Bioactive Dehydrotyrosyl and Dehydrodopyl Compounds of Marine Origin


Abstract
:1. Introduction
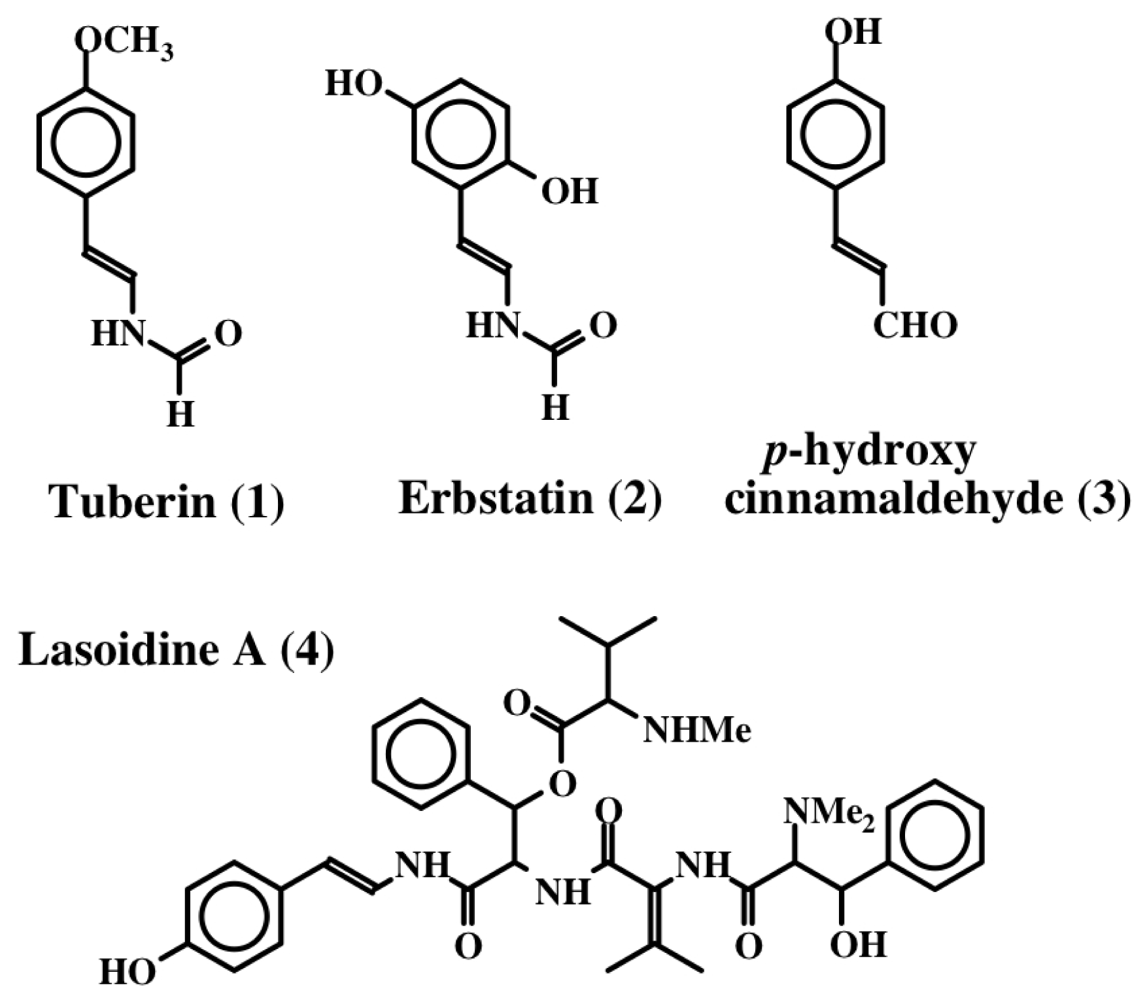
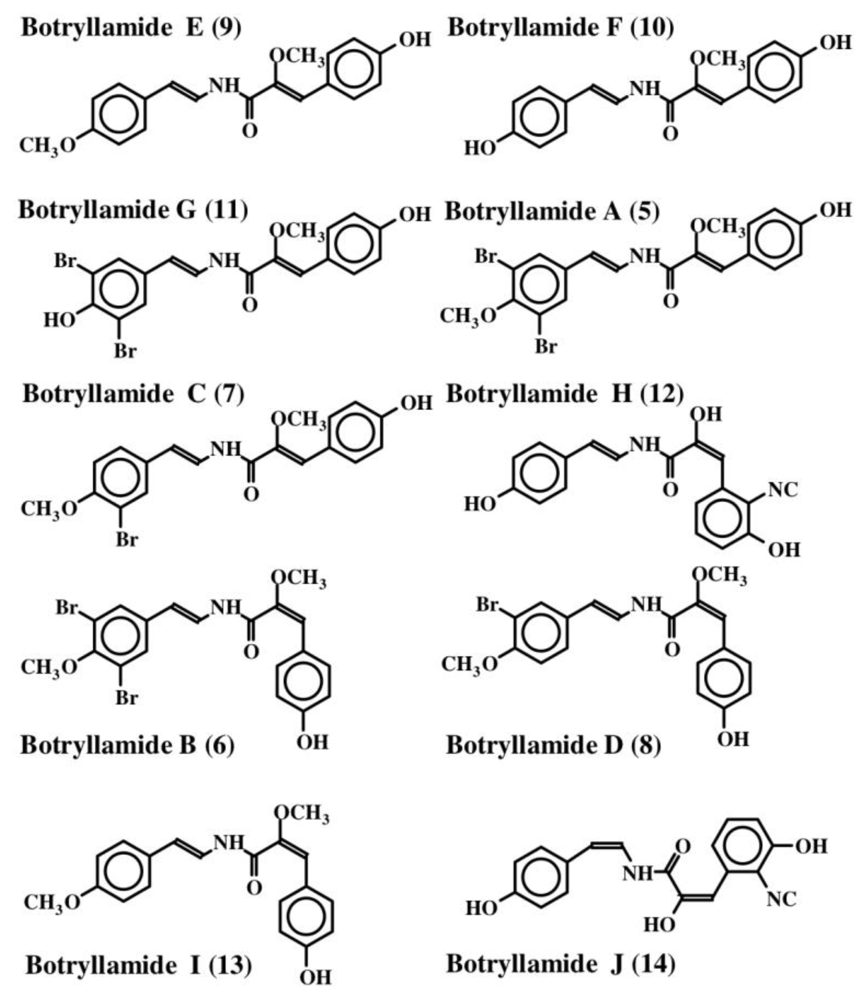
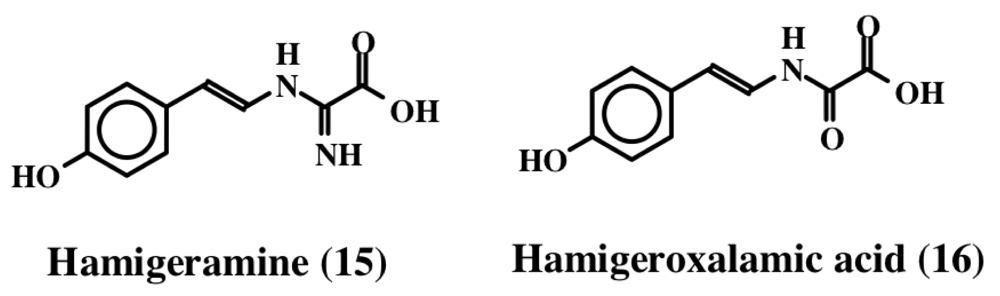
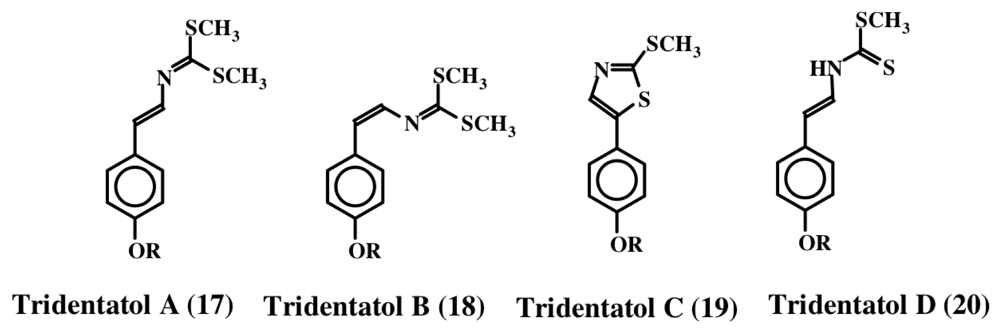
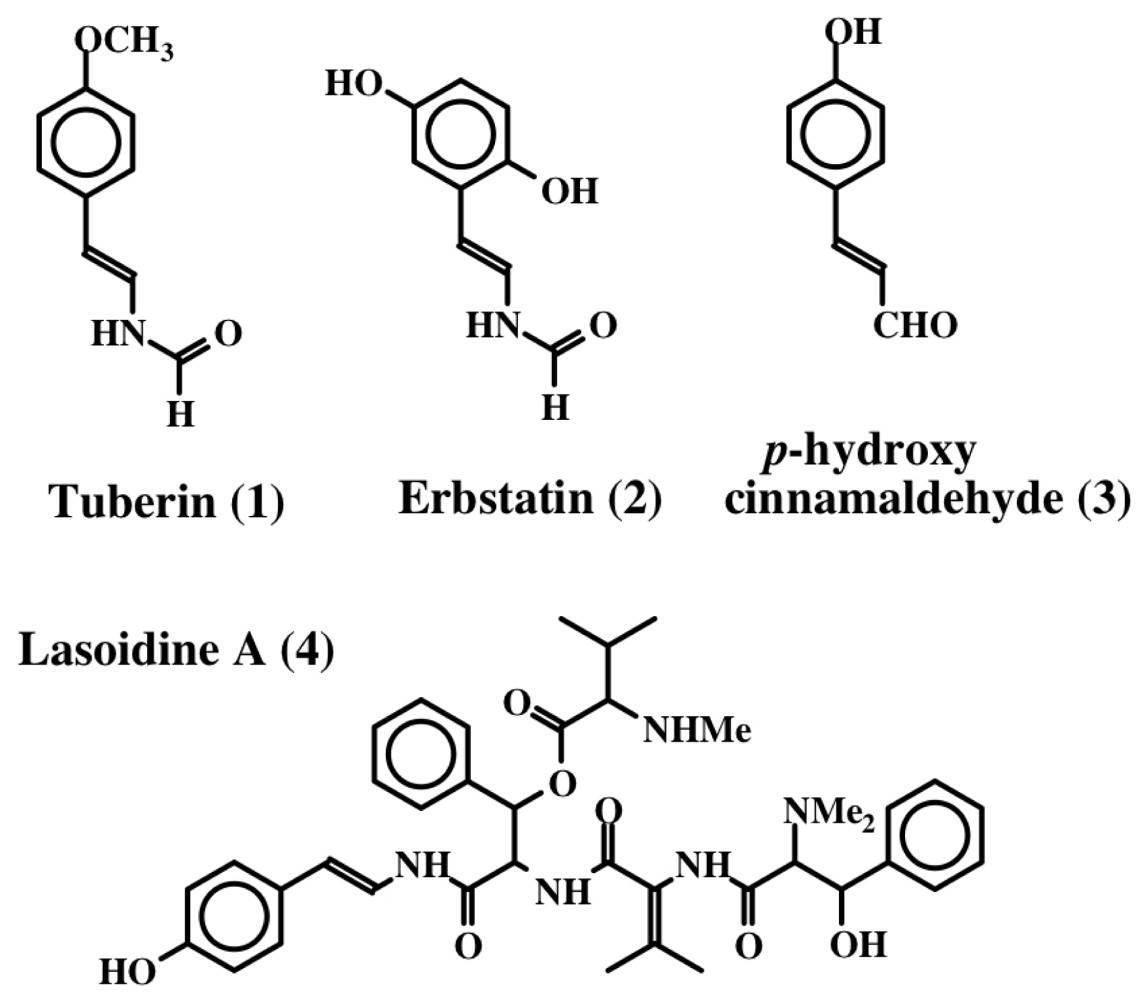
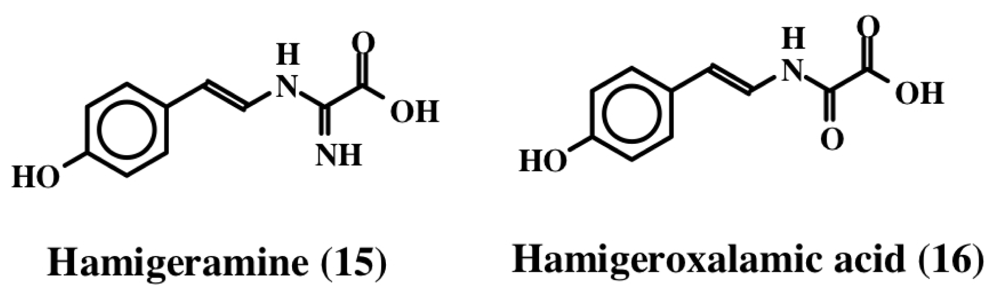
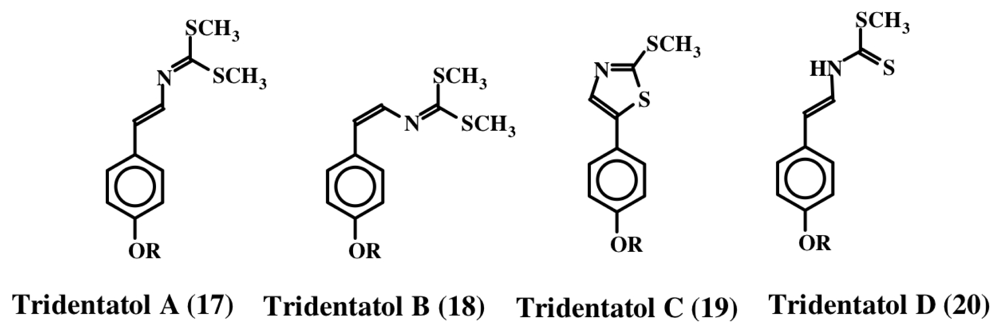
2. Simple Dehydrotyrosine Derivatives: Tuberine, Erbastatin, p-Hydroxycinnamaldehyde, Lasoidine, Botryllamide, Hamigeramine, Hamigeroxalamic Acid, Tridentatols, Anthosamines, Cyclo-Arg-Dehydrotyrosine and Rigidins
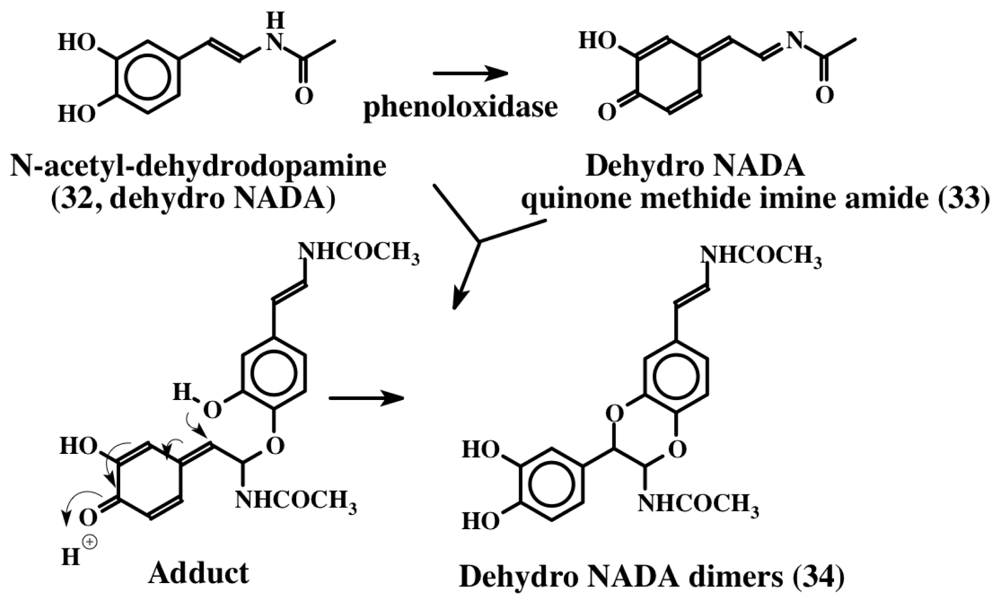
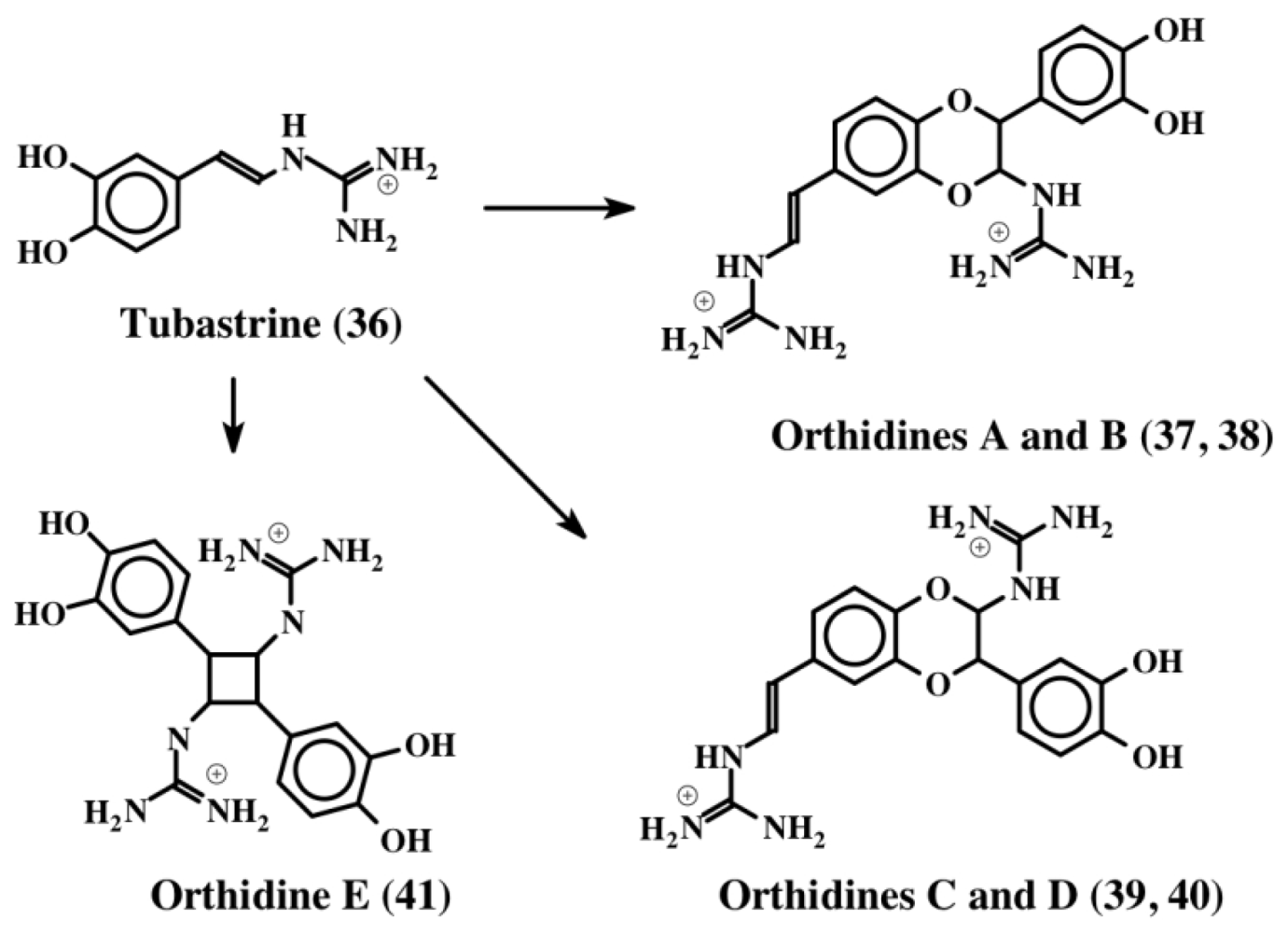
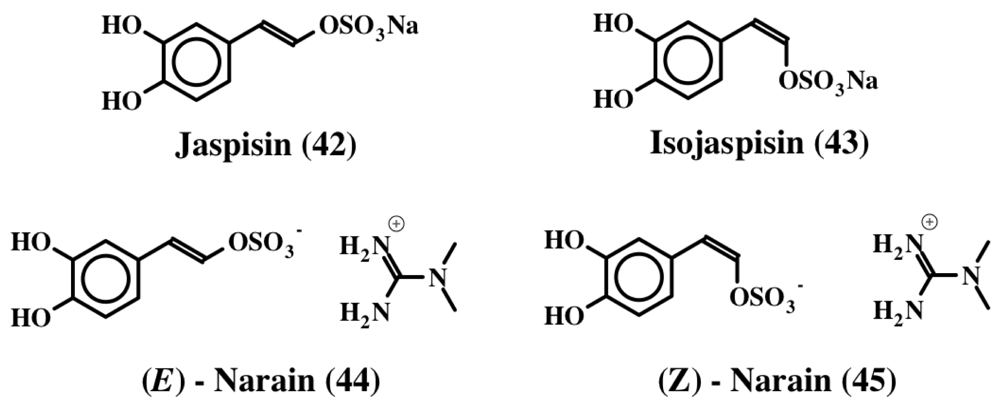
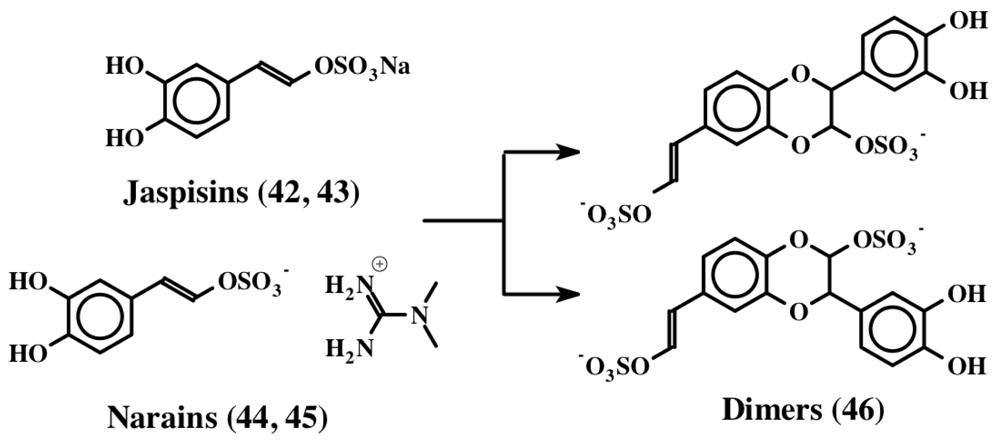
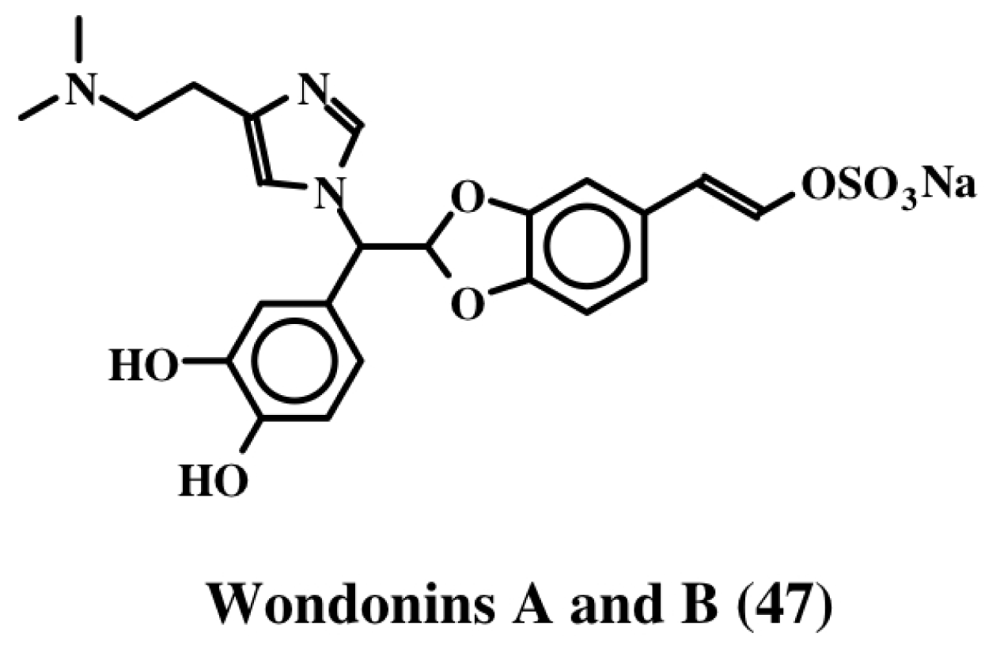
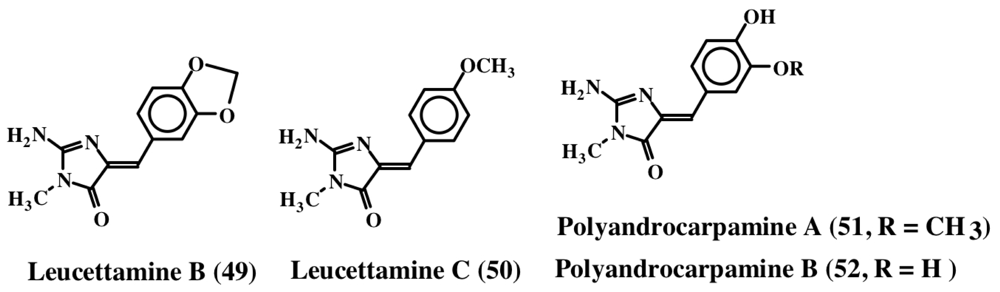
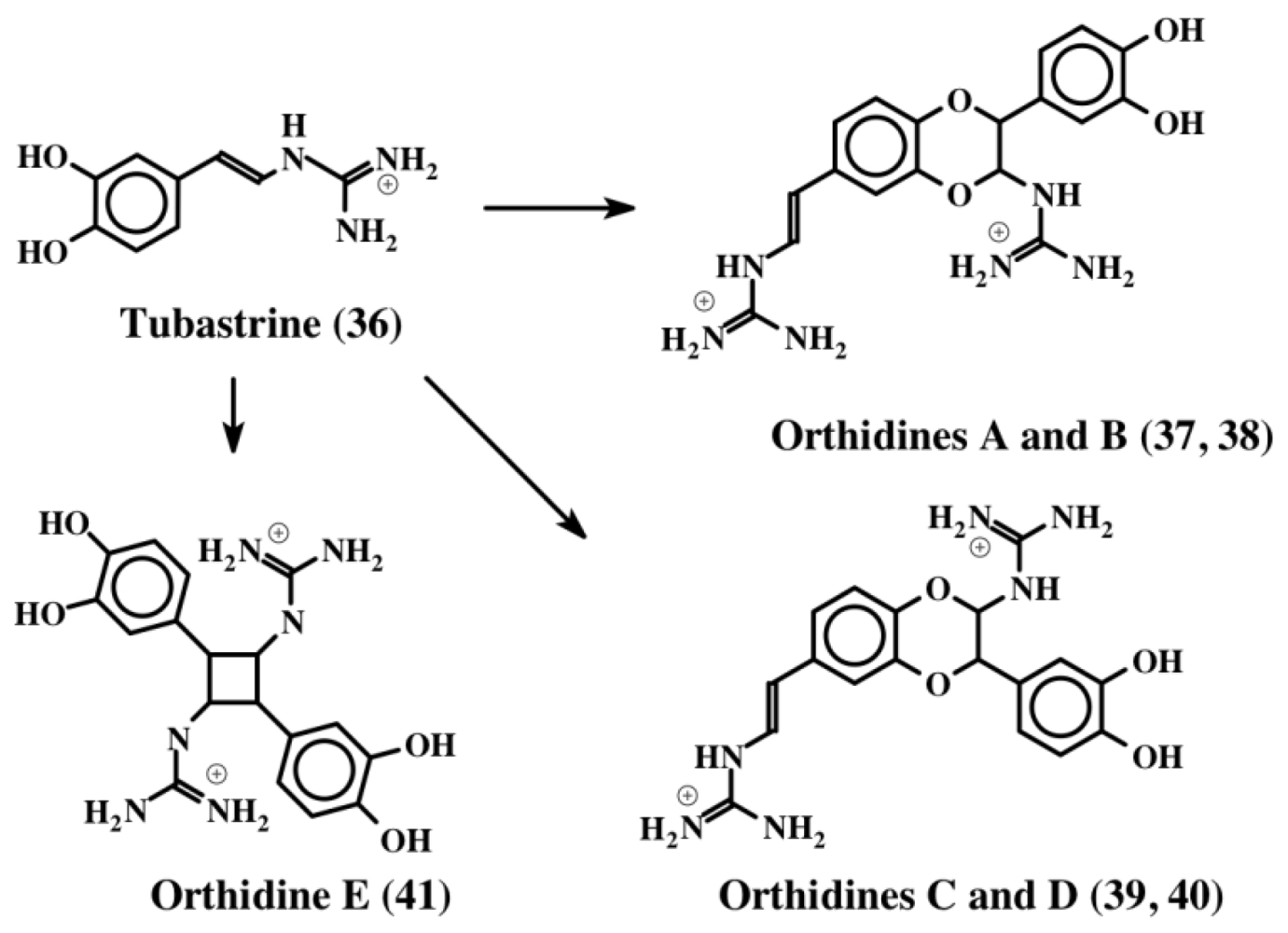
3. Simple Dehydrodopa Derivatives: 1,2-Dehydro-N-acetyldopamine, Narain, Jaspisin, Isojaspisin, Tubastrine, Orthidine and Wondonin A and B, Leucettamines
4. Simple Peptidyl Dehydrotyrosine and Dehydrodopa Derivatives: Clionamide, Morulin PM, and Plicatamide
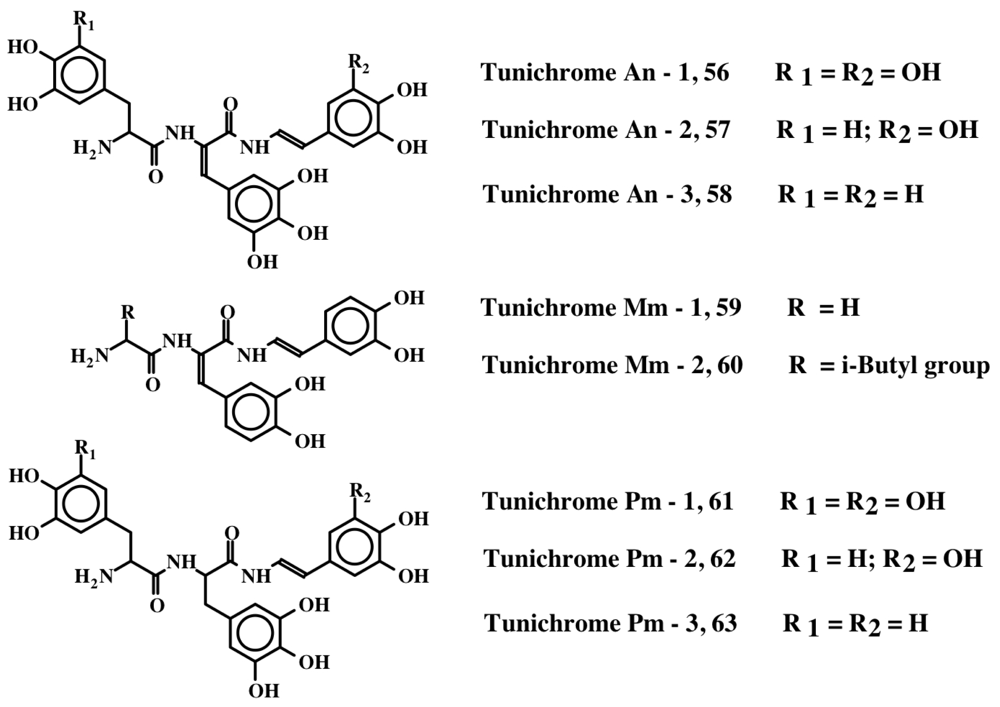
5. Peptides with Multiple Dehydrodopa Units: Tunichromes, Polyphenolic Protein and Celenamides
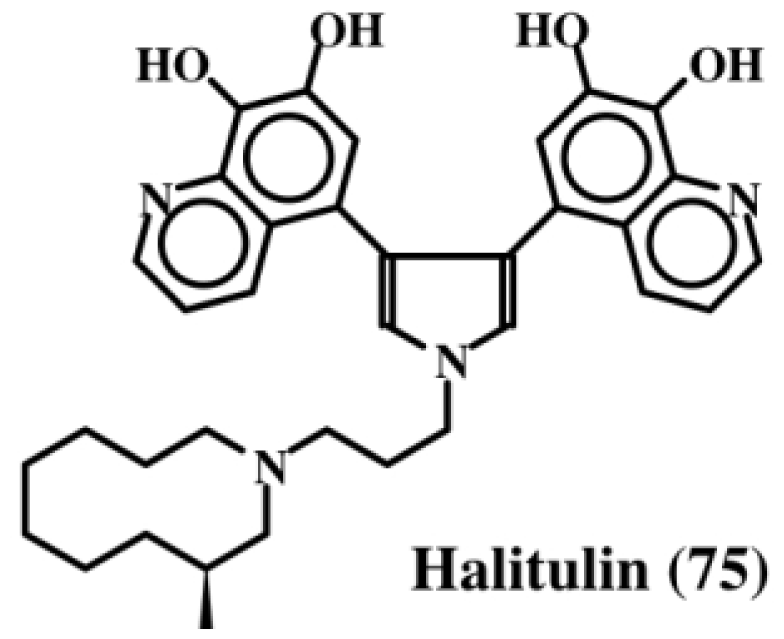
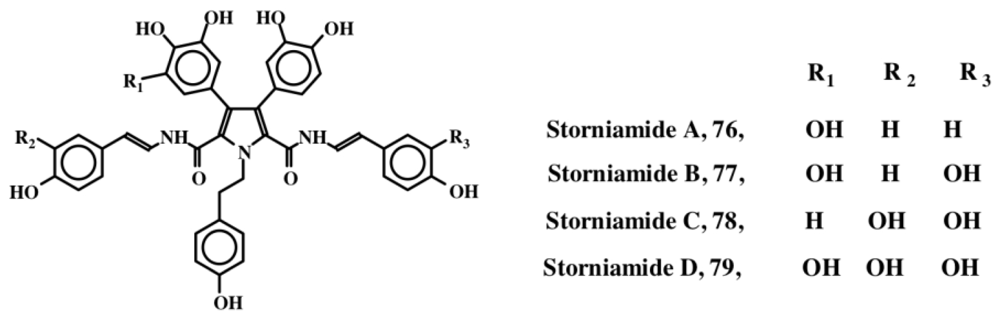
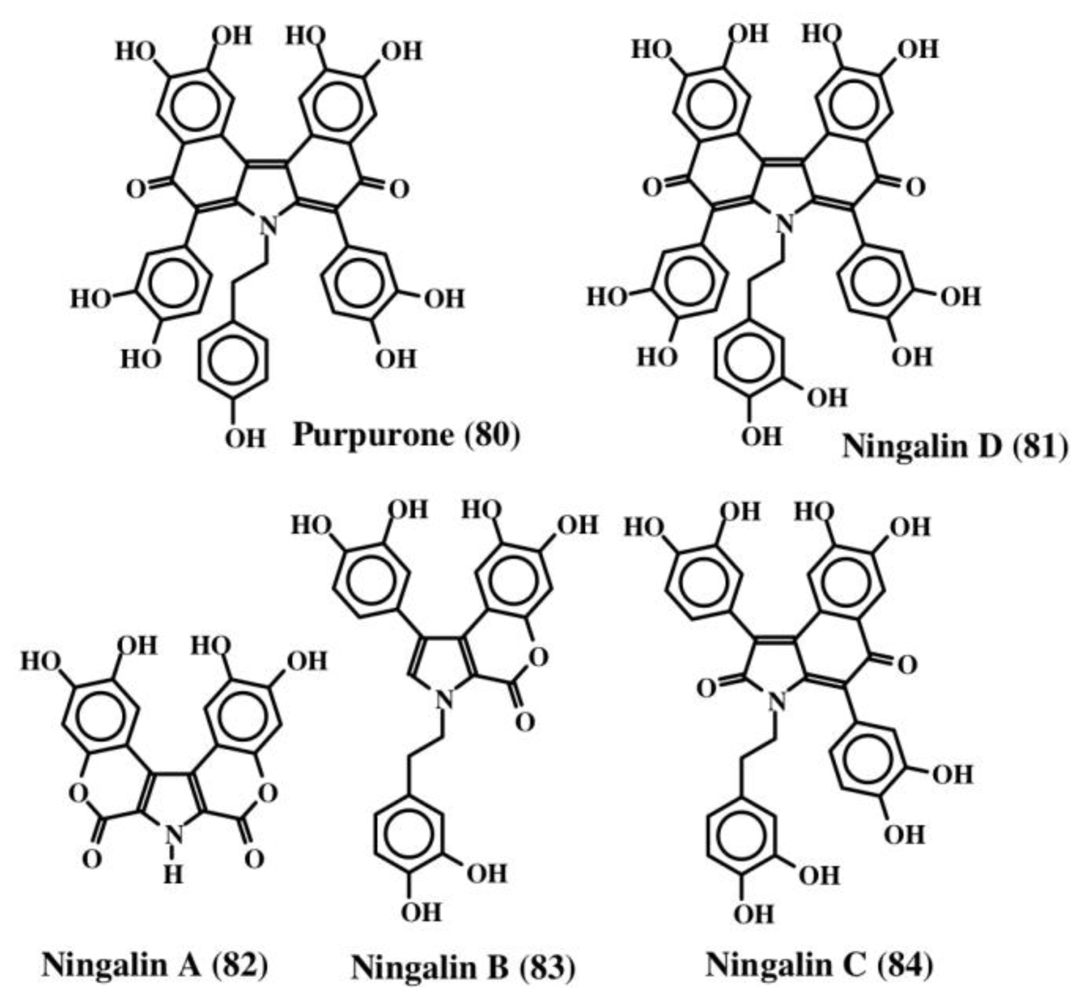
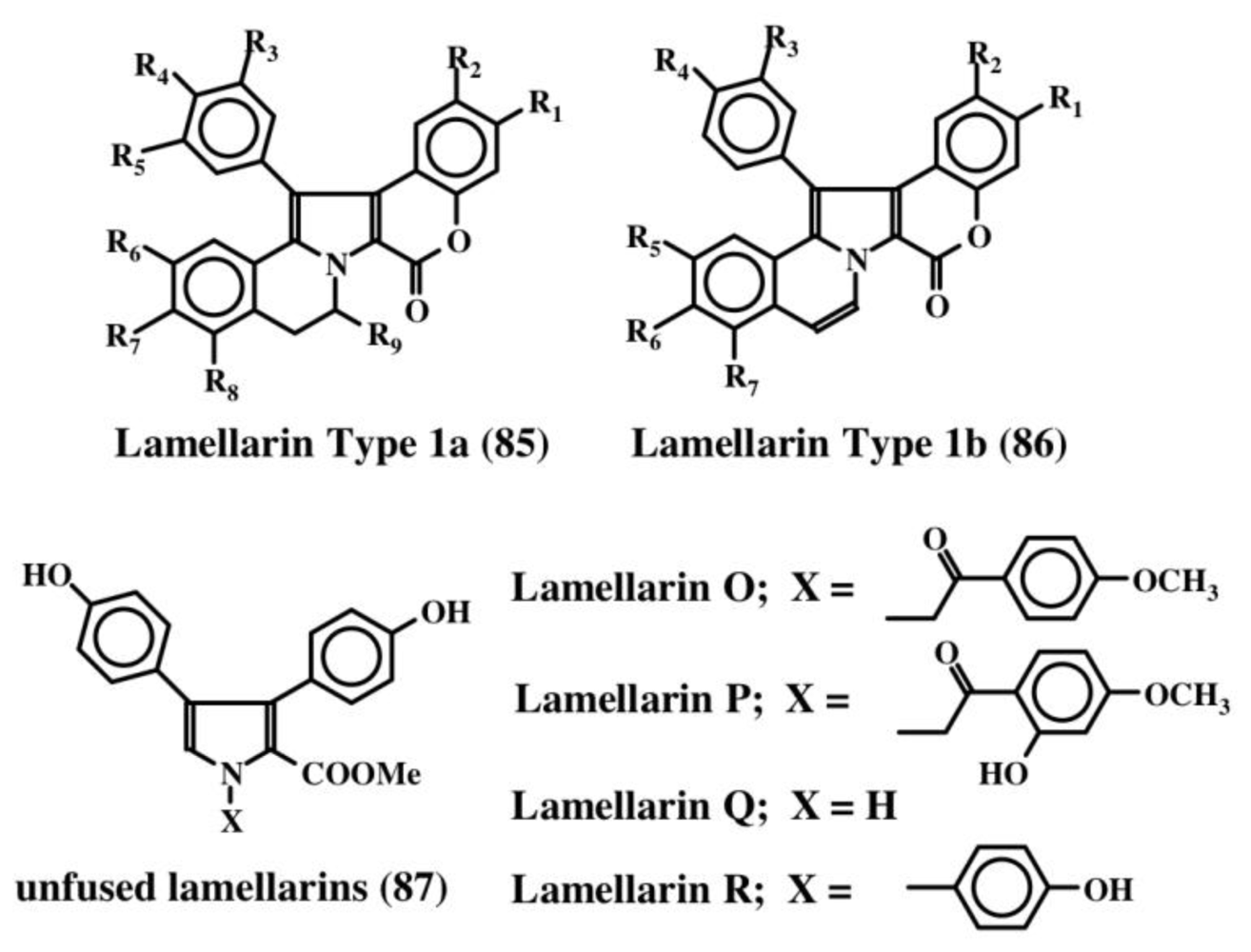
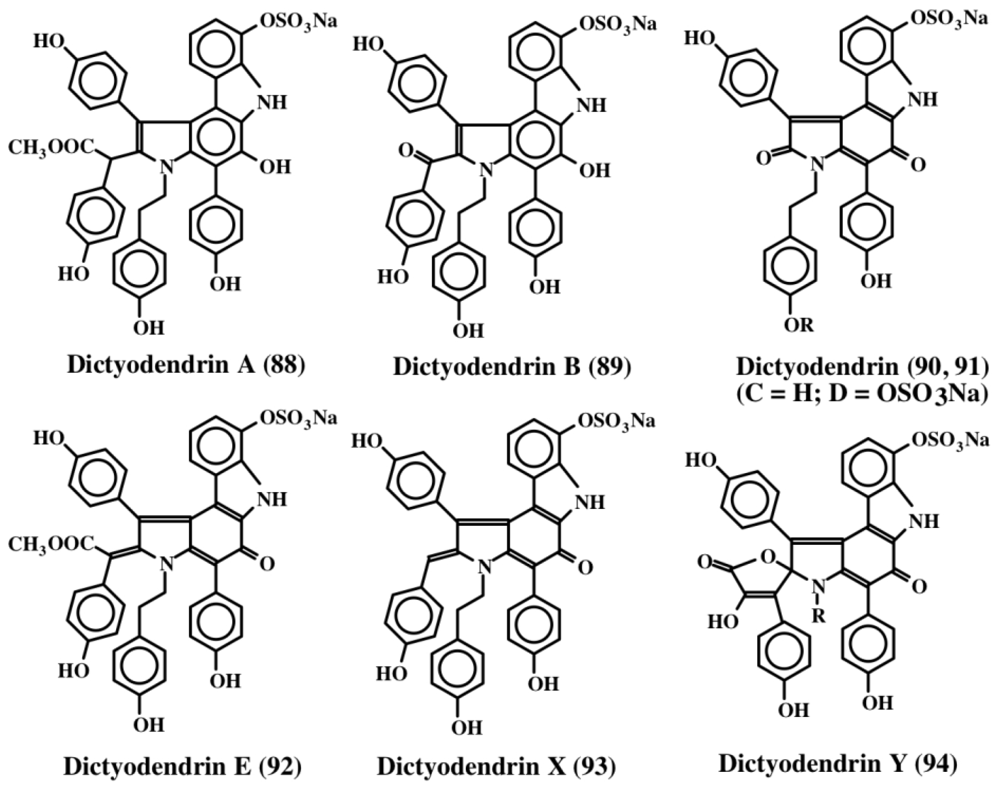
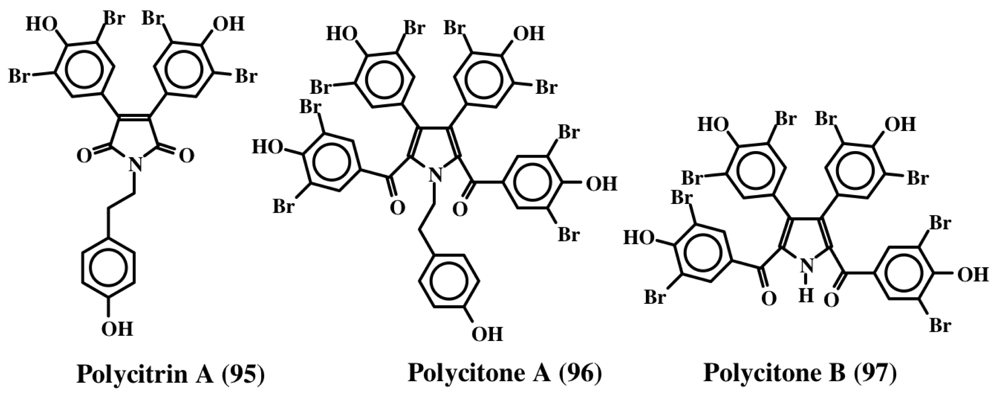
6. Polycyclic Condensed Dehydrodopa Derivatives: Halitulin, Storniamides, Purpurones, Ningalins, Baculiferins, Lamellarin, Dictyodendrins, Polycitrins, Polycitones and Lukainols
7. Biogenesis of Dehydro Compounds
8. Conclusions
References
- Von Nussbaum, F; Brands, M; Hinzen, B; Weigand, S; Häbich, D. Antibacterial natural products in medicinal chemistry—Exodus or revival. Angew Chem Int Ed Engl 2006, 45, 5072–5129. [Google Scholar]
- Slama, TG. Gram-negative antibiotic resistance: There is a price to pay. Crit Care 2008, 12, 1–7. [Google Scholar]
- Nikaido, H. Multidrug resistance in bacteria. Annu Rev Biochem 2009, 78, 119–146. [Google Scholar]
- McKinney, ML; Schoch, RM; Yonavjak, L. Environmental Science: Systems and Solutions, 4th ed; Jones and Bartlett Publishers: Sudbury, MA, USA, 2007. [Google Scholar]
- Brusca, RC; Brusca, GJ. Invertebrates; Sinauer Associates: Sunderland, MA, USA, 1990. [Google Scholar]
- Van Epps, HL. René Dubos: Unearthing antibiotics. J Exp Med 2006, 203, 259. [Google Scholar]
- Zeng, L; Swalla, BJ. Molecular phylogeny of the protochordates: Chordate evolution. Can J Zool 2005, 83, 24–33. [Google Scholar]
- Taylor, SW; Craig, AF; Fischer, WH; Park, M; Lehrer, RI. Styelin D, an extensively modified antimicrobial peptide from ascidian hemocytes. J Biol Chem 2000, 275, 38417–38426. [Google Scholar]
- Tincu, JA; Craig, AG; Taylor, SW. Plicatamide: A lead to the biosynthetic origins of the tunichromes. Biochem Biophys Res Commun 2000, 270, 421–424. [Google Scholar]
- Azumi, K; Yokosawa, H; Ishii, S. Halocyamines: Novel antibacterial tetrapeptide-like structures isolated from the hemocytes of the solitary ascidian halocynthia roretzi. Biochemistry 1990, 29, 159–165. [Google Scholar]
- Fan, H; Peng, J; Hamann, MT; Hu, JF. Lamellarins and related pyrrole derived alkaloids from marine organisms. Chem Rev 2008, 108, 264–287. [Google Scholar]
- Dorsett, LC; Hawkins, CJ; Grice, JA; Lavin, MF; Merefield, PM; Parry, DL; Ross, IL. Ferreascidin: A highly aromatic protein containing 3,4-dihydroxypphenylalanine from the blood cells of a stolidobranch ascidian. Biochemistry 1987, 26, 8078–8082. [Google Scholar]
- Macara, IG; McLeod, GC; Kustin, K. Isolation, properties and structural studies on a compound from tunicate blood cells that may be involved in vanadium accumulation. Biochem J 1979, 181, 457–465. [Google Scholar]
- Bruening, RC; Oltz, EM; Furukawa, J; Nakanishi, K. Isolation and structure of tunichrome B-1, a reducing blood pigment from the tunicate Ascidia nigra L. J Am Chem Soc 1985, 107, 5298–5300. [Google Scholar]
- Bruening, RC; Oltz, EM; Furukawa, J; Nakanishi, K; Kustin, K. Isolation of tunichrome B-1, a reducing blood pigment of the sea squirt Ascidia nigra. J Nat Prod 1986, 49, 193–204. [Google Scholar]
- Oltz, EM; Bruening, RC; Smith, MJ; Kustin, K; Nakanishi, K. The tunichromes. A class of reducing blood pigments from sea squirts: Isolation, structures and vanadium chemistry. J Am Chem Soc 1988, 110, 6162–6172. [Google Scholar]
- Bayer, E; Schiefer, G; Waidelich, D; Scippa, S; De Vincentiis, M. Structure of the tunichrome of tunicates and its role in concentration of vanadium. Angew Chem Int Ed Engl 1992, 31, 52–54. [Google Scholar]
- Parry, DL; Brand, SG; Kustin, K. Distribution of tunichromes in the Ascidiacea. Bull Mar Sci 1992, 50, 302–306. [Google Scholar]
- Tincu, JA; Taylor, SW. Tunichrome Sp-1: New pentapeptide tunichrome from the hemocytes of Styela plicata. J Nat Prod 2002, 65, 377–378. [Google Scholar]
- Okuma, K; Anzai, S; Suzuki, S. A new antibiotic, tuberine, V: Biological activities of tuberine analogs. J Antibiot 1962, 15, 202–209. [Google Scholar]
- Umezawa, H; Imoto, M; Sawa, T; Isshiki, K; Matsuda, N; Uchida, T; Iinuma, H; Hamada, M; Takeuchi, T. Studies on a new epidermal growth factor-receptor kinase inhibitor, erbstatin, produced by MH435-hF3. J Antibiot 1986, 39, 170–173. [Google Scholar]
- Leem, JY; Jeong, IM; Park, KT; Park, HY. Isolation of p-hydroxycinnamaldehyde as an antibacterial substance from the saw fly, Acantholyda parki S. FEBS Lett 1999, 442, 53–56. [Google Scholar]
- Marchand, J; Pais, M; Monseur, X; Jarreau, FX. Alcaloides peptidiques—VII. Les lasiodines A et B, alcoloides du Lasidiscus marmoratus. C.H. Wright (Rhamnacees). Tetrahedron 1969, 25, 937–954. [Google Scholar]
- McDonald, LA; Swersey, JC; Ireland, CM; Carroll, AR; Coll, JC; Bowden, BF; Fairchild, CR; Cornell, L. Botryllamides A–D. New brominated tyrosine derivatives from styelid ascidians of the genus. Botryllus Tetrahedron 1995, 51, 5237–5244. [Google Scholar]
- Henrich, CJ; Robey, RW; Takada, K; Bokesch, HR; Bates, SE; Shukla, S; Ambudkar, SV; McMohan, JB; Gustafson, KR. Botryllamides: Natural product inhibitors of ABCG2. ACS Chem Biol 2009, 4, 637–647. [Google Scholar]
- Takada, K; Imamura, N; Gustafson, KR; Henrich, CJ. Synthesis and structure-activity relationship of botryllamides that block the ABCG2 multidrug transporter. Bioorg Med Chem Lett 2010, 20, 1330–1333. [Google Scholar]
- Hassan, W; Edrada, R; Ebel, R; Wray, V; Proksch, P. New alkaloids from the Mediterranean sponge Hamigera hamigera. Mar Drugs 2004, 2, 88–100. [Google Scholar]
- Lindquist, N; Lobkovsky, E; Clardy, J. Tridentatols A–C, novel natural products of the marine hydroid Tridentata marginata. Tetrahedron Lett 1996, 37, 9131–9134. [Google Scholar]
- Johnson, MK; Alexander, KE; Lindquist, N; Loo, G. Potent antioxidant activity of a dithiocarbamate related compound from a marine hydroid. Biochem Pharmacol 1999, 58, 1313–1319. [Google Scholar]
- Stachowicz, JJ; Lindquist, N. Chemical defense among hydroids on pelagic Sargassum: Predator deterrence and absorption of solar UV radiation by secondary metabolites. Mar Ecol Prog Ser 1997, 155, 115–126. [Google Scholar]
- Lindquist, N. Tridentatols D–H, nematocyst metabolites and precursors of the activated chemical defense in the marine hydroid Tridentata marginata (Kirchenpauer 1864. J. Nat. Prod 2002, 65, 681–684. [Google Scholar]
- Tsukamoto, S; Kato, H; Hirota, H; Fusetani, N. Pipecolate derivates, anthosamines A and B inducers of larval metamorphosis in ascidians, from a marine sponge, Anthosigmella aff. raromicrosclera. Tetrahedron 1995, 51, 6687–6694. [Google Scholar]
- Kobayashi, J; Cheng, JF; Kikuchi, Y; Ishibashi, M; Yamamura, S; Ohizumi, Y; Ohta, T; Nozoe, S. Rigidin, a novel alkaloid with calmodulin antagonistic activity from the okinawan marine tunicate Eudistoma cf. rigida. Tetrahedron Lett 1990, 31, 4617–4620. [Google Scholar]
- Tsuda, M; Nozawa, K; Shimbo, K; Kobayashi, J. Rigidins B–D, New pyrrolopyrimidine alkaloids from a tunicate Cystodytes species. J Nat Prod 2003, 66, 292–294. [Google Scholar]
- Andersen, SO; Roepstorff, P. Sclerotization of insect cuticle—II Isolation and identification of phenolic dimers from sclerotized insect cuticle. Insect Biochem 1981, 11, 25–31. [Google Scholar]
- Andersen, SO; Jacobsen, JP; Roepstorff, P. Studies of the sclerotization of insect cuticle—The structure of dimeric products formed by incubation of N-acetyldopamine with locust cuticle. Tetrahedron 1980, 36, 3249–3252. [Google Scholar]
- Andersen, SO; Roepstorff, P. Sclerotization of insect cuticle—III. An unsaturated derivative of N-acetyldopamine and its role in sclerotization. Insect Biochem 1982, 12, 269–276. [Google Scholar]
- Sugumaran, M; Dali, H; Semensi, V; Hennigan, B. Tyrosinase catalyzed unusual oxidative dimerization of 1,2-dehydro-N-acetyldopamine. J Biol Chem 1987, 262, 10546–10549. [Google Scholar]
- Sugumaran, M; Hennigan, B; Semensi, V; Dali, H. On the nature of nonenzymatic and enzymatic oxidation of the putative sclerotizing precursor, 1,2-dehydro-N-acetyldopamine. Arch Insect Biochem Physiol 1988, 8, 89–100. [Google Scholar]
- Sugumaran, M; Schinkmann, K; Bedell-Hogan, D; Dali, H. Mechanism of activation of 1,2-dehydro-N-acetyldopamine for cuticular sclerotization. Arch Insect Biochem Physiol 1990, 14, 93–109. [Google Scholar]
- Sugumaran, M; Semensi, V; Kalyanaraman, B; Bruce, JM; Land, EJ. Evidence for the formation of a quinone methide during the oxidation of the insect cuticular sclerotizing precursor, 1,2-dehydro-N-acetyldopamine. J Biol Chem 1992, 267, 10355–10361. [Google Scholar]
- Sugumaran, M. Oxidation chemistry of 1,2-dehydro-N-acetyldopamines: Direct evidence for the formation of 1,2-dehydro-N-acetyldopamine quinone. Arch Biochem Biophys 2000, 378, 404–410. [Google Scholar]
- Abebe, A; Zheng, D; Evans, J; Sugumaran, M. Reexamination of the mechanisms of oxidative transformations of the insect cuticular sclerotization precursor, 1,2-dehydro-N-acetyldopamine. Insect Biochem Mol Biol 2010, 40, 650–659. [Google Scholar]
- Sugumaran, M. Unified mechanism for sclerotization of insect cuticle. Adv Insect Physiol 1998, 27, 229–334. [Google Scholar]
- Sugumaran, M. Chemistry of cuticular sclerotization. Adv Insect Physiol 2010, 39, 151–209. [Google Scholar]
- Noda, N; Kubota, S; Miyata, Y; Miyahara, K. Optically active N-acetyldopamine dimer of the crude drug “Zentai”, the cast-off shell of the Cicada Cryptotympana sp. Chem Pharm Bull 2000, 48, 1749–1752. [Google Scholar]
- Tada, T; Ohnishi, K; Suzuki, K; Tomita, H; Okamori, M; Katuzaki, H; Komiya, T; Imai, K. Potential cosmetic whitening agents from insect cuticle: Tyrosinase inhibitory activity of N-acetyldopamine dimers from the exuviae of Cicada, Cryptotympana tuslulata FABR. J Oleo Sci 2002, 51, 355–358. [Google Scholar]
- Xu, MZ; Lee, WS; Han, JM; Oh, HW; Park, DS; Tian, GR; Jeong, TS; Park, HY. Antioxidant and anti-inflammatory activities of N-acetyldopamine dimers from periostracum Cicadae. Bioorg Med Chem 2006, 14, 7826–7834. [Google Scholar]
- Sakai, R; Higa, T. Tubastrine, a new guanidinostyrene from the coral Tubastrea aurea. Chem Lett 1987, 127–128. [Google Scholar]
- Sperry, S; Crews, P. Dihydrotubastrines: phenethylguanidine analogues from the Indo-Pacific marine sponge, Petrosia cf. contignata. J Nat Prod 1998, 61, 859–861. [Google Scholar]
- Barenbrock, JS; Kock, M. Screening enzyme—inhibitory activity in several ascidian species from Orkney Islands using protein tyrosine kinase (PTK) bioassay—guided fractionation. J Biotechnol 2005, 117, 225–232. [Google Scholar]
- Pearce, AN; Chia, EW; Berridge, MV; Maas, EW; Page, MJ; Harper, JL; Webb, VL; Copp, BR. Orthidines A–E, tubastrine, 3,4-dimethoxyphenylethyl-β-guanidine, and 1,14-sperminedihomovanillamide: Potential anti-inflammatory alkaloids isolated from the New Zealand ascidian Aplidium orthium that act as inhibitors of neutrophil respiratory burst. Tetrahedron 2008, 64, 5748–5755. [Google Scholar]
- Ohta, S; Kobayashi, H; Ikegami, S. Jaspisin, a novel styryl sulfate from the marine sponge, Jaspis species. Biosci Biotechnol Biochem 1994, 58, 1752–1753. [Google Scholar]
- Ohta, S; Kobayashi, H; Ikegami, S. Isojaspisin, a novel styryl sulfate from a marine sponge, Jaspis sp., that inhibits hatching of sea urchin embryos. Tetrahedron Lett 1994, 35, 4579–4580. [Google Scholar]
- Ikegami, S; Kobayashi, H; Myotoishi, Y; Ohta, S; Kato, KH. Selective inhibition of exoplasmic membrane fusion in echinoderm gametes with jaspisin, a novel antihatching substance isolated from a marine sponge. J Biol Chem 1994, 269, 23262–23267. [Google Scholar]
- Tsukamoto, B; Kato, H; Hirota, H; Fusetani, N. Narains: N,N-dimethylguanidinium styryl sulfates, metamorphosis inducers of ascidian larvae from a marine sponge, Jaspis sp. Tetrahedron Lett 1994, 35, 5874–5874. [Google Scholar]
- Tsukamoto, B; Kato, H; Hirota, H; Fusetani, N. 3,4-Dihydroxystyrene dimers, inducers of larval metamorphosis in ascidians, from a marine sponge, Jaspis sp. Tetrahedron 1994, 48, 13583–13592. [Google Scholar]
- Shin, J; Rho, JR; Seo, Y; Lee, HS; Cho, KW; Kwon, HJ; Sim, CJ. Wondonins A and B, new bis(dihydroxystyryl)imidazoles from a two sponge association. Tetrahedron Lett 2001, 42, 1965–1968. [Google Scholar]
- Chan, GW; Mong, S; Hemling, ME; Freyer, AJ; Offen, PH; DeBrosse, CW; Sarau, HM; Westley, JW. New leukotriene B4 receptor antagonist: Leucettamine A and related imidazoles alkaloids from the marine sponge Leucetta microraphis. J Nat Prod 1993, 56, 116–121. [Google Scholar]
- Crews, P; Clark, DP; Tenney, K. Variation in the alkaloids among Indo-Pacific Leucetta sponges. J Nat Prod 2003, 66, 177–182. [Google Scholar]
- Debdab, M; Renault, S; Lozach, O; Meijer, L; Paquin, L; Carreaux, F; Bazuraeu, JP. Synthesis and preliminary biological evaluation of new derivatives of the marine alkaloid leucettamine B as kinase inhibitors. Eur J Med Chem 2010, 45, 805–810. [Google Scholar]
- Davis, RA; Aalbersberg, W; Meo, S; Moreira da Rocha, R; Ireland, CM. The isolation and synthesis of polyandrocarpamines A and B. Two new 2-aminoimidazolone compounds from the Fijian ascidian, Polyandrocarpa sp. Tetrahedron 2002, 58, 3263–3269. [Google Scholar]
- Davis, RA; Baron, PS; Neve, JE; Cullinane, C. A microwave-assisted stereoselective synthesis of polyandrocarpamines A and B. Tetrahedron Lett 2009, 50, 880–882. [Google Scholar]
- Andersen, RJ; Stonard, RJ. Clionamide, a major metabolite of the sponge Cliona celata Grant. Can J Chem 1979, 57, 2325–2328. [Google Scholar]
- Taylor, SW; Kammerer, B; Nicholson, GJ; Pusecker, K; Walk, T; Bayer, E; Scippa, S; De Vincentiis, M. Morulin Pm: A modified polypeptide containing TOPA and 6-bromotryptophan from the morula cells of the ascidian Phalusia mammillata. Arch Biochem Biophys 1997, 348, 278–288. [Google Scholar]
- Tincu, JA; Menzel, LP; Azimov, R; Sands, J; Hong, T; Waring, AJ; Taylor, SW; Lehrer, RI. Plicatamide, an antimicrobial octapeptide from Sytela plicata hemocytes. J Biol Chem 2003, 278, 13546–13553. [Google Scholar]
- Tincu, JA; Taylor, SW. Antimicrobial peptides from marine invertebrates. Antimicrob Agents Chemother 2004, 48, 3645–3654. [Google Scholar]
- Azumi, K; Yoshimizu, M; Suzuki, S; Ezura, Y; Yokosawa, H. Inhibitory effect of halocyamine, an antimicrobial substance from ascidian hemocytes, on the growth of fish viruses and marine bacteria. Experientia 1990, 46, 1066–1068. [Google Scholar]
- Stonard, RJ; Andersen, RJ. Celenamides A and B linear peptide alkaloids from the sponge Cliona celala. J Org Chem 1980, 45, 3687–3691. [Google Scholar]
- Stonard, RJ; Andersen, RJ. Linear peptide alkaloids from the sponge Cliona celata (Grant). Celenamides C and D. Can J Chem 1980, 58, 2121–2126. [Google Scholar]
- Palermo, JA; Brasco, MFR; Cabezas, E; Balzaretti, V; Seldes, AM. Celenamide E, a tripeptide alkaloid from the Patagonian sponge Cliona chilensis. J Nat Prod 1998, 61, 488–490. [Google Scholar]
- Lee, IH; Cho, Y; Lehrer, RI. Effects of pH and salinity on the antimicrobial properties of clavanins. Infect Immun 1997, 65, 2898–2903. [Google Scholar]
- Lee, IH; Cho, Y; Lehrer, RI. Styelins, broad-spectrum antimicrobial peptides from the solitary tunicate Styela clava. Comp Biochem Physiol B Biochem Mol Biol 1997, 118, 515–521. [Google Scholar]
- Lehrer, RI. Plicatamide, an antimicrobial octapeptide from Styela plicata hemocytes. J Biol Chem 2003, 278, 13546–13553. [Google Scholar]
- Lehrer, RI; Lee, IH; Menzel, L; Waring, A; Zhao, C. Clavanins and styelins, alpha-helical antimicrobial peptides from the hemocytes of Styela clava. Adv Exp Med Biol 2001, 484, 71–76. [Google Scholar]
- Lehrer, RI; Tincu, JA; Taylor, SW; Menzel, LP; Waring, AJ. Natural peptide antibiotics from tunicates: Structures, functions and potential uses. Integr Comp Biol 2003, 43, 313–322. [Google Scholar]
- Taylor, SW; Ross, MM; Waite, HJ. Novel 3,4-di- and 3,4,5-trihydroxyphenylalanine containing polypeptides from the blood cells of the ascidians Ascidia ceratodes and Molgula manhattensis. Arch Biochem Biophys 1995, 324, 228–240. [Google Scholar]
- Cai, M; Sugumaran, M; Robinson, WE. The crosslinking and antibacterial properties of tunichromes. Comp Biochem Physiol B Biochem Mol Biol 2008, 151, 110–117. [Google Scholar]
- Oltz, EM; Pollack, S; Delohery, T; Smith, MJ; Ojika, M; Lee, S; Kustin, K; Nakanishi, K. Distribution of tunichrome and vanadium in sea squirt blood cells sorted by flow cytometry. Experientia 1989, 45, 186–190. [Google Scholar]
- Smith, MJ; Kim, D; Horenstein, B; Nakanishi, K; Kustin, K. Unraveling the chemistry of tunichromes. Acc Chem Res 1991, 24, 117–124. [Google Scholar]
- Taylor, SW; Kammerer, B; Bayer, E. New perspectives in the chemistry and biochemistry of the tunichromes and related compounds. Chem Rev 1997, 97, 333–346. [Google Scholar]
- Michibata, H; Terada, T; Anada, N; Yamakawa, K; Numakunai, T. The accumulation and distribution of vanadium, iron, and manganese in some solitary ascidians. Biol Bull 1986, 171, 672–681. [Google Scholar]
- Kustin, K; Robinson, WE; Smith, MJ. Tunichromes, vanadium, and vacuolated blood cells in tunicates. Invert Reprod Develop 1990, 17, 129–139. [Google Scholar]
- Waite, HJ. Evidence for a repeating 3,4-dihydroxyphenylalanine- and hydroxyproline-containing decapeptide in the adhesive protein of the mussel, Mytilus edulis L. J Biol Chem 1983, 258, 2911–2915. [Google Scholar]
- Waite, HJ. Nature’s underwater adhesive specialist. Int J Adhes Adhes 1987, 7, 9–14. [Google Scholar]
- Waite, HJ. The phylogeny and chemical diversity of quinone tanned glues and varnishes. Comp Biochem Physiol B 1990, 97, 19–29. [Google Scholar]
- Kashman, Y; Koren-Goldshlager, G; Gravalos, MDG; Schleyer, M. Halitulin, a new cytotoxic alkaloid from the marine sponge Haliclona tulearensis. Tetrahedron Lett 1999, 40, 997–1000. [Google Scholar]
- Palermo, JA; Brasco, MFR; Seldes, AM. Storniamides A–D: Alkaloids from a Patagonian Sponge Cliona sp. Tetrahedron 1996, 52, 2727–2734. [Google Scholar]
- Boger, D; Boyce, C; Labroli, M; Sehon, C; Jin, Q. Total Syntheses of Ningalin A, Lamellarin O, Lukianol A, and Permethyl Storniamide A Utilizing Heterocyclic Azadiene Diels–Alder Reactions. J Am Chem Soc 1999, 121, 54–62. [Google Scholar]
- Furstner, A; Krause, H; Thiel, O. Efficient relay syntheses and assessment of the DNA-cleaving properties of the pyrrole alkaloid derivatives permethyl storniamide A, lycogalic acid A dimethyl ester, and the halitulin core. Tetrahedron 2002, 58, 6373–6380. [Google Scholar]
- Chan, GW; Francis, T; Thureen, DR; Offen, PO; Pierce, NJ; Westley, JW; Randall, K; Johnson, RK; Faulkner, JD. Purpurone, an inhibitor of ATP-citrate lyase: A novel alkaloid from the marine sponge Iotrochota sp. J Org Chem 1993, 58, 2544–2546. [Google Scholar]
- Liu, Y; Ji, H; Dong, J; Zhang, S; Lee, KJ; Mathew, S. Antioxidant alkaloid from the South China sea marine sponge, Iotrochota sp. Z Naturforsch C 2008, 63, 636–638. [Google Scholar]
- Kang, H; Fenical, W. Ningalins A–D: Novel aromatic alkaloids from a western Australian ascidian of the genus Didemnum. J Org Chem 1997, 62, 3254–3262. [Google Scholar]
- Boger, DL; Soenen, DR; Boyce, CW; Hedrick, MP; Jin, Q. Total synthesis of Ningalin B utilizing a heterocyclic azadiene Diels-Alder reaction and discovery of a new class of potent multidrug resistant (MDR) reversal agents. J Org Chem 2000, 65, 2479–2483. [Google Scholar]
- Tao, H; Hwang, I; Boger, DL. Multidrug resistance reversal activity of permethyl ningalin B amide derivatives. Bioorg Med Chem Lett 2004, 14, 5979–5981. [Google Scholar]
- Fan, H; Li, Z; Shen, S; Zeng, Y; Yang, Y; Xu, M; Bruhn, T; Nhrun, H; Morschhauser, J; Bringmann, G; Lin, W. Baculiferins A–O, O-sulfated pyrrole alkaloids with anti-HIV-1 activity, from the Chinese marine sponge Iotrochota Baculifera. Bioorg Med Chem 2010, 18, 5466–5474. [Google Scholar]
- Andersen, RJ; Faukner, DJ; He, C; Van Duyne, GD; Clardy, J. Metabolites of the marine prosobranch mollusk Lamellaria sp. J Am Chem Soc 1985, 107, 5492–5495. [Google Scholar]
- Lindquist, N; Fenical, W; Van Duyne, GD. Clardy, New alkaloids of the lamellarin class from the marine ascidian, Didemnum chartaceum (Sluiter, 1909. J. Org. Chem 1988, 53, 4570–4574. [Google Scholar]
- Warabi, K; Matsunaga, S; van Soest, RWM; Fusetani, N. Dictyodendrins A–E, the first telomerase inhibitory marine natural products from the sponge Dictyodendrilla verongiformis. J Org Chem 2003, 68, 2735–2770. [Google Scholar]
- Sato, A; Morishita, T; Shiraki, T; Yoshioka, S; Horikoshi, H; Kuwano, H; Hanakawa, H; Hata, T. Aldose reductase inhibitors from a marine sponge, Dictyodendrilla sp. J Org Chem 1993, 58, 7632–7634. [Google Scholar]
- Rudi, A; Goldberg, I; Stein, Z; Frolow, F; Benayahu, Y; Schleyer, M; Kashman, Y. Polycitone A and polycitrins A and B: New alkaloids from the marine ascidian, Polycitor sp. J Org Chem 1994, 59, 999–1003. [Google Scholar]
- Loya, S; Rudi, A; Kashman, Y; Hizi, A. Polycitone A, a novel and potent general inhibitor of retroviral reverse transcriptases and cellular DNA polymerases. Biochem J 1999, 344, 85–92. [Google Scholar]
- Rudi, A; Evan, T; Akin, M; Kashman, Y. Polycitone B and prepolycitrin A: Two novel alkaloids from the marine ascidian Polycitor africanus. J Nat Prod 2000, 63, 832–833. [Google Scholar]
- Yoshida, W; Lee, K; Carroll, A; Scheuer, P. A complex pyrrolo-oxazinone and its derivate isolated from tunicates. Helv Chim Acta 1992, 75, 1721–1725. [Google Scholar]
- Gupton, J; Krumpe, K; Burnham, B; Webb, T; Shuford, J; Siberoski, J. The application of vinylogous iminium salt derivatives to a regiocontrolled and efficient relay synthesis of lukianol A and related marine natural products. Tetrahedron 1999, 55, 14515–14522. [Google Scholar]
- Gupton, JT. Pyrrole natural products with antitumor properties. Top Heterocycl Chem 2006, 2, 53–92. [Google Scholar]
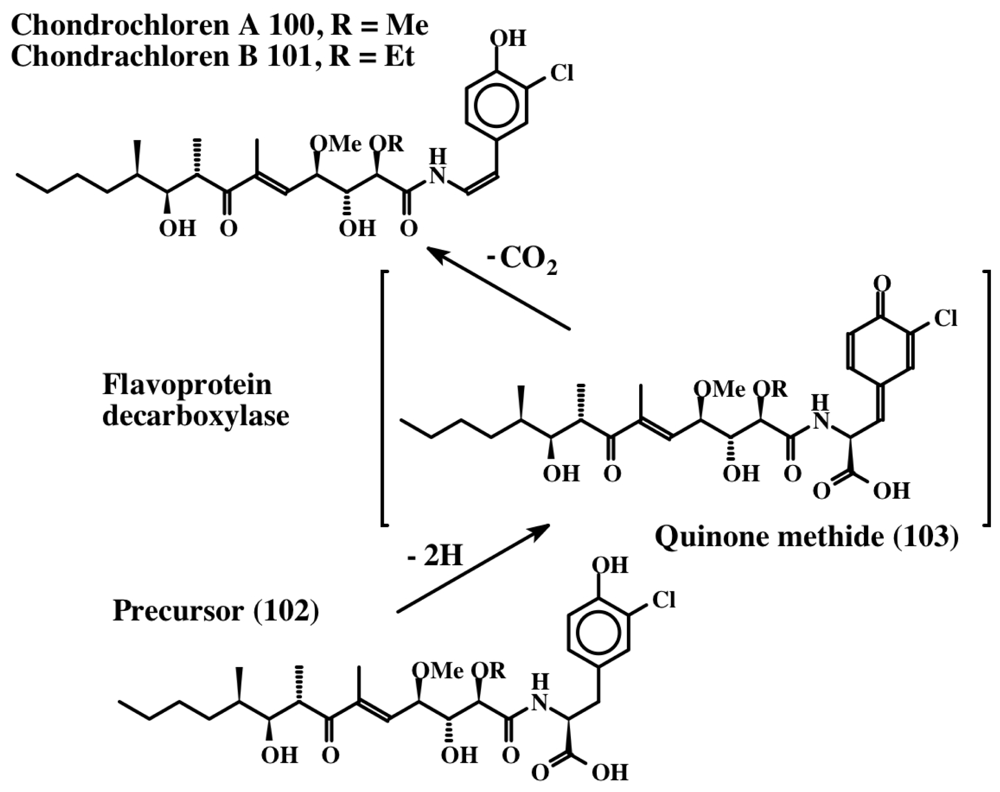
- Rachid, S; Revemann, O; Dauth, C; Kazmaier, U; Muller, R. Characterization of a novel type of oxidative decarboxylase involved in the biosynthesis of the styryl moiety of chondrochloren from an acylated tyrosine. J Biol Chem 2010, 285, 12482–12489. [Google Scholar]
- Sugumaran, M. Comparative biochemistry of eumelanogenesis and the protective roles of phenoloxidase and melanin in insects. Pigment Cell Res 2002, 15, 2–9. [Google Scholar]
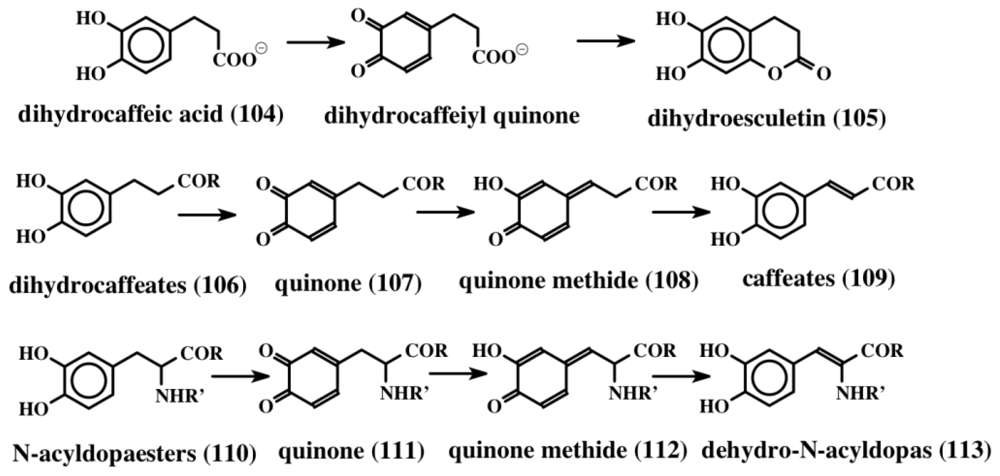
- Sugumaran, M; Dali, H; Kundzicz, H; Semensi, V. Unusual intramolecular cyclization and side chain desaturation of carboxyethyl-o-benzoquinone derivatives. Bioorg Chem 1989, 17, 443–453. [Google Scholar]
- Sugumaran, M; Semensi, V; Dali, H; Saul, SJ. Nonenzymatic transformations of enzymatically generated N-acetyl dopamine quinone and isomeric dihydrocaffeiyl methylamide quinone. FEBS Lett 1989, 255, 345–349. [Google Scholar]
- Rzepecki, LM; Waite, JH. α,β-dehydro-3,4-dihydroxyphenylalanine derivatives: Rate and mechanism of formation. Arch Biochem Biophys 1991, 285, 27–36. [Google Scholar]
- Sugumaran, M; Ricketts, D. Model sclerotization studies. 3. Cuticular enzyme catalyzed oxidation of peptidyl model tyrosine and dopa derivatives. Arch Insect Biochem Physiol 1995, 28, 17–32. [Google Scholar]
- Saul, SJ; Sugumaran, M. A novel quinone: Quinone methide isomerase generates quinone methides in insect cuticle. FEBS Lett 1988, 237, 155–158. [Google Scholar]
- Saul, SJ; Sugumaran, M. 4-Alkyl-o-quinone/2-hydroxy-p-quinone methide isomerase from the larval hemolymph of Sarcophaga bullata. I. Purification and characterization of enzyme-catalyzed reaction. J Biol Chem 1990, 265, 16992–16999. [Google Scholar]
- Saul, SJ; Sugumaran, M. N-acetyldopamine quinone methide/1,2-dehydro-N-acetyldopamine tautomerase. A new enzyme involved in sclerotization of insect cuticle. FEBS Lett 1989, 255, 340–344. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | Structure | References |
|---|---|---|---|
| 1 | Tunichrome An-1, 56 | Topa-DeTopa-DeTopamine | [14,15] |
| 2 | Tunichrome An-2, 57 | Dopa-DeTopa-DeTopamine | [14,15] |
| 3 | Tunichrome An-3, 58 | Dopa-DeTopa-DeDopamine | [14,15] |
| 4 | Tunichrome Mm-1, 59 | Gly-DeDopa-DeDopamine | [16] |
| 5 | Tunichrome Mm-2, 60 | Leu-DeDopa-DeDopamine | [16] |
| 6 | Tunichrome Pm-1, 61 | Topa-Topa-DeTopamine | [17] |
| 7 | Tunichrome Pm-2, 62 | Dopa-Topa-DeTopamine | [17] |
| 8 | Tunichrome Pm-3, 63 | Dopa-Topa-DeDopamine | [17] |
| 9 | Tunichrome Sp-1, 64 | Dopa-Dopa-Gly-Pro-DeDopamine | [19] |
| 10 | Plicatamide, 55 | Phe-Phe-His-Leu-His-Phe-His-DeDopamine | [9,66] |
| 11 | Morulin Pm, 54 | Polypeptide with 6-BrTrp & DeDopamine | [65] |
| 12 | Clionamide, 53 | 6-BrTrp-DeTopamine | [64] |
| 13 | Celenamide A, 65 | Leu-DeTopa-6-BrTrp-DeDopamine | [69–71] |
| 14 | Celenamide B, 66 | Val-DeTopa-6-BrTrp-DeDopamine | [69–71] |
| 15 | Celenamide C, 67 | Leu-DeTopa-6-BrTrp-DeTyramine | [69–71] |
| 16 | Celenamide D, 68 | Leu-DeTopa-DeTopa-DeDopamine | [69–71] |
| 17 | Celenamide E, 69 | DeTopa-6-BrTrp-DeDopamine | [69–71] |
| Category | Sponges | Coelenterates | Molluscs | Ascidians |
|---|---|---|---|---|
| 1 | Hamigeramine (15); Hamigeroxalamic acid (16); | Tridentatols A–D (17–20) | Botryllamide (5–14); Rigidin A–D (28–31) | |
| Anthosamine A & B (25, 26); | ||||
| Cyclo-l-arginine-dehydrotyrosine (27) | ||||
| 2 | Tubastrine (36); | Tubastrine (36) | Tubastrine (36); | |
| Jaspisin (42); | Orthidines (37–41); | |||
| Isojaspisin (43); | Polyandrocarpamine A & B | |||
| Narains (44, 45); | (51, 52) | |||
| Wondonins (47); | ||||
| Leucettamine A–C (48–50) | ||||
| 3 | Clionamide (53) | Morulin PM (54); | ||
| Plicatamide (55); | ||||
| Styelin A–D | ||||
| 4 | Celenamides (65–69) | Polyphenolic protein | Tunichromes (56–64) | |
| 5 | Purpurone (80); | Lamellarin | Ningalin D (81); | |
| Halitulin (75); | A–D | Ningalin A–C (82–84); | ||
| Storniamide A–D (76–79); | Lamellarin E–H; | |||
| Dictyodendrin A–E (88–92) | Polycitrin A (95); | |||
| Polycitone A & B (96, 97); | ||||
| Lukianol A & B (98, 99) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sugumaran, M.; Robinson, W.E. Bioactive Dehydrotyrosyl and Dehydrodopyl Compounds of Marine Origin. Mar. Drugs 2010, 8, 2906-2935. https://doi.org/10.3390/md8122906
Sugumaran M, Robinson WE. Bioactive Dehydrotyrosyl and Dehydrodopyl Compounds of Marine Origin. Marine Drugs. 2010; 8(12):2906-2935. https://doi.org/10.3390/md8122906
Chicago/Turabian StyleSugumaran, Manickam, and William E. Robinson. 2010. "Bioactive Dehydrotyrosyl and Dehydrodopyl Compounds of Marine Origin" Marine Drugs 8, no. 12: 2906-2935. https://doi.org/10.3390/md8122906
APA StyleSugumaran, M., & Robinson, W. E. (2010). Bioactive Dehydrotyrosyl and Dehydrodopyl Compounds of Marine Origin. Marine Drugs, 8(12), 2906-2935. https://doi.org/10.3390/md8122906