Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands



Abstract
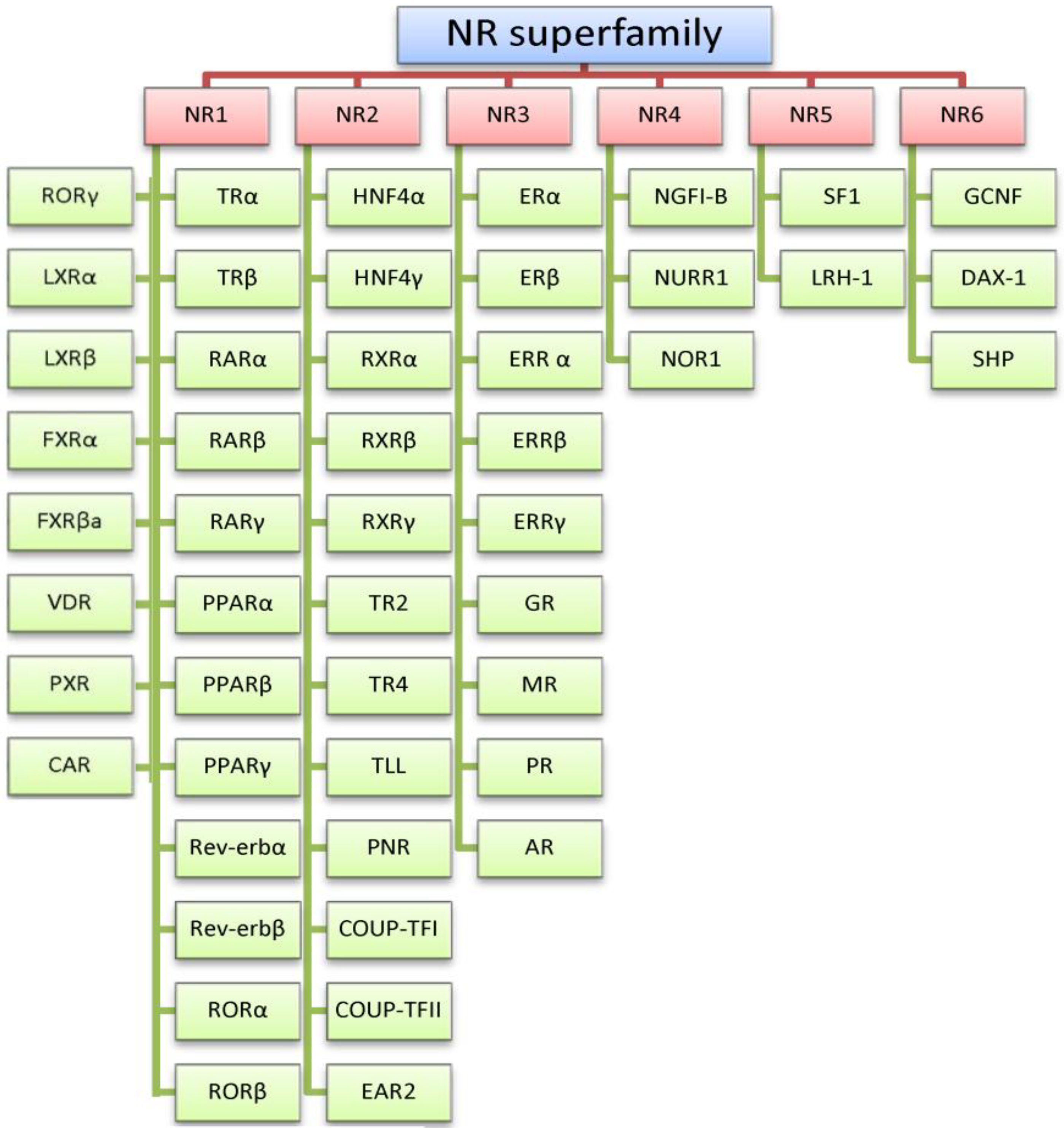
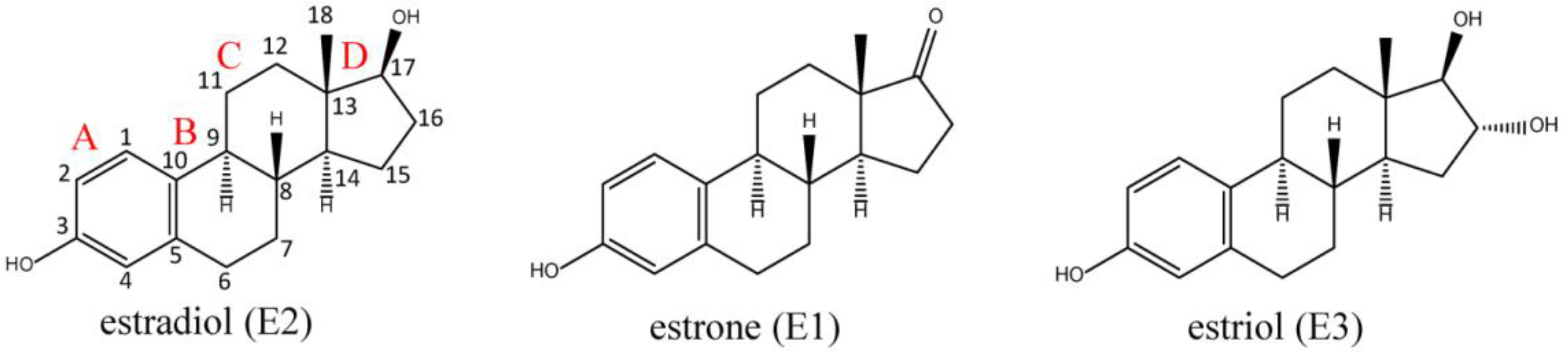
:1. The Estrogen Receptors
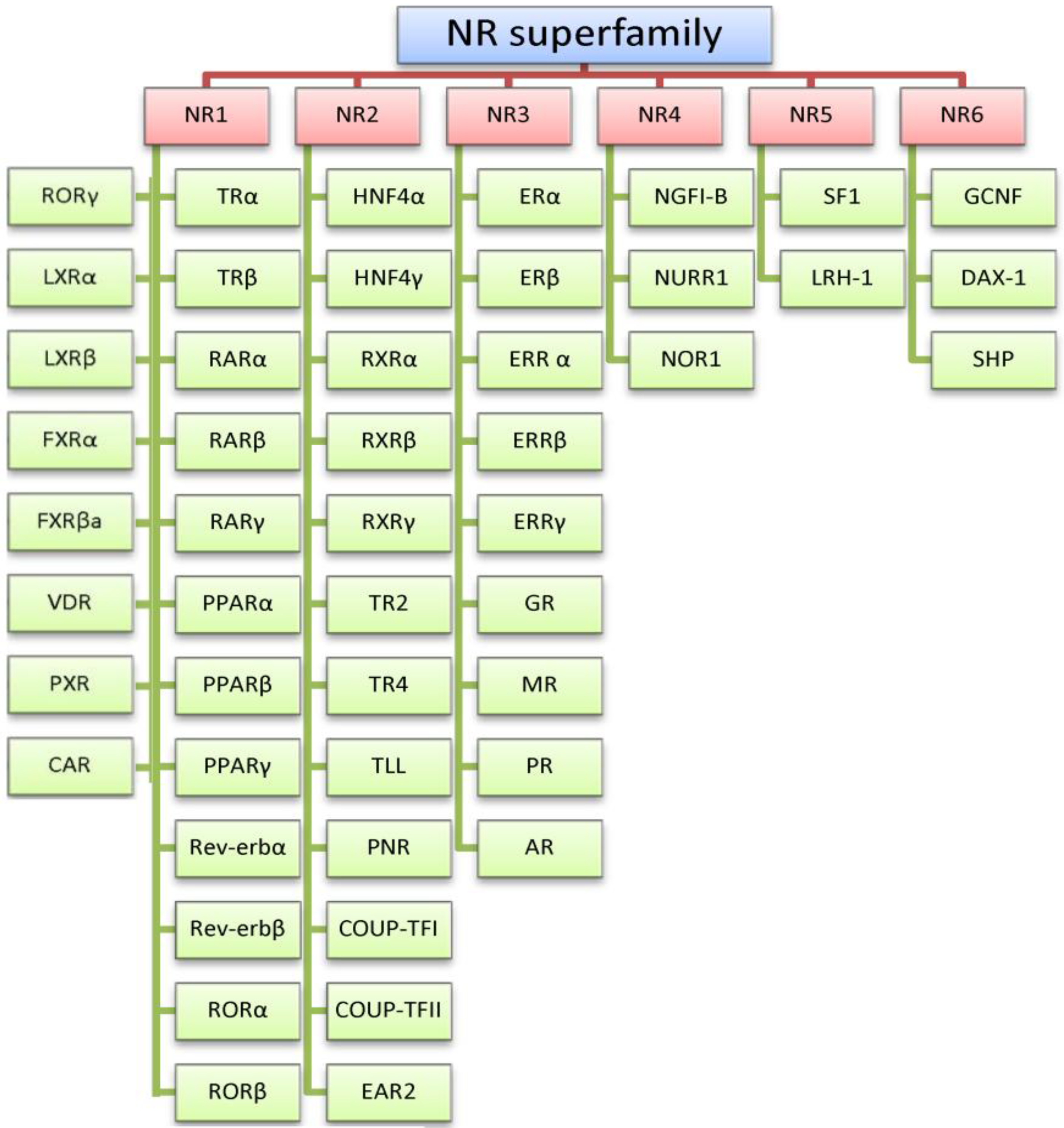
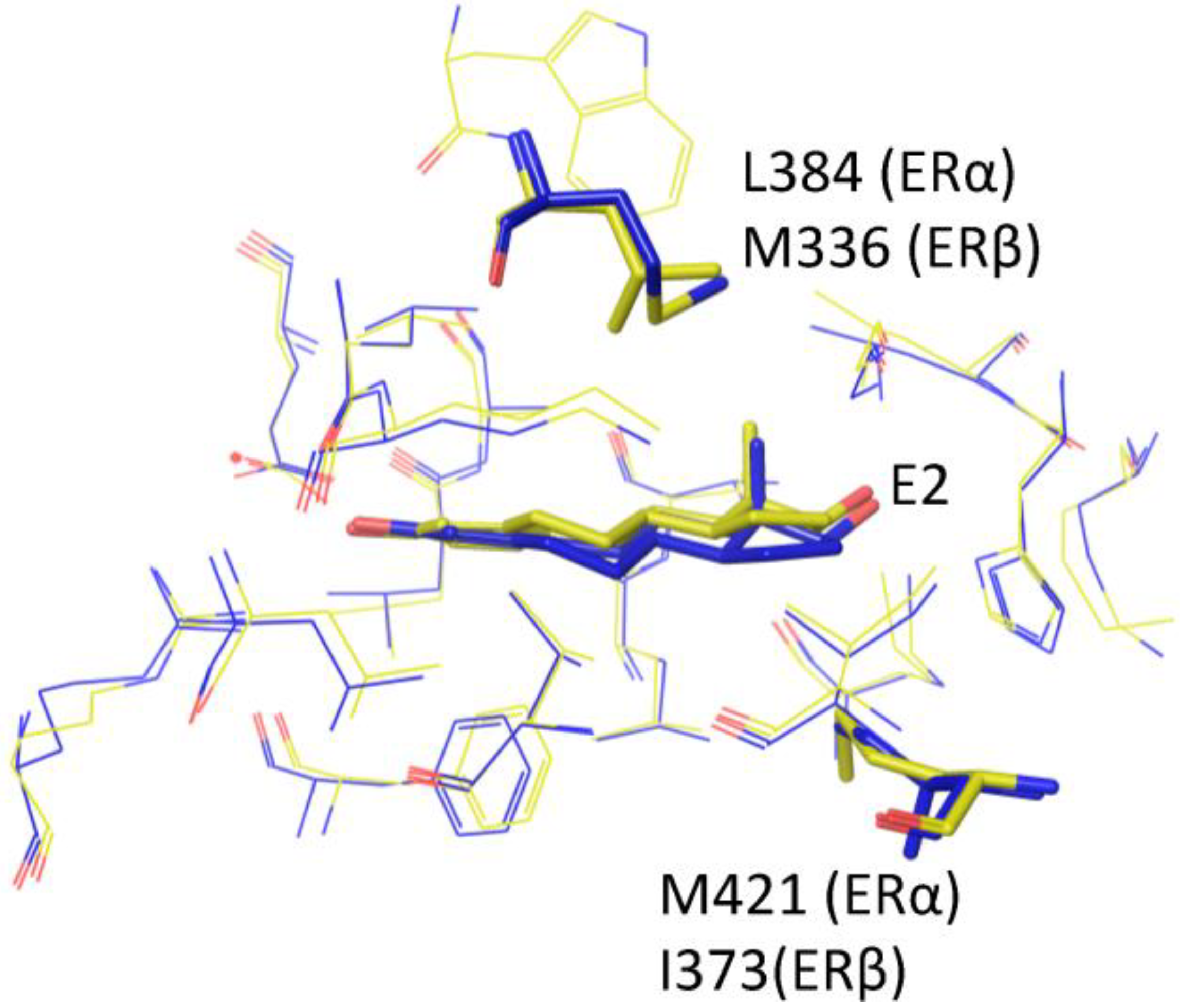
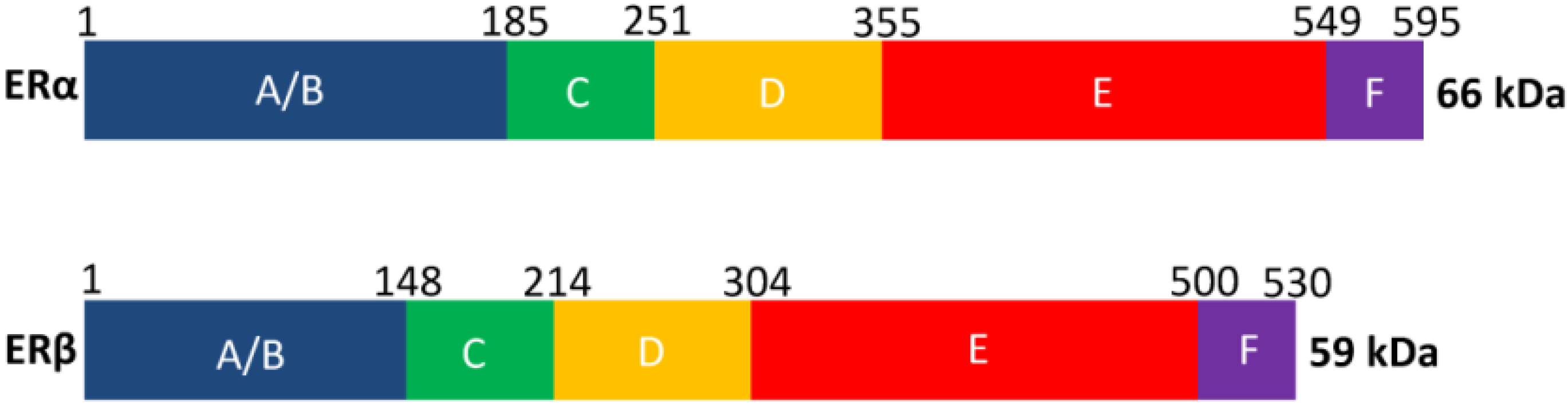
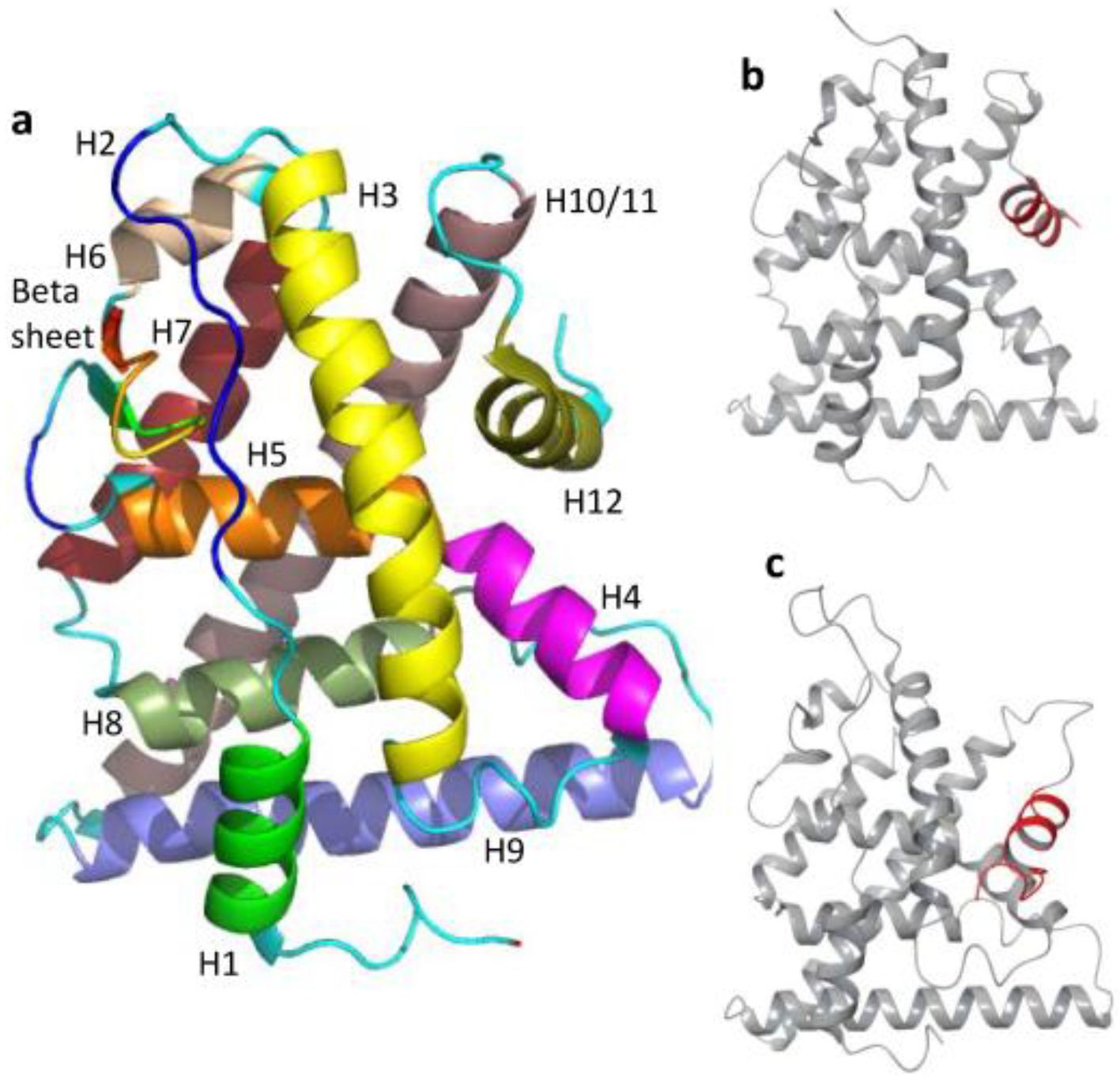
2. Estrogen Receptor Architecture
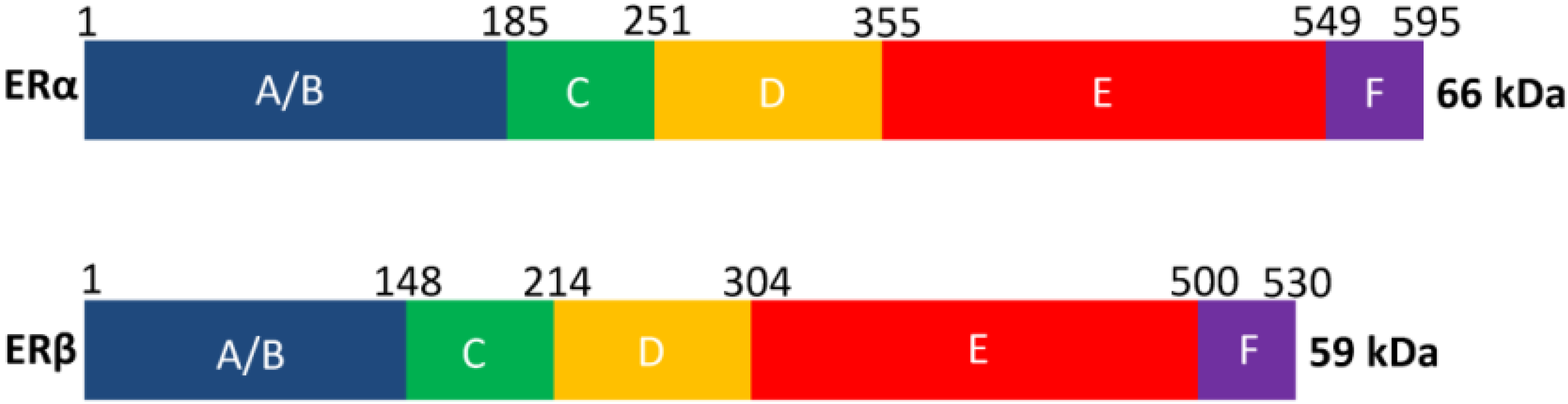

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Structure-Type | Ligand | Res.(Å) | Ref |
|---|---|---|---|---|
| 1A52 | Dimer | Estradiol | 2.8 | [29] |
| 1ERE | Hexamer | Estradiol | 3.1 | [30] |
| 1ERR | Dimer | Raloxifene | 2.6 | [30] |
| 3ERD | Dimer | Diethylstilbestrol | 2.03 | [31] |
| 3ERT | Monomer | 4-Hydroxytamoxifen | 1.9 | [31] |
| 1QKT | Monomer | Estradiol | 2.2 | [32] |
| 1QKU | Trimer | Estradiol | 3.2 | [32] |
| 1G50 | Trimer | Estradiol | 2.9 | [33] |
| 1GWQ | Dimer | Raloxifene core | 2.45 | [34] |
| 1GWR | Dimer | Estradiol | 2.4 | [34] |
| 1L2I | Dimer | (R,R)-5,11-cis-Diethyl-5,6,11,12-tetrahydrochrysene- 2,8-diol | 1.95 | [35] |
| 1PCG | Dimer | Estradiol | 2.7 | [36] |
| 1UOM | Monomer | 2-Phenyl-1-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-1,2,3, 4-tetrahydroisoquinolin-6-ol | 2.28 | [37] |
| 1R5K | Trimer | (2E)-3-{4-[(1E)-1,2-Diphenylbut-1-enyl]phenyl}acrylic acid | 2.7 | [38] |
| 1SJ0 | Monomer | (2S,3R)-2-(4-(2-(Piperidin-1-yl)ethoxy)phenyl)-2,3-dihydro-3-(4-hydroxyphenyl)benzo[b][1,4]oxathiin-6-ol | 1.9 | [39] |
| 1XP1 | Monomer | (2S,3R)-2-(4-{2-[(3R,4R)-3,4-Dimethylpyrrolidin-1-yl]ethoxy}phenyl)-3-(4-hydroxyphenyl)-2,3-dihydro-1,4-benzoxathiin-6-ol | 1.8 | [40] |
| 1XP6 | Monomer | (2S,3R)-2-(4-{2-[(3S,4S)-3,4-Dimethylpyrrolidin-1-yl]ethoxy}phenyl)-3-(4-hydroxyphenyl)-2,3-dihydro-1,4-benzoxathiin-6-ol | 1.7 | [40] |
| 1XP9 | Monomer | (2S,3R)-3-(4-Hydroxyphenyl)-2-(4-{[(2S)-2-pyrrolidin-1-ylpropyl]oxy}phenyl)-2,3-dihydro-1,4-benzoxathiin-6-ol | 1.8 | [40] |
| 1XPC | Monomer | (2S,3R)-3-(4-Hydroxyphenyl)-2-(4-{[(2R)-2-pyrrolidin-1-ylpropyl]oxy}phenyl)-2,3-dihydro-1,4-benzoxathiin-6-ol | 1.6 | [40] |
| 1X7E | Dimer | [5-Hydroxy-2-(4-hydroxyphenyl)-1-benzofuran-7-yl]acetonitrile | 2.8 | [41] |
| 1X7R | Monomer | Genistein | 2 | [42] |
| 1XQC | Tetramer | (1S)-1-{4-[(9aR)-Octahydro-2H-pyrido[1,2-a]pyrazin-2-yl]phenyl}-2-phenyl-1,2,3,4-tetrahydroisoquinolin-6-ol | 2.05 | [43] |
| 1YIM | Monomer | (2R,3R,4S)-3-(4-Hydroxyphenyl)-4-methyl-2-[4-(2-pyrrolidin-1-ylethoxy)phenyl]chroman-6-ol | 1.9 | [44] |
| 1YIN | Monomer | (2R,3R,4S)-5-Fluoro-3-(4-hydroxyphenyl)-4-methyl-2-[4-(2-piperidin-1-ylethoxy)phenyl]chroman-6-ol | 2.2 | [44] |
| 2AYR | Monomer | 6-(4-Methylsulfonyl-phenyl)-5-[4-(2-piperidin-1-ylethoxy)phenoxy]naphthalen-2-ol | 1.9 | [45] |
| 2B23 | Dimer | 2.1 | [46] | |
| 2BJ4 | Dimer | 4-Hydroxytamoxifen | 2 | [47] |
| 1ZKY | Dimer | 4-[(1S,2S,5S)-5-(Hydroxymethyl)-6,8,9-trimethyl-3-oxabicyclo[3.3.1]non-7-en-2-yl]phenol | 2.25 | [48] |
| 2B1V | Dimer | 4-[(1S,2S,5S)-5-(Hydroxymethyl)-8-methyl-3-oxabicyclo[3.3.1]non-7-en-2-yl]phenol | 1.8 | [48] |
| 2B1Z | Dimer | 17-Methyl-17-α-dihydroequilenin | 1.78 | [49] |
| 2FAI | Dimer | 4-[(1S,2S,5S,9R)-5-(Hydroxymethyl)-8,9-dimethyl-3-oxabicyclo[3.3.1]non-7-en-2-yl]phenol | 2.1 | [48] |
| 2I0J | Tetramer | (3aS,4R,9bR)-4-(4-Hydroxyphenyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-8-ol | 2.9 | [50] |
| 2G44 | Dimer | 4-[(1S,2R,5S)-4,4,8-Trimethyl-3-oxabicyclo[3.3.1]non-7-en-2-yl]phenol | 2.65 | - |
| 2G5O | Dimer | (9α,13β,17β)-2-[(1Z)-But-1-en-1-yl]estra-1,3,5(10)-triene-3,17-diol | 2.3 | - |
| 2IOG | Monomer | N-[(1R)-3-(4-Hydroxyphenyl)-1-methylpropyl]-2-[2-phenyl-6-(2-piperidin-1-ylethoxy)-1h-indol-3-yl]acetamide | 1.6 | [51] |
| 2IOK | Dimer | N-[(1R)-3-(4-Hydroxyphenyl)-1-methylpropyl]-2-(2-phenyl-1H-indol-3-yl)acetamide | 2.4 | [51] |
| 2JF9 | Trimer | 4-Hydroxytamoxifen | 2.1 | [52] |
| 2JFA | Dimer | Raloxifene | 2.55 | [52] |
| 2OCF | Monomer | Estradiol | 2.95 | [53] |
| 2OUZ | Monomer | (5R,6S)-6-Phenyl-5-[4-(2-pyrrolidin-1-ylethoxy)phenyl]-5,6,7,8-tetrahydronaphthalen-2-ol | 2 | [54] |
| 2P15 | Dimer | (17β)-17-{(E)-2-[2-(Trifluoromethyl)phenyl]vinyl}estra-1(10),2,4-triene-3,17-diol | 1.94 | [55] |
| 2POG | Dimer | (3aS,4R,9bR)-4-(4-Hydroxyphenyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-9-ol | 1.84 | [56] |
| 2Q6J | Dimer | 4-[(Dimesitylboryl)(2,2,2-trifluoroethyl)amino]phenol | 2.7 | [57] |
| 2Q70 | Dimer | (3aS,4R,9bR)-2,2-Difluoro-4-(4-hydroxyphenyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-8-ol | 1.95 | [58] |
| 2QE4 | Dimer | (3aS,4R,9bR)-4-(4-Hydroxyphenyl)-6-(methoxymethyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-8-ol | 2.4 | [50] |
| 2QA6 | Dimer | 4-(6-Hydroxy-1H-indazol-3-yl)benzene-1,3-diol | 2.6 | [46] |
| 2QA8 | Dimer | Genistein | 1.85 | [46] |
| 2QAB | Dimer | 3-Ethyl-2-(4-hydroxyphenyl)-2H-indazol-5- ol | 1.89 | [46] |
| 2QGT | Dimer | (9β,11α,13α,14β,17α)-11-(methoxymethyl)estra-1(10),2,4-triene-3,17-diol | 2.15 | [46] |
| 2QGW | Dimer | 3-Chloro-2-(4-hydroxyphenyl)-2H-indazol-5-ol | 2.39 | [46] |
| 2QH6 | Dimer | Diethyl (1R,2S,3R,4S)-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate | 2.7 | [46] |
| 2QR9 | Dimer | Dimethyl (1R,4S)-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hepta-2,5-diene-2,3-dicarboxylate | 2 | [46] |
| 2QSE | Dimer | 4-(2-Amino-1-methyl-1H-imidazo[4,5-b]pyridin-6-yl)phenol | 1.85 | [46] |
| 2QXM | Dimer | 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyrid | 2.3 | [46] |
| 2QXS | Dimer | Raloxifene | 1.7 | [59] |
| 2QZO | Dimer | 4-[1-Allyl-7-(trifluoromethyl)-1h-indazol- 3-yl]bezene-1,3-diol | 1.72 | [59] |
| 2R6W | Dimer | [6-Hydroxy-2-(4-hydroxyphenyl)-1-benzothien-3-yl]{4-[2-(4-methylpiperidin-1-yl)ethoxy]phenyl}methanone | 2 | [60] |
| 2R6Y | Dimer | [6-Hydroxy-2-(4-hydroxyphenyl)-1-benzothien-3-yl][4-(2-pyrrolidin-1-ylethoxy)phenyl]methanone | 2 | [60] |
| 3DT3 | Dimer | 5-(4-Hydroxyphenoxy)-6-(3-hydroxyphenyl)- 7-methylnapthalen-2-ol | 2.4 | [61] |
| 3HLV | Dimer | (9β,13α,16β)-3,16-Dihydroxyestra- 1,3,5(10)-trien-17-one | 3 | - |
| 3HM1 | Dimer | (9β,13α)-3-Hydroxyestra-1,3,5(10)-trien-17-one | 2.33 | - |
| 3L03 | Dimer | (14β,15α,16α,17α)-Estra-1,3,5(10)-triene-3,15,16,17-tetrol | 1.9 | - |
| 3OS8 | Tetramer | 4-[1-Benzyl-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1, 3-diol | 2.03 | [59] |
| 3OS9 | Tetramer | 4-[1-Allyl-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1,3-diol | 2.3 | [59] |
| 3OSA | Tetramer | 4-[1-(3-Methylbut-2-en-1-yl)-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1,3-diol | 2.3 | [59] |
| 2YAT | Monomer | Estradiol-pyridinium tetraacetic acid | 2.6 | [62] |
| 2YJA | Monomer | Estradiol | 1.82 | [63] |
| 3Q95 | Dimer | Estriol | 2.05 | - |
| 3Q97 | Dimer | 4,4’-[(1Z)-1-(4-Ethoxyphenyl)but-1-ene-1,2-diyl]diphenol; 4,4’-[2-(4-Ethoxyphenyl)but-1-ene-1,1-diyl]diphenol | 2.1 | - |
| 3UU7 | Dimer | 4,4’-Propane-2,2-diyldiphenol | 2.2 | [64] |
| 3UUA | Dimer | 4,4’-(1,1,1,3,3,3-Hexafluoropropane-2,2-diyl)diphenol | 2.05 | [64] |
| 3UUC | Tetramer | 4,4’-(2,2-Dichloroethene-1,1-diyl)diphenol | 2.1 | [64] |
| 3UUD | Dimer | Estradiol | 1.6 | [64] |
| 4DMA | Dimer | 2’-Bromo-6’-(furan-3-yl)-4’-(hydroxymethyl)biphenyl-4-ol | 2.3 | [65] |
| 4IU7 | Dimer | 4-[2-Ethyl-7-(trifluoromethyl)-2H-indazol-3-yl]benzene-1, 3-diol | 2.29 | [66] |
| 4IUI | Dimer | 4-[1-Butyl-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1, 3-diol | 2.3 | [66] |
| 4IV2 | Dimer | 4-[1-(2-Methylpropyl)-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1,3-diol | 2.14 | [66] |
| 4IV4 | Dimer | 4-[2-(2-Methylpropyl)-7-(trifluoromethyl)- 2h-indazol-3-yl]benzene-1,3-diol | 2.3 | [66] |
| 4IVW | Dimer | 4-[2-Benzyl-7-(trifluoromethyl)-2H-indazol-3-yl]benzene-1,3-diol | 2.06 | [66] |
| 4IVY | Dimer | 4-[1-(But-3-en-1-yl)-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1,3-diol | 1.95 | [66] |
| 4IW6 | Dimer | 4-[2-(But-3-en-1-yl)-7-(trifluoromethyl)-2H-indazol-3-yl]benzene-1,3-diol | 1.98 | [66] |
| 4IW8 | Dimer | 4-[1-(3-Methylbut-2-en-1-yl)-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1,3-diol | 2.04 | [66] |
| 4IWC | Dimer | 4,4’-Thiene-2,5-diylbis(3-methylphenol) | 2.24 | [66] |
| 4IWF | Dimer | 2-Chloro-3’-fluoro-3-[(E)-(hydroxyimino)methyl]biphenyl- 4,4’-diol | 1.93 | [66] |
| PDB ID | Structure-Type | Ligand | Res. (Å) | Ref |
|---|---|---|---|---|
| 1QKM | Monomer | Genistein | 1.8 | [67] |
| 1QKN | Monomer | Raloxifene | 2.25 | [67] |
| 1HJ1 | Monomer | ICI164384 or N-Butyl-11-[(7r,8r,9s,13s,14s,17s)-3,17-dihydroxy-13-methyl-7,8,9,11,12,13,14,15,16,17- decahydro- 6 H-cyclopenta[a]phenanthren-7-yl]-n-methylundecanamide | 2.3 | [68] |
| 1L2J | Dimer | (R,R)-5,11-cis-Diethyl-5,6,11,12-tetrahydrochrysene-2,8-diol | 2.95 | [35] |
| 1NDE | Monomer | 4-(2-{[4-{[3-(4-Chlorophenyl)propyl]sulfanyl}-6-(1-piperazinyl)-1,3,5-triazin-2-yl]amino}ethyl)phenol | 3 | [69] |
| 1U3Q | Tetramer | 4-(6-Hydroxybenzo[d]isoxazol-3-yl)benzene-1,3-diol | 2.4 | [70] |
| 1U3R | Dimer | 2-(5-Hydroxynaphthalen-1-yl)-1,3-benzooxazol-6-ol | 2.21 | [70] |
| 1U3S | Dimer | 3-(6-Hydroxynaphthalen-2-yl)-benzo[d]isooxazol-6-ol | 2.5 | [70] |
| 1U9E | Dimer | 2-(4-Hydroxyphenyl)benzofuran-5-ol | 2.4 | [41] |
| 1X76 | Dimer | 5-Hydroxy-2-(4-hydroxyphenyl)-1-benzofuran-7-carbonitrile | 2.2 | [41] |
| 1X78 | Dimer | [5-Hydroxy-2-(4-hydroxyphenyl)-1-benzofuran-7-yl]acetonitrile | 2.3 | [41] |
| 1X7B | Dimer | 2-(3-Fluoro-4-hydroxyphenyl)-7-vinyl-1,3-benzoxazol-5-ol | 2.3 | [41] |
| 1X7J | Dimer | Genistein | 2.3 | [41] |
| 1YY4 | Dimer | 1-Chloro-6-(4-hydroxyphenyl)-2-naphthol | 2.7 | [71] |
| 1YYE | Dimer | 3-(3-Fluoro-4-hydroxyphenyl)-7-hydroxy-1-naphthonitrile | 2.03 | [71] |
| 1ZAF | Dimer | 3-Bromo-6-hydroxy-2-(4-hydroxyphenyl)-1H-inden-1-one | 2.2 | [72] |
| 2FSZ | Dimer | 4-Hydroxytamoxifen | 2.2 | [73] |
| 2GIU | Monomer | (9aS)-4-bromo-9a-butyl-7-hydroxy-1,2,9,9a-tetrahydro-3H-fluoren-3-one | 2.2 | [74] |
| 2I0G | Dimer | (3aS,4R,9bR)-4-(4-hydroxyphenyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-8-ol | 2.5 | [75] |
| 2J7X | Monomer | Estradiol | 2.1 | - |
| 2J7Y | Monomer | (16α,17α)-Estra-1,3,5(10)-triene- 3,16,17-triol | 1.8 | - |
| 2JJ3 | Dimer | (3aS,4R,9bR)-4-(4-Hydroxyphenyl)-6-(methoxymethyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-8-ol | 2.28 | [50] |
| 2NV7 | Dimer | 4-(4-Hydroxyphenyl)-1-naphthaldehyde oxime | 2.1 | [76] |
| 2QTU | Dimer | (3aS,4R,9bR)-2,2-Difluoro-4-(4-hydroxyphenyl)-6-(methoxymethyl)-1,2,3,3a,4,9b-hexahydrocyclopental[c]chromen-8-ol | 2.53 | [77] |
| 2Z4B | Dimer | (3aS,4R,9bR)-2,2-Difluoro-4-(4-hydroxyphenyl)-1,2,3,3a,4,9b-hexahydrocyclopenta[c]chromen-8-ol | 2.34 | [58] |
| 3OLL | Dimer | Estradiol | 1.5 | [78] |
| 2YJD | Dimer | 4-(2-Propan-2-yloxybenzimidazol-1-yl)phenol | 1.93 | [63] |
| 3OLS | Dimer | Estradiol | 2.2 | [78] |
| 3OMO | Dimer | 2-(Trifluoroacetyl)-1,2,3,4-tetrahydroisoquinolin-6-ol | 2.21 | [79] |
| 3OMP | Dimer | 2-(Trifluoroacetyl)-1,2,3,4-tetrahydroisoquinolin-7-ol | 2.05 | [79] |
| 3OMQ | Dimer | 2-[(Trifluoromethyl)sulfonyl]-1,2,3,4-tetrahydroisoquinolin-6-ol | 1.97 | [79] |
| 2YLY | Dimer | N-Cyclopropyl-4-oxidanyl-N-[(2R)-2-oxidanyl-2-phenylpropyl]benzenesulfonamide | 3.2 | [80] |
| 4J24 | Tetramer | Estradiol | 2.1 | [81] |
| 4J26 | Dimer | Estradiol | 2.3 | [81] |

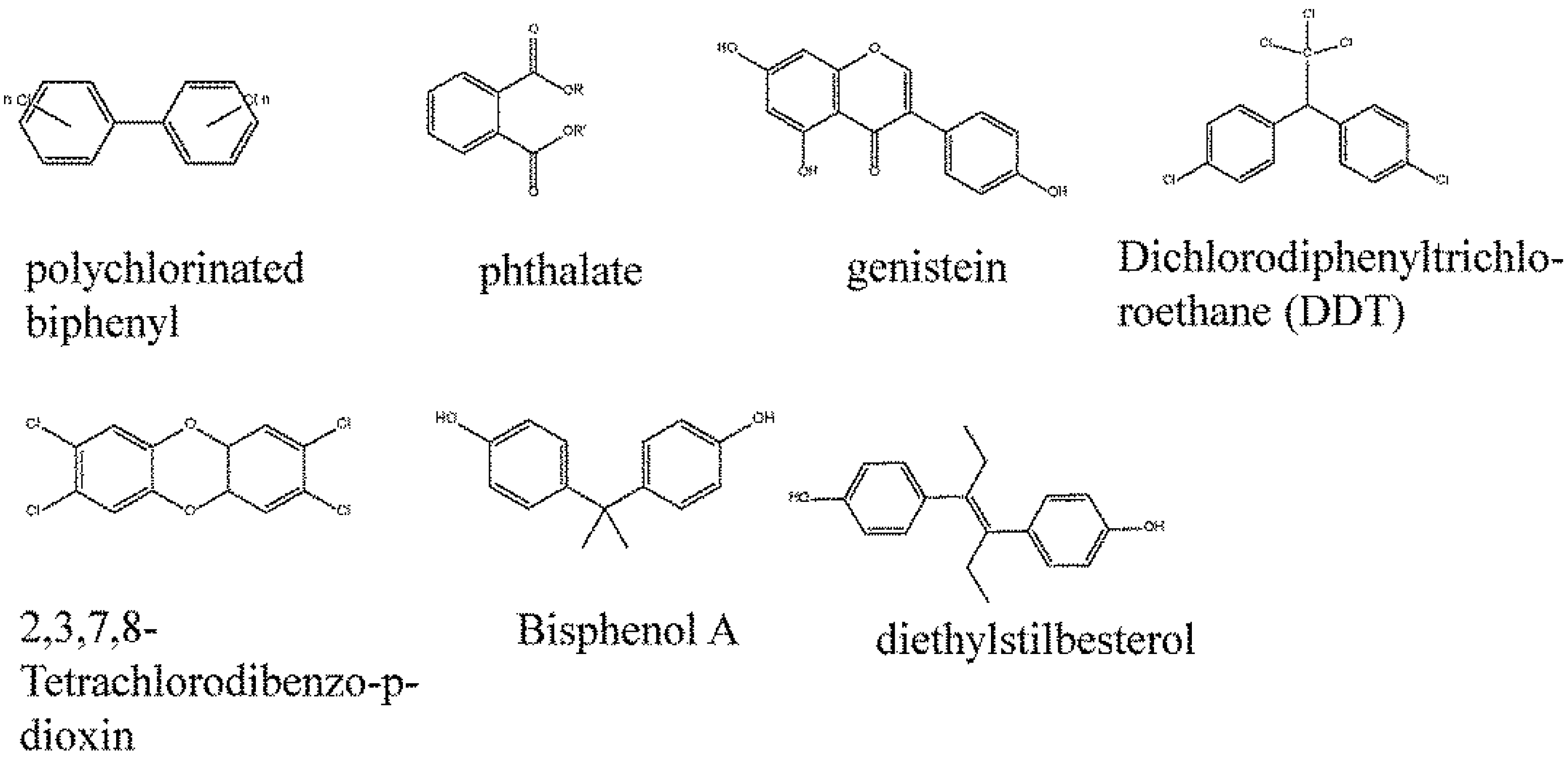
3. Promiscuity of Estrogen Receptors

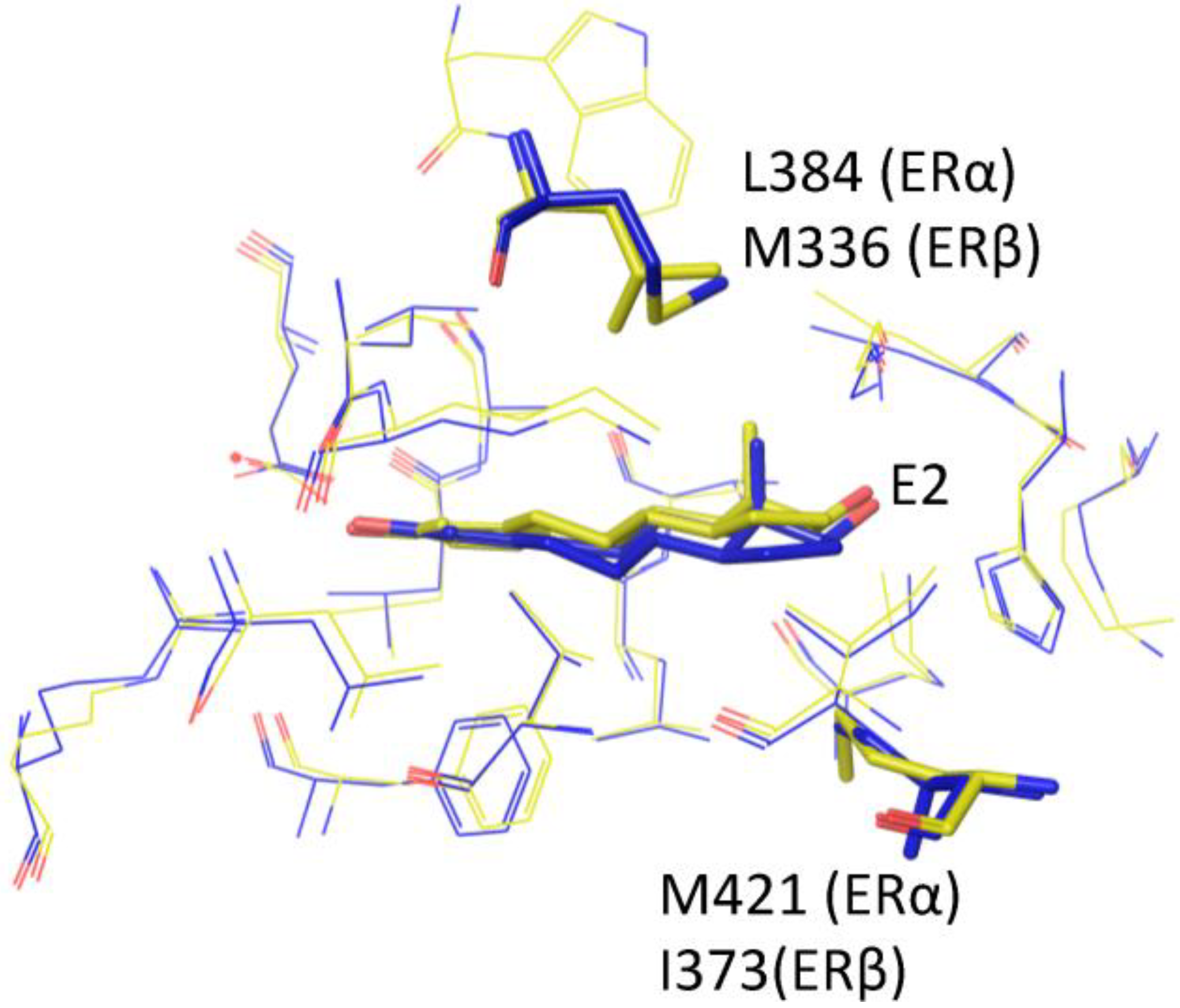
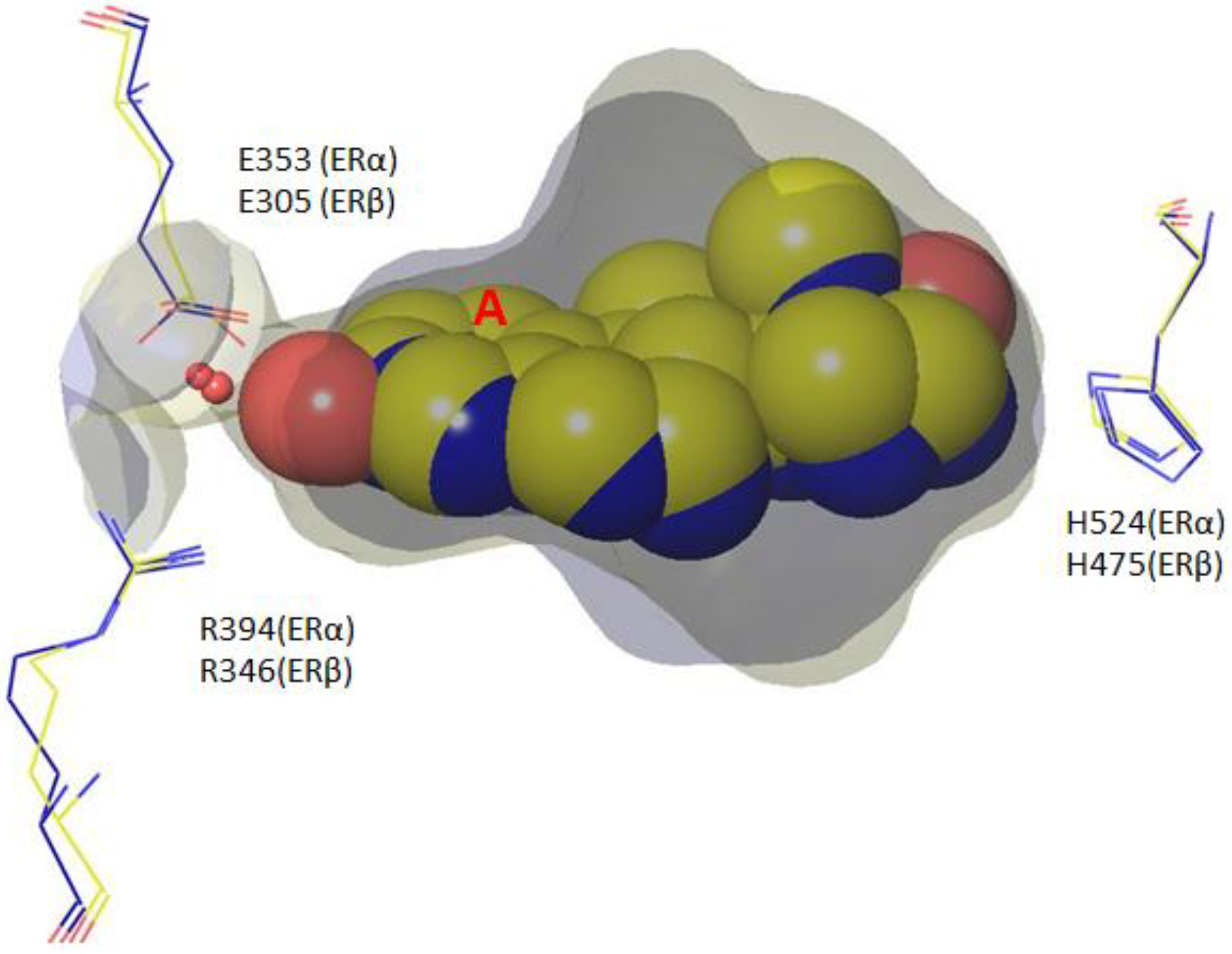
4. Root of ER Promiscuity
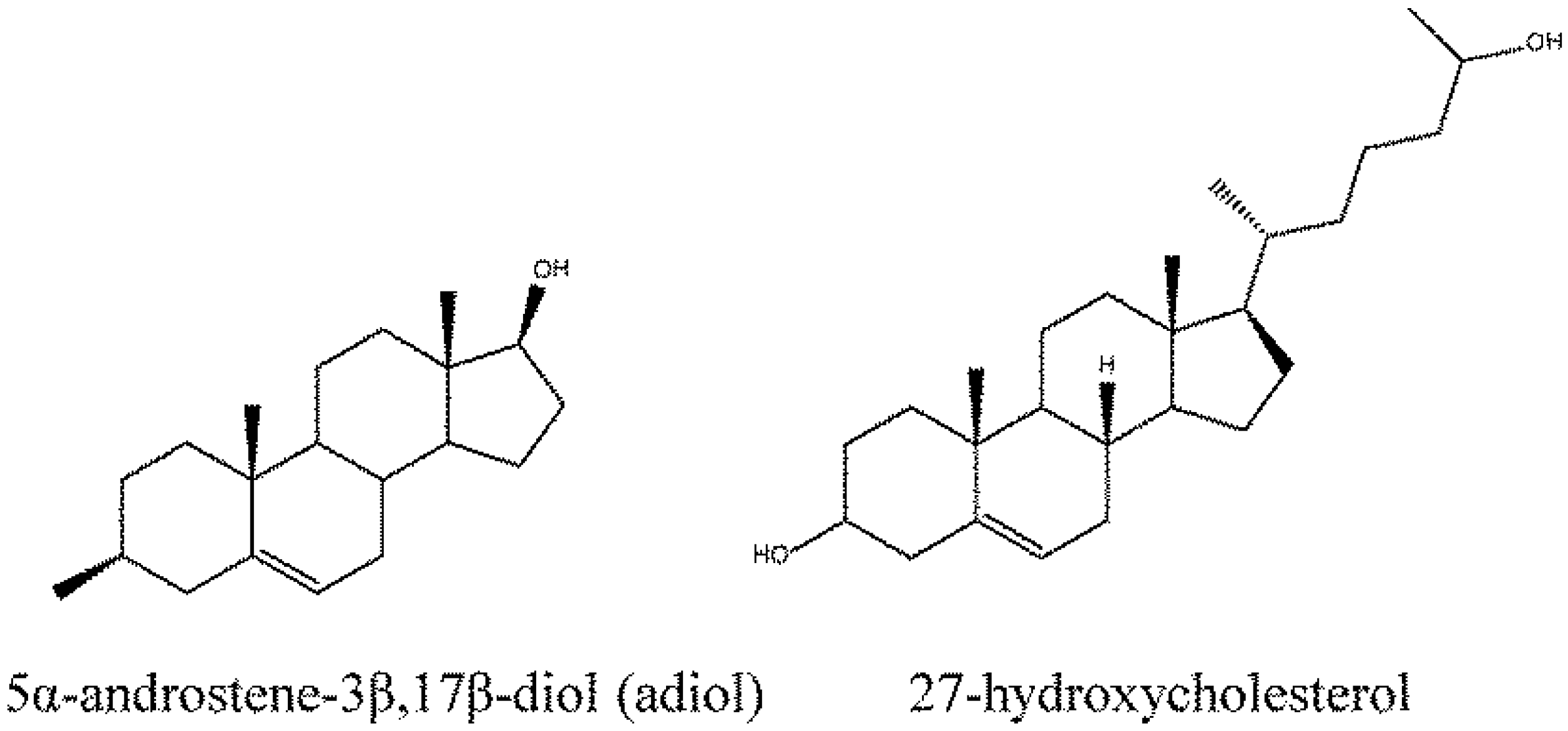
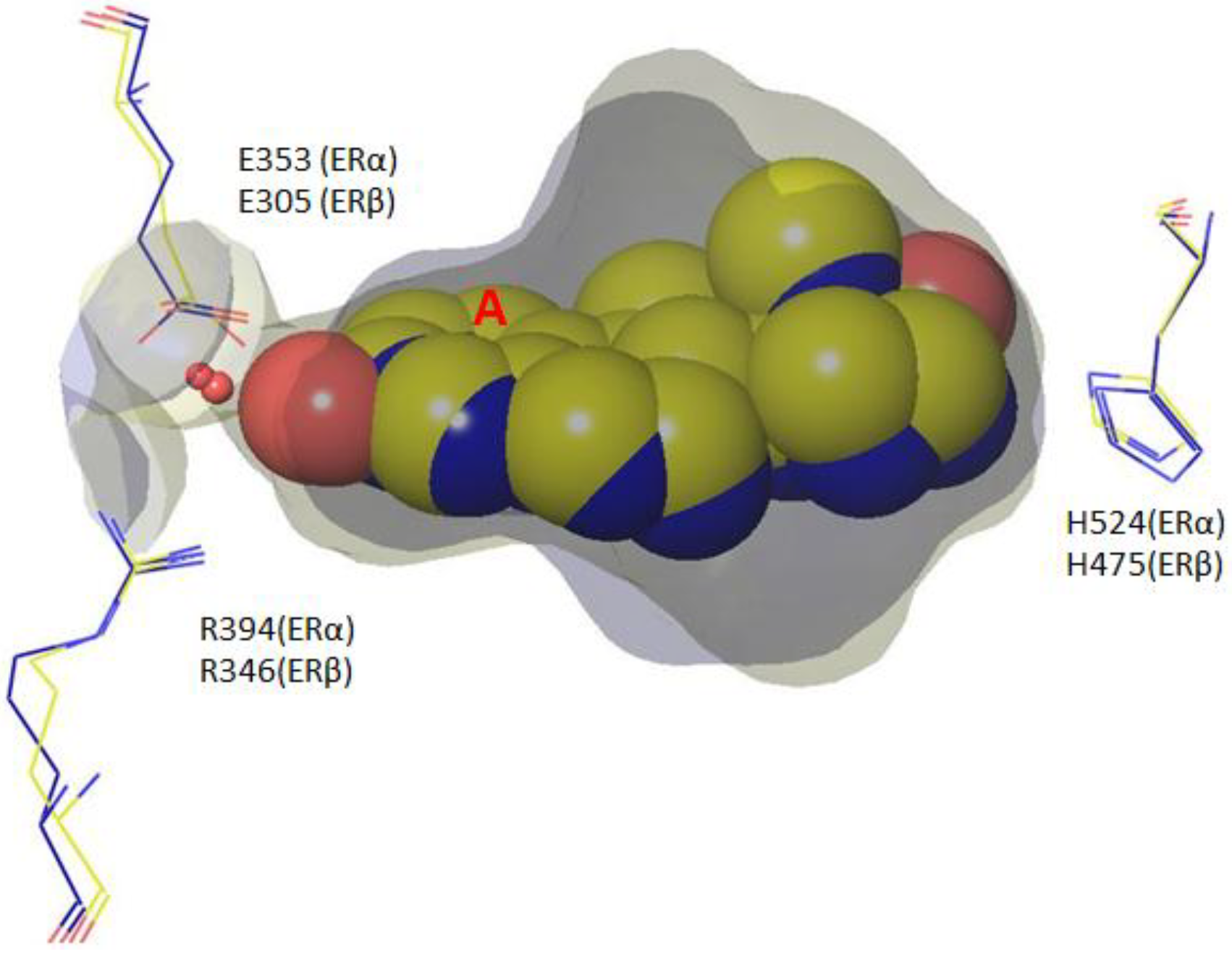
5. ER Specificity and Achieving Subtype Selectivity
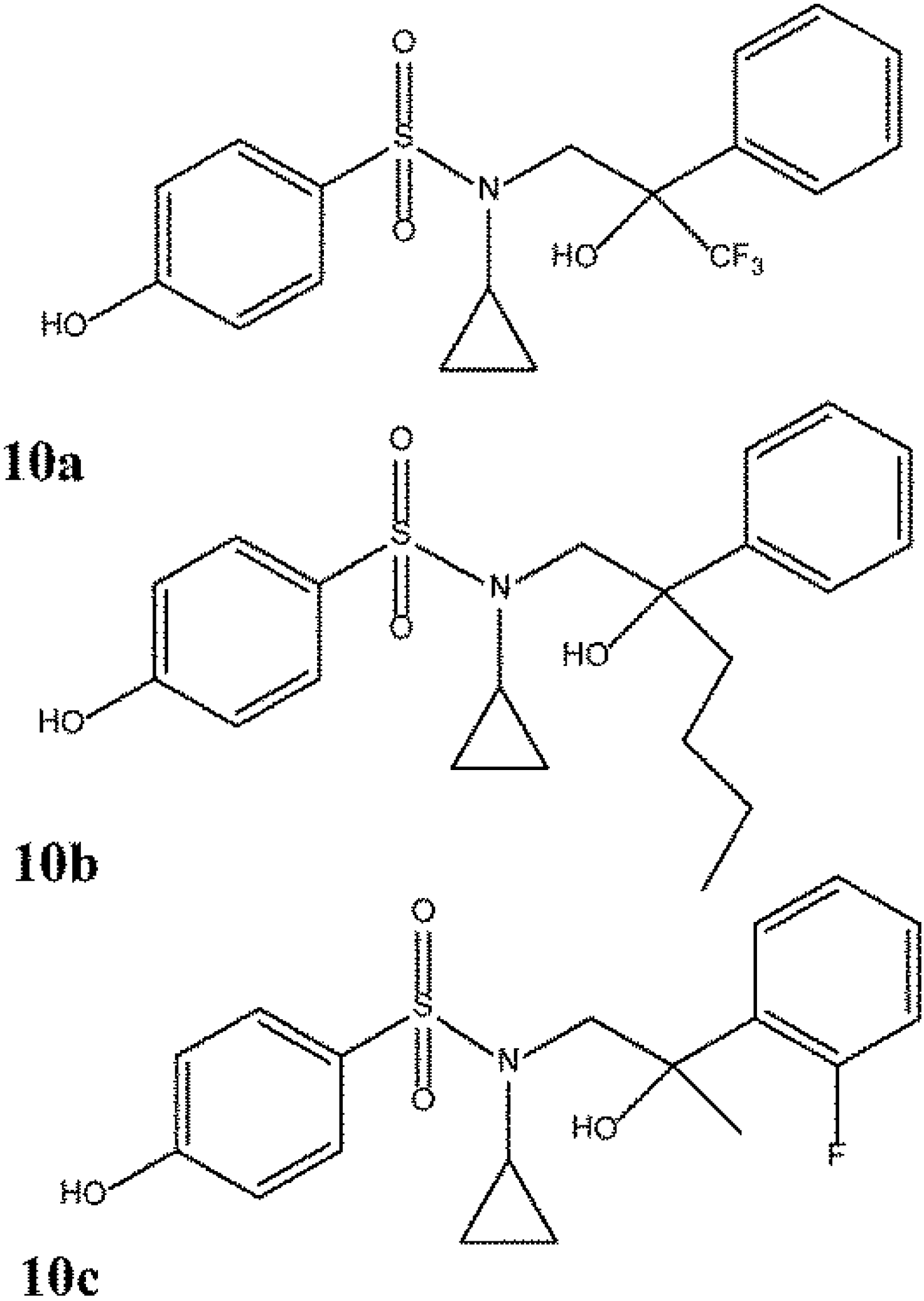
6. Updated Overview of Subtype Selective Compounds
6.1. Updated Overview of ERα Selective Compounds
| ID | Structure | Ref. | Fold Selectivity | Data * |
|---|---|---|---|---|
| 1 | 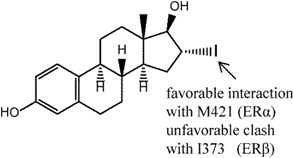 | [113] | 20–30 B | NA |
| 2 | 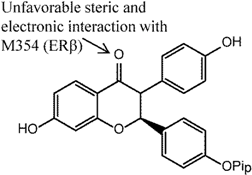 | [114] | 66 B | (31 nM/2049 nM) B |
| 3 | 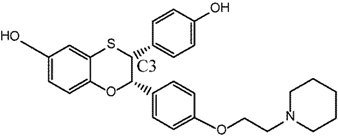 | [115] | 46 B 5.4 R | (3.1 ± 1.4 nM/143 ± 72 nM)B (9.6 nM/52 nM) R |
| 4 | 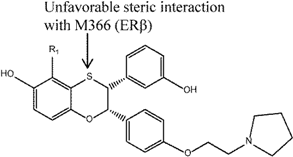 | [116] | 40 B 23.6 R | (0.9 nM/37 nM) B (1.7 nM/40.1 nM) R |
| 5 | 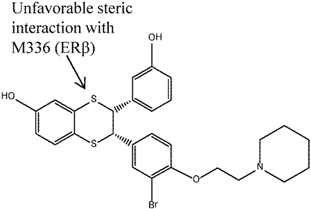 | [117] | 40 B | (4 nM/161 nM) B |
| 6 | 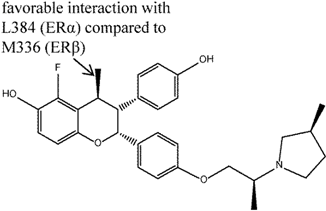 | [44] | 29 B | (0.9 nM/26 nM) B |
| 7 | 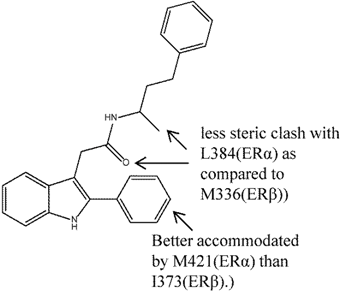 | [51] | 445 B | (11 nM/4900 nM) B |
| 8 | 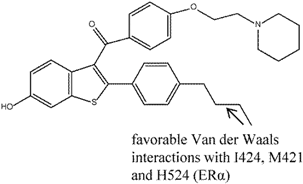 | [118] | 140 B | (0.25 ± 0.15 nM/35 ± 14.3 nM) B |
| 9 |  | [119] | 64 B | (RBA: 0.64/0.01) B |
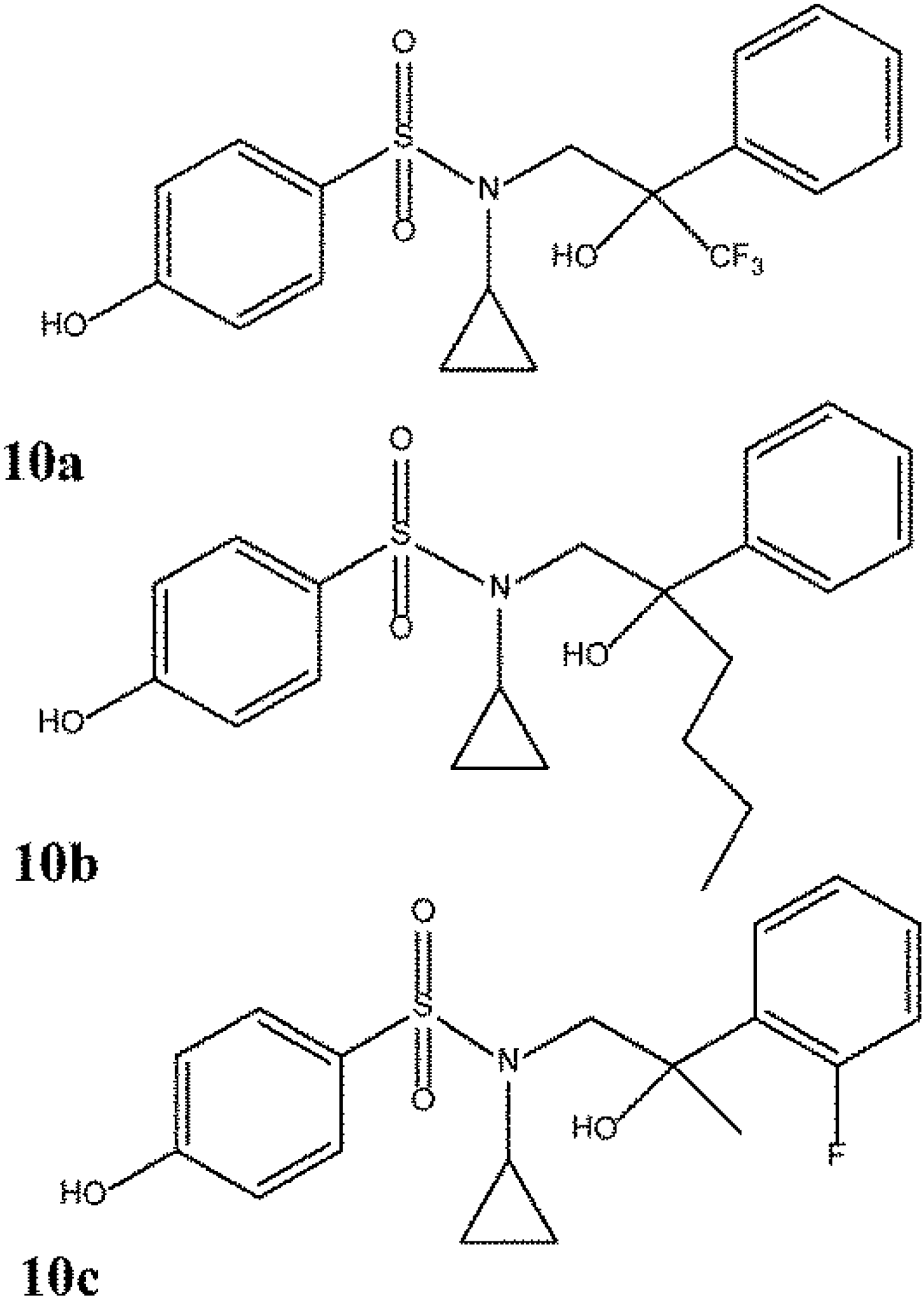
6.2. Updated Overview of ERβ Selective Compounds
| ID | Structure | Ref | Fold Selectivity | Data * |
|---|---|---|---|---|
| 10 | 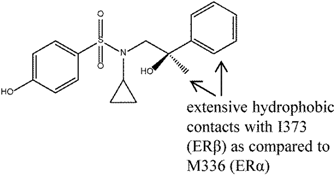 | [80] | 2.2 B 68.4 R | (RBA: 107/48) B (>5400 nM/79 nM) R |
| 11 |  | [125] | 0.54 B | (RBA: 0.038/0.07) B |
| 12 |  | [125] | 1.56 B | (RBA:0.056/0.036) B |
| 13 |  | [124] | NA | NA |
| 14 | 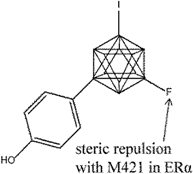 | [126] | 8.2 B | (RBA: 61.1/7.8) B |
| 15 | 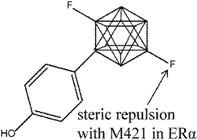 | [126] | 10.1 B | (RBA: 49.9/4.9) B |
| 16 |  | [127] | 7.4 B | (87/11.8) B |
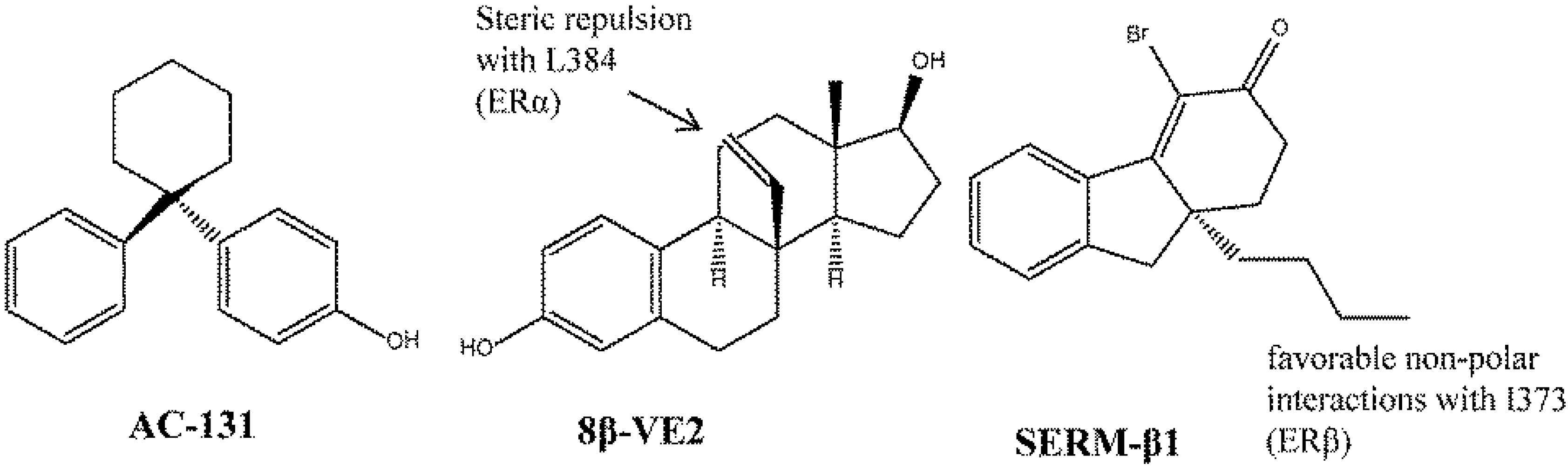
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Paris, M.; Pettersson, K.; Schubert, M.; Bertrand, S.; Pongratz, I.; Escriva, H.; Laudet, V. An amphioxus orthologue of the estrogen receptor that does not bind estradiol: Insights into estrogen receptor evolution. BMC Evol. Biol. 2008, 8. [Google Scholar] [CrossRef]
- Kampa, M.; Pelekanou, V.; Notas, G.; Stathopoulos, E.N.; Castanas, E. The estrogen receptor: Two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones (Athens) 2013, 12, 69–85. [Google Scholar]
- Beato, M.; Klug, J. Steroid hormone receptors: An update. Hum. Reprod. Update 2000, 6, 225–236. [Google Scholar] [CrossRef]
- Gouva, L.; Tsatsoulis, A. The role of estrogens in cardiovascular disease in the aftermath of clinical trials. Hormones (Athens) 2004, 3, 171–183. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Aspects Med. 2006, 27, 299–402. [Google Scholar] [CrossRef]
- Kavlock, R.J.; Daston, G.P.; DeRosa, C.; Fenner-Crisp, P.; Gray, L.E.; Kaattari, S.; Lucier, G.; Luster, M.; Mac, M.J.; Maczka, C.; et al. Research needs for the risk assessment of health and environmental effects of endocrine disruptors: A report of the U.S. EPA-sponsored workshop. Environ. Health Perspect. 1996, 104, 715–740. [Google Scholar] [CrossRef]
- Arnal, J.F.; Valera, M.C.; Payrastre, B.; Lenfant, F.; Gourdy, P. Structure-function relationship of estrogen receptors in cardiovascular pathophysiological models. Thromb. Res. 2012, 130, S7–S11. [Google Scholar]
- Lobo, R.A. Menopause and stroke and the effects of hormonal therapy. Climacteric 2007, 10, 27–31. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the women’s health initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef]
- Muramatsu, M.; Inoue, S. Estrogen receptors: How do they control reproductive and nonreproductive functions? Biochem. Biophys. Res. Commun. 2000, 270, 1–10. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Hawkins, M.B.; Thornton, J.W.; Crews, D.; Skipper, J.K.; Dotte, A.; Thomas, P. Identification of a third distinct estrogen receptor and reclassification of estrogen receptors in teleosts. Proc. Natl. Acad. Sci. USA 2000, 97, 10751–10756. [Google Scholar]
- Xu, X.; Yang, W.; Li, Y.; Wang, Y. Discovery of estrogen receptor modulators: A review of virtual screening and SAR efforts. Expert Opin. Drug Discov. 2010, 5, 21–31. [Google Scholar] [CrossRef]
- Koehler, K.F.; Helguero, L.A.; Haldosen, L.A.; Warner, M.; Gustafsson, J.A. Reflections on the discovery and significance of estrogen receptor beta. Endocr. Rev. 2005, 26, 465–478. [Google Scholar] [CrossRef]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitman, D.C. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef]
- Maggiolini, M.; Bonofiglio, D.; Marsico, S.; Panno, M.L.; Cenni, B.; Picard, D.; Ando, S. Estrogen receptor alpha mediates the proliferative but not the cytotoxic dose-dependent effects of two major phytoestrogens on human breast cancer cells. Mol. Pharmacol. 2001, 60, 595–602. [Google Scholar]
- Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Lanzino, M.; De Amicis, F.; Mauro, L.; Sisci, D.; Catalano, S.; Wright, K.D.; et al. Estrogen receptor beta as a novel target of androgen receptor action in breast cancer cell lines. Breast Cancer Res. 2014, 16. [Google Scholar] [CrossRef]
- Ruff, M.; Gangloff, M.; Marie Wurtz, J.; Moras, D. Estrogen receptor transcription and transactivation: Structure-function relationship in DNA- and ligand-binding domains of estrogen receptors. Breast Cancer Res. 2000, 2, 353–359. [Google Scholar] [CrossRef]
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef]
- Benoit, G.; Cooney, A.; Giguere, V.; Ingraham, H.; Lazar, M.; Muscat, G.; Perlmann, T.; Renaud, J.P.; Schwabe, J.; Sladek, F.; et al. International Union of Pharmacology. LXVI. Orphan nuclear receptors. Pharmacol. Rev. 2006, 58, 798–836. [Google Scholar] [CrossRef]
- Gearhart, M.D.; Holmbeck, S.M.; Evans, R.M.; Dyson, H.J.; Wright, P.E. Monomeric complex of human orphan estrogen related receptor-2 with DNA: A pseudo-dimer interface mediates extended half-site recognition. J. Mol. Biol. 2003, 327, 819–832. [Google Scholar] [CrossRef]
- Forman, B.M.; Samuels, H.H. Dimerization among nuclear hormone receptors. New. Biol. 1990, 2, 587–594. [Google Scholar]
- Sotoca, A.M.; Vervoort, J.; Rietjens, I.M.C.M.; Gustafsson, J. Human ERα and ERβ Splice Variants: Understanding Their Domain Structure in Relation to Their Biological Roles in Breast Cancer Cell Proliferation; InTech: Rijeka, Croatia, 2012; p. 452. [Google Scholar]
- Poola, I.; Koduri, S.; Chatra, S.; Clarke, R. Identification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approach. J. Steroid Biochem. Mol. Boil. 2000, 72, 249–258. [Google Scholar] [CrossRef]
- Lewandowski, S.; Kalita, K.; Kaczmarek, L. Estrogen receptor beta. Potential functional significance of a variety of mRNA isoforms. FEBS Lett. 2002, 524, 1–5. [Google Scholar] [CrossRef]
- Figtree, G.A.; McDonald, D.; Watkins, H.; Channon, K.M. Truncated estrogen receptor alpha 46-kDa isoform in human endothelial cells: Relationship to acute activation of nitric oxide synthase. Circulation 2003, 107, 120–126. [Google Scholar] [CrossRef]
- Leung, Y.K.; Mak, P.; Hassan, S.; Ho, S.M. Estrogen receptor (ER)-beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef]
- Pink, J.J.; Wu, S.Q.; Wolf, D.M.; Bilimoria, M.M.; Jordan, V.C. A novel 80 kDa human estrogen receptor containing a duplication of exons 6 and 7. Nucl. Acids Res. 1996, 24, 962–969. [Google Scholar] [CrossRef]
- Tanenbaum, D.M.; Wang, Y.; Williams, S.P.; Sigler, P.B. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5998–6003. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Gangloff, M.; Ruff, M.; Eiler, S.; Duclaud, S.; Wurtz, J.M.; Moras, D. Crystal structure of a mutant hERalpha ligand-binding domain reveals key structural features for the mechanism of partial agonism. J. Biol. Chem. 2001, 276, 15059–15065. [Google Scholar]
- Eiler, S.; Gangloff, M.; Duclaud, S.; Moras, D.; Ruff, M. Overexpression, purification, and crystal structure of native ER alpha LBD. Protein Exp. Purif. 2001, 22, 165–173. [Google Scholar] [CrossRef]
- Warnmark, A.; Treuter, E.; Gustafsson, J.A.; Hubbard, R.E.; Brzozowski, A.M.; Pike, A.C. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. J. Biol. Chem. 2002, 277, 21862–21868. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Radek, J.T.; Meyers, M.J.; Nettles, K.W.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Agard, D.A.; Greene, G.L. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 2002, 9, 359–364. [Google Scholar]
- Leduc, A.M.; Trent, J.O.; Wittliff, J.L.; Bramlett, K.S.; Briggs, S.L.; Chirgadze, N.Y.; Wang, Y.; Burris, T.P.; Spatola, A.F. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor-coactivator interactions. Proc. Natl. Acad. Sci. USA 2003, 100, 11273–11278. [Google Scholar] [CrossRef]
- Renaud, J.; Bischoff, S.F.; Buhl, T.; Floersheim, P.; Fournier, B.; Halleux, C.; Kallen, J.; Keller, H.; Schlaeppi, J.M.; Stark, W. Estrogen receptor modulators: Identification and structure-activity relationships of potent ERalpha-selective tetrahydroisoquinoline ligands. J. Med. Chem. 2003, 46, 2945–2957. [Google Scholar] [CrossRef]
- Wu, Y.L.; Yang, X.; Ren, Z.; McDonnell, D.P.; Norris, J.D.; Willson, T.M.; Greene, G.L. Structural basis for an unexpected mode of serm-mediated ER antagonism. Mol. Cell 2005, 18, 413–424. [Google Scholar] [CrossRef]
- Kim, S.; Wu, J.Y.; Birzin, E.T.; Frisch, K.; Chan, W.; Pai, L.Y.; Yang, Y.T.; Mosley, R.T.; Fitzgerald, P.M.; Sharma, N.; et al. Estrogen receptor ligands. II. Discovery of benzoxathiins as potent, selective estrogen receptor alpha modulators. J. Med. Chem. 2004, 47, 2171–2175. [Google Scholar] [CrossRef]
- Blizzard, T.A.; Dininno, F.; Morgan, J.D., 2nd; Chen, H.Y.; Wu, J.Y.; Kim, S.; Chan, W.; Birzin, E.T.; Yang, Y.T.; Pai, L.Y.; et al. Estrogen receptor ligands. Part 9: Dihydrobenzoxathiin serams with alkyl substituted pyrrolidine side chains and linkers. Bioorg. Med. Chem. Lett. 2005, 15, 107–113. [Google Scholar] [CrossRef]
- Manas, E.S.; Unwalla, R.J.; Xu, Z.B.; Malamas, M.S.; Miller, C.P.; Harris, H.A.; Hsiao, C.; Akopian, T.; Hum, W.T.; Malakian, K.; et al. Structure-based design of estrogen receptor-beta selective ligands. J. Am. Chem. Soc. 2004, 126, 15106–15119. [Google Scholar] [CrossRef]
- Manas, E.S.; Xu, Z.B.; Unwalla, R.J.; Somers, W.S. Understanding the selectivity of genistein for human estrogen receptor-beta using X-ray crystallography and computational methods. Structure 2004, 12, 2197–2207. [Google Scholar] [CrossRef]
- Renaud, J.; Bischoff, S.F.; Buhl, T.; Floersheim, P.; Fournier, B.; Geiser, M.; Halleux, C.; Kallen, J.; Keller, H.; Ramage, P. Selective estrogen receptor modulators with conformationally restricted side chains. Synthesis and structure-activity relationship of ERalpha-selective tetrahydroisoquinoline ligands. J. Med. Chem. 2005, 48, 364–379. [Google Scholar] [CrossRef]
- Tan, Q.; Blizzard, T.A.; Morgan, J.D., 2nd; Birzin, E.T.; Chan, W.; Yang, Y.T.; Pai, L.Y.; Hayes, E.C.; DaSilva, C.A.; Warrier, S.; et al. Estrogen receptor ligands. Part 10: Chromanes: Old scaffolds for new SERAMs. Bioorg. Med. Chem. Lett. 2005, 15, 1675–1681. [Google Scholar] [CrossRef]
- Hummel, C.W.; Geiser, A.G.; Bryant, H.U.; Cohen, I.R.; Dally, R.D.; Fong, K.C.; Frank, S.A.; Hinklin, R.; Jones, S.A.; Lewis, G.; et al. A selective estrogen receptor modulator designed for the treatment of uterine leiomyoma with unique tissue specificity for uterus and ovaries in rats. J. Med. Chem. 2005, 48, 6772–6775. [Google Scholar] [CrossRef]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; Nowak, J.; Sharma, S.K.; Hahm, J.B.; Kulp, K.; Hochberg, R.B.; Zhou, H.; Katzenellenbogen, J.A.; et al. Nfkappab selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat. Chem. Biol. 2008, 4, 241–247. [Google Scholar]
- Kong, E.H.; Heldring, N.; Gustafsson, J.A.; Treuter, E.; Hubbard, R.E.; Pike, A.C. Delineation of a unique protein-protein interaction site on the surface of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 3593–3598. [Google Scholar]
- Hsieh, R.W.; Rajan, S.S.; Sharma, S.K.; Guo, Y.; DeSombre, E.R.; Mrksich, M.; Greene, G.L. Identification of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype selectivity. J. Biol. Chem. 2006, 281, 17909–17919. [Google Scholar]
- Hsieh, R.W.; Rajan, S.S.; Sharma, S.K.; Greene, G.L. Molecular characterization of a B-ring unsaturated estrogen: Implications for conjugated equine estrogen components of premarin. Steroids 2008, 73, 59–68. [Google Scholar] [CrossRef]
- Norman, B.H.; Richardson, T.I.; Dodge, J.A.; Pfeifer, L.A.; Durst, G.L.; Wang, Y.; Durbin, J.D.; Krishnan, V.; Dinn, S.R.; Liu, S.; et al. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 4: Functionalization of the benzopyran A-ring. Bioorg. Med. Chem. Lett. 2007, 17, 5082–5085. [Google Scholar] [CrossRef]
- Dykstra, K.D.; Guo, L.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Hayes, E.C.; DaSilva, C.A.; Pai, L.Y.; Mosley, R.T.; Kraker, B.; et al. Estrogen receptor ligands. Part 16: 2-aryl indoles as highly subtype selective ligands for ERalpha. Bioorg. Med. Chem. Lett. 2007, 17, 2322–2328. [Google Scholar] [CrossRef]
- Heldring, N.; Pawson, T.; McDonnell, D.; Treuter, E.; Gustafsson, J.A.; Pike, A.C. Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J. Biol. Chem. 2007, 282, 10449–10455. [Google Scholar]
- Koide, A.; Abbatiello, S.; Rothgery, L.; Koide, S. Probing protein conformational changes in living cells by using designer binding proteins: Application to the estrogen receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 1253–1258. [Google Scholar] [CrossRef]
- Vajdos, F.F.; Hoth, L.R.; Geoghegan, K.F.; Simons, S.P.; LeMotte, P.K.; Danley, D.E.; Ammirati, M.J.; Pandit, J. The 2.0 a crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Sci. 2007, 16, 897–905. [Google Scholar] [CrossRef]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; O’Neill, E.E.; Nowak, J.; Guo, Y.; Kim, Y.; DeSombre, E.R.; Dilis, R.; Hanson, R.N.; et al. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007, 8, 563–568. [Google Scholar] [CrossRef]
- Richardson, T.I.; Norman, B.H.; Lugar, C.W.; Jones, S.A.; Wang, Y.; Durbin, J.D.; Krishnan, V.; Dodge, J.A. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 2: Structure-activity relationship studies on the benzopyran scaffold. Bioorg. Med. Chem. Lett. 2007, 17, 3570–3574. [Google Scholar] [CrossRef]
- Zhou, H.B.; Nettles, K.W.; Bruning, J.B.; Kim, Y.; Joachimiak, A.; Sharma, S.; Carlson, K.E.; Stossi, F.; Katzenellenbogen, B.S.; Greene, G.L.; et al. Elemental isomerism: A boron-nitrogen surrogate for a carbon-carbon double bond increases the chemical diversity of estrogen receptor ligands. Chem. Biol. 2007, 14, 659–669. [Google Scholar] [CrossRef]
- Richardson, T.I.; Dodge, J.A.; Durst, G.L.; Pfeifer, L.A.; Shah, J.; Wang, Y.; Durbin, J.D.; Krishnan, V.; Norman, B.H. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 3: Synthesis of cyclopentanone and cyclohexanone intermediates for C-ring modification. Bioorg. Med. Chem. Lett. 2007, 17, 4824–4828. [Google Scholar] [CrossRef]
- Bruning, J.B.; Parent, A.A.; Gil, G.; Zhao, M.; Nowak, J.; Pace, M.C.; Smith, C.L.; Afonine, P.V.; Adams, P.D.; Katzenellenbogen, J.A.; et al. Coupling of receptor conformation and ligand orientation determine graded activity. Nat. Chem. Biol. 2010, 6, 837–843. [Google Scholar]
- Dai, S.Y.; Chalmers, M.J.; Bruning, J.; Bramlett, K.S.; Osborne, H.E.; Montrose-Rafizadeh, C.; Barr, R.J.; Wang, Y.; Wang, M.; Burris, T.P.; et al. Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc. Natl. Acad. Sci. USA 2008, 105, 7171–7176. [Google Scholar] [CrossRef]
- Fang, J.; Akwabi-Ameyaw, A.; Britton, J.E.; Katamreddy, S.R.; Navas, F., 3rd.; Miller, A.B.; Williams, S.P.; Gray, D.W.; Orband-Miller, L.A.; Shearin, J.; et al. Synthesis of 3-alkyl naphthalenes as novel estrogen receptor ligands. Bioorg. Med. Chem. Lett. 2008, 18, 5075–5077. [Google Scholar] [CrossRef]
- Li, M.J.; Greenblatt, H.M.; Dym, O.; Albeck, S.; Pais, A.; Gunanathan, C.; Milstein, D.; Degani, H.; Sussman, J.L. Structure of estradiol metal chelate and estrogen receptor complex: The basis for designing a new class of selective estrogen receptor modulators. J. Med. Chem. 2011, 54, 3575–3580. [Google Scholar] [CrossRef]
- Phillips, C.; Roberts, L.R.; Schade, M.; Bazin, R.; Bent, A.; Davies, N.L.; Moore, R.; Pannifer, A.D.; Pickford, A.R.; Prior, S.H.; et al. Design and structure of stapled peptides binding to estrogen receptors. J. Am. Chem. Soc. 2011, 133, 9696–9699. [Google Scholar] [CrossRef]
- Delfosse, V.; Grimaldi, M.; Pons, J.L.; Boulahtouf, A.; le Maire, A.; Cavailles, V.; Labesse, G.; Bourguet, W.; Balaguer, P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol a substitutes. Proc. Natl. Acad. Sci. USA 2012, 109, 14930–14935. [Google Scholar] [CrossRef]
- Osz, J.; Brelivet, Y.; Peluso-Iltis, C.; Cura, V.; Eiler, S.; Ruff, M.; Bourguet, W.; Rochel, N.; Moras, D. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proc. Natl. Acad. Sci. USA 2012, 109, E588–E594. [Google Scholar] [CrossRef]
- Srinivasan, S.; Nwachukwu, J.C.; Parent, A.A.; Cavett, V.; Nowak, J.; Hughes, T.S.; Kojetin, D.J.; Katzenellenbogen, J.A.; Nettles, K.W. Ligand-binding dynamics rewire cellular signaling via estrogen receptor-alpha. Nat. Chem. Biol. 2013, 9, 326–332. [Google Scholar]
- Pike, A.C.; Brzozowski, A.M.; Hubbard, R.E.; Bonn, T.; Thorsell, A.G.; Engstrom, O.; Ljunggren, J.; Gustafsson, J.A.; Carlquist, M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. Embo J. 1999, 18, 4608–4618. [Google Scholar] [CrossRef]
- Pike, A.C.; Brzozowski, A.M.; Walton, J.; Hubbard, R.E.; Thorsell, A.G.; Li, Y.L.; Gustafsson, J.A.; Carlquist, M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, 145–153. [Google Scholar] [CrossRef]
- Henke, B.R.; Consler, T.G.; Go, N.; Hale, R.L.; Hohman, D.R.; Jones, S.A.; Lu, A.T.; Moore, L.B.; Moore, J.T.; Orband-Miller, L.A.; et al. A new series of estrogen receptor modulators that display selectivity for estrogen receptor beta. J. Med. Chem. 2002, 45, 5492–5505. [Google Scholar] [CrossRef]
- Malamas, M.S.; Manas, E.S.; McDevitt, R.E.; Gunawan, I.; Xu, Z.B.; Collini, M.D.; Miller, C.P.; Dinh, T.; Henderson, R.A.; Keith, J.C., Jr.; et al. Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-beta ligands. J. Med. Chem. 2004, 47, 5021–5040. [Google Scholar] [CrossRef]
- Mewshaw, R.E.; Edsall, R.J., Jr.; Yang, C.; Manas, E.S.; Xu, Z.B.; Henderson, R.A.; Keith, J.C., Jr.; Harris, H.A. Erbeta ligands. 3. Exploiting two binding orientations of the 2-phenylnaphthalene scaffold to achieve ERbeta selectivity. J. Med. Chem. 2005, 48, 3953–3979. [Google Scholar] [CrossRef]
- McDevitt, R.E.; Malamas, M.S.; Manas, E.S.; Unwalla, R.J.; Xu, Z.B.; Miller, C.P.; Harris, H.A. Estrogen receptor ligands: Design and synthesis of new 2-arylindene-1-ones. Bioorg. Med. Chem. Lett. 2005, 15, 3137–3142. [Google Scholar] [CrossRef]
- Wang, Y.; Chirgadze, N.Y.; Briggs, S.L.; Khan, S.; Jensen, E.V.; Burris, T.P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2006, 103, 9908–9911. [Google Scholar] [CrossRef]
- Wilkening, R.R.; Ratcliffe, R.W.; Tynebor, E.C.; Wildonger, K.J.; Fried, A.K.; Hammond, M.L.; Mosley, R.T.; Fitzgerald, P.M.; Sharma, N.; McKeever, B.M.; et al. The discovery of tetrahydrofluorenones as a new class of estrogen receptor beta-subtype selective ligands. Bioorg. Med. Chem. Lett. 2006, 16, 3489–3494. [Google Scholar] [CrossRef]
- Norman, B.H.; Dodge, J.A.; Richardson, T.I.; Borromeo, P.S.; Lugar, C.W.; Jones, S.A.; Chen, K.; Wang, Y.; Durst, G.L.; Barr, R.J.; et al. Benzopyrans are selective estrogen receptor beta agonists with novel activity in models of benign prostatic hyperplasia. J. Med. Chem. 2006, 49, 6155–6157. [Google Scholar] [CrossRef]
- Mewshaw, R.E.; Bowen, S.M.; Harris, H.A.; Xu, Z.B.; Manas, E.S.; Cohn, S.T. ERbeta ligands. Part 5: Synthesis and structure-activity relationships of a series of 4’-hydroxyphenyl-aryl-carbaldehyde oxime derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 902–906. [Google Scholar] [CrossRef]
- Richardson, T.I.; Dodge, J.A.; Wang, Y.; Durbin, J.D.; Krishnan, V.; Norman, B.H. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 5: Combined A- and C-ring structure-activity relationship studies. Bioorg. Med. Chem. Lett. 2007, 17, 5563–5566. [Google Scholar] [CrossRef]
- Mocklinghoff, S.; Rose, R.; Carraz, M.; Visser, A.; Ottmann, C.; Brunsveld, L. Synthesis and crystal structure of a phosphorylated estrogen receptor ligand binding domain. Chembiochem 2010, 11, 2251–2254. [Google Scholar] [CrossRef]
- Mocklinghoff, S.; van Otterlo, W.A.; Rose, R.; Fuchs, S.; Zimmermann, T.J.; Dominguez Seoane, M.; Waldmann, H.; Ottmann, C.; Brunsveld, L. Design and evaluation of fragment-like estrogen receptor tetrahydroisoquinoline ligands from a scaffold-detection approach. J. Med. Chem. 2011, 54, 2005–2011. [Google Scholar] [CrossRef]
- Roberts, L.R.; Armor, D.; Barker, C.; Bent, A.; Bess, K.; Brown, A.; Favor, D.A.; Ellis, D.; Irving, S.L.; MacKenny, M.; et al. Sulfonamides as selective oestrogen receptor beta agonists. Bioorg. Med. Chem. Lett. 2011, 21, 5680–5683. [Google Scholar] [CrossRef]
- Fuchs, S.; Nguyen, H.D.; Phan, T.T.; Burton, M.F.; Nieto, L.; de Vries-van Leeuwen, I.J.; Schmidt, A.; Goodarzifard, M.; Agten, S.M.; Rose, R.; et al. Proline primed helix length as a modulator of the nuclear receptor-coactivator interaction. J. Am. Chem. Soc. 2013, 135, 4364–4371. [Google Scholar] [CrossRef]
- Pike, A.C. Lessons learnt from structural studies of the oestrogen receptor. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 1–14. [Google Scholar] [CrossRef]
- Hickey, M.; Hart, R.; Keelan, J.A. The relationship between umbilical cord estrogens and perinatal characteristics: Implications for early life origins of reproductive cancers. Cancer Epidemiol. Biomarkers Prev. 2014. [Google Scholar]
- Ojasoo, T.; Raynaud, J.P.; Dore, J.C. Correspondence factor analysis of steroid libraries. Steroids 1995, 60, 458–469. [Google Scholar] [CrossRef]
- Ding, D.; Xu, L.; Fang, H.; Hong, H.; Perkins, R.; Harris, S.; Bearden, E.D.; Shi, L.; Tong, W. The EDKB: An established knowledge base for endocrine disrupting chemicals. BMC Bioinformatics 2010, 11. [Google Scholar] [CrossRef]
- Shen, J.; Xu, L.; Fang, H.; Richard, A.M.; Bray, J.D.; Judson, R.S.; Zhou, G.; Colatsky, T.J.; Aungst, J.L.; Teng, C.; et al. EADB: An estrogenic activity database for assessing potential endocrine activity. Toxicol. Sci. 2013, 135, 277–291. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An endocrine society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Jiang, Y.; Gong, P.; Madak-Erdogan, Z.; Martin, T.; Jeyakumar, M.; Carlson, K.; Khan, I.; Smillie, T.J.; Chittiboyina, A.G.; Rotte, S.C.; et al. Mechanisms enforcing the estrogen receptor beta selectivity of botanical estrogens. FASEB J. 2013, 27, 4406–4418. [Google Scholar] [CrossRef]
- Anstead, G.M.; Carlson, K.E.; Katzenellenbogen, J.A. The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids 1997, 62, 268–303. [Google Scholar] [CrossRef]
- Delettre, J.; Mornon, J.P.; Lepicard, G.; Ojasoo, T.; Raynaud, J.P. Steroid flexibility and receptor specificity. J. Steroid Biochem. 1980, 13, 45–59. [Google Scholar] [CrossRef]
- Hong, H.; Tong, W.; Fang, H.; Shi, L.; Xie, Q.; Wu, J.; Perkins, R.; Walker, J.D.; Branham, W.; Sheehan, D.M. Prediction of estrogen receptor binding for 58,000 chemicals using an integrated system of a tree-based model with structural alerts. Environ. Health Perspect. 2002, 110, 29–36. [Google Scholar] [CrossRef]
- Shi, L.; Tong, W.; Fang, H.; Xie, Q.; Hong, H.; Perkins, R.; Wu, J.; Tu, M.; Blair, R.M.; Branham, W.S.; et al. An integrated “4-phase” approach for setting endocrine disruption screening priorities—Phase I and II predictions of estrogen receptor binding affinity. SAR QSAR Environ. Res. 2002, 13, 69–88. [Google Scholar] [CrossRef]
- Birnbaum, L.S. State of the science of endocrine disruptors. Environ. Health Perspect. 2013, 121. [Google Scholar] [CrossRef]
- Malone, K.E. Diethylstilbestrol (DES) and breast cancer. Epidemiol. Rev. 1993, 15, 108–109. [Google Scholar]
- Palmer, J.R.; Hatch, E.E.; Rosenberg, C.L.; Hartge, P.; Kaufman, R.H.; Titus-Ernstoff, L.; Noller, K.L.; Herbst, A.L.; Rao, R.S.; Troisi, R.; et al. Risk of breast cancer in women exposed to diethylstilbestrol in utero: Preliminary results (United States). Cancer Causes Control 2002, 13, 753–758. [Google Scholar]
- Noller, K.L.; Fish, C.R. Diethylstilbestrol usage: Its interesting past, important present, and questionable future. Med. Clin. North Am. 1974, 58, 793–810. [Google Scholar]
- Piver, M.S.; Lele, S.B.; Baker, T.R.; Sandecki, A. Cervical and vaginal cancer detection at a regional diethylstilbestrol (DES) screening clinic. Cancer Detect. Prev. 1988, 11, 197–202. [Google Scholar]
- Verloop, J.; van Leeuwen, F.E.; Helmerhorst, T.J.; van Boven, H.H.; Rookus, M.A. Cancer risk in DES daughters. Cancer Causes Control 2010, 21, 999–1007. [Google Scholar] [CrossRef]
- State of the Art Assessment of Endocrine Disrupters Final Report. Available online: http://ec.europa.eu/environment/chemicals/endocrine/pdf/sota_edc_final_report.pdf (accessed on 23 December 2011).
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef]
- Lathe, R.; Kotelevtsev, Y. Steroid signaling: Ligand-binding promiscuity, molecular symmetry, and the need for gating. Steroids 2014, 82c, 14–22. [Google Scholar] [CrossRef]
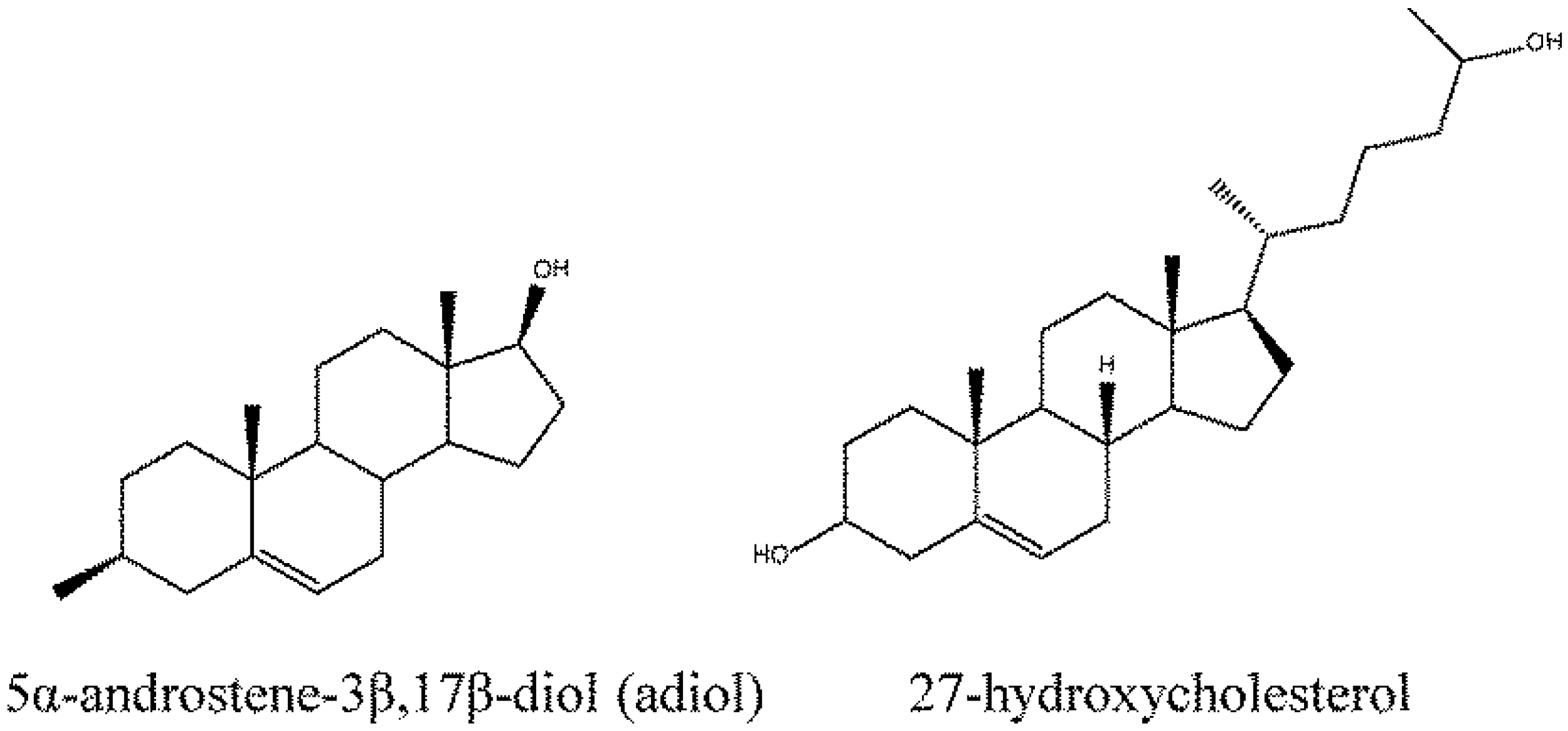
- DuSell, C.D.; Umetani, M.; Shaul, P.W.; Mangelsdorf, D.J.; McDonnell, D.P. 27-hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol. Endocrinol. 2008, 22, 65–77. [Google Scholar] [CrossRef]
- Saijo, K.; Collier, J.G.; Li, A.C.; Katzenellenbogen, J.A.; Glass, C.K. An adiol-ERbeta-CTBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 2011, 145, 584–595. [Google Scholar] [CrossRef]
- Umetani, M.; Shaul, P.W. 27-hydroxycholesterol: The first identified endogenous SERM. Trends Endocrinol. Metab. 2011, 22, 130–135. [Google Scholar] [CrossRef]
- Fleming, F.J.; Hill, A.D.K.; McDermott, E.W.; O’Higgins, N.J.; Young, L.S. Differential recruitment of coregulator proteins steroid receptor coactivator-1 and silencing mediator for retinoid and thyroid receptors to the estrogen receptor-estrogen response element by β-estradiol and 4-hydroxytamoxifen in human breast cancer. J. Clin. Endocrinol. Metab. 2004, 89, 375–383. [Google Scholar] [CrossRef]
- Nettles, K.W.; Sun, J.; Radek, J.T.; Sheng, S.; Rodriguez, A.L.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; Greene, G.L. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: Implications for other nuclear receptors. Mol. Cell. 2004, 13, 317–327. [Google Scholar] [CrossRef]
- Redden, P.R. Selective oestrogen receptor modulators, pure antioestrogens and related oestrogen receptor ligands. Expert Opin. Ther. Patents 2004, 14, 337–353. [Google Scholar] [CrossRef]
- Zhao, L.; O’Neill, K.; Diaz Brinton, R. Selective estrogen receptor modulators (SERMs) for the brain: Current status and remaining challenges for developing NeuroSERMs. Brain Res. Brain Res. Rev. 2005, 49, 472–493. [Google Scholar] [CrossRef]
- Henke, B.R.; Heyer, D. Recent advances in estrogen receptor modulators. Curr. Opin. Drug Discov. Devel. 2005, 8, 437–448. [Google Scholar]
- Veeneman, G.H. Non-steroidal subtype selective estrogens. Curr. Med. Chem 2005, 12, 1077–1136. [Google Scholar] [CrossRef]
- Blizzard, T.A. Selective estrogen receptor modulator medicinal chemistry at Merck. A review. Curr. Top. Med. Chem. 2008, 8, 792–812. [Google Scholar]
- Minutolo, F.; Macchia, M.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor beta ligands: Recent advances and biomedical applications. Med. Res. Rev. 2011, 31, 364–442. [Google Scholar] [CrossRef]
- Bhat, R.A.; Stauffer, B.; Unwalla, R.J.; Xu, Z.; Harris, H.A.; Komm, B.S. Molecular determinants of ER alpha and ER beta involved in selectivity of 16 alpha-iodo-7 beta estradiol. J. Steroid Biochem. Mol. Biol. 2004, 88, 17–26. [Google Scholar]
- Chen, H.Y.; Dykstra, K.D.; Birzin, E.T.; Frisch, K.; Chan, W.; Yang, Y.T.; Mosley, R.T.; DiNinno, F.; Rohrer, S.P.; Schaeffer, J.M.; et al. Estrogen receptor ligands. Part 1: The discovery of flavanoids with subtype selectivity. Bioorg. Med. Chem. Lett. 2004, 14, 1417–1421. [Google Scholar] [CrossRef]
- Chen, H.Y.; Kim, S.; Wu, J.Y.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Dahllund, J.; DiNinno, F.; Rohrer, S.P.; Schaeffer, J.M.; et al. Estrogen receptor ligands. Part 3: The SAR of dihydrobenzoxathiin SERMs. Bioorg. Med. Chem. Lett. 2004, 14, 2551–2554. [Google Scholar] [CrossRef]
- Kim, S.; Wu, J.; Chen, H.Y.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Colwell, L.; Li, S.; Dahllund, J.; DiNinno, F.; et al. Estrogen receptor ligands. Part 4: The SAR of the syn-dihydrobenzoxathiin SERAMs. Bioorg. Med. Chem. Lett. 2004, 14, 2741–2745. [Google Scholar] [CrossRef]
- Tan, Q.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Pai, L.Y.; Hayes, E.C.; DaSilva, C.A.; DiNinno, F.; Rohrer, S.P.; Schaeffer, J.M.; et al. Estrogen receptor ligands. Part 6: Synthesis and binding affinity of dihydrobenzodithiins. Bioorg. Med. Chem. Lett. 2004, 14, 3753–3755. [Google Scholar] [CrossRef]
- Chalmers, M.J.; Wang, Y.; Novick, S.; Sato, M.; Bryant, H.U.; Montrose-Rafizdeh, C.; Griffin, P.R.; Dodge, J.A. Hydrophobic interactions improve selectivity to ERalpha for ben-zothiophene serms. ACS Med. Chem. Lett. 2012, 3, 207–210. [Google Scholar] [CrossRef]
- Yeo, H.L.; Song, Y.S.; Ryu, J.H.; Kim, H.D. Design, synthesis, and biological evaluation of cyclopropyl analogues of stilbene with raloxifene side chain as subtype-selective ligands for estrogen receptor. Arch. Pharm. Res. 2013, 36, 1096–1103. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar]
- Unwalla, R.J.; Manas, E.S.; Miller, C.P.; Xu, Z. Computational approaches to understand selectivity between receptors alpha and beta. In Proceedings of the 226 ACS National Meeting, New York, NY, USA, 7–11 September 2003.
- Tan, Q.; Birzin, E.T.; Chan, W.; Tien Yang, Y.; Pai, L.Y.; Hayes, E.C.; DaSilva, C.A.; DiNinno, F.; Rohrer, S.P.; Schaeffer, J.M.; et al. Estrogen receptor ligands. Part 5: The SAR of dihydrobenzoxathiins containing modified basic side chains. Bioorg. Med. Chem. Lett. 2004, 14, 3747–3751. [Google Scholar] [CrossRef]
- Sunden, H.; Ma, J.N.; Hansen, L.K.; Gustavsson, A.L.; Burstein, E.S.; Olsson, R. Design of a highly selective and potent class of non-planar estrogen receptor beta agonists. ChemMedChem 2013, 8, 1283–1294. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Filipiak, K.; Maslyk, M.; Ciepielski, J.; Demkowicz, S.; de Pascual-Teresa, S.; Martin-Santamaria, S.; de Pascual-Teresa, B.; Ramos, A. Towards beta-selectivity in functional estrogen receptor antagonists. Org. Biomol. Chem. 2012, 10, 7334–7346. [Google Scholar] [CrossRef]
- Ohta, K.; Ogawa, T.; Kaise, A.; Endo, Y. Enhanced estrogen receptor beta (ERbeta) selectivity of fluorinated carborane-containing ER modulators. Bioorg. Med. Chem. Lett. 2013, 23, 6555–6558. [Google Scholar] [CrossRef]
- Ohta, K.; Ogawa, T.; Kaise, A.; Oda, A.; Endo, Y. Aliphatic substitution of o-carboranyl phenols enhances estrogen receptor beta selectivity. Chem. Pharm. Bull. (Tokyo) 2014, 62, 386–391. [Google Scholar] [CrossRef]
- Wilkening, R.R.; Ratcliffe, R.W.; Fried, A.K.; Meng, D.; Sun, W.; Colwell, L.; Lambert, S.; Greenlee, M.; Nilsson, S.; Thorsell, A.; et al. Estrogen receptor beta-subtype selective tetrahydrofluorenones: Use of a fused pyrazole as a phenol bioisostere. Bioorg. Med. Chem. Lett. 2006, 16, 3896–3901. [Google Scholar] [CrossRef]
- Hegele-Hartung, C.; Siebel, P.; Peters, O.; Kosemund, D.; Muller, G.; Hillisch, A.; Walter, A.; Kraetzschmar, J.; Fritzemeier, K.H. Impact of isotype-selective estrogen receptor agonists on ovarian function. Proc. Natl. Acad. Sci. USA 2004, 101, 5129–5134. [Google Scholar] [CrossRef]
- Harrington, W.R.; Sheng, S.; Barnett, D.H.; Petz, L.N.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol. Cell. Endocrinol. 2003, 206, 13–22. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ng, H.W.; Perkins, R.; Tong, W.; Hong, H. Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands. Int. J. Environ. Res. Public Health 2014, 11, 8709-8742. https://doi.org/10.3390/ijerph110908709
Ng HW, Perkins R, Tong W, Hong H. Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands. International Journal of Environmental Research and Public Health. 2014; 11(9):8709-8742. https://doi.org/10.3390/ijerph110908709
Chicago/Turabian StyleNg, Hui Wen, Roger Perkins, Weida Tong, and Huixiao Hong. 2014. "Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands" International Journal of Environmental Research and Public Health 11, no. 9: 8709-8742. https://doi.org/10.3390/ijerph110908709
APA StyleNg, H. W., Perkins, R., Tong, W., & Hong, H. (2014). Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands. International Journal of Environmental Research and Public Health, 11(9), 8709-8742. https://doi.org/10.3390/ijerph110908709