Marine Metagenomics: New Tools for the Study and Exploitation of Marine Microbial Metabolism

 and
and
Abstract
:1. Introduction
2. Molecular Approaches to Study Marine Microbial Biodiversity
2.1. Who is there?
2.2. Resources for marine metagenomic analysis
3. Functional Metagenomic Based Approaches
4. Marine Microbes and Their Potential for Functional Metagenomics
4.1. Biotechnological uses of marine enzymes
4.2. Novel enzyme discoveries
4.3. Phenotypic screens
5. Novel Approaches
Acknowledgements
References and Notes
- Sogin, ML; Morrison, HG; Huber, JA; Mark Welch, D; Huse, SM; Neal, PR; Arrieta, JM; Herndl, GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA 2006, 103, 12115–12120. [Google Scholar]
- Dufresne, A; Garczarek, L; Partensky, F. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol 2005, 6, R14. [Google Scholar]
- Giovannoni, SJ; Tripp, HJ; Givan, S; Podar, M; Vergin, KL; Baptista, D; Bibbs, L; Eads, J; Richardson, TH; Noordewier, M; Rappe, MS; Short, JM; Carrington, JC; Mathur, EJ. Genome streamlining in a cosmopolitan oceanic bacterium. Science 2005, 309, 1242–1245. [Google Scholar]
- Beatty, JT; Overmann, J; Lince, MT; Manske, AK; Lang, AS; Blankenship, RE; Van Dover, CL; Martinson, TA; Plumley, FG. An obligately photosynthetic bacterial anaerobe from a deep-sea hydrothermal vent. Proc Natl Acad Sci USA 2005, 102, 9306–9310. [Google Scholar]
- Jorgensen, BB; Boetius, A. Feast and famine--microbial life in the deep-sea bed. Nat Rev Microbiol 2007, 5, 770–781. [Google Scholar]
- Handelsman, J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 2004, 68, 669–685. [Google Scholar]
- Edwards, RA; Rodriguez-Brito, B; Wegley, L; Haynes, M; Breitbart, M; Peterson, DM; Saar, MO; Alexander, S; Alexander, EC, Jr; Rohwer, F. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 2006, 7, 57. [Google Scholar]
- Wommack, KE; Bhavsar, J; Ravel, J. Metagenomics: read length matters. Appl Environ Microbiol 2008, 74, 1453–1463. [Google Scholar]
- Horton, M; Bodenhausen, N; Bergelson, J. MARTA: A suite of Java-based tools for assigning taxonomic status to DNA sequences. Bioinformatics 2009, 6, 568–569. [Google Scholar]
- Yu, F; Sun, Y; Liu, L; Farmerie, W. GSTaxClassifier: a genomic signature based taxonomic classifier for metagenomic data analysis. Bioinformation 2010, 4, 46–49. [Google Scholar]
- Finn, RD; Tate, J; Mistry, J; Coggill, PC; Sammut, SJ; Hotz, HR; Ceric, G; Forslund, K; Eddy, SR; Sonnhammer, EL; Bateman, A. The Pfam protein families database. Nucleic Acids Res 2008, 36, D281–288. [Google Scholar]
- Tatusov, RL; Koonin, EV; Lipman, DJ. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar]
- Selengut, JD; Haft, DH; Davidsen, T; Ganapathy, A; Gwinn-Giglio, M; Nelson, WC; Richter, AR; White, O. TIGRFAMs and Genome Properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res 2007, 35, D260–264. [Google Scholar]
- Markowitz, VM; Ivanova, N; Palaniappan, K; Szeto, E; Korzeniewski, F; Lykidis, A; Anderson, I; Mavromatis, K; Kunin, V; Garcia Martin, H; Dubchak, I; Hugenholtz, P; Kyrpides, NC. An experimental metagenome data management and analysis system. Bioinformatics 2006, 22, e359–e367. [Google Scholar]
- Rusch, DB; Halpern, AL; Sutton, G; Heidelberg, KB; Williamson, S; Yooseph, S; Wu, D; Eisen, JA; Hoffman, JM; Remington, K; Beeson, K; Tran, B; Smith, H; Baden-Tillson, H; Stewart, C; Thorpe, J; Freeman, J; Andrews-Pfannkoch, C; Venter, JE; Li, K; Kravitz, S; Heidelberg, JF; Utterback, T; Rogers, YH; Falcon, LI; Souza, V; Bonilla-Rosso, G; Eguiarte, LE; Karl, DM; Sathyendranath, S; Platt, T; Bermingham, E; Gallardo, V; Tamayo-Castillo, G; Ferrari, MR; Strausberg, RL; Nealson, K; Friedman, R; Frazier, M; Venter, JC. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 2007, 5, e77. [Google Scholar]
- Venter, JC; Remington, K; Heidelberg, JF; Halpern, AL; Rusch, D; Eisen, JA; Wu, D; Paulsen, I; Nelson, KE; Nelson, W; Fouts, DE; Levy, S; Knap, AH; Lomas, MW; Nealson, K; White, O; Peterson, J; Hoffman, J; Parsons, R; Baden-Tillson, H; Pfannkoch, C; Rogers, YH; Smith, HO. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar]
- Giovannoni, SJ; Rappe, MS; Vergin, KL; Adair, NL. 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the Green Non-Sulfur bacteria. Proc Natl Acad Sci USA 1996, 93, 7979–7984. [Google Scholar]
- Martin-Cuadrado, AB; Lopez-Garcia, P; Alba, JC; Moreira, D; Monticelli, L; Strittmatter, A; Gottschalk, G; Rodriguez-Valera, F. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS One 2007, 2, e914. [Google Scholar]
- Zaballos, M; Lopez-Lopez, A; Ovreas, L; Bartual, SG; D’Auria, G; Alba, JC; Legault, B; Pushker, R; Daae, FL; Rodriguez-Valera, F. Comparison of prokaryotic diversity at offshore oceanic locations reveals a different microbiota in the Mediterranean Sea. FEMS Microbiol Ecol 2006, 56, 389–405. [Google Scholar]
- DeLong, EF; Preston, CM; Mincer, T; Rich, V; Hallam, SJ; Frigaard, NU; Martinez, A; Sullivan, MB; Edwards, R; Brito, BR; Chisholm, SW; Karl, DM. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar]
- Biddle, JF; Fitz-Gibbon, S; Schuster, SC; Brenchley, JE; House, CH. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc Natl Acad Sci USA 2008, 105, 10583–10588. [Google Scholar]
- Cooney, RP; Pantos, O; Le Tissier, MD; Barer, MR; O’Donnell, AG; Bythell, JC. Characterization of the bacterial consortium associated with black band disease in coral using molecular microbiological techniques. Environ Microbiol 2002, 4, 401–413. [Google Scholar]
- Frias-Lopez, J; Klaus, JS; Bonheyo, GT; Fouke, BW. Bacterial community associated with black band disease in corals. Appl Environ Microbiol 2004, 70, 5955–5962. [Google Scholar]
- Kennedy, J; Codling, CE; Jones, BV; Dobson, AD; Marchesi, JR. Diversity of microbes associated with the marine sponge, Haliclona simulans, isolated from Irish waters and identification of polyketide synthase genes from the sponge metagenome. Environ Microbiol 2008, 10, 1888–1902. [Google Scholar]
- Lafi, FF; Fuerst, JA; Fieseler, L; Engels, C; Goh, WW; Hentschel, U. Widespread distribution of poribacteria in demospongiae. Appl Environ Microbiol 2009, 75, 5695–5699. [Google Scholar]
- Webster, NS; Taylor, MW; Behnam, F; Lucker, S; Rattei, T; Whalan, S; Horn, M; Wagner, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ Microbiol 2009. [Google Scholar]
- Moreira, D; Rodriguez-Valera, F; Lopez-Garcia, P. Metagenomic analysis of mesopelagic Antarctic plankton reveals a novel deltaproteobacterial group. Microbiology 2006, 152, 505–517. [Google Scholar]
- Fieseler, L; Horn, M; Wagner, M; Hentschel, U. Discovery of the novel candidate phylum “Poribacteria” in marine sponges. Appl Environ Microbiol 2004, 70, 3724–3732. [Google Scholar]
- Holmes, B; Blanch, H. Genus-specific associations of marine sponges with group 1 crenarchaeotes. Mar Biol 2007, 150, 759–771. [Google Scholar]
- Mohamed, NM; Cicirelli, EM; Kan, J; Chen, F; Fuqua, C; Hill, RT. Diversity and quorum-sensing signal production of Proteobacteria associated with marine sponges. Environ Microbiol 2008, 10, 75–86. [Google Scholar]
- Pape, T; Hoffmann, F; Queric, N.-V; von Juterzenka, K; Reitner, J; Michaelis, W. Dense populations of Archaea associated with the demosponge Tentorium semisuberites Schmidt, 1870 from Artic deep-waters. Polar Biol 2006, 29, 662–667. [Google Scholar]
- Cragg, GM; Grothaus, PG; Newman, DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev 2009, 109, 3012–3043. [Google Scholar]
- Wegley, L; Edwards, R; Rodriguez-Brito, B; Liu, H; Rohwer, F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 2007, 9, 2707–2719. [Google Scholar]
- Seshardi, R; Kravitz, SA; Smarr, L; Gilna, P; Frazier, M. CAMERA: A Community Resource for Metagenomics. PLoS Biol 2007, 5, e75. [Google Scholar]
- Markowitz, VM; Ivanova, NN; Szeto, E; Palaniappan, K; Chu, K; Dalevi, D; Chen, IM; Grechkin, Y; Dubchak, I; Anderson, I; Lykidis, A; Mavromatis, K; Hugenholtz, P; Kyrpides, NC. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res 2008, 36, D534–538. [Google Scholar]
- Sharma, VK; Kumar, N; Prakash, T; Taylor, TD. MetaBioME: a database to explore commercially useful enzymes in metagenomic datasets. Nucleic Acids Res 2009, 38, D468–472. [Google Scholar]
- Hoff, KJ; Lingner, T; Meinicke, P; Tech, M. Orphelia: Predicting genes in metagenomic sequencing reads. Nucleic Acids Res 2009, 37, W101–105. [Google Scholar]
- Meyer, F; Paarmann, D; D’Souza, M; Olson, R; Glass, EM; Kubal, M; Paczian, T; Rodriguez, A; Stevens, R; Wilke, A; Wilkening, J; Edwards, RA. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf 2008, 9, 386. [Google Scholar]
- Liang, C; Schmid, A; Lopez-Sanchez, MJ; Moya, A; Gross, R; Bernhardt, J; Dandekar, T. JANE: efficient mapping of prokaryotic ESTs and variable length sequence reads on related template genomes. BMC Bioinf 2009, 10, 391. [Google Scholar]
- Gerlach, W; Junemann, S; Tille, F; Goesmann, A; Stoye, J. WebCARMA: a web application for the functional and taxonomic classification of unassembled metagenomic reads. BMC Bioinf 2009, 10, 430. [Google Scholar]
- Huson, DH; Auch, AF; Qi, J; Schuster, SC. MEGAN analysis of metagenomic data. Genome Res 2007, 17, 377–386. [Google Scholar]
- Mitra, S; Klar, B; Huson, DH. Visual and statistical comparison of metagenomes. Bioinformatics 2009, 25, 1849–1855. [Google Scholar]
- Kennedy, J; Marchesi, JR; Dobson, AD. Metagenomic approaches to exploit the biotechnological potential of the microbial consortia of marine sponges. Appl Microbiol Biotechnol 2007, 75, 11–20. [Google Scholar]
- D’Costa, VM; Griffiths, E; Wright, GD. Expanding the soil antibiotic resistome: exploring environmental diversity. Curr Opin Microbiol 2007, 10, 481–489. [Google Scholar]
- Lammle, K; Zipper, H; Breuer, M; Hauer, B; Buta, C; Brunner, H; Rupp, S. Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 2007, 127, 575–592. [Google Scholar]
- Suenaga, H; Ohnuki, T; Miyazaki, K. Functional screening of a metagenomic library for genes involved in microbial degradation of aromatic compounds. Environ Microbiol 2007, 9, 2289–2297. [Google Scholar]
- Azam, F. OCEANOGRAPHY: Microbial Control of Oceanic Carbon Flux: The Plot Thickens. Science 1998, 280, 694–696. [Google Scholar]
- Azam, F; Long, RA. Sea snow microcosms. Nature 2001, 414, 497–498. [Google Scholar]
- Whitman, WB; Coleman, DC; Wiebe, WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 1998, 95, 6578–6583. [Google Scholar]
- Amann, RI; Ludwig, W; Schleifer, KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 1995, 59, 143–169. [Google Scholar]
- Yooseph, S; Sutton, G; Rusch, DB; Halpern, AL; Williamson, SJ; Remington, K; Eisen, JA; Heidelberg, KB; Manning, G; Li, W; Jaroszewski, L; Cieplak, P; Miller, CS; Li, H; Mashiyama, ST; Joachimiak, MP; van Belle, C; Chandonia, JM; Soergel, DA; Zhai, Y; Natarajan, K; Lee, S; Raphael, BJ; Bafna, V; Friedman, R; Brenner, SE; Godzik, A; Eisenberg, D; Dixon, JE; Taylor, SS; Strausberg, RL; Frazier, M; Venter, JC. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 2007, 5, e16. [Google Scholar]
- Kennedy, J; Marchesi, JR; Dobson, AD. Marine metagenomics: strategies for the discovery of novel enzymes with biotechnological applications from marine environments. Microb Cell Fact 2008, 7, 27. [Google Scholar]
- Martinez, A; Kolvek, SJ; Yip, CL; Hopke, J; Brown, KA; MacNeil, IA; Osburne, MS. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 2004, 70, 2452–2463. [Google Scholar]
- Wexler, M; Bond, PL; Richardson, DJ; Johnston, AW. A wide host-range metagenomic library from a waste water treatment plant yields a novel alcohol/aldehyde dehydrogenase. Environ Microbiol 2005, 7, 1917–1926. [Google Scholar]
- Wang, GY; Graziani, E; Waters, B; Pan, W; Li, X; McDermott, J; Meurer, G; Saxena, G; Andersen, RJ; Davies, J. Novel natural products from soil DNA libraries in a streptomycete host. Org Lett 2000, 2, 2401–2404. [Google Scholar]
- Makrides, SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 1996, 60, 512–538. [Google Scholar]
- Ferrer, M; Chernikova, TN; Yakimov, MM; Golyshin, PN; Timmis, KN. Chaperonins govern growth of Escherichia coli at low temperatures. Nat Biotechnol 2003, 21, 1266–1267. [Google Scholar]
- Uchiyama, T; Abe, T; Ikemura, T; Watanabe, K. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol 2005, 23, 88–93. [Google Scholar]
- Yun, J; Ryu, S. Screening for novel enzymes from metagenome and SIGEX, as a way to improve it. Microb Cell Fact 2005, 4, 8. [Google Scholar]
- Uchiyama, T; Watanabe, K. Substrate-induced gene expression (SIGEX) screening of metagenome libraries. Nat Protoc 2008, 3, 1202–1212. [Google Scholar]
- Yamada, K; Terahara, T; Kurata, S; Yokomaku, T; Tsuneda, S; Harayama, S. Retrieval of entire genes from environmental DNA by inverse PCR with pre-amplification of target genes using primers containing locked nucleic acids. Environ Microbiol 2008, 10, 978–987. [Google Scholar]
- Boubakri, H; Beuf, M; Simonet, P; Vogel, TM. Development of metagenomic DNA shuffling for the construction of a xenobiotic gene. Gene 2006, 375, 87–94. [Google Scholar]
- Chalfie, M; Tu, Y; Euskirchen, G; Ward, WW; Prasher, DC. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar]
- Shimomura, O; Johnson, FH; Saiga, Y. Microdetermination of Calcium by Aequorin Luminescence. Science 1963, 140, 1339–1340. [Google Scholar]
- Engebrecht, J; Simon, M; Silverman, M. Measuring gene expression with light. Science 1985, 227, 1345–1347. [Google Scholar]
- Mathur, EJ; Toledo, G; Green, BD; Podar, M; Richardson, TH; Kulwiec, M; Chang, HW. A biodiversity-based approach to development of performance enzymes. Ind Biotechnol 2006, 1, 283–287. [Google Scholar]
- Hardeman, F; Sjoling, S. Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol Ecol 2007, 59, 524–534. [Google Scholar]
- Xu, M; Xiao, X; Wang, F. Isolation and characterization of alkane hydroxylases from a metagenomic library of Pacific deep-sea sediment. Extremophiles 2008, 12, 255–262. [Google Scholar]
- Chu, X; He, H; Guo, C; Sun, B. Identification of two novel esterases from a marine metagenomic library derived from South China Sea. Appl Microbiol Biotechnol 2008, 80, 615–625. [Google Scholar]
- Lee, DG; Jeon, JH; Jang, MK; Kim, NY; Lee, JH; Lee, JH; Kim, SJ; Kim, GD; Lee, SH. Screening and characterization of a novel fibrinolytic metalloprotease from a metagenomic library. Biotechnol Lett 2007, 29, 465–472. [Google Scholar]
- Aurilia, V; Parracino, A; D’Auria, S. Microbial carbohydrate esterases in cold adapted environments. Gene 2008, 410, 234–240. [Google Scholar]
- Ferrer, M; Golyshina, OV; Chernikova, TN; Khachane, AN; Martins Dos Santos, VA; Yakimov, MM; Timmis, KN; Golyshin, PN. Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem Biol 2005, 12, 895–904. [Google Scholar]
- de Pascale, D; Cusano, AM; Autore, F; Parrilli, E; di Prisco, G; Marino, G; Tutino, ML. The cold-active Lip1 lipase from the Antarctic bacterium Pseudoalteromonas haloplanktis TAC125 is a member of a new bacterial lipolytic enzyme family. Extremophiles 2008, 12, 311–323. [Google Scholar]
- Lee, MH; Lee, CH; Oh, TK; Song, JK; Yoon, JH. Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: evidence for a new family of bacterial lipases. Appl Environ Microbiol 2006, 72, 7406–7409. [Google Scholar]
- Jeon, JH; Kim, JT; Kim, YJ; Kim, HK; Lee, HS; Kang, SG; Kim, SJ; Lee, JH. Cloning and characterization of a new cold-active lipase from a deep-sea sediment metagenome. Appl Microbiol Biotechnol 2009, 81, 865–874. [Google Scholar]
- Shanmughapriya, S; Kiran, GS; Selvin, J; Thomas, TA; Rani, C. Optimization, purification, and characterization of extracellular mesophilic alkaline cellulase from sponge-associated Marinobacter sp. MSI032. Appl Biochem Biotechnol 2009. [Google Scholar] [CrossRef]
- Zeng, R; Xiong, P; Wen, J. Characterization and gene cloning of a cold-active cellulase from a deep-sea psychrotrophic bacterium Pseudoalteromonas sp. DY3. Extremophiles 2006, 10, 79–82. [Google Scholar]
- Garsoux, G; Lamotte, J; Gerday, C; Feller, G. Kinetic and structural optimization to catalysis at low temperatures in a psychrophilic cellulase from the Antarctic bacterium Pseudoalteromonas haloplanktis. Biochem J 2004, 384, 247–253. [Google Scholar]
- Ekborg, NA; Morrill, W; Burgoyne, AM; Li, L; Distel, DL. CelAB, a multifunctional cellulase encoded by Teredinibacter turnerae T7902T, a culturable symbiont isolated from the wood-boring marine bivalve Lyrodus pedicellatus. Appl Environ Microbiol 2007, 73, 7785–7788. [Google Scholar]
- Cottrell, MT; Moore, JA; Kirchman, DL. Chitinases from uncultured marine microorganisms. Appl Environ Microbiol 1999, 65, 2553–2557. [Google Scholar]
- Hobel, CF; Hreggvidsson, GO; Marteinsson, VT; Bahrani-Mougeot, F; Einarsson, JM; Kristjansson, JK. Cloning, expression, and characterization of a highly thermostable family 18 chitinase from Rhodothermus marinus. Extremophiles 2005, 9, 53–64. [Google Scholar]
- Wasmund, K; Burns, KA; Kurtboke, DI; Bourne, DG. Novel alkane hydroxylase gene (alkB) diversity in sediments associated with hydrocarbon seeps in the Timor Sea, Australia. Appl Environ Microbiol 2009, 75, 7391–7398. [Google Scholar]
- Ferrer, M; Beloqui, A; Timmis, KN; Golyshin, PN. Metagenomics for mining new genetic resources of microbial communities. J Mol Microbiol Biotechnol 2009, 16, 109–123. [Google Scholar]
- Jones, BV; Sun, F; Marchesi, JR. Using skimmed milk agar to functionally screen a gut metagenomic library for proteases may lead to false positives. Lett Appl Microbiol 2007, 45, 418–420. [Google Scholar]
- Lee, SW; Won, K; Lim, HK; Kim, JC; Choi, GJ; Cho, KY. Screening for novel lipolytic enzymes from uncultured soil microorganisms. Appl Microbiol Biotechnol 2004, 65, 720–726. [Google Scholar]
- Guan, C; Ju, J; Borlee, BR; Williamson, LL; Shen, B; Raffa, KF; Handelsman, J. Signal mimics derived from a metagenomic analysis of the gypsy moth gut microbiota. Appl Environ Microbiol 2007, 73, 3669–3676. [Google Scholar]
- Stepanauskas, R; Sieracki, ME. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc Natl Acad Sci USA 2007, 104, 9052–9057. [Google Scholar]
- Woyke, T; Xie, G; Copeland, A; Gonzalez, JM; Han, C; Kiss, H; Saw, JH; Senin, P; Yang, C; Chatterji, S; Cheng, JF; Eisen, JA; Sieracki, ME; Stepanauskas, R. Assembling the marine metagenome, one cell at a time. PLoS One 2009, 4, e5299. [Google Scholar]
- Gilbert, JA; Field, D; Swift, P; Newbold, L; Oliver, A; Smyth, T; Somerfield, PJ; Huse, S; Joint, I. The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol 2009, 11, 3132–3139. [Google Scholar]
- Frias-Lopez, J; Shi, Y; Tyson, GW; Coleman, ML; Schuster, SC; Chisholm, SW; Delong, EF. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA 2008, 105, 3805–3810. [Google Scholar]
- Gilbert, JA; Field, D; Huang, Y; Edwards, R; Li, W; Gilna, P; Joint, I. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS One 2008, 3, e3042. [Google Scholar]
- Gilbert, JA; Thomas, S; Cooley, NA; Kulakova, A; Field, D; Booth, T; McGrath, JW; Quinn, JP; Joint, I. Potential for phosphonoacetate utilization by marine bacteria in temperate coastal waters. Environ Microbiol 2009, 11, 111–125. [Google Scholar]
- Wilmes, P; Bond, PL. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ Microbiol 2004, 6, 911–920. [Google Scholar]
- Schweder, T; Markert, S; Hecker, M. Proteomics of marine bacteria. Electrophoresis 2008, 29, 2603–2616. [Google Scholar]
- Toyoda, A; Iio, W; Mitsumori, M; Minato, H. Isolation and identification of cellulose-binding proteins from sheep rumen contents. Appl Environ Microbiol 2009, 75, 1667–1673. [Google Scholar]
- Beloqui, A; Guazzaroni, ME; Pazos, F; Vieites, JM; Godoy, M; Golyshina, OV; Chernikova, TN; Waliczek, A; Silva-Rocha, R; Al-Ramahi, Y; La Cono, V; Mendez, C; Salas, JA; Solano, R; Yakimov, MM; Timmis, KN; Golyshin, PN; Ferrer, M. Reactome array: forging a link between metabolome and genome. Science 2009, 326, 252–257. [Google Scholar]
- Park, HJ; Jeon, JH; Kang, SG; Lee, JH; Lee, SA; Kim, HK. Functional expression and refolding of new alkaline esterase, EM2L8 from deep-sea sediment metagenome. Protein Expr Purif 2007, 52, 340–347. [Google Scholar]
- Jeon, JH; Kim, JT; Kang, SG; Lee, JH; Kim, SJ. Characterization and its potential application of two esterases derived from the arctic sediment metagenome. Mar Biotechnol (NY) 2009, 11, 307–316. [Google Scholar]
- Park, SY; Kim, JT; Kang, SG; Woo, JH; Lee, JH; Choi, HT; Kim, SJ. A new esterase showing similarity to putative dienelactone hydrolase from a strict marine bacterium, Vibrio sp. GMD509. Appl Microbiol Biotechnol 2007, 77, 107–115. [Google Scholar]
- Liu, Z; Li, X; Chi, Z; Wang, L; Li, J; Wang, X. Cloning, characterization and expression of the extracellular lipase gene from Aureobasidium pullulans HN2-3 isolated from sea saltern. Antonie Van Leeuwenhoek 2008, 94, 245–255. [Google Scholar]
- Lonhienne, T; Mavromatis, K; Vorgias, CE; Buchon, L; Gerday, C; Bouriotis, V. Cloning, sequences, and characterization of two chitinase genes from the Antarctic Arthrobacter sp. strain TAD20: isolation and partial characterization of the enzymes. J Bacteriol 2001, 183, 1773–1779. [Google Scholar]
- Gabor, EM; de Vries, EJ; Janssen, DB. Construction, characterization, and use of small-insert gene banks of DNA isolated from soil and enrichment cultures for the recovery of novel amidases. Environ Microbiol 2004, 6, 948–958. [Google Scholar]
- Li, H; Chi, Z; Wang, X; Duan, X; Ma, L; Gao, L. Purification and characterization of extracellular amylase from the marine yeast Aureobasidium pullulans N13d and its raw potato starch digestion. Enzyme Microb Technol 2007, 40, 1006–1012. [Google Scholar]
- Li, X; Chi, Z; Liu, Z; Li, J; Wang, X; Hirimuthugoda, NY. Purification and characterization of extracellular phytase from a marine yeast Kodamaea ohmeri BG3. Mar Biotechnol (NY) 2008, 10, 190–197. [Google Scholar]
- Zeng, R; Zhang, R; Zhao, J; Lin, N. Cold-active serine alkaline protease from the psychrophilic bacterium Pseudomonas strain DY-A: enzyme purification and characterization. Extremophiles 2003, 7, 335–337. [Google Scholar]
- Acevedo, JP; Reyes, F; Parra, LP; Salazar, O; Andrews, BA; Asenjo, JA. Cloning of complete genes for novel hydrolytic enzymes from Antarctic sea water bacteria by use of an improved genome walking technique. J Biotechnol 2008, 133, 277–286. [Google Scholar]
- Chavez Croocker, P; Sako, Y; Uchida, A. Purification and characterization of an intracellular heat-stable proteinase (pernilase) from the marine hyperthermophilic archaeon Aeropyrum pernix K1. Extremophiles 1999, 3, 3–9. [Google Scholar]
- Collins, T; Meuwis, MA; Stals, I; Claeyssens, M; Feller, G; Gerday, C. A novel family 8 xylanase, functional and physicochemical characterization. J Biol Chem 2002, 277, 35133–35139. [Google Scholar]
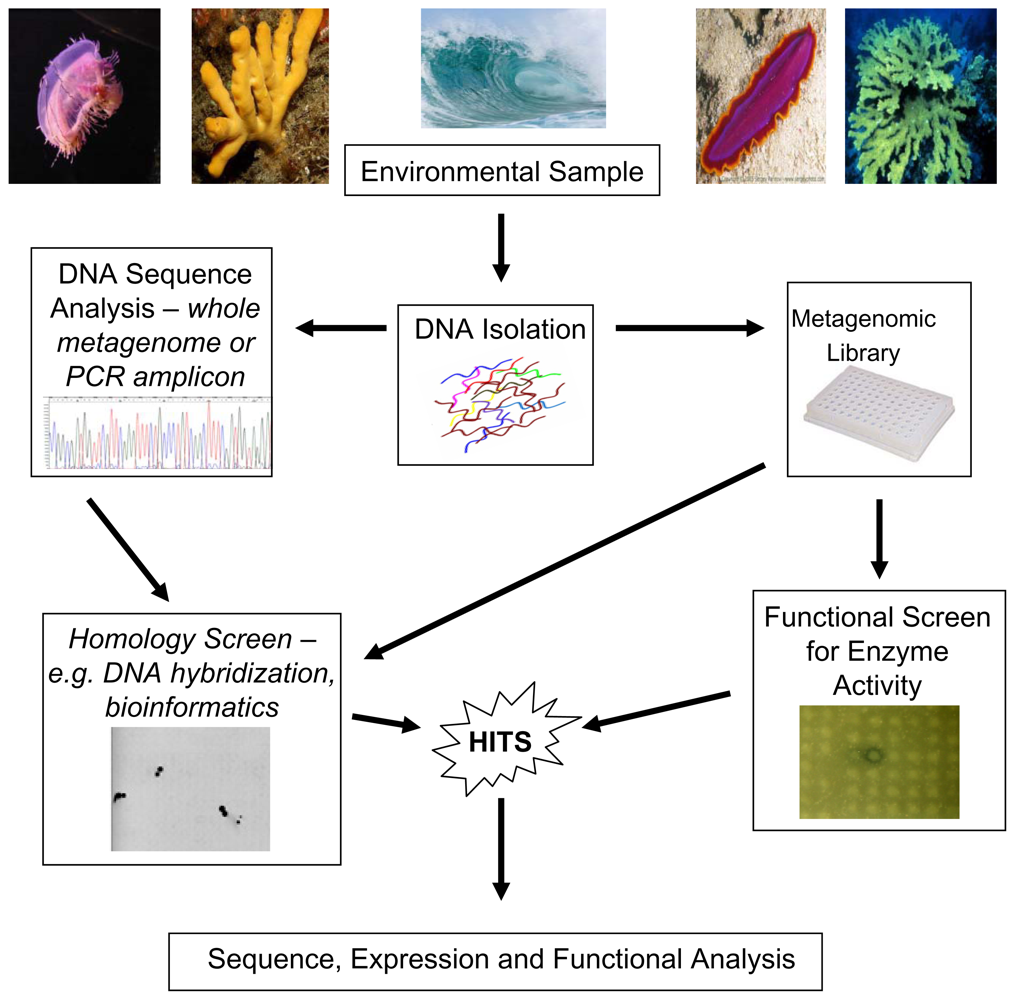


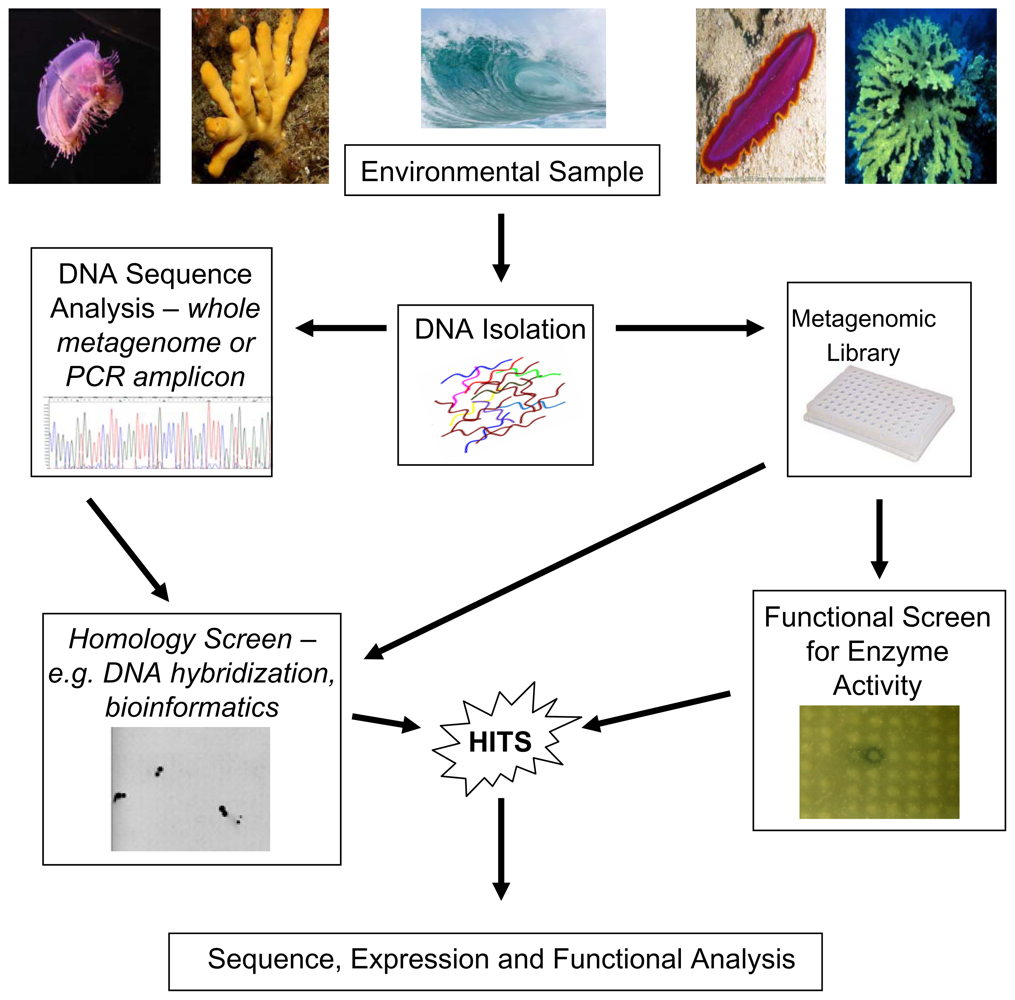{kind=link}
{kind=link}
| Activity | Source | Habitat | Reference |
|---|---|---|---|
| Esterase | Metagenome | Deep-sea sediment | [97] |
| Metagenome | Deep-sea basin | [72] | |
| Metagenome | Surface seawater | [69] | |
| Metagenome | Arctic sediment | [98] | |
| Vibrio sp. | Sea Hare Eggs | [99] | |
| Pseudoalteromonas haloplanktis | Antarctic Seawater | [71] | |
| Lipase | Metagenome | Tidal Flat | [74] |
| Metagenome | Deep Sea sediment | [75] | |
| Metagenome | Baltic Sea sediment | [67] | |
| Pseudoalteromonas haloplanktis TAC125 | Antarctic Seawater | [73] | |
| Aureobasidium pullulans HN2.3 | Sea saltern | [100] | |
| Cellulase | Pseudoalteromonas sp. DY3 | Deep-sea sediment | [77] |
| Pseudoalteromonas haloplanktis | Antarctic Seawater | [78] | |
| Teredinibacter turnerae T7902T | Shipworm | [79] | |
| Marinobacter sp. MSI032. | Marine sponge | [76] | |
| Chitinase | Metagenome | Estuary | [80] |
| Arthrobacter sp. TAD20 | Antarctic ice | [101] | |
| Rhodothermus marinus | Marine hot spring | [81] | |
| Amidase | Metagenome | Marine sediments/sludges | [102] |
| Amylase | Aureobasidium pullulans N13d | Deep-sea sediment | [103] |
| Metagenome | Deep sea hydrothermal vent | [66] | |
| Phytase | Kodomaea ohmeri BG3 | Fish gut | [104] |
| Protease | Pseudomonas strain DYA | Deep-sea sediment | [105] |
| Marine bacterium | Antarctic Seawater | [106] | |
| Aerpyrum pernix K1 | Coastal solfataric vent | [107] | |
| Alkane hydroxylase | Metagenome | Hydrocarbon seep | [82] |
| Metagenome | Deep sea sediment | [68] | |
| Xylanase | Pseudoalteromonas haloplanktis | Antarctic Seawater | [108] |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kennedy, J.; Flemer, B.; Jackson, S.A.; Lejon, D.P.H.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D.W. Marine Metagenomics: New Tools for the Study and Exploitation of Marine Microbial Metabolism. Mar. Drugs 2010, 8, 608-628. https://doi.org/10.3390/md8030608
Kennedy J, Flemer B, Jackson SA, Lejon DPH, Morrissey JP, O’Gara F, Dobson ADW. Marine Metagenomics: New Tools for the Study and Exploitation of Marine Microbial Metabolism. Marine Drugs. 2010; 8(3):608-628. https://doi.org/10.3390/md8030608
Chicago/Turabian StyleKennedy, Jonathan, Burkhardt Flemer, Stephen A. Jackson, David P. H. Lejon, John P. Morrissey, Fergal O’Gara, and Alan D. W. Dobson. 2010. "Marine Metagenomics: New Tools for the Study and Exploitation of Marine Microbial Metabolism" Marine Drugs 8, no. 3: 608-628. https://doi.org/10.3390/md8030608
APA StyleKennedy, J., Flemer, B., Jackson, S. A., Lejon, D. P. H., Morrissey, J. P., O’Gara, F., & Dobson, A. D. W. (2010). Marine Metagenomics: New Tools for the Study and Exploitation of Marine Microbial Metabolism. Marine Drugs, 8(3), 608-628. https://doi.org/10.3390/md8030608