Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity Through ATP-Competitive Inhibition

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
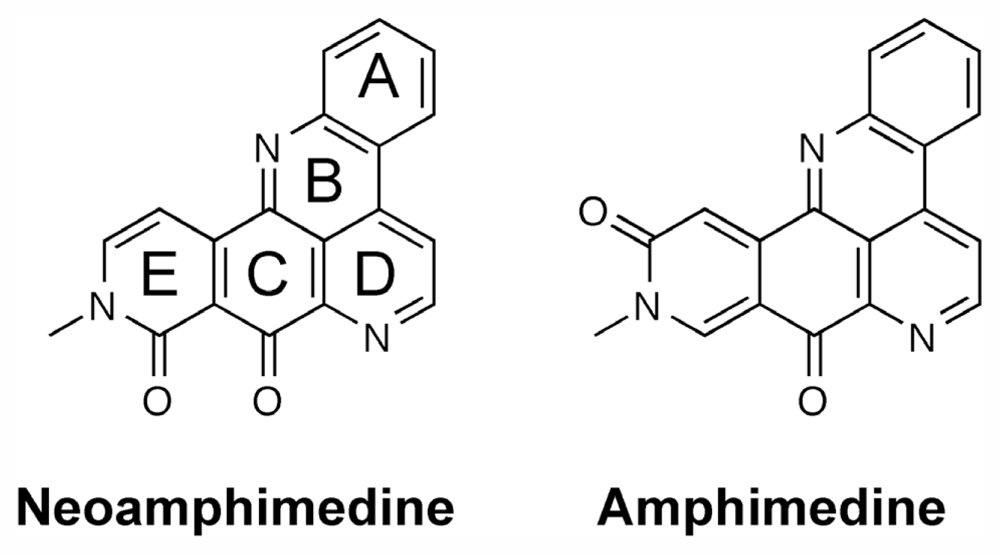
1. Introduction
2. Results and Discussion
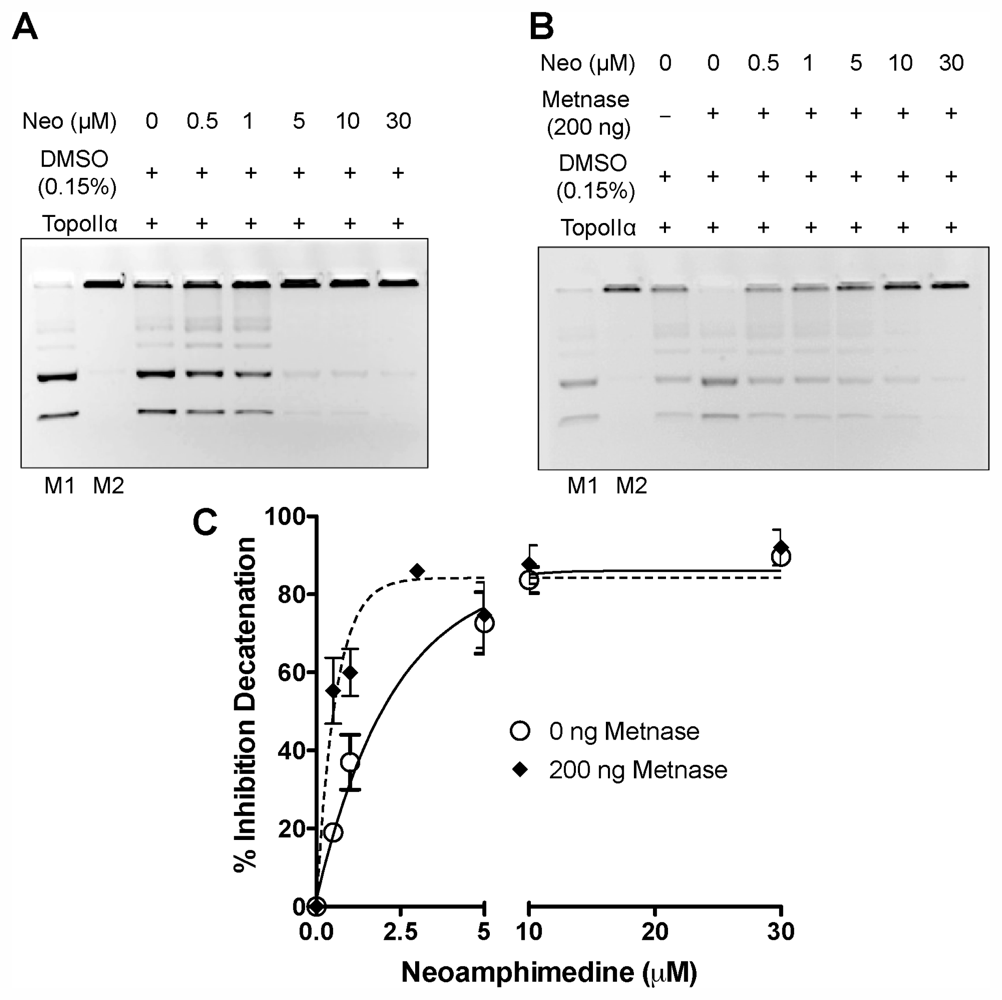
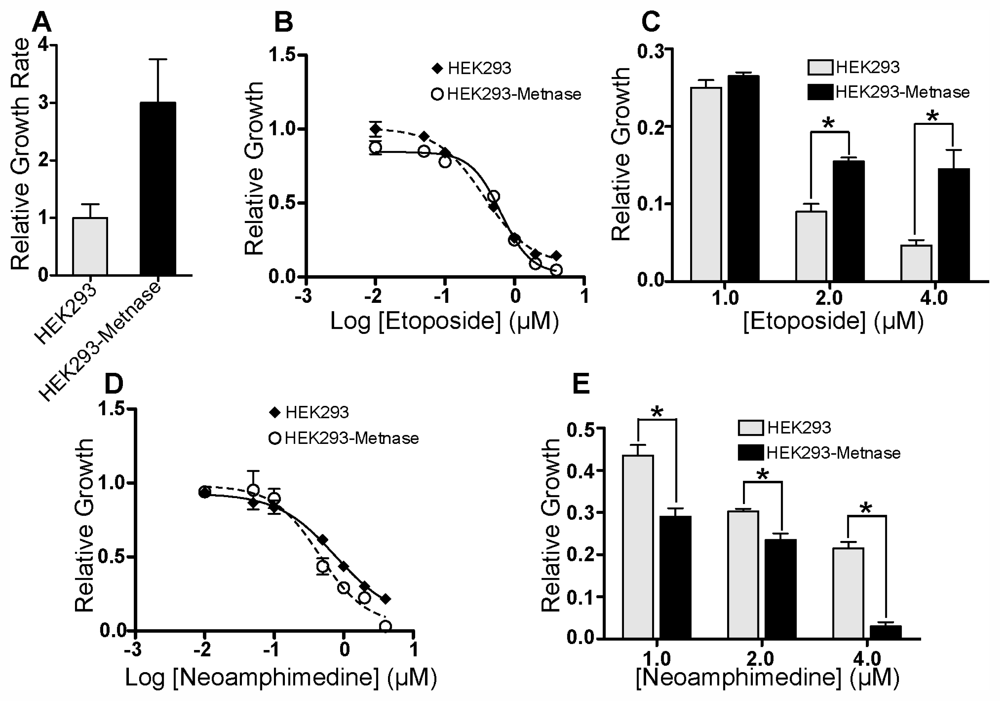
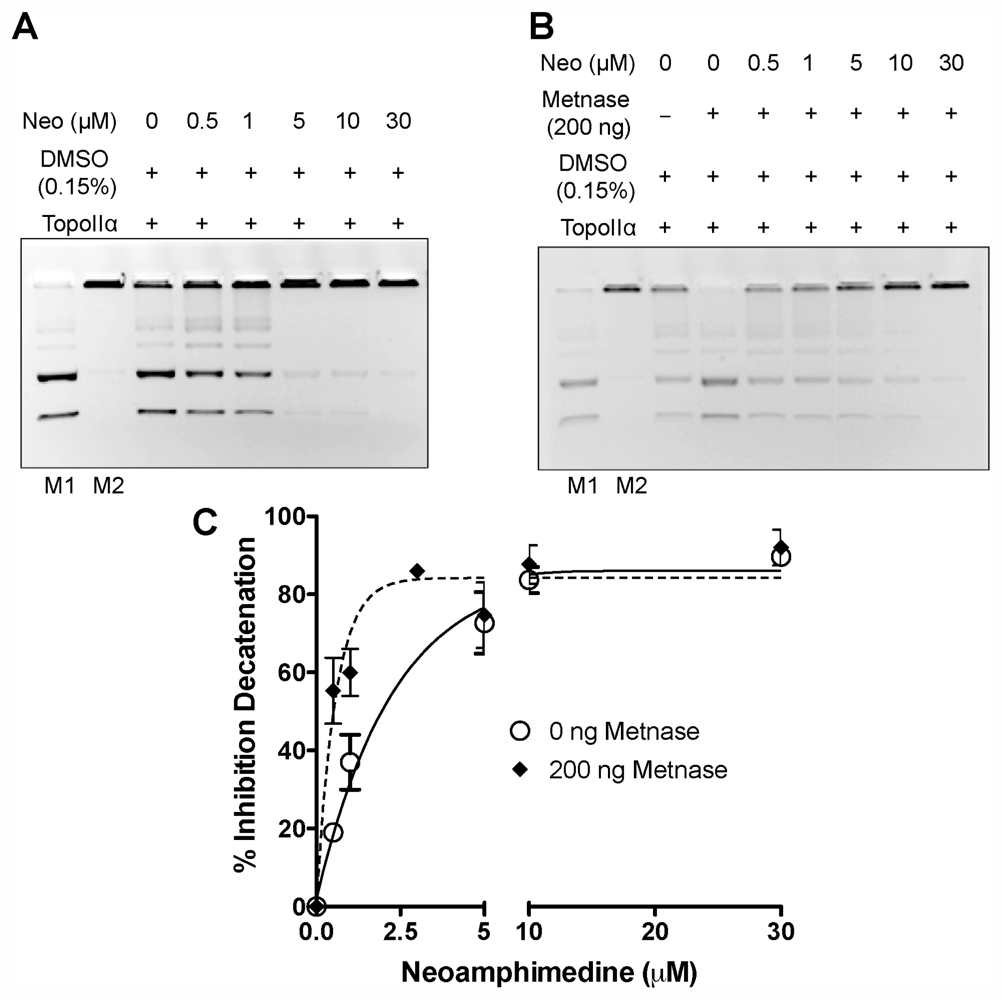
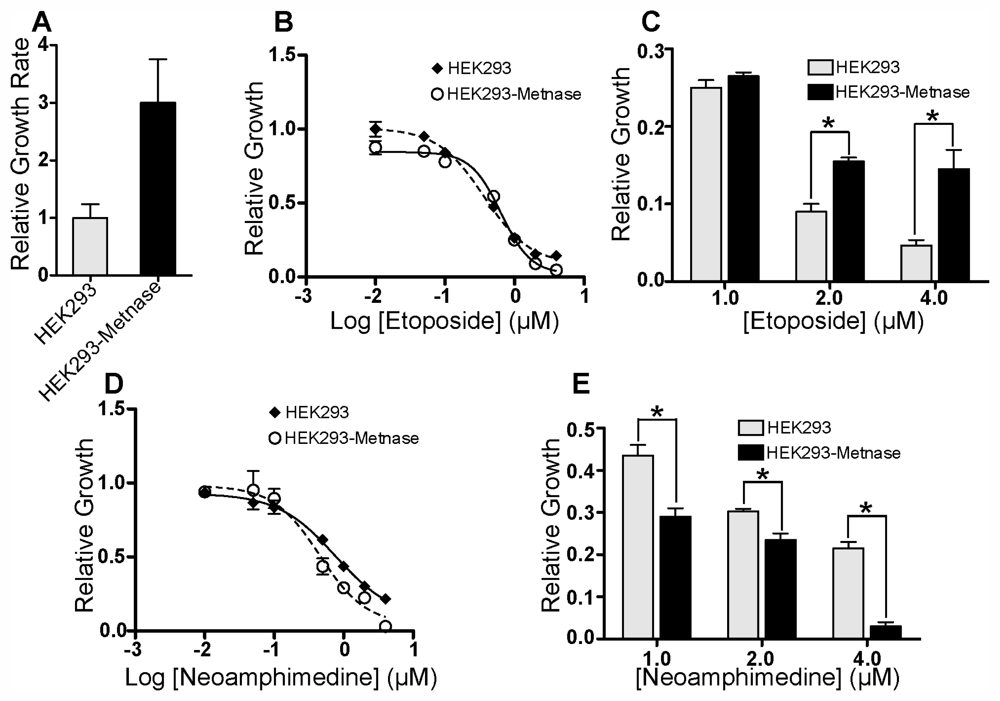
2.1. In Vitro DNA Decatenation and Cell Proliferation Assays
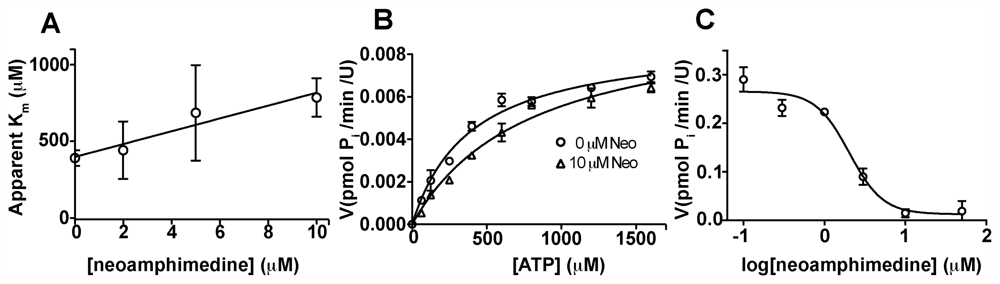
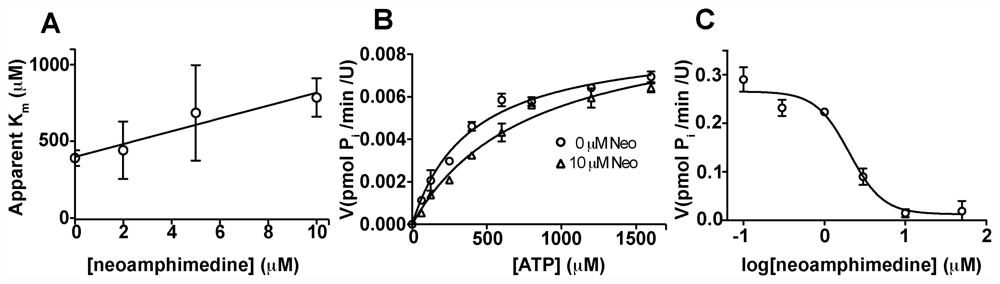
2.2. Competitive Inhibition Studies with Neoamphimedine and TopoIIα
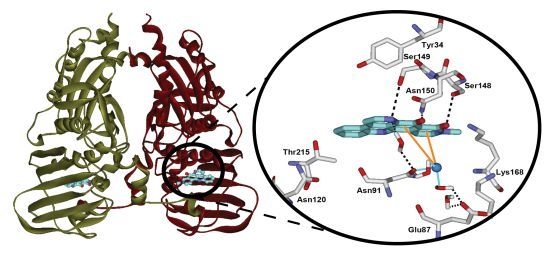
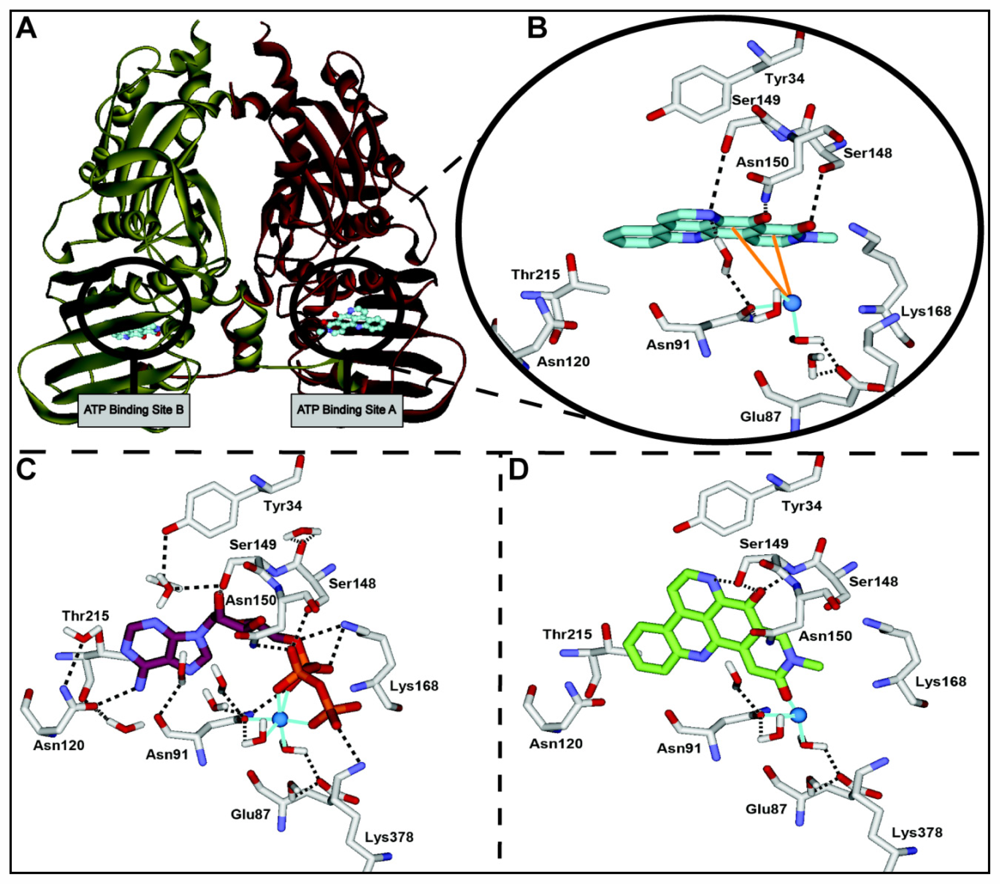
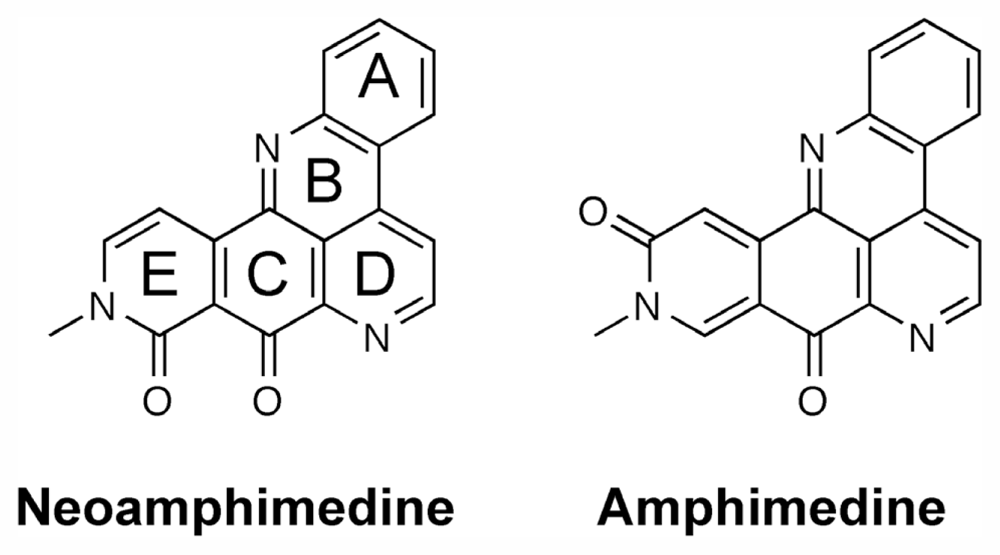
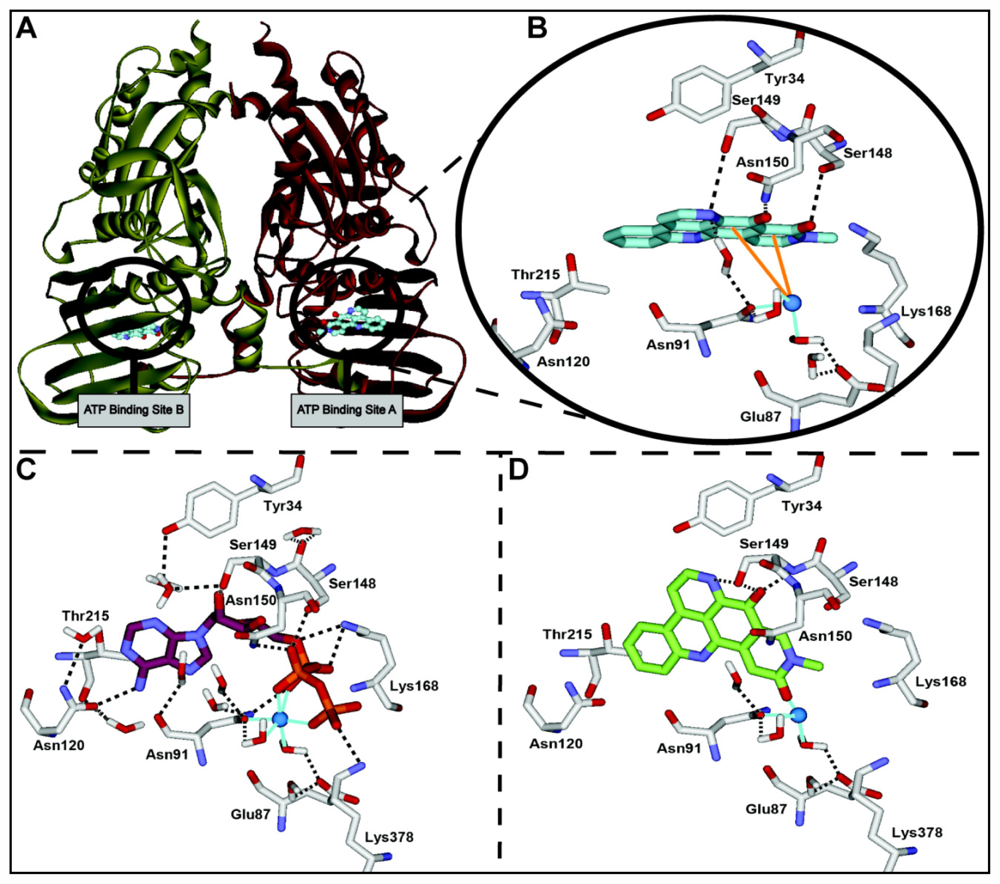
2.3. Computational Modeling of Neoamphimedine with TopoIIα
2.4. Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. DNA Decatenation Assay
3.3. Cell Proliferation Assay
3.4. Competitive Inhibition Studies
3.5. Molecular Docking of Neoamphimedine and Amphimedine with TopoIIα
4. Conclusions
Acknowledgments
References
- Champoux, J.J. DNA Topoisomerases: Structure, Function and Mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [Google Scholar]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev.Cancer 2009, 9, 338–350. [Google Scholar]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J. Biol. Chem 2005, 280, 37041–37047. [Google Scholar]
- Osheroff, N.; Zechiedrich, E.L.; Gale, K.C. Catalytic function of DNA topoisomerase II. BioEssays 1991, 13, 269–273. [Google Scholar]
- Williamson, E.A.; Rasila, K.K.; Corwin, L.K.; Wray, J.; Beck, B.D.; Severns, V.; Mobarak, C.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. The SET and transposase domain protein Metnase enhances chromosome decatenation: Regulation by automethylation. Nucleic Acids Res 2008, 36, 5822–5831. [Google Scholar]
- Wray, J.; Williamson, E.A.; Royce, M.; Shaheen, M.; Beck, B.D.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Metnase mediates resistance to topoisomerase II inhibitors in breast cancer cells. PLoS One 2009, 4, e5323. [Google Scholar]
- Wray, J.; Williamson, E.A.; Sheema, S.; Lee, S.H.; Libby, E.; Willman, C.L.; Nickoloff, J.A.; Hromas, R. Metnase mediates chromosome decatenation in acute leukemia cells. Blood 2009, 114, 1852–1858. [Google Scholar]
- Marshall, K.M.; Matumoto, S.S.; Holden, J.A.; Concepcion, G.P.; Tasdemir, D.; Ireland, C.M.; Barrows, L.R. The anti-neoplastic and novel topoisomerase II-mediated cytotoxicity of neoamphimedine, a marine pyridoacridine. Biochem. Pharmacol 2003, 66, 447–458. [Google Scholar]
- Guzman, F.S.D.; Carte, B.; Troupe, N.; Faulkner, D.J.; Harper, M.K.; Concepcion, G.P.; Mangalindan, G.C.; Matsumoto, S.S.; Barrows, L.R.; Ireland, C.M. Neoamphimideine: A New Pyridoacridine Topoismerase II Inhibitor Which Cantenates DNA. J. Org. Chem 1999, 64, 1400–1402. [Google Scholar]
- Bender, R.P.; Jablonksy, M.J.; Shadid, M.; Romaine, I.; Dunlap, N.; Anklin, C.; Graves, D.E.; Osheroff, N. Substituents on etoposide that interact with human topoisomerase IIalpha in the binary enzyme-drug complex: contributions to etoposide binding and activity. Biochemistry 2008, 47, 4501–4509. [Google Scholar]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar]
- Classen, S. Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc. Natl. Acad. Sci. USA 2003, 100, 10629–10634. [Google Scholar]
- Baird, C.; Harkins, T.; Morris, S.; Lindsley, J. Topoisomerase II drives DNA transport by hydrolyzing one ATP. Proc. Natl. Acad. Sci. USA 1999, 96, 13685–13690. [Google Scholar]
- Maxwell, A.; Costenaro, L.; Mitelheiser, S.; Bates, A.D. Coupling ATP hydrolysis to DNA strand passage in type IIA DNA topoisomerases. Biochem. Soc. Trans 2005, 33, 1460–1464. [Google Scholar]
- Lanzetta, P.A.; Alvarez, L.J.; Reinach, P.S.; Candia, O.A. An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem 1979, 100, 95–97. [Google Scholar]
- Chene, P.; Rudloff, J.; Schoepfer, J.; Furet, P.; Meier, P.; Qian, Z.; Schlaeppi, J.M.; Schmitz, R.; Radimerski, T. Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem. Biol 2009, 9, 1. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov 2006, 5, 219–234. [Google Scholar]
- Beck, W.T.; Danks, M.K.; Wolverton, J.S.; Kim, R.; Chen, M. Drug resistance associated with altered DNA topoisomerase II. Adv. Enzyme Regul 1993, 33, 113–127. [Google Scholar]
- Matsumoto, Y.; Kunishio, K.; Nagao, S. Increased phosphorylation of DNA topoisomerase II in etoposide resistant mutants of human glioma cell line. J. Neurooncol 1999, 45, 37–46. [Google Scholar]
- Shapiro, P.S.; Whalen, A.M.; Tolwinski, N.S.; Wilsbacher, J.; Froelich-Ammon, S.J.; Garcia, M.; Osheroff, N.; Ahn, N.G. Extracellular signal-regulated kinase activates topoisomerase IIalpha through a mechanism independent of phosphorylation. Mol. Cell. Biol 1999, 19, 3551–3560. [Google Scholar]
- Marshall, K.M.; Andjelic, C.D.; Tasdemir, D.; Concepcion, G.P.; Ireland, C.M.; Barrows, L.R. Deoxyamphimedine, a pyridoacridine alkaloid, damages DNA via the production of reactive oxygen species. Mar. Drugs 2009, 7, 196–209. [Google Scholar]
- Matsumoto, S.S.; Haughey, H.M.; Schmehl, D.M.; Venables, D.A.; Ireland, C.M.; Holden, J.A.; Barrows, L.R. Makaluvamines vary in ability to induce dose-dependent DNA cleavage via topoisomerase II interaction. Anticancer Drugs 1999, 10, 39–45. [Google Scholar]
- Rybenkov, V.V.; Ullsperger, C.; Vologodskii, A.V.; Cozzarelli, N.R. Simplification of DNA topology below equilibrium values by type II topoisomerases. Science 1997, 277, 690–693. [Google Scholar]
- Discovery Studio, version 2.5.5; Accelrys Inc: San Diego, CA, USA, 2009.
- LaBarbera, D.V.; Bugni, T.S.; Ireland, C.M. The Total Synthesis of Neoamphimedine. J. Org. Chem 2007, 72, 8501–8505. [Google Scholar]
- Lee, S.H.; Oshige, M.; Durant, S.T.; Rasila, K.K.; Williamson, E.A.; Ramsey, H.; Kwan, L.; Nickoloff, J.A.; Hromas, R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18075–18080. [Google Scholar]
- Quantity One; BioRad Laboratories Inc: Hercules, CA, USA, 2009.
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst 1990, 82, 1107–1112. [Google Scholar]
- SoftMax Pro, version 4.8; Molecular Devices LLC: Sunnyvale, CA, USA, 2004.
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [Google Scholar]
- Prism, version 5.0.3; GraphPad Software Inc: La Jolla, CA, USA, 2007.
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [Google Scholar]
- Montes, M.; Miteva, M.A.; Villoutreix, B.O. Structure-based virtual ligand screening with LigandFit: pose prediction and enrichment of compound collections. Proteins 2007, 68, 712–725. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ponder, J.; Yoo, B.H.; Abraham, A.D.; Li, Q.; Ashley, A.K.; Amerin, C.L.; Zhou, Q.; Reid, B.G.; Reigan, P.; Hromas, R.; et al. Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity Through ATP-Competitive Inhibition. Mar. Drugs 2011, 9, 2397-2408. https://doi.org/10.3390/md9112397
Ponder J, Yoo BH, Abraham AD, Li Q, Ashley AK, Amerin CL, Zhou Q, Reid BG, Reigan P, Hromas R, et al. Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity Through ATP-Competitive Inhibition. Marine Drugs. 2011; 9(11):2397-2408. https://doi.org/10.3390/md9112397
Chicago/Turabian StylePonder, Jessica, Byong Hoon Yoo, Adedoyin D. Abraham, Qun Li, Amanda K. Ashley, Courtney L. Amerin, Qiong Zhou, Brian G. Reid, Philip Reigan, Robert Hromas, and et al. 2011. "Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity Through ATP-Competitive Inhibition" Marine Drugs 9, no. 11: 2397-2408. https://doi.org/10.3390/md9112397
APA StylePonder, J., Yoo, B. H., Abraham, A. D., Li, Q., Ashley, A. K., Amerin, C. L., Zhou, Q., Reid, B. G., Reigan, P., Hromas, R., Nickoloff, J. A., & LaBarbera, D. V. (2011). Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity Through ATP-Competitive Inhibition. Marine Drugs, 9(11), 2397-2408. https://doi.org/10.3390/md9112397