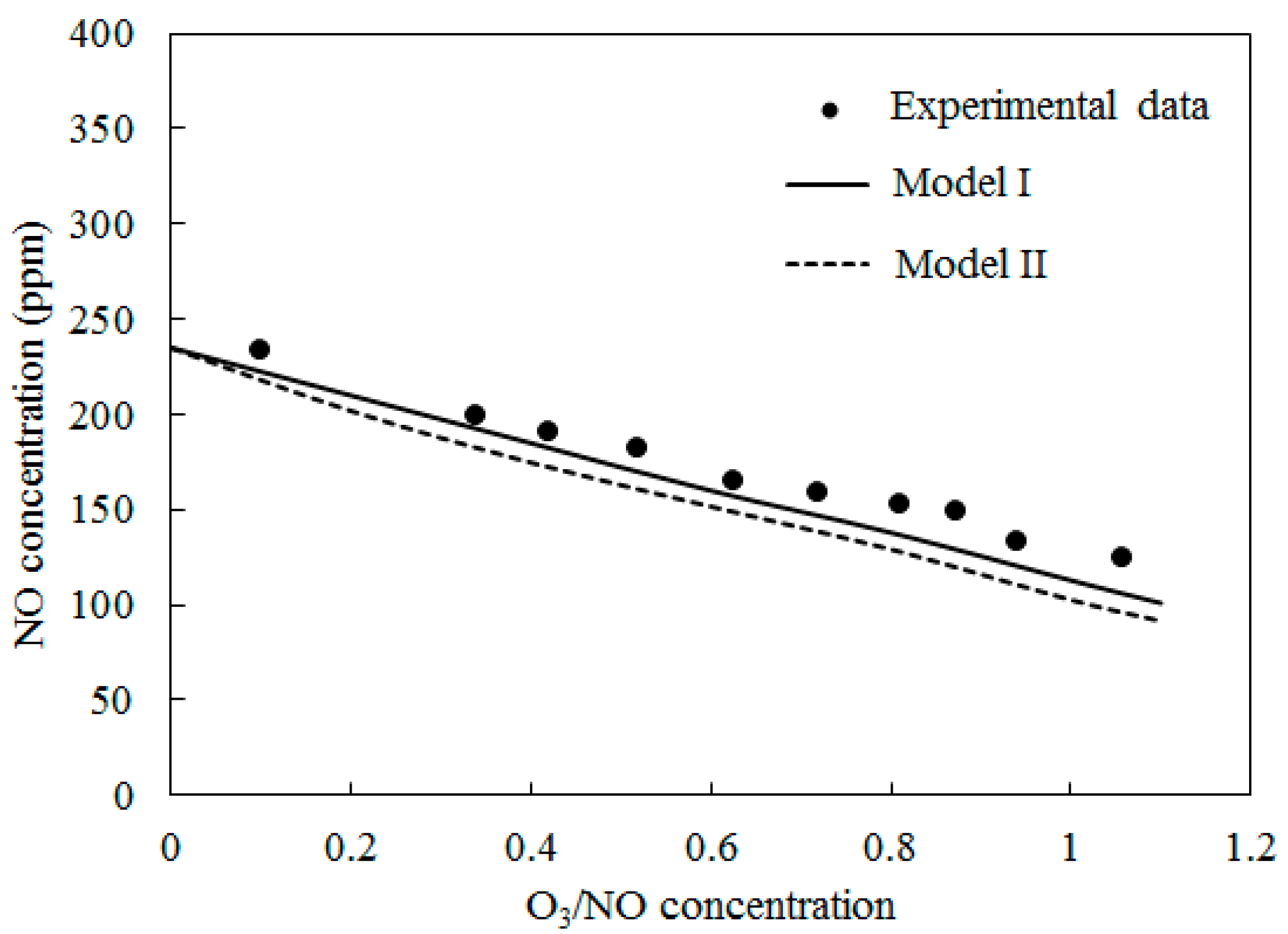
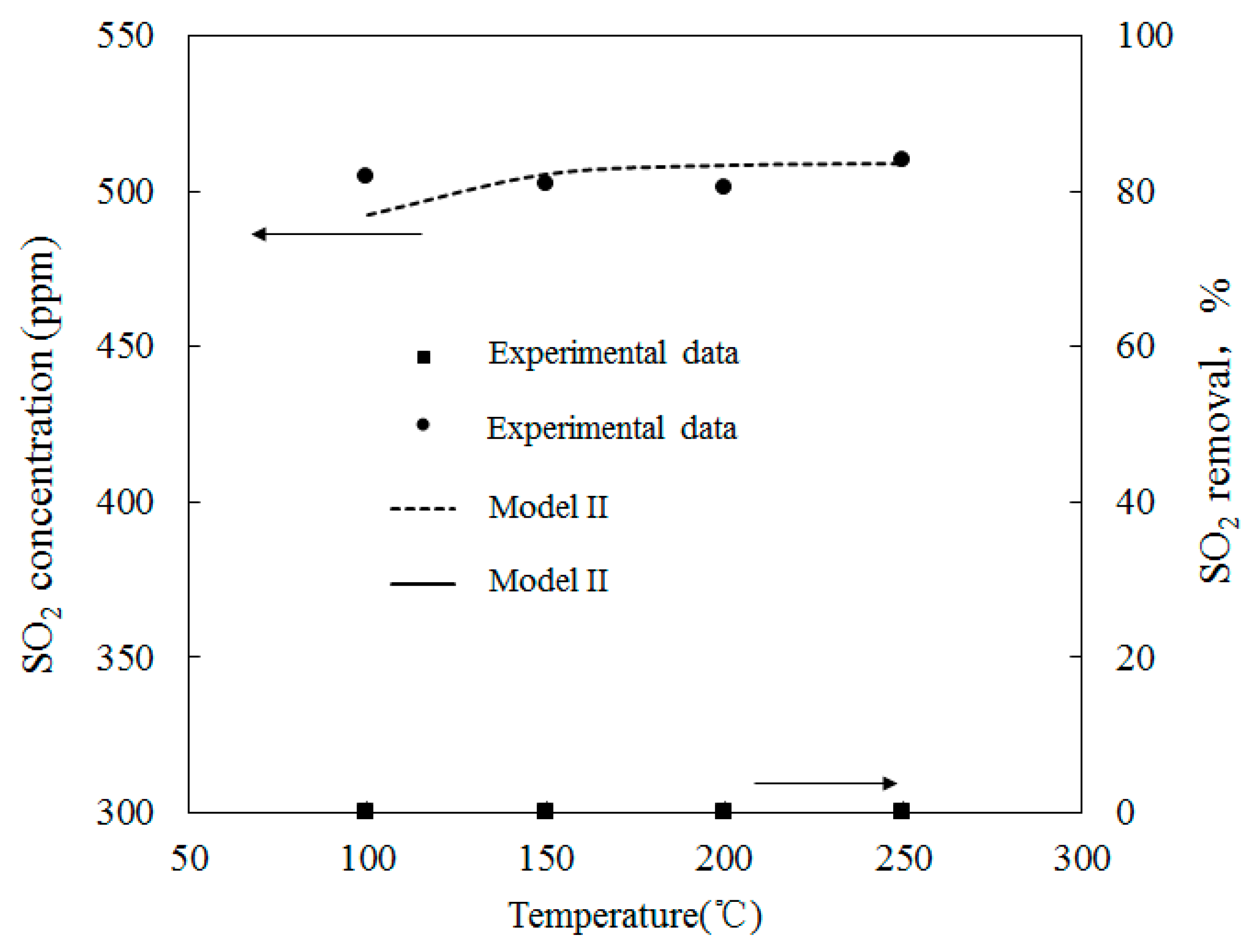
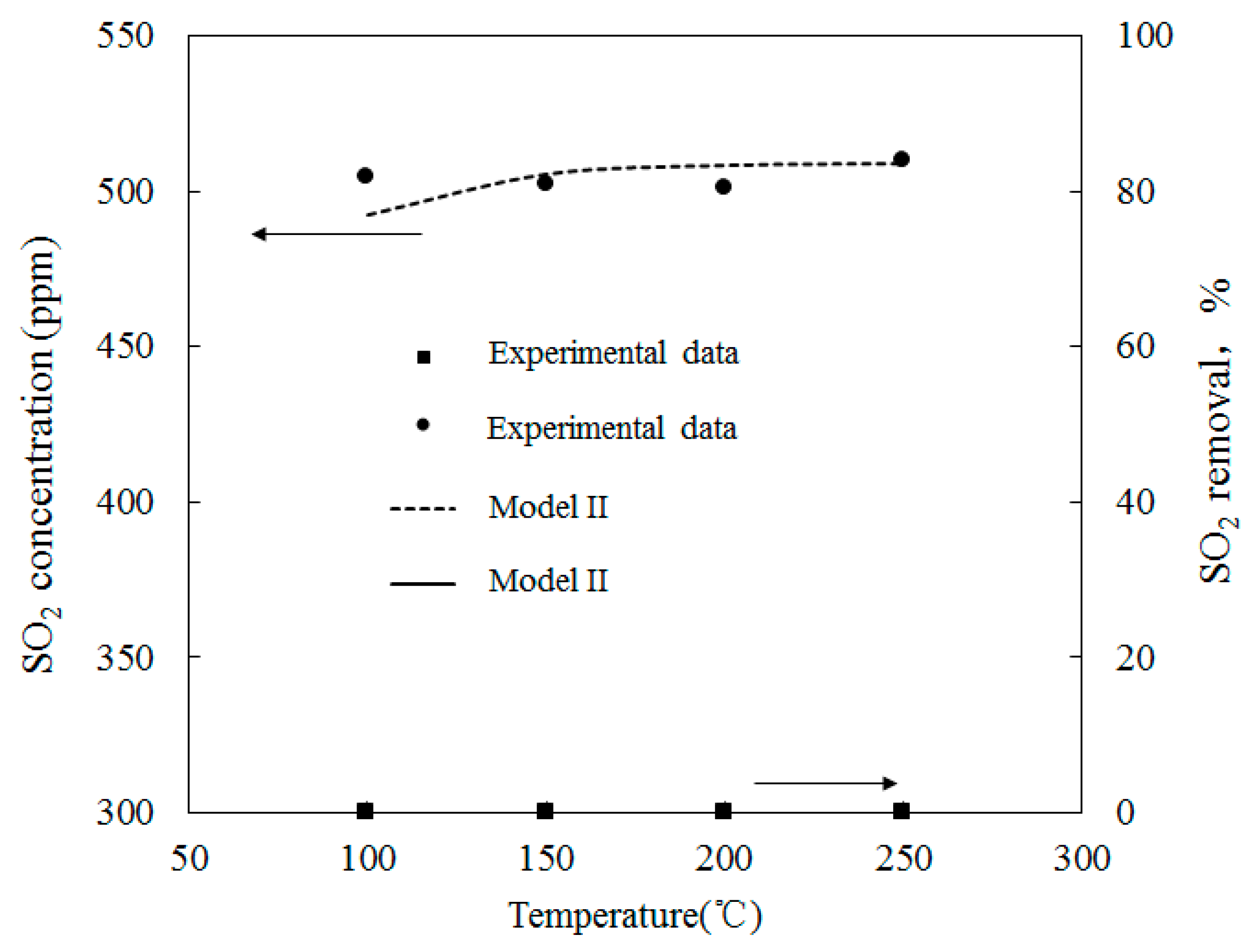
4.1. Model Validation
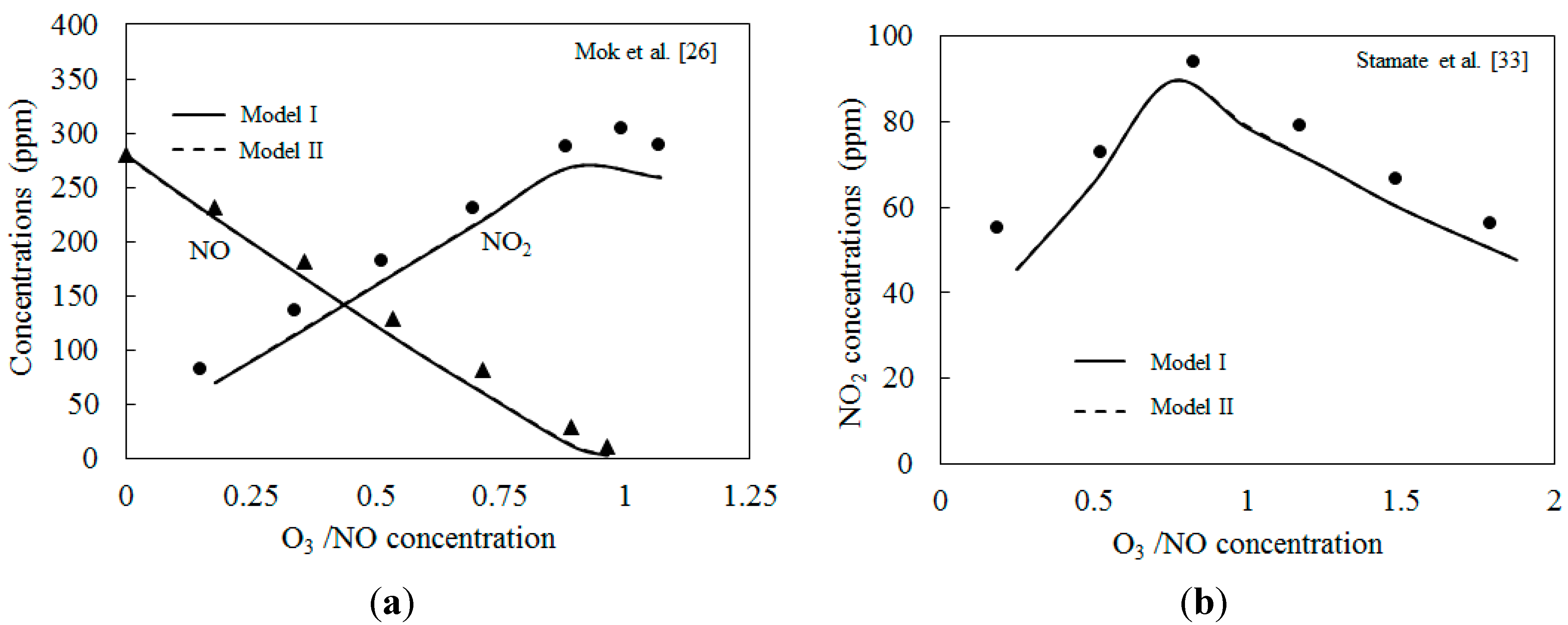
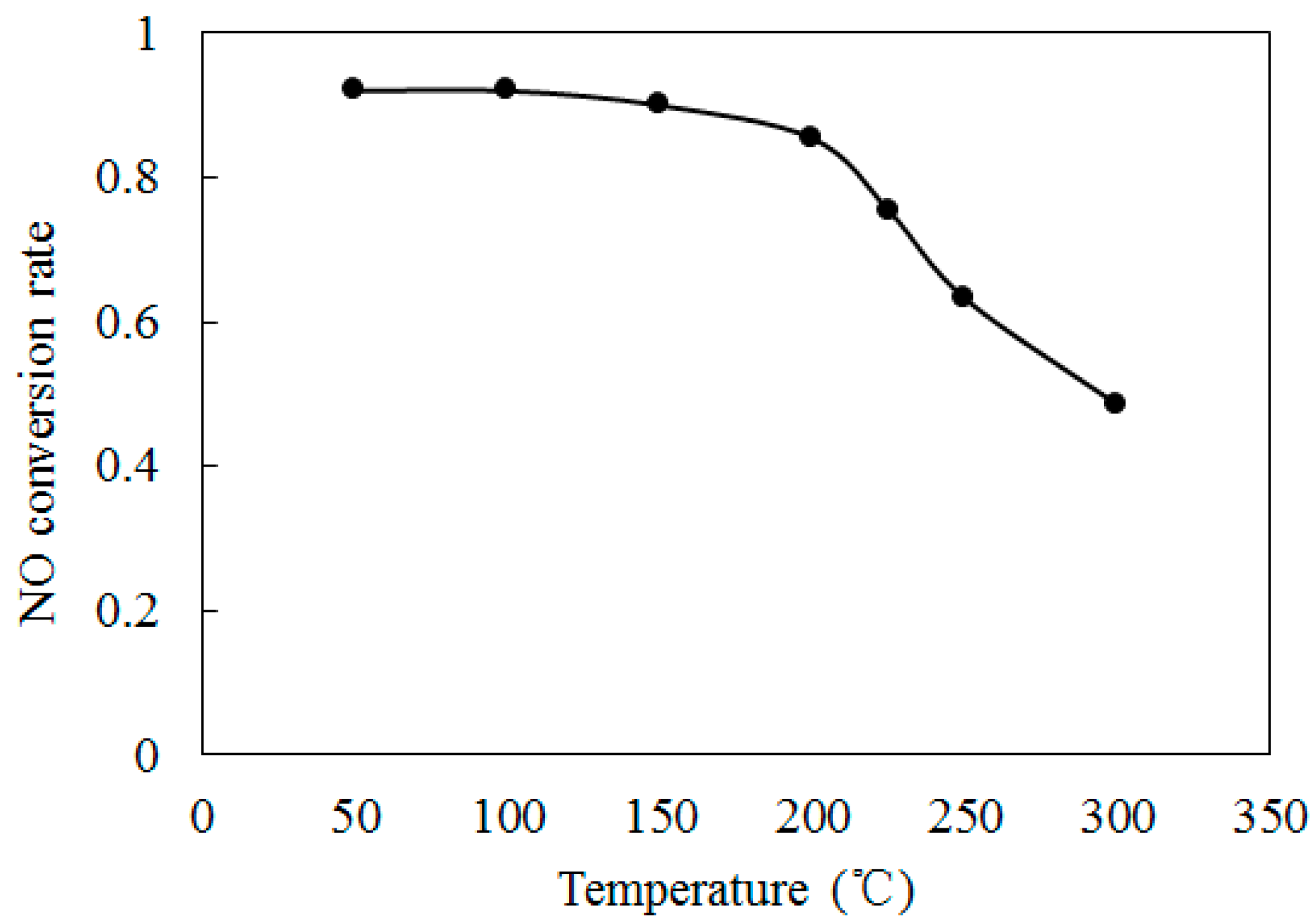
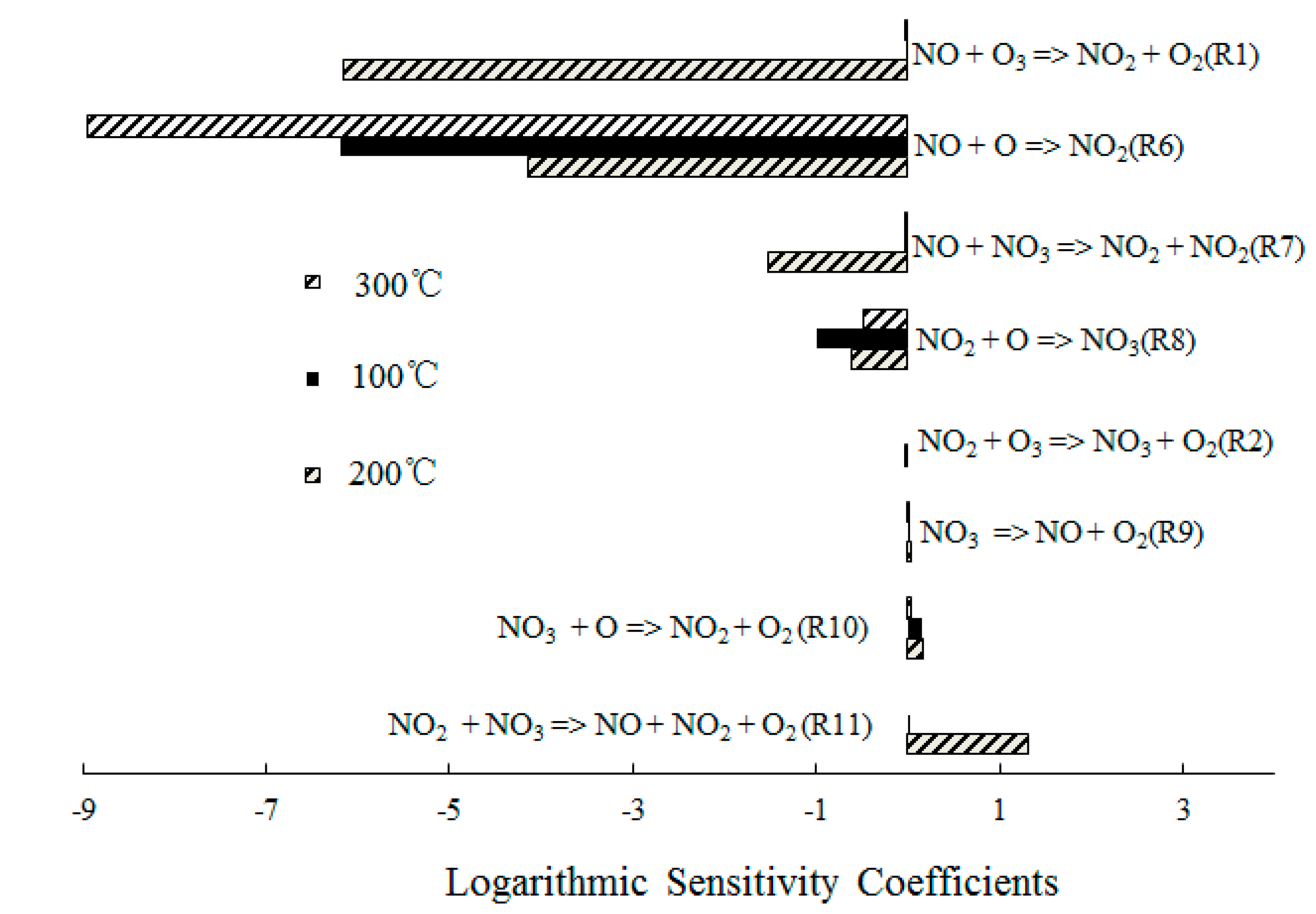
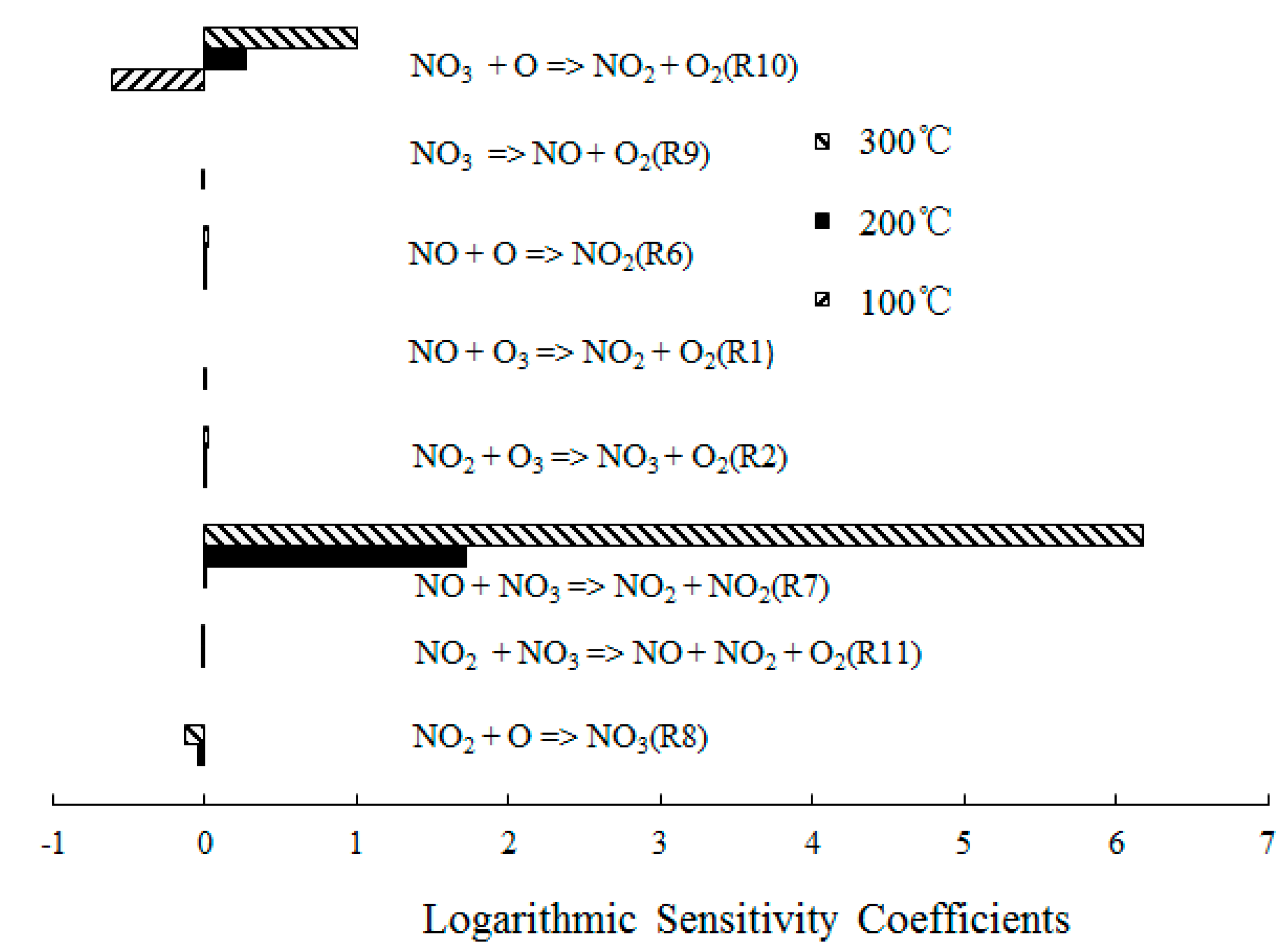
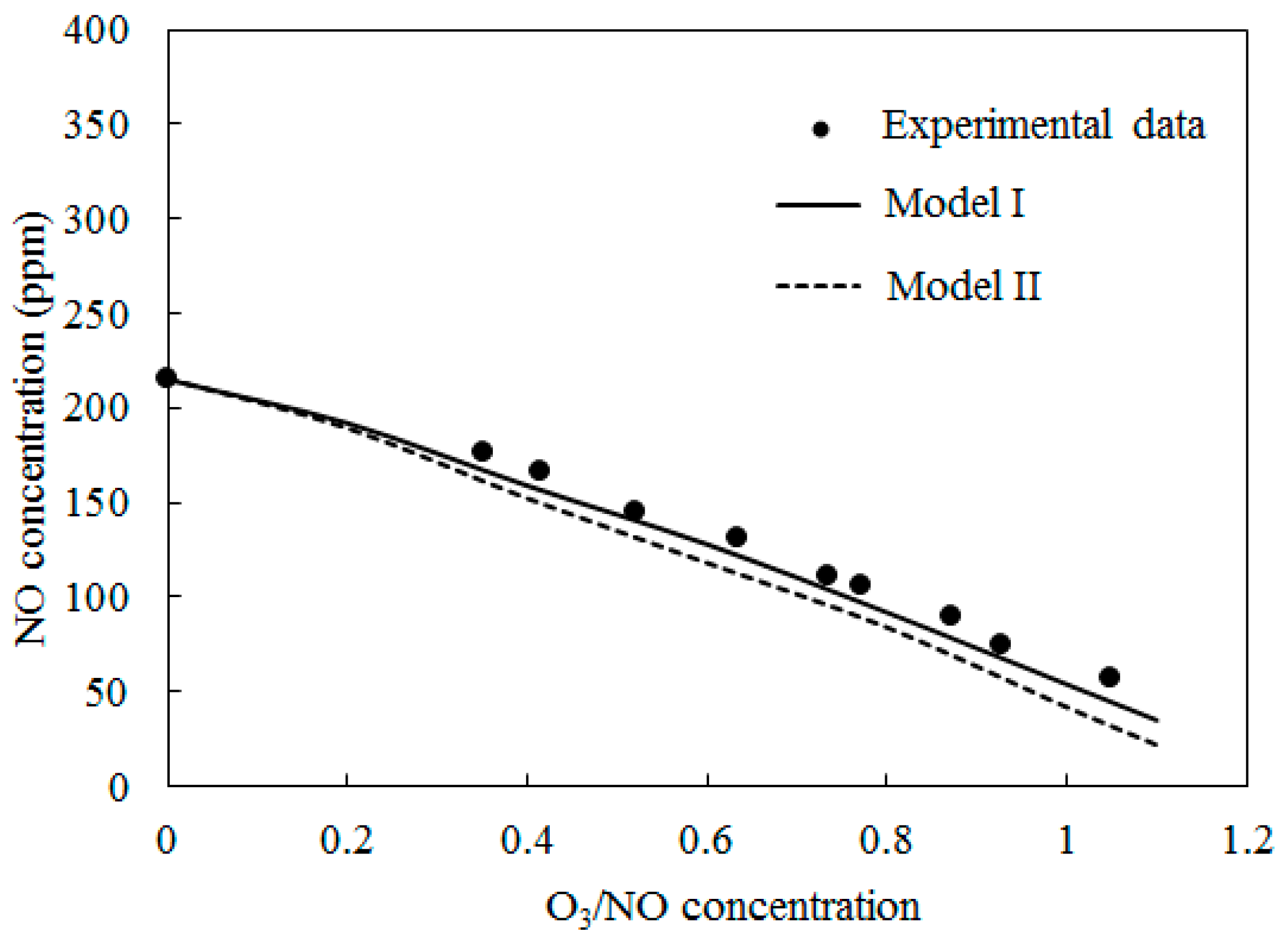
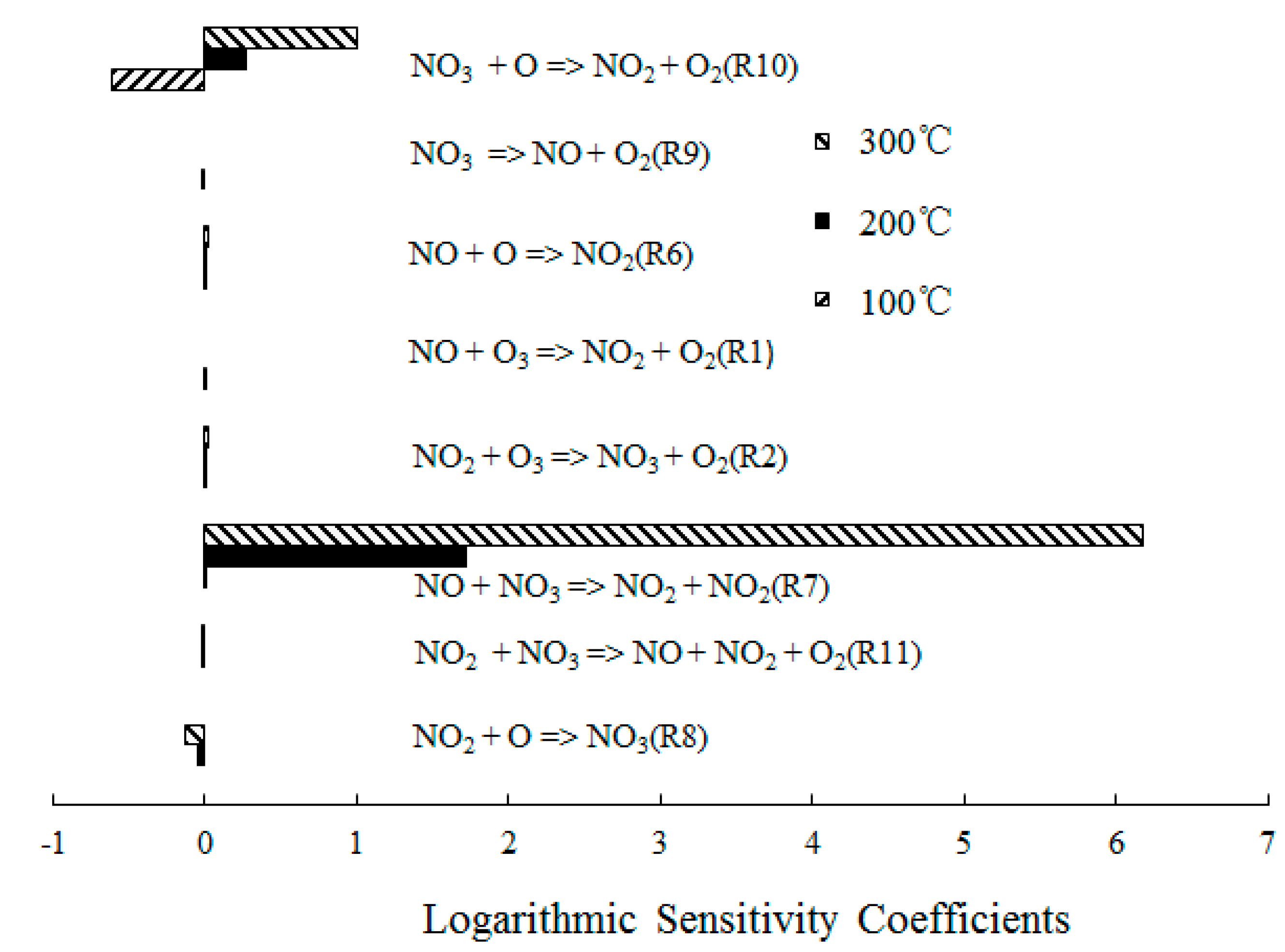
4.2. NO Oxidation by O3
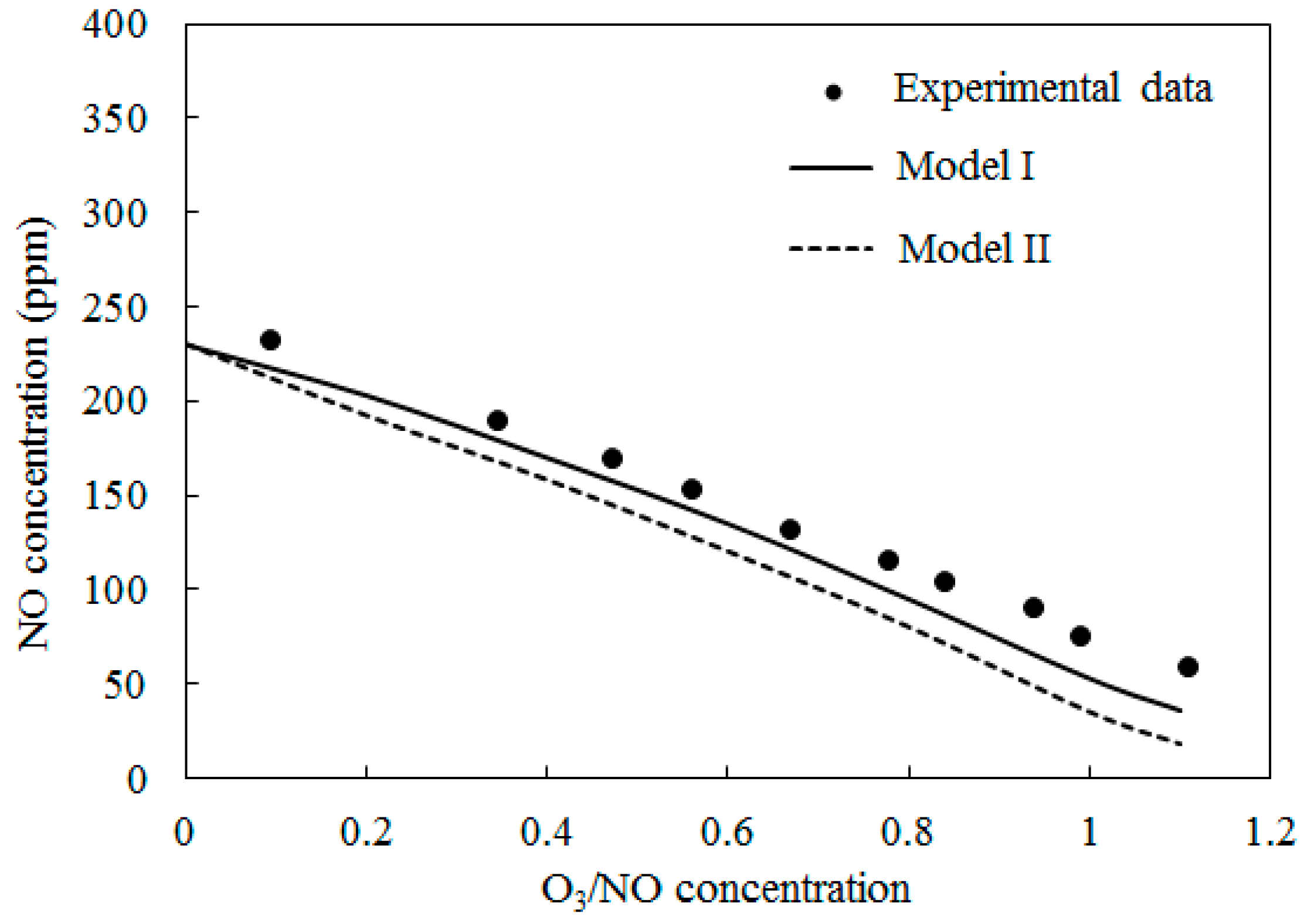
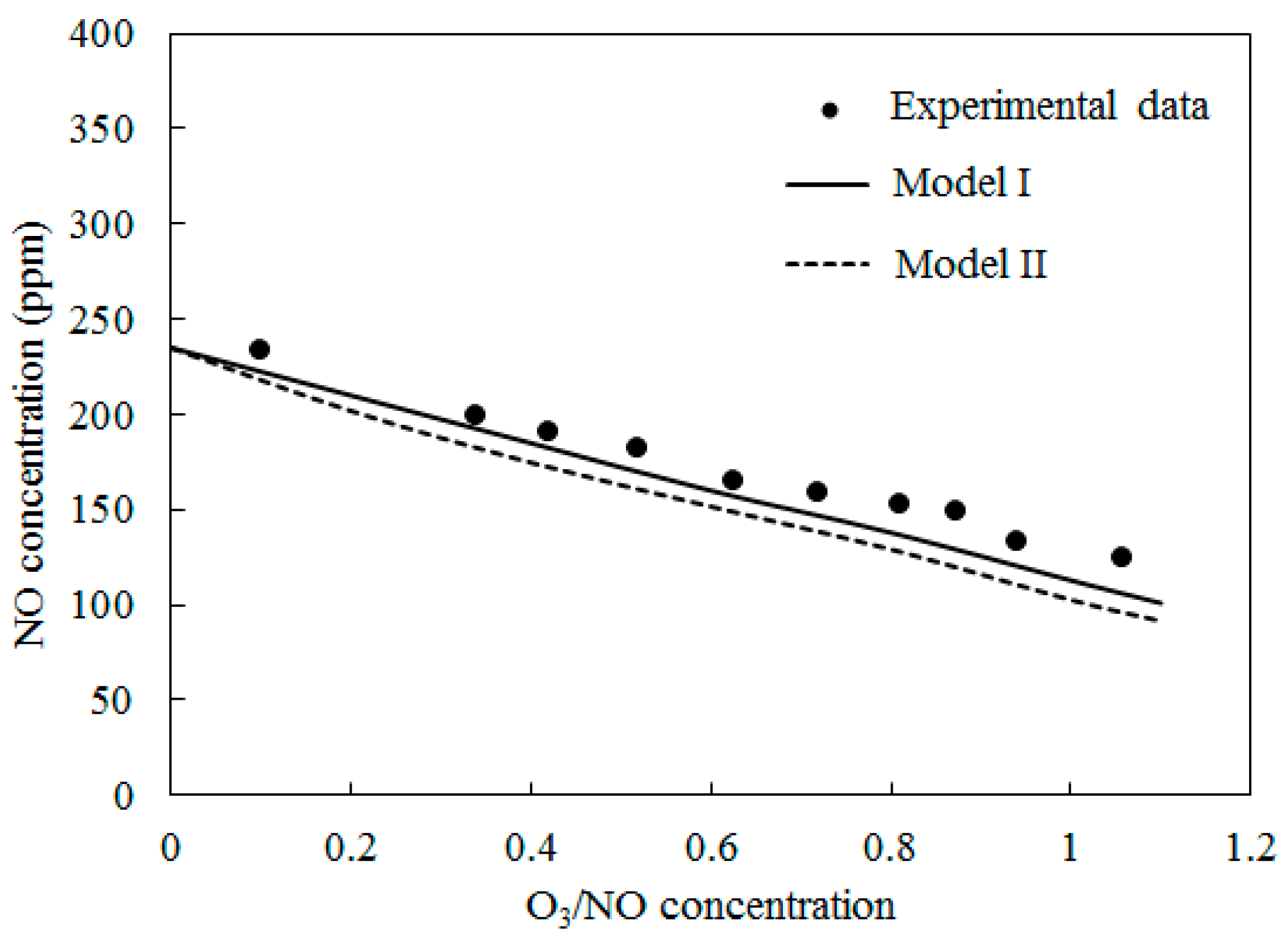
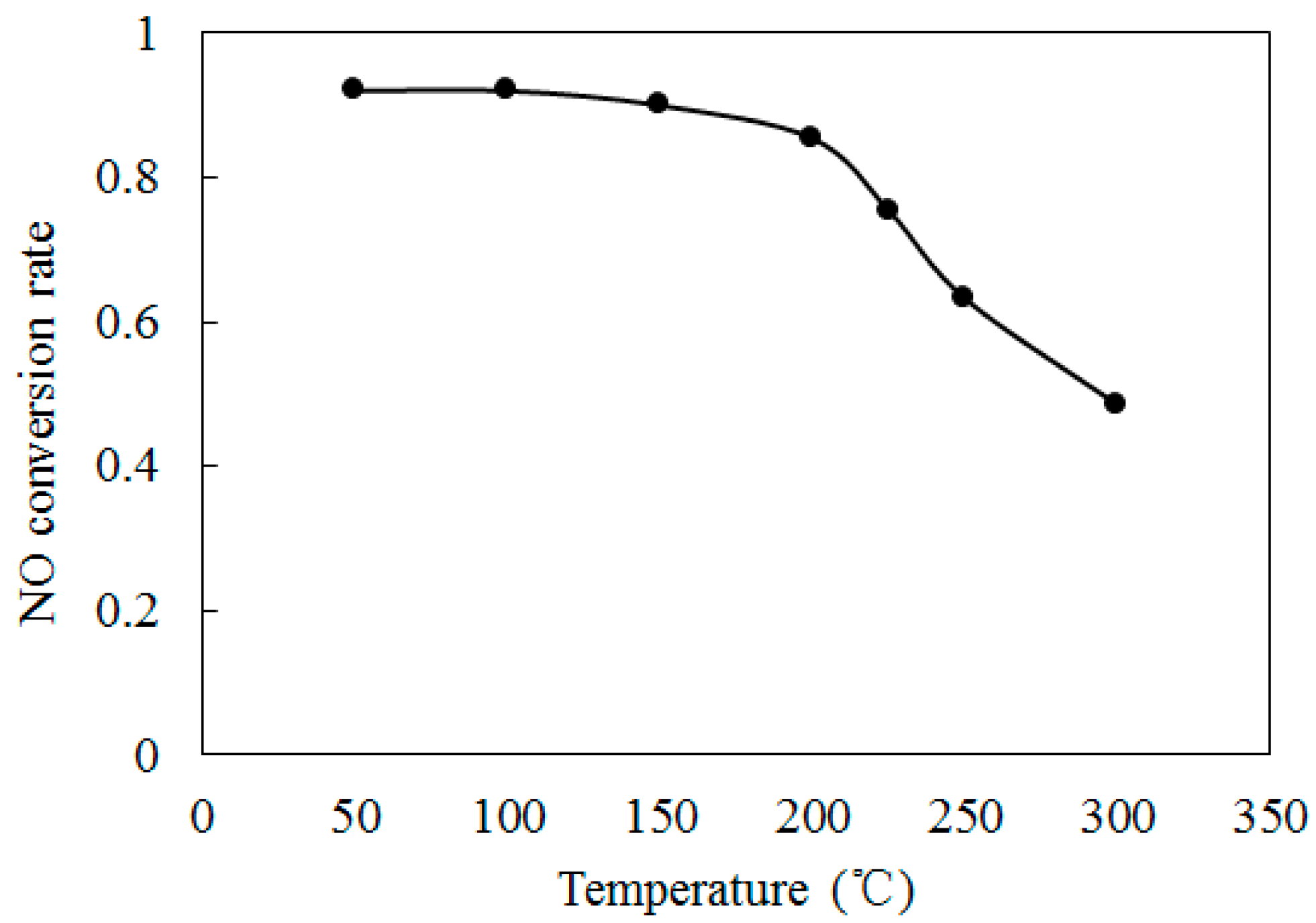
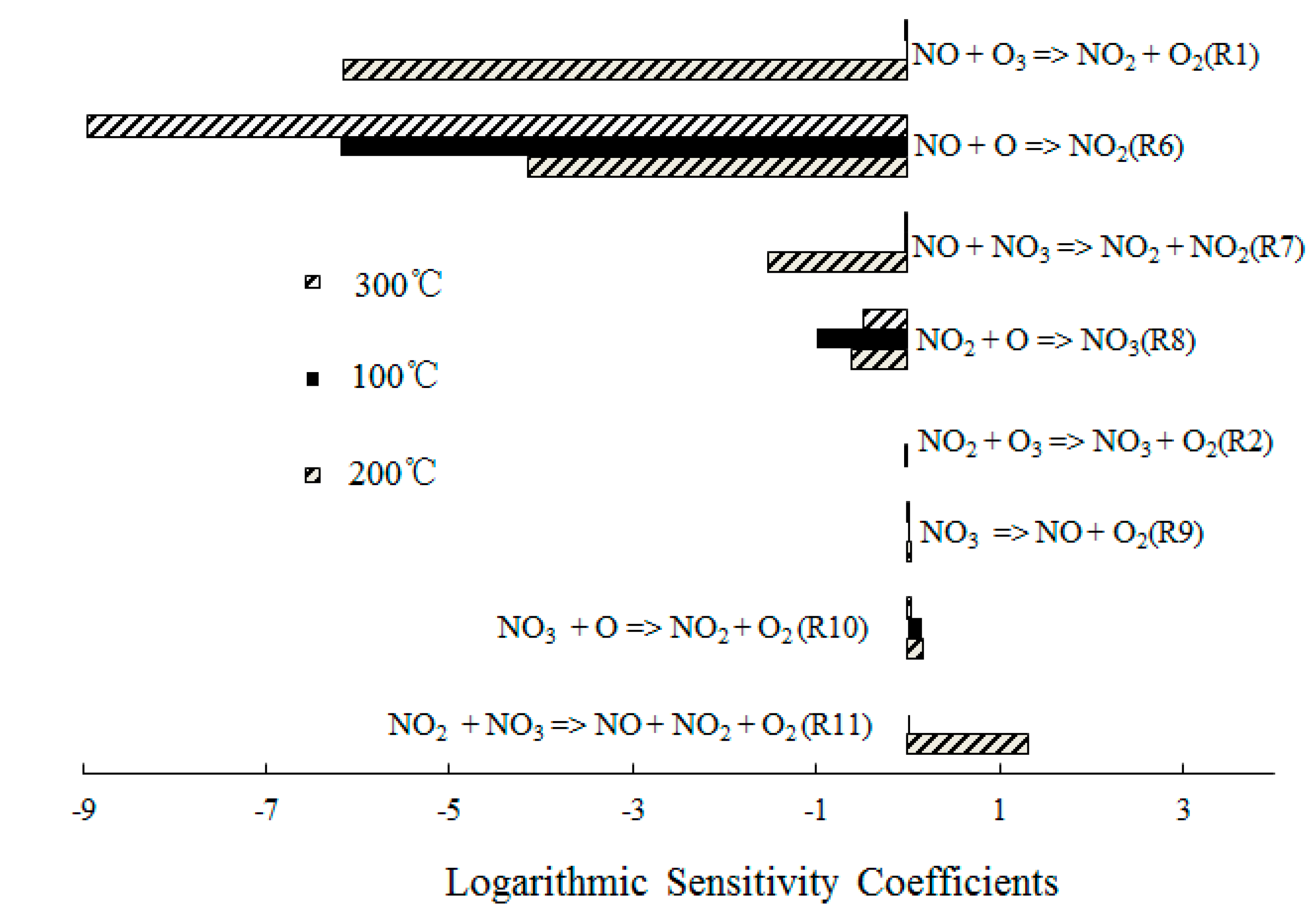

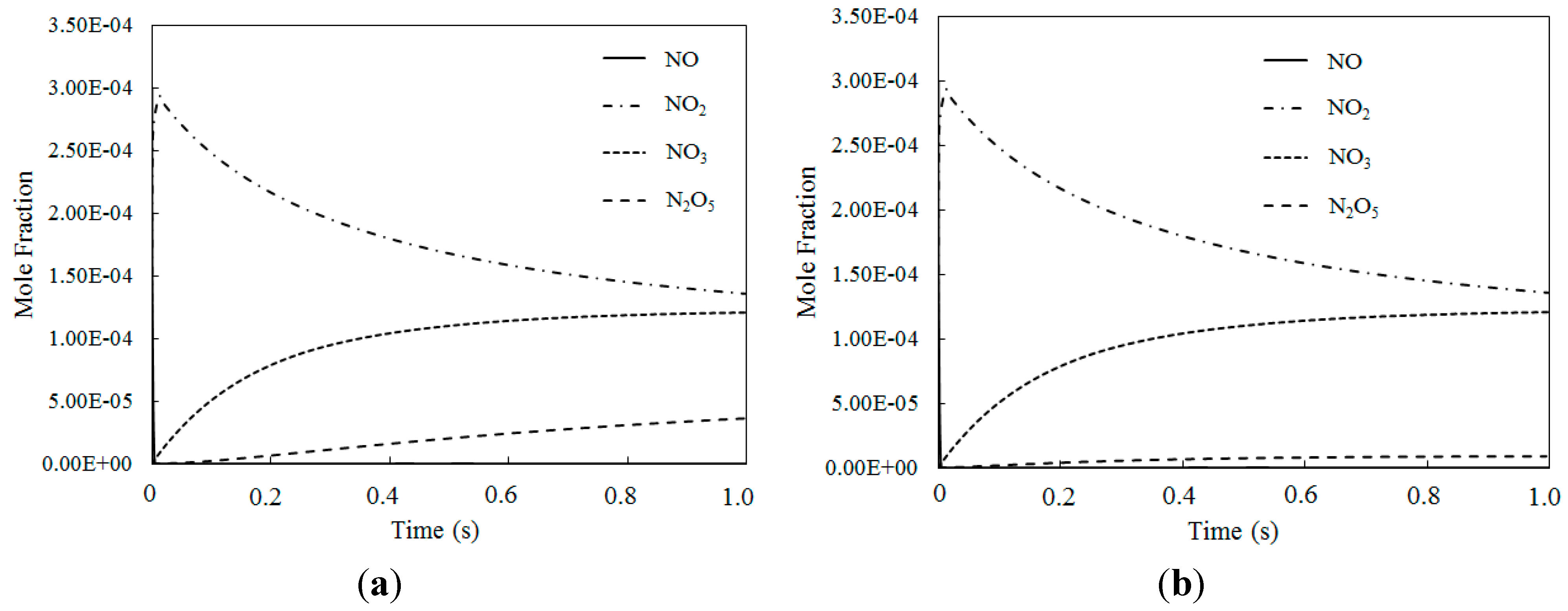
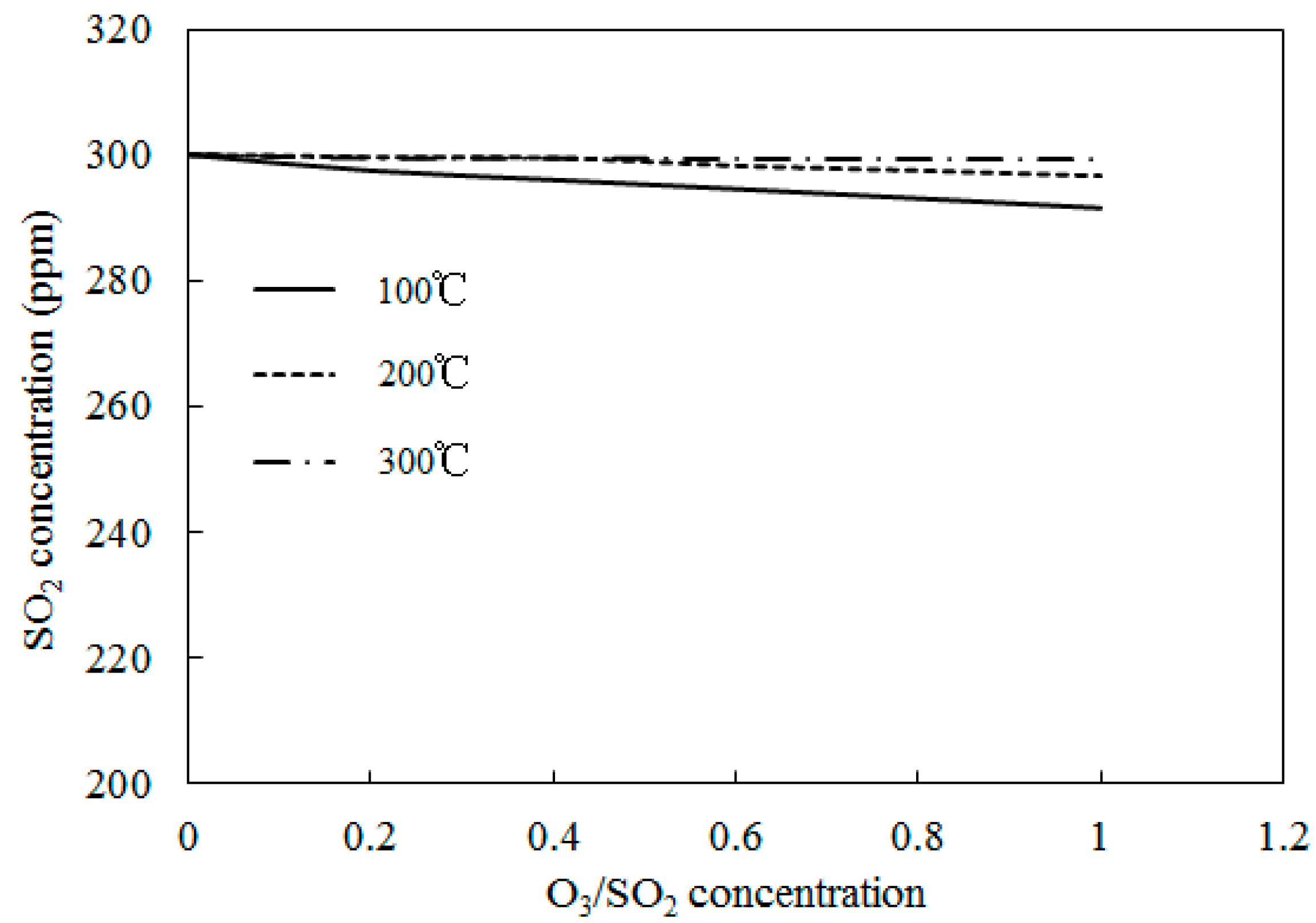
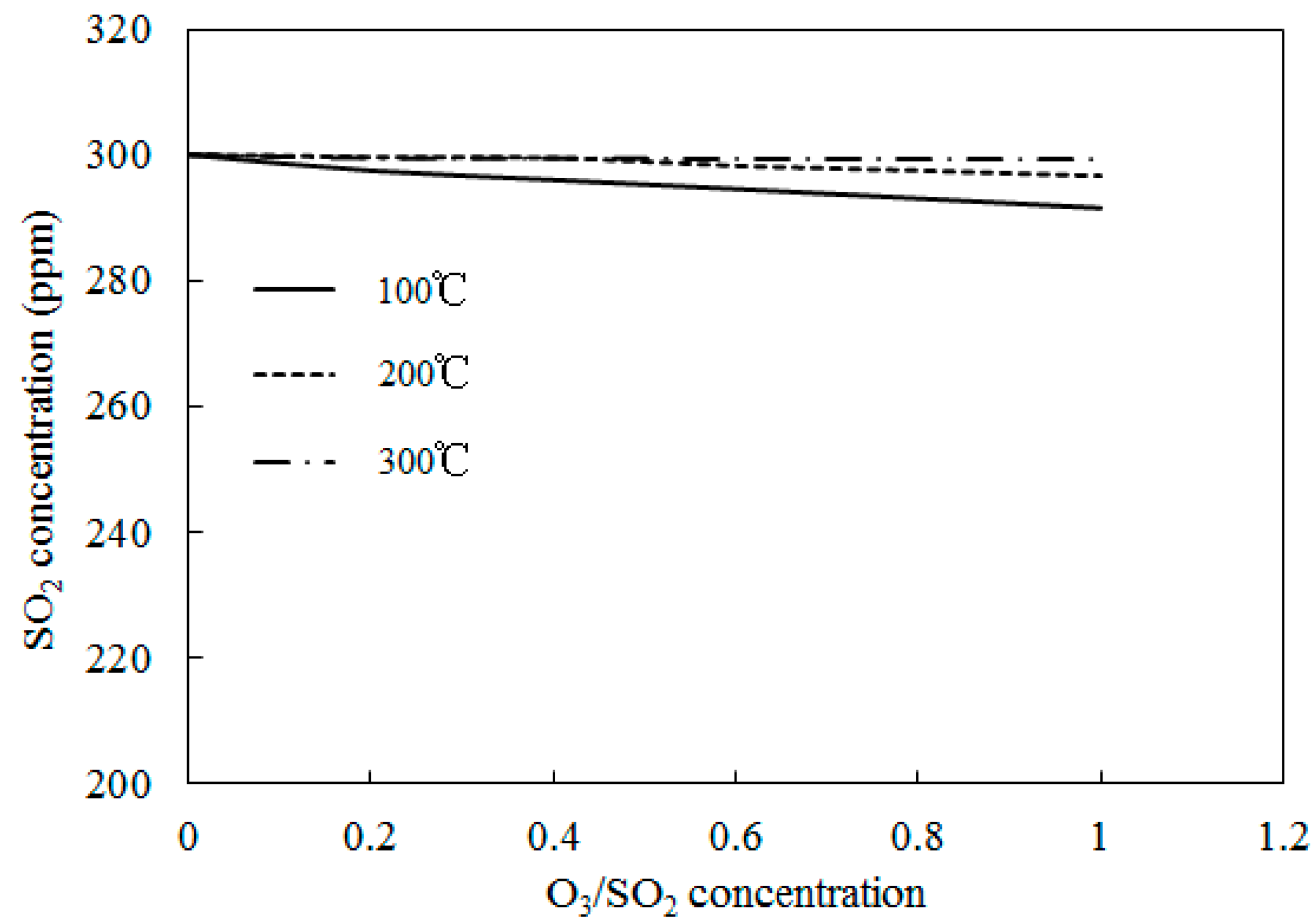
4.3. SO2 Oxidation by O3
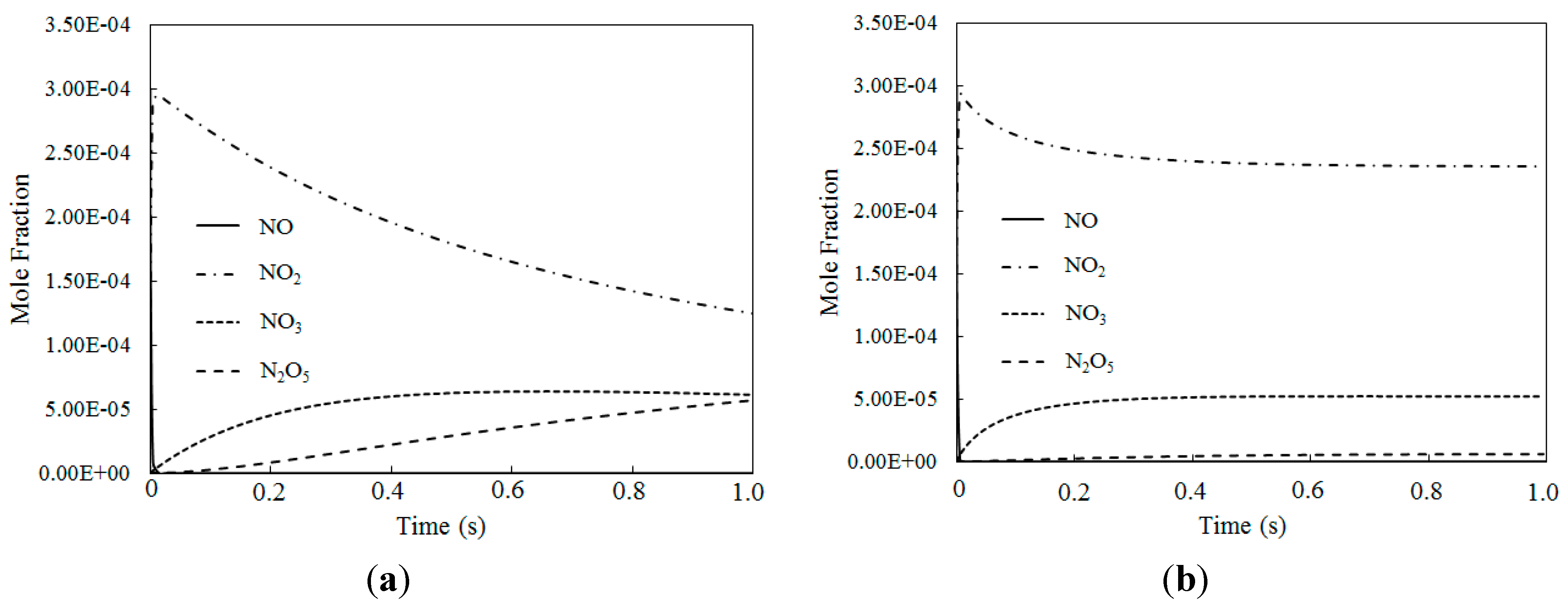
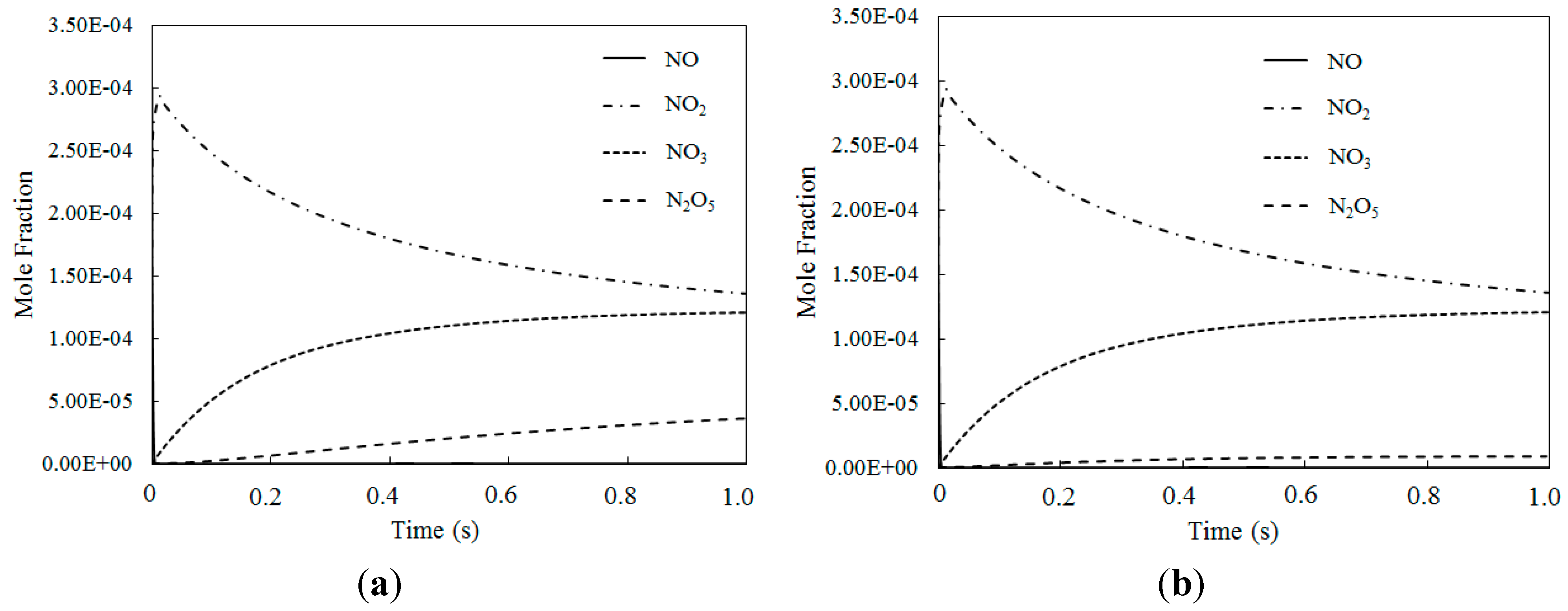
4.4. Simultaneous Oxidation of NO and SO2 by O3
5. Conclusions
Supplementary Files
Supplementary File 1Author Contributions
Conflicts of Interest
References
- Zhong, L.P.; Cao, Y.; Li, W.Y.; Pan, W.P.; Xie, K.C. Effect of the existing air pollutant control devices on mercury emission in coal-fired power plants. J. Fuel Chem. Technol. 2010, 38, 641–646. [Google Scholar] [CrossRef]
- Schopp, W.; Amann, M.; Cofala, J.; Heyes, C.; Klimont, Z. Integrated assessment of European air pollution emission control strategies. Environ. Model. Softw. 1999, 14, 1–9. [Google Scholar] [CrossRef]
- Schreifels, J.J.; Fu, Y.; Wilson, E.J. Sulfur dioxide control in China, policy evolution during the 10th and 11th Five-year Plans and lessons for the future. Energy Policy 2012, 48, 779–789. [Google Scholar] [CrossRef]
- Wang, F.; Du, Y.; Liu, Y.; Wang, X. The present development of flue gas denitrification technologies in domestic coal-fired power plants. Electr. Power Environ. Prot. 2007, 23, 20–23. [Google Scholar]
- Song, X.; Yan, M. Influence fctor analysis of the flue gas denitration of SCR design in the coal-fired power plant. Saf. Environ. Eng. 2013, 20, 68–71. [Google Scholar]
- Bao, J.; Yang, L.; Song, S.; Xiong, G. Separation of fine particles from gassed in wet flue gas desulfurization system using a cascade of double tower. Energy Fuel 2012, 26, 2090–2097. [Google Scholar] [CrossRef]
- Ma, Y. The Selection of Flue Gas DeNOx Technology in the Power Plant of China. Master’s Thesis, North China Electric Power University, Beijing, China, December 2005. [Google Scholar]
- Zhang, M.; Chen, J. The present situation and the development of the nitrogen oxides control in China’s coal-fired power plant. Sichuan Chem. Eng. 2009, 5, 44–52. (In Chinese) [Google Scholar]
- Carpenter, A.M. Advances in Multi-Pollutant Control; IEA: London, UK, 2013. [Google Scholar]
- Yan, J.Y.; Zheng, Z.; Yu, G.F. Progress in study on multi-pollutant control technology for coal-fired flue gas. Therm. Power Gener. 2011, 40, 9–13. [Google Scholar]
- Tavoulareas, E.S.; Jozewicz, W. Multi-Pollutant Emission Control Technology Options for Coal-Fired Power Plants; EPA Report EPA-600/R-05/034; EPA: McLean, VA, USA, 2005. [Google Scholar]
- Ghorishi, S.B.; Keeney, R.M.; Serre, S.D.; Gullett, B.K.; Jozewicz, W.S. Development of a Cl-impregnated activated carbon for entrained-flow capture of element mercury. Environ. Sci. Technol. 2002, 36, 4454–4459. [Google Scholar] [CrossRef] [PubMed]
- Callen, M.S.; Cruz, M.T.; Marinov, S.; Murillo, R.; Stefanova, M.; Mastral, A.M. Flue gas cleaning in power stations by using electron beam technology influence on PAH emission. Fuel Process. Technol. 2007, 88, 251–258. [Google Scholar] [CrossRef]
- Zhao, Y. Experiments and reaction characteristics of liquid phased simultaneous removal of SO2 and NO. Sci. China 2009, 52, 1768–1775. [Google Scholar] [CrossRef]
- Jin, D.S.; Deshwal, B.R.; Park, Y.S.; Lee, H.K. Simultaneous removal of SO2 and NO by wet scrubbing using aqueous chlorine dioxide solution. J. Hazard. Mater. 2006, 135, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L. Aqueous absorption of nitric oxide induced by sodium chlorite oxidation in the presence of sulphur dioxide. Environ. Prog. 1998, 17, 80–85. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Sheng, C. Kinetic model of NO removal from SO2-containing simulated flue gas by wet UV/H2O2 advanced oxidation process. J. Chem. Eng. 2011, 168, 183–189. [Google Scholar] [CrossRef]
- Sun, W.Y.; Ding, S.L.; Zeng, S.S.; Su, S.; Jiang, W. Simultaneous absorption of NOx and SO2 from flue gas with pyrolusite slurry combined with gas-phase oxidation of NO using ozone. J. Hazard. Mater. 2011, 192, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.S.; Zhou, J.H.; Wang, Z.H.; Cen, K.F. Kinetic modelling of homogeneous low temperature multi-pollutant oxidation by ozone, the importance of SO and HCl in predicting oxidation. J. Zhejiang Uni. Sci. A 2006, 7, 335–339. [Google Scholar] [CrossRef]
- Wang, Z.H.; Zhou, J.; Zhu, Y.; Wen, Z.; Liu, J.; Cen, K. Simultaneous removal of NOx, SO2 and Hg in nitrogen flow in a narrow reactor by ozone injection, experimental results. Fuel Process. Technol. 2007, 88, 817–823. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, N.; Zhuang, Z.K.; Wang, H.; Liu, Y.; Weng, X.; Wu, Z. Mechanisms and reaction pathways for simultaneous oxidation of NOx and SO2 by ozone determined by in situ IR measurements. J. Hazard. Mater. 2014, 274, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Skalska, K.; Miller, J.S.; Ledakowicz, S. Intensification of NOx absorption process by means of ozone injection into exhaust gas stream. Chem. Eng. Process. Process Intensif. 2012, 61, 69–74. [Google Scholar] [CrossRef]
- Mok, Y.S. Absorption-reduction technique assisted by ozone injection and sodium sulfide for NOx removal from exhaust gas. Chem. Eng. J. 2006, 118, 63–67. [Google Scholar] [CrossRef]
- Sun, W.; Wang, Q.Y.; Ding, S.L.; Su, S. Simultaneous absorption of SO2 and NOx with pyrolusite slurry combined with gas-phase oxidation of NO using ozone, Effect of molar ratio of O2/(SO2 + 0.5 NOx) in flue gas. Chem. Eng. J. 2013, 228, 700–707. [Google Scholar] [CrossRef]
- Stamate, E.; Chen, W.; Jorgensen, L.; Jensen, T.K.; Fateev, A.; Michelsen, P.K. IR and UV gas absorption measurements during NOx reduction on an industrial nature gas fired power plant. Fuel 2010, 89, 978–985. [Google Scholar] [CrossRef]
- Mok, Y.S.; Lee, H.J. Removal of sulfur dioxide and nitrogen oxides by using ozone injection and absorption-reduction technique. Fuel Process. Technol. 2006, 87, 591–597. [Google Scholar] [CrossRef]
- Wang, Z.H. Mechanism Study on Multi-Pollution Control Simultaneously during Coal Combustion and Direct Numerical Simulation of Reaction Jet Flow. Ph.D. Thesis, Zhejiang University, Hangzhou, China, 2005. [Google Scholar]
- Wang, J. The Experimental Study of Oxidative-Desulfurization in the O3 Multi-Pollutant Control Technology. Master’s Thesis, Zhejiang University, Hangzhou, China, 2008. [Google Scholar]
- Wen, Z.C. Mechanism Investigation on the Oxidation and Degradation of Multiple Pollutants in Flue Gas by Ozone. Ph.D. Thesis, Zhejiang University, Hangzhou, China, 2009. [Google Scholar]
- Leeds NOx & SO2 Mechanism. Available online: http://garfield.chem.elte.hu/combustion/nox.htm (accessed on 17 December 2014).
- Kee, R.J.; Rupley, F.M.; Miller, J.A. Chemkin II: A Fortran Chemical Kinetics Package for the analysis of Gas-Phase Chemical Kinetics; Sandia Report, SAND 89-8009; Sandia National Laboratories: Livermore, CA, USA, 1989. [Google Scholar]
- Mallard, W.G.; Westley, F.; Herron, J.T.; Hampson, R.F.; Frizzell, D.H. NIST Chemical Kinetics Database; NIST: Gaithersburg, MD, USA, 1998. [Google Scholar]
- Skalska, K.; Miller, J.S.; Wilk, M.; Ledakowicz, S. Nitrogen oxidizes ozonation as a method for NOx emission abatement. Ozone Sci. Eng. 2012, 34, 252–258. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Zhao, J.; Lu, J. Numerical Study of the Simultaneous Oxidation of NO and SO2 by Ozone. Int. J. Environ. Res. Public Health 2015, 12, 1595-1611. https://doi.org/10.3390/ijerph120201595
Li B, Zhao J, Lu J. Numerical Study of the Simultaneous Oxidation of NO and SO2 by Ozone. International Journal of Environmental Research and Public Health. 2015; 12(2):1595-1611. https://doi.org/10.3390/ijerph120201595
Chicago/Turabian StyleLi, Bo, Jinyang Zhao, and Junfu Lu. 2015. "Numerical Study of the Simultaneous Oxidation of NO and SO2 by Ozone" International Journal of Environmental Research and Public Health 12, no. 2: 1595-1611. https://doi.org/10.3390/ijerph120201595
APA StyleLi, B., Zhao, J., & Lu, J. (2015). Numerical Study of the Simultaneous Oxidation of NO and SO2 by Ozone. International Journal of Environmental Research and Public Health, 12(2), 1595-1611. https://doi.org/10.3390/ijerph120201595