Arsenic-Induced Genotoxicity and Genetic Susceptibility to Arsenic-Related Pathologies




Abstract
:1. Introduction
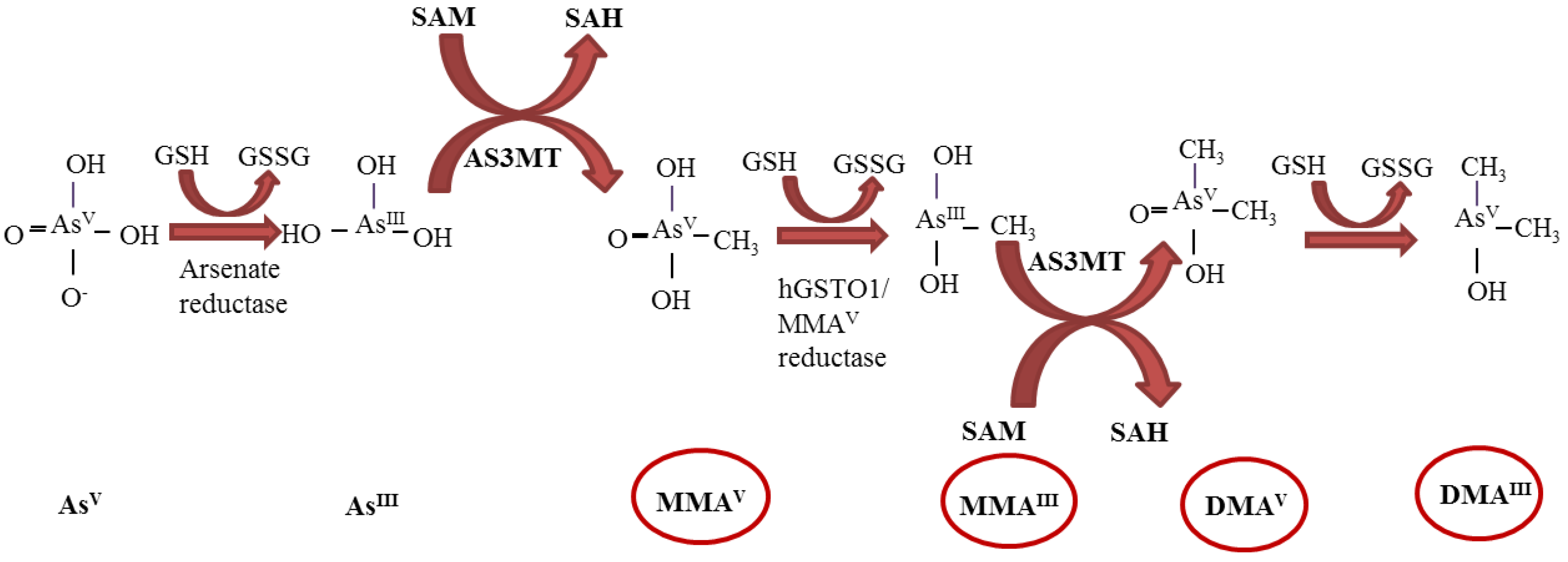
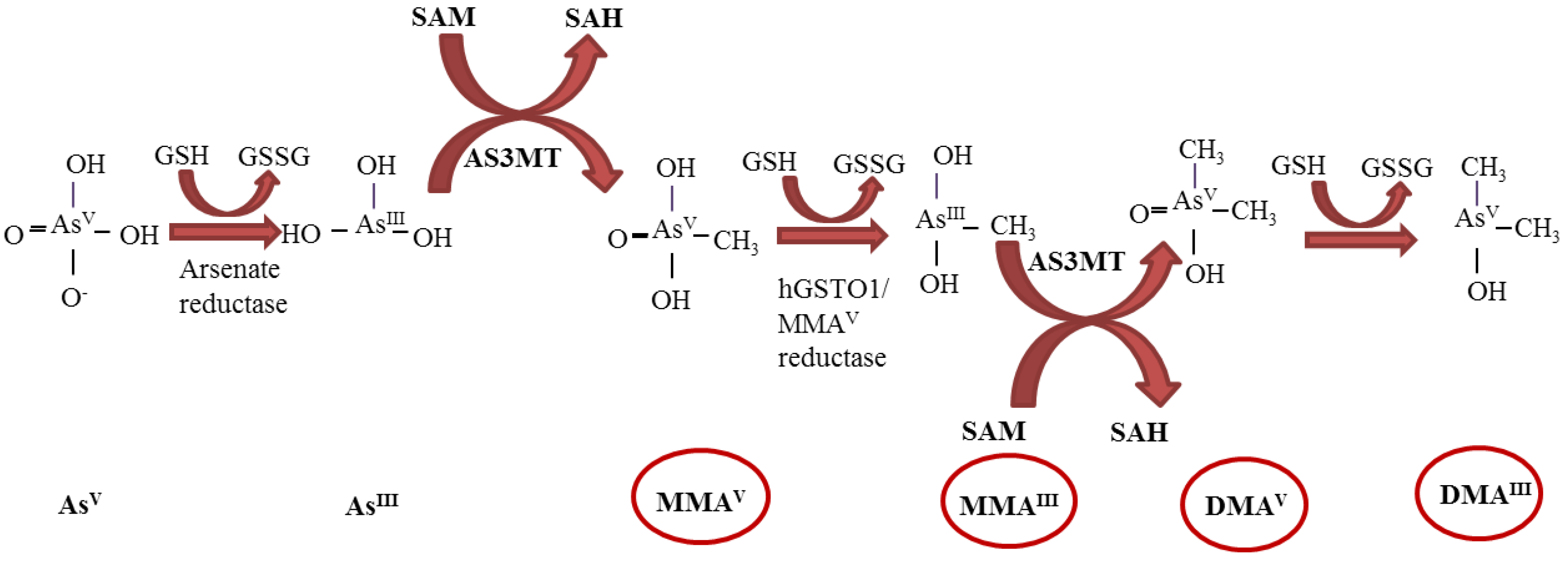
2. Genotoxicity
2.1. DNA Damage
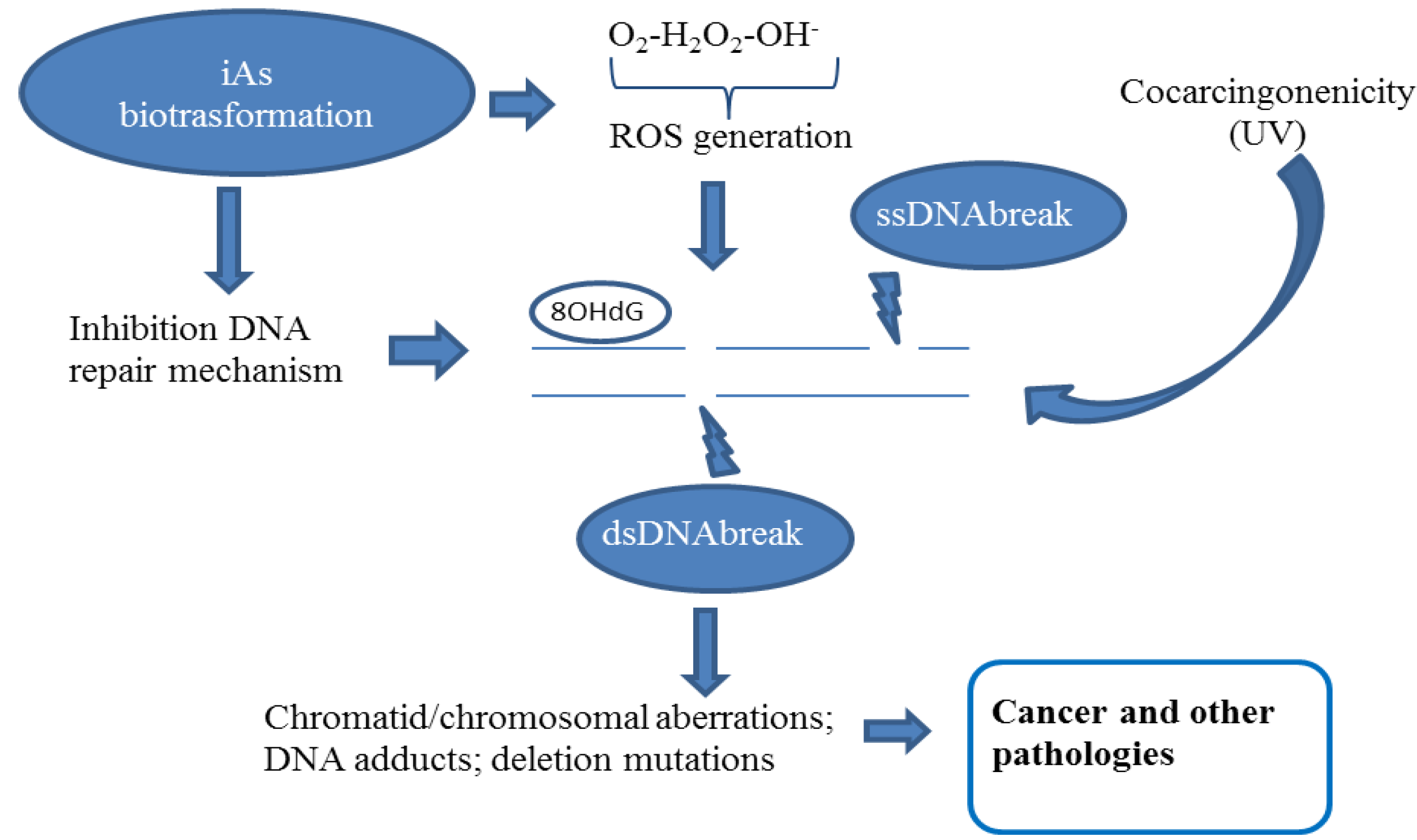
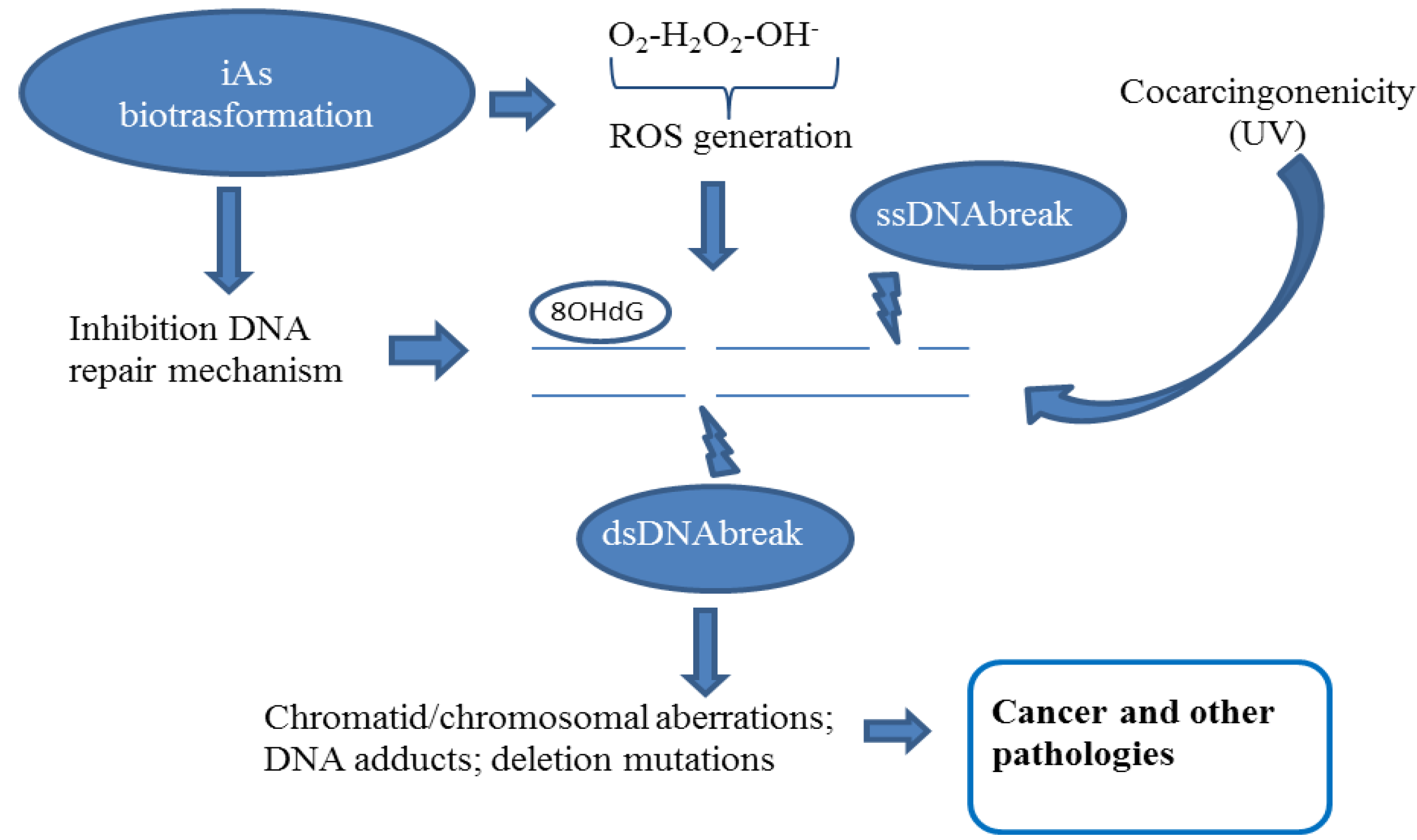
2.2. Chromatid and Chromosomal and Telomere Damage

{kind=link}
{kind=link}
| Study population, n | Mean arsenic water drinking exposure | Country | Main endpoint result | References |
|---|---|---|---|---|
| 18 exposed subjects 18 controls | 1,312 μg/L 16 μg/L | Nevada | 1.8 fold increase in bladder cells MN correlation/iAs urinary level | Warner et al. [54] |
| 31 exposed subjects 27 controls | 408.7 μg/L 29.88 μg/L | Mexico | CA increase in lymphocytes MN increase in oral and urinary cells | Gonsebatt et al. [44] |
| 42 exposed subjects 8 controls | 410 μg/L <1 μg/L | Finland | CA correlation/urinary As exposure, among current users | Maki-Paakanen [50] |
| 19 exposed subjects 13 controls | 527.5 μg/L 4.4 μg/L | USA | 3.4 fold increase of MN in buccal cells 2.7 fold increase in bladder cells | Tian [45] |
| 32 cancer cases of risk area 32 controls of risk area | n.d. | Taiwan | No difference in spontaneous and mitomycin C-induced SCE | Liou et al. [49] |
| 45 exposed subjects 21 controls | 368.11 μg/L 5.49 μg/L | India | MN increase | Basu et al. [42] |
| 59 exposed subjects 36 controls | 211.70 μg/L 6.35 μg/L | India | CA and SCE increases | Mahata et al. [39] |
| 106 exposed subjects 111 controls | >750 μg/L >2 μg/L | Chile | MN increase | Martinez et al. [40] |
| 163 exposed subjects 154 controls | 214.7 μg/L 9.2 μg/L | India | 5.3 fold MN increase in lymphocytes 4.6 fold MN increase in oral cells 4.7 fold MN increase in urothelial cells | Basu et al. [41] |
| 45 exposed subjects 25 controls | 66.75 μg/L 6.4 μg/L | India | CA and MN increases | Chakraborty et al. [43] |
| 422 exposed subjects (244 skin symptomatic) 120 controls | 202.33 μg/L 7.16 μg/L | India | CA and MN increases | Ghosh et al. [37,38] |
| 200 subjects exposed 165 controls | 56.76 μg/L * 117.4 μg/L * | India | MN increase in buccal cells DNA increase in lymphocytes | Vuyyuri [44] |
| 27 exposed subjects 30 controls | >50 μg/L (water drinking) <50 μg/L (water drinking) | Argentina | MN increase in buccal cells | Bartolotta [46] |
2.3. DNA Repair Inhibition
3. Genetic Susceptibility to Arsenic Toxicity
| Gene symbol | Biological function | SNP | Main associated effect | References |
|---|---|---|---|---|
| ASIIIMT | As metabolism | G7395A (intronic) T35587C(intronic) G12390C(intronic) C14215T(intronic) Met287Thr A35991G (intronic) | Arsenic metabolite levels | Meza et al. [72] Schläwicke Engström et al. [69] Hernandez et al.[73] Lindberg et al. [74] Agusa et al. [75] Gong et al. [76] |
| GST-O2 | As detoxification | Asn142Asp Ala140Asp | iAs and arsenic metabolites levels Major risk of carotid atherosclerosis | Chung et al. [68] Chen et al. [77] Hsieh et al. [78] |
| GST-P1 | As detoxification | Ile105Val | Arsenic metabolite levels Major risk of TCC Major risk of bladder cancer Major risk of carotid atherosclerosis | Agusa et al. [79] Hsu et al. [80,81] Lesseur et al. [82] Wang et al. [83] |
| GST-M1 | As detoxification | Null genotype | iAs and arsenic metabolite levels | Chiou et al. [84] Steinmaus et al. [85] |
| GST-T1 | As detoxification | null genotype | Arsenic metabolite levels | Chiou et al. [86] |
| MTHFR | As metabolism | Ala222Val | iAs and arsenic metabolite levels | Lindberg et al. [74] Schläwicke Engström et al. [69] |
| hOGG1 | DNA repair | Ser326 Cys | 8-oxoguanine levels | Fujihara et al. [70] |
| APE1 | DNA repair | Asp148Glu | 8-oxoguanine levels | Fujihara et al. [70] |
| XRCC3 | DNA repair | Thr241Met | Arsenic-induced skin lesions; Chromosomal aberrations | Kundu et al. [71] |
| HO1 | Inducible antioxidant enzyme | short GT-repeat | BP regulation and cardiovascular mortality risk | Wu et al. [87,88] |
| P53 | Tumor suppressor | Arg72Pro | Risk for arsenic-induced kerastosis Risk for renal cell carcinoma | De Chaudhuri et al. [89] Huang et al. [90] |
3.1. ASIIIMT Genetic Polymorphisms
3.2. Polymorphisms in GSTs Genes and Other Detoxification Genes
3.3. SNPs in Genes of DNA Repair
3.4. Genetic Variants in Other Genes Implicated in Arsenic Susceptibility
4. Conclusions and Future Perspectives
Conflicts of Interest
References
- Pierce, B.L.; Kibriya, M.G.; Tong, L.; Jasmine, F.; Argos, M.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Rakibuz-Zaman, M.; Parvez, F.; et al. Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLoS Genet. 2012, 8, e1002522. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some drinking-water disinfectants and contaminants, including arsenic. IARC Monogr. Eval. Carcinog. Risks. Hum. 2004, 84, 1–477.
- Flora, S.J. Arsenic-induced oxidative stress and its reversibility. Free Radic. Biol. Med. 2011, 51, 257–281. [Google Scholar] [CrossRef]
- Basu, A.; Som, A.; Ghoshal, S.; Mondal, L.; Chaubey, R.C.; Bhilwade, H.N.; Rahman, M.M.; Giri, A.K. Assessment of DNA damage in peripheral blood lymphocytes of individuals susceptible to arsenic induced toxicity in West Bengal, India. Toxicol. Lett. 2005, 159, 100–112. [Google Scholar] [CrossRef]
- Méndez-Gómez, J.; García-Vargas, G.G.; López-Carrillo, L.; Calderón-Aranda, E.S.; Gómez, A.; Vera, E.; Valverde, M.; Cebrián, M.E.; Rojas, E. Genotoxic effects of environmental exposure to arsenic and lead on children in region Lagunera, Mexico. Ann. New York Acad. Sci. 2008, 1140, 358–367. [Google Scholar] [CrossRef]
- Sampayo-Reyes, A.; Hernández, A.; El-Yamani, N.; López-Campos, C.; Mayet-Machado, E.; Rincón-Castañeda, C.B.; Limones-Aguilar Mde, L.; López-Campos, J.E.; de León, M.B.; González-Hernández, S. Arsenic induces DNA damage in environmentally exposed Mexican children and adults. Influence of GSTO1 and AS3MT polymorphisms. Toxicol. Sci. 2010, 117, 63–71. [Google Scholar] [CrossRef]
- Schoen, A.; Beck, B.; Sharma, R.; Dubé, E. Arsenic toxicity at low doses: Epidemiological and mode of action considerations. Toxicol. Appl. Pharmacol. 2004, 198, 253–267. [Google Scholar] [CrossRef]
- Wu, M.M.; Chiou, H.Y.; Wang, T.W.; Hsueh, Y.M.; Wang, I.H.; Chen, C.J.; Lee, T.C. Association of blood arsenic levels with increased reactive oxidants and decreased antioxidant capacity in a human population of northeastern Taiwan. Environ. Health Perspect. 2001, 109, 1011–1017. [Google Scholar]
- Tapio, S.; Grosche, B. Arsenic in the aetiology of cancer. Mutat. Res. 2006, 612, 215–246. [Google Scholar] [CrossRef]
- Pei, Q.; Ma, N.; Zhang, J.; Xu, W.; Li, Y.; Ma, Z.; Li, Y.; Tian, F.; Zhang, W.; Mu, J.; et al. Oxidative DNA damage of peripheral blood polymorphonuclear leukocytes, selectively induced by chronic arsenic exposure, is associated with extent of arsenic-related skin lesions. Toxicol. Appl. Pharmacol. 2013, 266, 143–149. [Google Scholar] [CrossRef]
- Ahmed, S.; Ahsan, K.B.; Kippler, M.; Mily, A.; Wagatsuma, Y.; Hoque, A.M.; Ngom, P.T.; El Arifeen, S.; Raqib, R.; Vahter, M. In utero arsenic exposure is associated with impaired thymic function in newborns possibly via oxidative stress and apoptosis. Toxicol. Sci. 2012, 129, 305–314. [Google Scholar] [CrossRef]
- Vahter, M.; Concha, G. Role of metabolism in arsenic toxicity. Pharmacol. Toxicol. 2001, 89, 1–5. [Google Scholar] [CrossRef]
- Styblo, M.; Del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Reed, W.; Wang, C.; Cullen, W.R.; Thomas, D.J. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000, 74, 289–299. [Google Scholar] [CrossRef]
- Petrick, J.S.; Ayala-Fierro, F.; Cullen, W.R.; Carter, D.E.; Vasken Aposhian, H. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol. Appl. Pharmacol. 2000, 163, 203–207. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Adonis, M.; Gil, L.; Lam, W.L. Arsenic biotransformation as a cancer promoting factor by inducing DNA damage and disruption of repair mechanisms. Mol. Biol. Int. 2011, 2011, 718974. [Google Scholar] [CrossRef]
- Basu, A.; Mahata, J.; Gupta, S.; Giri, A.K. Genetic toxicology of a paradoxical human carcinogen, arsenic: A review. Mutat. Res. 2001, 488, 171–194. [Google Scholar] [CrossRef]
- Rossman, T.G. Mechanism of arsenic carcinogenesis: An integrated approach. Mutat. Res. 2003, 533, 37–65. [Google Scholar] [CrossRef]
- Li, J.H.; Rossman, T.G. Comutagenesis of sodium arsenite with ultraviolet radiation in Chinese hamster V79 cells. Biol. Met. 1991, 4, 197–200. [Google Scholar] [CrossRef]
- Lee, T.C.; Oshimura, M.; Barrett, J.C. Comparison of arsenic-induced cell transformation, cytotoxicity, mutation and cytogenetic effects in Syrian hamster embryo cells in culture. Carcinogenesis. 1985, 6, 1421–1426. [Google Scholar] [CrossRef]
- Kessel, M.; Liu, S.X.; Xu, A.; Santella, R.; Hei, T.K. Arsenic induces oxidative DNA damage in mammalian cells. Mol. Cell. Biochem. 2002, 234–235, 301–308. [Google Scholar] [CrossRef]
- Nesnow, S.; Roop, B.C.; Lambert, G.; Kadiiska, M.; Mason, R.P.; Cullen, W.R.; Mass, M.J. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. Chem. Res. Toxicol. 2002, 15, 1627–1634. [Google Scholar] [CrossRef]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar]
- Kitchin, K.T.; Wallace, K. Evidence against the nuclear in situ binding of arsenicals—Oxidative stress theory of arsenic carcinogenesis. Toxicol. Appl. Pharmacol. 2008, 232, 252–257. [Google Scholar] [CrossRef]
- Bau, D.T.; Wang, T.S.; Chung, C.H.; Wang, A.S.; Wang, A.S.; Jan, K.Y. Oxidative DNA adducts and DNA-protein cross-links are the major DNA lesions induced by arsenite. Environ. Health Perspect. 2002, 110, 753–756. [Google Scholar] [CrossRef]
- Hwang, E.S.; Kim, G.H. Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research. Toxicology 2007, 229, 1–10. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Georgakilas, A.G.; Georgakila, S.; Malamou-Mitsi, V.; Schoneveld, O.; Pappa, A.; Panayiotidis, M.I. The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem. Biol. Interact. 2010, 188, 334–339. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Chung, C.J.; Huang, C.J.; Pu, Y.S.; Su, C.T.; Huang, Y.K.; Chen, Y.T.; Hsueh, Y.M. Polymorphisms in cell cycle regulatory genes, urinary arsenic profile and urothelial carcinoma. Toxicol. Appl. Pharmacol. 2008, 232, 203–209. [Google Scholar] [CrossRef]
- De Vizcaya-Ruiz, A.; Barbier, O.; Ruiz-Ramos, R.; Cebrian, M.E. Biomarkers of oxidative stress and damage in human populations exposed to arsenic. Mutat. Res. 2009, 674, 85–92. [Google Scholar] [CrossRef]
- Huang, C.Y.; Su, C.T.; Chung, C.J.; Pu, Y.S.; Chu, J.S.; Yang, H.Y.; Wu, C.C.; Hsueh, Y.M. Urinary total arsenic and 8-hydroxydeoxyguanosine are associated with renal cell carcinoma in an area without obvious arsenic exposure. Toxicol. Appl. Pharmacol. 2012, 262, 349–354. [Google Scholar] [CrossRef]
- Kligerman, A.D.; Malik, S.I.; Campbell, J.A. Cytogenetic insights into DNA damage and repair of lesions induced by a monomethylated trivalent arsenical. Mutat. Res. 2010, 695, 2–8. [Google Scholar] [CrossRef]
- Mourón, S.A.; Grillo, C.A.; Dulout, F.N.; Golijow, C.D. Induction of DNA strand breaks, DNA-protein crosslinks and sister chromatid exchanges by arsenite in a human lung cell line. Toxicol In Vitro 2006, 20, 279–285. [Google Scholar] [CrossRef]
- Liu, F.; Jan, K.Y. DNA damage in arsenite- and cadmium-treated bovine aortic endothelial cells. Free Radic. Biol. Med. 2000, 28, 55–63. [Google Scholar] [CrossRef]
- Qin, X.J.; Hudson, L.G.; Liu, W.; Timmins, G.S.; Liu, K.J. Low concentration of arsenite exacerbates UVR-induced DNA strand breaks by inhibiting PARP-1 activity. Toxicol. Appl. Pharmacol. 2008, 232, 41–50. [Google Scholar] [CrossRef]
- Colognato, R.; Coppedè, F.; Ponti, J.; Sabbioni, E.; Migliore, L. Genotoxicity induced by arsenic compounds in peripheral human lymphocytes analysed by cytokinesis-block micronucleus assay. Mutagenesis 2007, 22, 255–261. [Google Scholar] [CrossRef]
- Ghosh, P.; Basu, A.; Mahata, J.; Basu, S.; Sengupta, M.; Das, J.; Mukherjee, A.; Sarkar, A.K.; Mondal, L.; Ray, K.; Giri, A.K. Cytogenetic damage and genetic variants in the individuals susceptible to arsenic-induced cancer through drinking water. Int. J. Canc. 2006, 118, 2470–2478. [Google Scholar] [CrossRef]
- Ghosh, P.; Banerjee, M.; de Chaudhuri, S.; Das, J.K.; Sarma, N.; Basu, A.; Giri, A.K. Increased chromosome aberration frequencies in the Bowen’s patients compared to non-cancerous skin lesions individuals exposed to arsenic. Mutat. Res. 2007, 632, 104–110. [Google Scholar] [CrossRef]
- Mahata, J.; Basu, A.; Ghoshal, S.; Sarkar, J.N.; Roy, A.K.; Poddar, G.; Nandy, A.K.; Banerjee, A.; Ray, K.; Natarajan, A.T.; et al. Chromosomal aberrations and sister chromatid exchanges in individuals exposed to arsenic through drinking water in West Bengal, India. Mutat. Res. 2003, 533, 133–143. [Google Scholar]
- Martínez, V.; Creus, A.; Venegas, W.; Arroyo, A.; Beck, J.P.; Gebel, T.W.; Surrallés, J.; Marcos, R. Evaluation of micronucleus induction in a Chilean population environmentally exposed to arsenic. Mutat. Res. 2004, 564, 65–74. [Google Scholar] [CrossRef]
- Basu, A.; Ghosh, P.; Das, J.K.; Banerjee, A.; Ray, K.; Giri, A.K. Micronuclei as biomarkers of carcinogen exposure in populations exposed to arsenic through drinking water in West Bengal, India: A comparative study in three cell types. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 820–827. [Google Scholar]
- Basu, A.; Mahata, J.; Roy, A.K.; Sarkar, J.N.; Poddar, G.; Nandy, A.K.; Sarkar, P.K.; Dutta, P.K.; Banerjee, A.; Das, M.; Ray, K.; et al. Enhanced frequency of micronuclei in individuals exposed to arsenic through drinking water in West Bengal, India. Mutat. Res. 2002, 516, 29–40. [Google Scholar] [CrossRef]
- Chakraborty, T.; Das, U.; Poddar, S.; Sengupta, B.; De, M. Micronuclei and chromosomal aberrations as biomarkers: A study in an arsenic exposed population in West Bengal, India. Bull. Environ. Contam. Toxicol. 2006, 76, 970–976. [Google Scholar] [CrossRef]
- Gonsebatt, M.E.; Vega, L.; Salazar, A.M.; Montero, R.; Guzmán, P.; Blas, J.; del Razo, L.M.; García-Vargas, G.; Albores, A.; Cebrián, M.E.; Kelsh, M.; Ostrosky-Wegman, P. Cytogenetic effects in human exposure to arsenic. Mutat. Res. 1997, 386, 219–228. [Google Scholar] [CrossRef]
- Tian, D.; Ma, H.; Feng, Z.; Xia, Y.; Le, X.C.; Ni, Z.; Allen, J.; Collins, B.; Schreinemachers, D.; Mumford, J.L. Analyses of micronuclei in exfoliated epithelial cells from individuals chronically exposed to arsenic via drinking water in inner Mongolia, China. J. Toxicol. Environ. Health A 2001, 64, 473–484. [Google Scholar] [CrossRef]
- Bartolotta, S.A.; Pacskowski, M.G.; Hick, A.; Carballo, M.A. Micronuclei assay in exfoliated buccal cells from individuals exposed to arsenic in Argentina. Arch. Environ. Contam. Toxicol. 2011, 61, 337–343. [Google Scholar]
- Vuyyuri, S.B.; Ishaq, M.; Kuppala, D.; Grover, P.; Ahuja, Y.R. Evaluation of micronucleus frequencies and DNA damage in glass workers exposed to arsenic. Environ. Mol. Mutagen. 2006, 47, 562–570. [Google Scholar] [CrossRef]
- Marchiset-Ferlay, N.; Savanovitch, C.; Sauvant-Rochat, M.P. What is the best biomarker to assess arsenic exposure via drinking water? Environ. Int. 2012, 39, 150–171. [Google Scholar] [CrossRef]
- Liou, S.H.; Chen, Y.H.; Loh, C.H.; Yang, T.; Wu, T.N.; Chen, C.J.; Hsieh, L.L. The association between frequencies of mitomycin C-induced sister chromatid exchange and cancer risk in arseniasis. Toxicol. Lett. 2002, 129, 237–243. [Google Scholar] [CrossRef]
- Mäki-Paakkanen, J.; Kurttio, P.; Paldy, A.; Pekkanen, J. Association between the clastogenic effect in peripheral lymphocytes and human exposure to arsenic through drinking water. Environ. Mol. Mutagen. 1998, 32, 301–313. [Google Scholar] [CrossRef]
- Hagmar, L.; Bonassi, S.; Stromberg, U.; Brogger, A.; Knudsen, L.S.; Norppa, H.; Reuterwall, C. Chromosomal aberrations in lymphocytes predict human cancer: A report from the European Study Group on Cytogenetic Biomarkers and Health (ESCH). Cancer Res. 1998, 58, 4117–4121. [Google Scholar]
- Bonassi, S.; Znaor, A.; Ceppi, M.; Lando, C.; Chang, W.P.; Holland, N.; Kirsch-Volders, M.; Zeiger, E.; Ban, S.; Barale, R.; et al. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 2007, 28, 625–631. [Google Scholar]
- Federici, C.; Botto, N.; Manfredi, S.; Rizza, A.; Del Fiandra, M.; Andreassi, M.G. Relation of increased chromosomal damage to future adverse cardiac events in patients with known coronary artery disease. Am. J. Cardiol. 2008, 102, 1296–1300. [Google Scholar]
- Warner, M.L.; Moore, L.E.; Smith, M.T.; Kalman, D.A.; Fanning, E.; Smith, A.H. Increased micronuclei in exfoliated bladder cells of individuals who chronically ingest arsenic-contaminated water in Nevada. Cancer Epidemiol. Biomarkers Prev. 1994, 3, 583–590. [Google Scholar]
- Ferrario, D.; Collotta, A.; Carfi, M.; Bowe, G.; Vahter, M.; Hartung, T.; Gribaldo, L. Arsenic induces telomerase expression and maintains telomere length in human cord blood cells. Toxicology 2009, 260, 132–141. [Google Scholar] [CrossRef]
- Li, H.; Engström, K.; Vahter, M.; Broberg, K. Arsenic exposure through drinking water is associated with longer telomeres in peripheral blood. Chem. Res. Toxicol. 2012, 25, 2333–2339. [Google Scholar] [CrossRef]
- Lai, Y.; Zhao, W.; Chen, C.; Wu, M.; Zhang, Z. Role of DNA polymerase beta in the genotoxicity of arsenic. Environ. Mol. Mutagen. 2011, 52, 460–468. [Google Scholar] [CrossRef]
- Hartwig, A.; Groblinghoff, U.D.; Beyersmann, D.; Natarajan, A.T.; Filon, R.; Mullenders, L.H. Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis 1997, 18, 399–405. [Google Scholar] [CrossRef]
- Curnow, A.; Salter, L.; Morley, N.; Gould, D. A preliminary investigation of the effects of arsenate on irradiation-induced DNA damage in cultured human lung fibroblasts. J. Toxicol. Environ. Health A 2001, 63, 605–616. [Google Scholar] [CrossRef]
- Andrew, A.S.; Karagas, M.R.; Hamilton, J.W. Decreased DNA repair gene expression among individuals exposed to arsenic in United States drinking water. Int. J. Cancer 2003, 104, 263–268. [Google Scholar] [CrossRef]
- Andrew, A.S.; Burgess, J.L.; Meza, M.M.; Demidenko, E.; Waugh, M.G.; Hamilton, J.W.; Karagas, M.R. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environ. Health Perspect. 2006, 114, 1193–1198. [Google Scholar] [CrossRef]
- Schwerdtle, T.; Walter, I.; Mackiw, I.; Hartwig, A. Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA. Carcinogenesis 2003, 24, 967–974. [Google Scholar] [CrossRef]
- Ebert, F.; Weiss, A.; Bültemeyer, M.; Hamann, I.; Hartwig, A.; Schwerdtle, T. Arsenicals affect base excision repair by several mechanisms. Mutat. Res. 2011, 715, 32–41. [Google Scholar] [CrossRef]
- Sykora, P.; Snow, E.T. Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite. Toxicol. Appl. Pharmacol. 2008, 228, 385–394. [Google Scholar] [CrossRef]
- Osmond, M.J.; Kunz, B.A.; Snow, E.T. Age and exposure to arsenic alter base excision repair transcript levels in mice. Mutagenesis 2010, 25, 517–522. [Google Scholar] [CrossRef]
- Snow, E.T.; Hu, Y.; Klein, C.; McCluskey, K.; Schuliga, M.; Sykora, P. Regulation of redox and DNA repair genes by arsenic: Low dose protection against oxidative stress? In Arsenic Exposure and Health Effects V; Chappel W.R. Abernathy, C.O., Calderon R.L. Thomas, D.J., Eds.; Elsevier Science: San Diego, CA, USA, 2003; pp. 305–319. [Google Scholar]
- Valenzuela, O.L.; Drobná, Z.; Hernández-Castellanos, E.; Sánchez-Peña, L.C.; García-Vargas, G.G.; Borja-Aburto, V.H.; Stýblo, M.; del Razo, L.M. Association of AS3MT polymorphisms and the risk of premalignant arsenic skin lesions. Toxicol. Appl. Pharmacol. 2009, 239, 200–207. [Google Scholar] [CrossRef]
- Chung, C.J.; Hsueh, Y.M.; Bai, C.H.; Huang, Y.K.; Huang, Y.L.; Yang, M.H.; Chen, C.J. Polymorphisms in arsenic metabolism genes, urinary arsenic methylation profile and cancer. Canc. Causes Contr. 2009, 20, 1653–1661. [Google Scholar] [CrossRef]
- Schläwicke Engström, K.; Broberg, K.; Concha, G.; Nermell, B.; Warholm, M.; Vahter, M. Genetic polymorphisms influencing arsenic metabolism: Evidence from Argentina. Environ. Health Perspect. 2007, 115, 599–605. [Google Scholar] [CrossRef]
- Fujihara, J.; Soejima, M.; Yasuda, T.; Koda, Y.; Kunito, T.; Iwata, H.; Tanabe, S.; Takeshita, H. Polymorphic trial in oxidative damage of arsenic exposed Vietnamese. Toxicol. Appl. Pharmacol. 2011, 256, 174–178. [Google Scholar] [CrossRef]
- Kundu, M.; Ghosh, P.; Mitra, S.; Das, J.K.; Sau, T.J.; Banerjee, S.; States, J.C.; Giri, A.K. Precancerous and non-cancer disease endpoints of chronic arsenic exposure: The level of chromosomal damage and XRCC3 T241M polymorphism. Mutat. Res. 2011, 706, 7–12. [Google Scholar] [CrossRef]
- Meza, M.M.; Yu, L.; Rodriguez, Y.Y.; Guild, M.; Thompson, D.; Gandolfi, A.J.; Klimecki, W.T. Developmentally restricted genetic determinants of human arsenic metabolism: Association between urinary methylated arsenic and CYT19 polymorphisms in children. Environ. Health Perspect. 2005, 113, 775–781. [Google Scholar] [CrossRef]
- Gomez-Rubio, P.; Meza-Montenegro, M.M.; Cantu-Soto, E.; Klimecki, W.T. Genetic association between intronic variants in AS3MT and arsenic methylation efficiency is focused on a large linkage disequilibrium cluster in chromosome 10. J. Appl. Toxicol. 2010, 30, 260–270. [Google Scholar]
- Lindberg, A.L.; Kumar, R.; Goessler, W.; Thirumaran, R.; Gurzau, E.; Koppova, K.; Rudnai, P.; Leonardi, G.; Fletcher, T.; Vahter, M. Metabolism of low-dose inorganic arsenic in a central European population: Influence of sex and genetic polymorphisms. Environ. Health Perspect. 2007, 115, 1081–1086. [Google Scholar] [CrossRef]
- Agusa, T.; Iwata, H.; Fujihara, J.; Kunito, T.; Takeshita, H.; Minh, T.B.; Trang, P.T.; Viet, P.H.; Tanabe, S. Genetic polymorphisms in AS3MT and arsenic metabolism in residents of the Red River Delta, Vietnam. Toxicol. Appl. Pharmacol. 2009, 236, 131–141. [Google Scholar] [CrossRef]
- Gong, G.; O’Bryant, S.E. Low-level arsenic exposure, AS3MT gene polymorphism and cardiovascular diseases in rural Texas counties. Environ. Res. 2012, 113, 52–57. [Google Scholar] [CrossRef]
- Chen, J.W.; Wang, S.L.; Wang, Y.H.; Sun, C.W.; Huang, Y.L.; Chen, C.J.; Li, W.F. Arsenic methylation, GSTO1 polymorphisms, and metabolic syndrome in an arseniasis endemic area of southwestern Taiwan. Chemosphere 2012, 88, 432–438. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Lien, L.M.; Chung, W.T.; Hsieh, F.I.; Hsieh, P.F.; Wu, M.M.; Tseng, H.P.; Chiou, H.Y.; Chen, C.J. Significantly increased risk of carotid atherosclerosis with arsenic exposure and polymorphisms in arsenic metabolism genes. Environ. Res. 2011, 111, 804–810. [Google Scholar] [CrossRef]
- Agusa, T.; Kunito, T.; Tue, N.M.; Lan, V.T.; Fujihara, J.; Takeshita, H.; Minh, T.B.; Trang, P.T.; Takahashi, S.; Viet, P.H.; et al. Individual variations in arsenic metabolism in Vietnamese: The association with arsenic exposure and GSTP1 genetic polymorphism. Metallomics 2012, 4, 91–100. [Google Scholar] [CrossRef]
- Hsu, L.I.; Chiu, A.W.; Huan, S.K.; Chen, C.L.; Wang, Y.H.; Hsieh, F.I.; Chou, W.L.; Wang, L.H.; Chen, C.J. SNPs of GSTM1, T1, P1, epoxide hydrolase and DNA repair enzyme XRCC1 and risk of urinary transitional cell carcinoma in southwestern Taiwan. Toxicol. Appl. Pharmacol. 2008, 228, 144–155. [Google Scholar] [CrossRef]
- Hsu, L.I.; Chen, W.P.; Yang, T.Y.; Chen, Y.H.; Lo, W.C.; Wang, Y.H.; Liao, Y.T.; Hsueh, Y.M.; Chiou, H.Y.; Wu, M.M.; Chen, C.J. Genetic polymorphisms in glutathione S-transferase (GST) superfamily and risk of arsenic-induced urothelial carcinoma in residents of southwestern Taiwan. J. Biomed. Sci. 2011, 18. [Google Scholar] [CrossRef]
- Lesseur, C.; Gilbert-Diamond, D.; Andrew, A.S.; Ekstrom, R.M.; Li, Z.; Kelsey, K.T.; Marsit, C.J.; Karagas, M.R. A case-control study of polymorphisms in xenobiotic and arsenic metabolism genes and arsenic-related bladder cancer in New Hampshire. Toxicol. Lett. 2012, 210, 100–106. [Google Scholar] [CrossRef]
- Wang, Y.H.; Wu, M.M.; Hong, C.T.; Lien, L.M.; Hsieh, Y.C.; Tseng, H.P.; Chang, S.F.; Su, C.L.; Chiou, H.Y.; Chen, C.J. Effects of arsenic exposure and genetic polymorphisms of p53, glutathione S-transferase M1, T1, and P1 on the risk of carotid atherosclerosis in Taiwan. Atherosclerosis 2007, 192, 305–312. [Google Scholar] [CrossRef]
- Chiou, H.Y.; Hsueh, Y.M.; Hsieh, L.L.; Hsu, L.I.; Hsu, Y.H.; Hsieh, F.I.; Wei, M.L.; Chen, H.C.; Yang, H.T.; Leu, L.C.; et al. Arsenic methylation capacity, body retention, and null genotypes of glutathione S-transferase M1 and T1 among current arsenic-exposed residents in Taiwan. Mutat. Res. 1997, 386, 197–207. [Google Scholar] [CrossRef]
- Steinmaus, C.; Moore, L.E.; Shipp, M.; Kalman, D.; Rey, O.A.; Biggs, M.L.; Hopenhayn, C.; Bates, M.N.; Zheng, S.; Wiencke, J.K.; Smith, A.H. Genetic polymorphisms in MTHFR 677 and 1298, GSTM1 and T1, and metabolism of arsenic. J. Toxicol. Environ. Health A 2007, 70, 159–170. [Google Scholar] [CrossRef]
- Agusa, T.; Iwata, H.; Fujihara, J.; Kunito, T.; Takeshita, H.; Minh, T.B.; Trang, P.T.; Viet, P.H.; Tanabe, S. Genetic polymorphisms in glutathione S-transferase (GST) superfamily and arsenic metabolism in residents of the Red River Delta, Vietnam. Toxicol. Appl. Pharmacol. 2010, 242, 352–362. [Google Scholar] [CrossRef]
- Wu, M.M.; Chiou, H.Y.; Chen, C.L.; Hsu, L.I.; Lien, L.M.; Wang, C.H.; Hsieh, Y.C.; Wang, Y.H.; Hsueh, Y.M.; Lee, T.C.; et al. Association of heme oxygenase-1 GT-repeat polymorphism with blood pressure phenotypes and its relevance to future cardiovascular mortality risk: An observation based on arsenic-exposed individuals. Atherosclerosis 2011, 219, 704–708. [Google Scholar] [CrossRef]
- Wu, M.M.; Chiou, H.Y.; Lee, T.C.; Chen, C.L.; Hsu, L.I.; Wang, Y.H.; Huang, W.L.; Hsieh, Y.C.; Yang, T.Y.; Lee, C.Y.; et al. GT-repeat polymorphism in the heme oxygenase-1 gene promoter and the risk of carotid atherosclerosis related to arsenic exposure. J. Biomed. Sci. 2010, 17. [Google Scholar] [CrossRef]
- De Chaudhuri, S.; Mahata, J.; Das, J.K.; Mukherjee, A.; Ghosh, P.; Sau, T.J.; Mondal, L.; Basu, S.; Giri, A.K.; Roychoudhury, S. Association of specific p53 polymorphisms with keratosis in individuals exposed to arsenic through drinking water in West Bengal, India. Mutat. Res. 2006, 601, 102–112. [Google Scholar] [CrossRef]
- Huang, C.Y.; Su, C.T.; Chu, J.S.; Huang, S.P.; Pu, Y.S.; Yang, H.Y.; Chung, C.J.; Wu, C.C.; Hsueh, Y.M. The polymorphisms of P53 codon 72 and MDM2 SNP309 and renal cell carcinoma risk in a low arsenic exposure area. Toxicol. Appl. Pharmacol. 2011, 257, 349–355. [Google Scholar] [CrossRef]
- Wood, T.C.; Salavagionne, O.E.; Mukherjee, B.; Wang, L.; Klumpp, A.F.; Thomae, B.A.; Eckloff, B.W.; Schaid, D.J.; Wieben, E.D.; Weinshilboum, R.M. Human arsenic methyltransferase (AS3MT) pharmacogenetics: Gene resequencing and functional genomics studies. J. Biol. Chem. 2006, 281, 7364–7373. [Google Scholar] [CrossRef]
- Drobná, Z.; Waters, S.B.; Walton, F.S.; LeCluyse, E.L.; Thomas, D.J.; Stýblo, M. Interindividual variation in the metabolism of arsenic in cultured primary human hepatocytes. Toxicol. Appl. Pharmacol. 2004, 201, 166–177. [Google Scholar] [CrossRef]
- Hernández, A.; Xamena, N.; Surrallés, J.; Sekaran, C.; Tokunaga, H.; Quinteros, D.; Creus, A.; Marcos, R. Role of the Met(287)Thr polymorphism in the AS3MT gene on the metabolic arsenic profile. Mutat. Res. 2008, 637, 80–92. [Google Scholar] [CrossRef]
- Agusa, T.; Fujihara, J.; Takeshita, H.; Iwata, H. Individual variations in inorganic Arsenic metabolism associated with AS3MT genetic polymorphisms. Int. J. Mol. Sci. 2011, 12, 2351–2382. [Google Scholar] [CrossRef]
- Zakharyan, R.A.; Sampayo-Reyes, A.; Healy, S.M.; Tsaprailis, G.; Board, P.G.; Liebler, D.C.; Aposhian, H.V. Human monomethylarsonic acid (MMA(V)) reductase is a member of the glutathione-S-transferase superfamily. Chem. Res. Toxicol. 2001, 14, 1051–1057. [Google Scholar] [CrossRef]
- Whitbread, A.K.; Tetlow, N.; Eyre, H.J.; Sutherland, G.R.; Board, P.G. Characterization of the human Omega class glutathione transferase genes and associated polymorphisms. Pharmacogenetics 2003, 13, 131–144. [Google Scholar] [CrossRef]
- Paiva, L.; Marcos, R.; Creus, A.; Coggan, M.; Oakley, A.J.; Board, P.G. Polymorphism of glutathione transferase Omega 1 in a population exposed to a high environmental arsenic burden. Pharmacogenet. Genom. 2008, 18, 1–10. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Miller, D.S.; Saavedra, J.E.; Keefer, L.K.; Johnson, D.R.; Klaassen, C.D.; Waalkes, M.P. Overexpression of glutathione S-transferase II and multidrug resistance transport proteins is associated with acquired tolerance to inorganic arsenic. Mol. Pharmacol. 2001, 60, 302–309. [Google Scholar]
- Brambila, E.M.; Achanzar, W.E.; Qu, W.; Webber, M.M.; Waalkes, M.P. Chronic arsenic-exposed human prostate epithelial cells exhibit stable arsenic tolerance: Mechanistic implications of altered cellular glutathione and glutathione S-transferase. Toxicol. Appl. Pharmacol. 2002, 183, 99–107. [Google Scholar]
- Zimniak, P.; Nanduri, B.; Pikuła, S.; Bandorowicz-Pikuła, J.; Singhal, S.S.; Srivastava, S.K.; Awasthi, S.; Awasthi, Y.C. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. Eur. J. Biochem. 1994, 224, 893–899. [Google Scholar] [CrossRef]
- McCarty, K.M.; Chen, Y.C.; Quamruzzaman, Q.; Rahman, M.; Mahiuddin, G.; Hsueh, Y.M.; Su, L.; Smith, T.; Ryan, L.; Christiani, D.C. Arsenic methylation, GSTT1, GSTM1, GSTP1 polymorphisms, and skin lesions. Environ. Health Perspect. 2007, 115, 341–345. [Google Scholar] [CrossRef]
- Beebe-Dimmer, J.L.; Iyer, P.T.; Nriagu, J.O.; Keele, G.; Mehta, S.; Meliker, J.R.; Lange, E.M.; Schwartz, A.G.; Zuhlke, K.A.; Schottenfeld, D.; Cooney, K.A. Genetic variation in Glutathione S-transferase omega-1, arsenic methyltransferase and methylene-tetrahydrofolate reductase, arsenic exposure and bladder cancer: A case-control study. Environ. Health. 2012, 11. [Google Scholar] [CrossRef]
- Hill, J.W.; Evans, M.K. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006, 34, 1620–1632. [Google Scholar] [CrossRef]
- Vodicka, P.; Stetina, R.; Polakova, V.; Tulupova, E.; Naccarati, A.; Vodickova, L.; Kumar, R.; Hanova, M.; Pardini, B.; Slyskova, J.; Musak, L.; de Palma, G.; Soucek, P.; Hemminki, K. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis 2007, 28, 657–664. [Google Scholar]
- States, J.C.; Srivastava, S.; Chen, Y.; Barchowsky, A. Arsenic and cardiovascular disease. Toxicol. Sci. 2009, 107, 312–323. [Google Scholar]
- Morita, T. Heme oxygenase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1786–1795. [Google Scholar] [CrossRef]
- Brydun, A.; Watari, Y.; Yamamoto, Y.; Okuhara, K.; Teragawa, H.; Kono, F.; Chayama, K.; Oshima, T.; Ozono, R. Reduced expression of heme oxygenase-1 in patients with coronary atherosclerosis. Hypertens. Res. 2007, 30, 341–348. [Google Scholar] [CrossRef]
- Del Razo, L.M.; Quintanilla-Vega, B.; Brambila-Colombres, E.; Calderón-Aranda, E.S.; Manno, M.; Albores, A. Stress proteins induced by arsenic. Toxicol. Appl. Pharmacol. 2001, 177, 132–148. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Faita, F.; Cori, L.; Bianchi, F.; Andreassi, M.G. Arsenic-Induced Genotoxicity and Genetic Susceptibility to Arsenic-Related Pathologies. Int. J. Environ. Res. Public Health 2013, 10, 1527-1546. https://doi.org/10.3390/ijerph10041527
Faita F, Cori L, Bianchi F, Andreassi MG. Arsenic-Induced Genotoxicity and Genetic Susceptibility to Arsenic-Related Pathologies. International Journal of Environmental Research and Public Health. 2013; 10(4):1527-1546. https://doi.org/10.3390/ijerph10041527
Chicago/Turabian StyleFaita, Francesca, Liliana Cori, Fabrizio Bianchi, and Maria Grazia Andreassi. 2013. "Arsenic-Induced Genotoxicity and Genetic Susceptibility to Arsenic-Related Pathologies" International Journal of Environmental Research and Public Health 10, no. 4: 1527-1546. https://doi.org/10.3390/ijerph10041527
APA StyleFaita, F., Cori, L., Bianchi, F., & Andreassi, M. G. (2013). Arsenic-Induced Genotoxicity and Genetic Susceptibility to Arsenic-Related Pathologies. International Journal of Environmental Research and Public Health, 10(4), 1527-1546. https://doi.org/10.3390/ijerph10041527