Cytotoxic Terpene Quinones from Marine Sponges

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
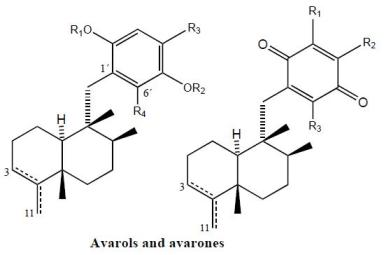
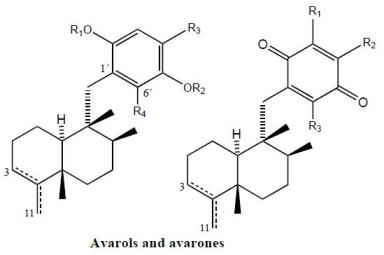
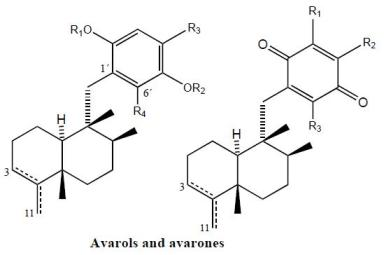
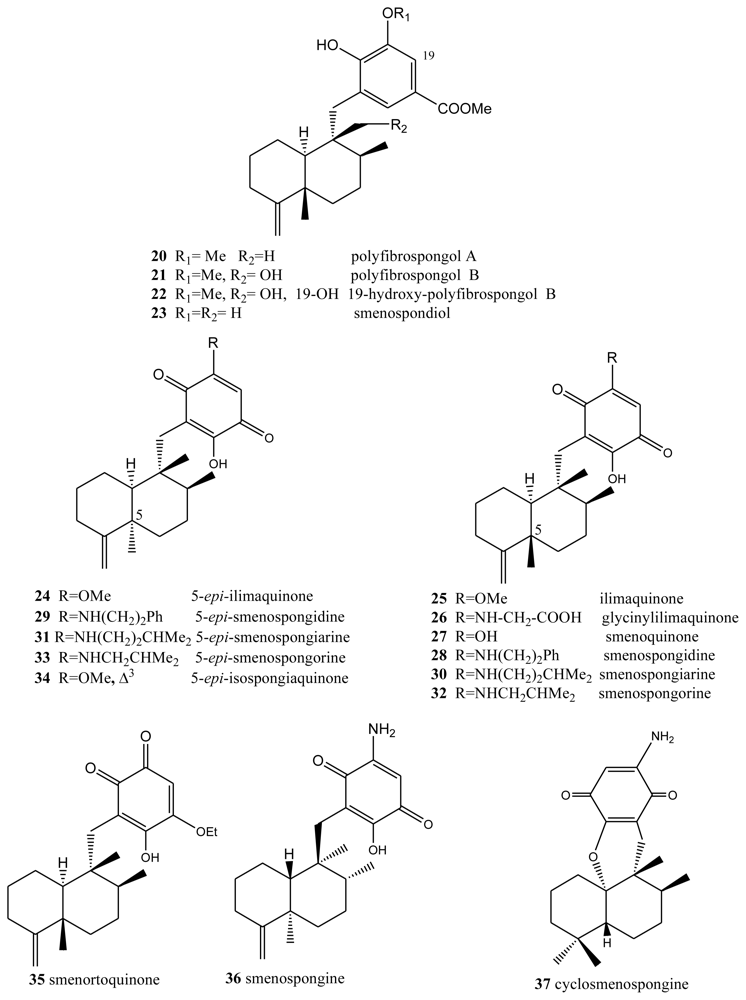
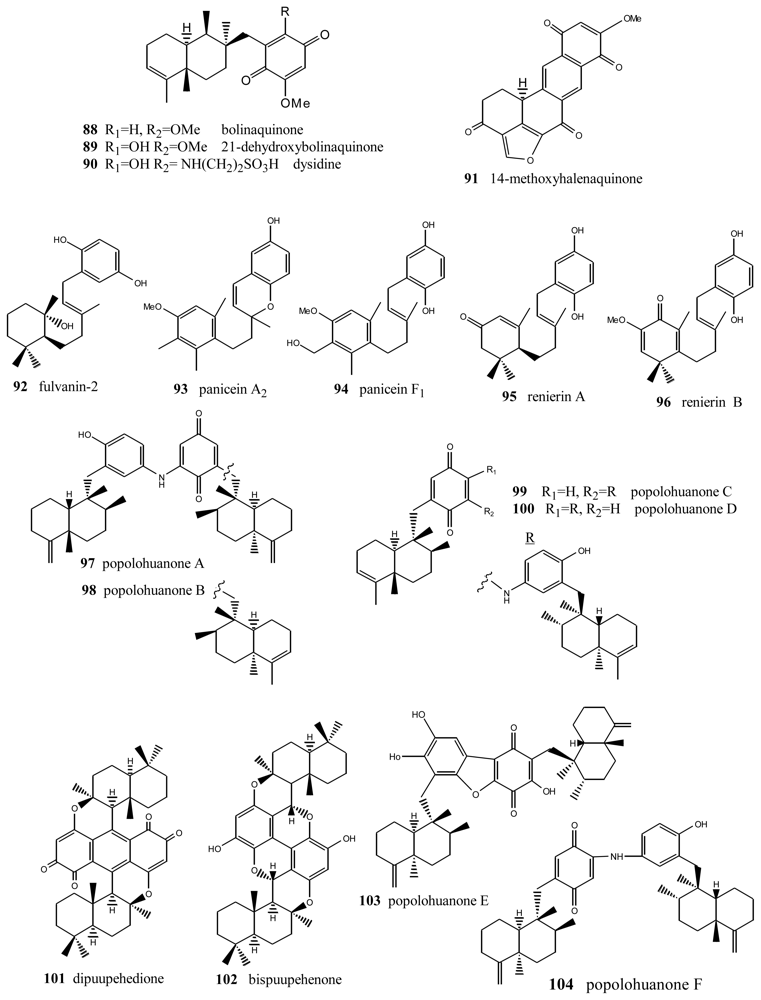
2. Terpenylquinones with a Clerodane-Type Decalin Ring
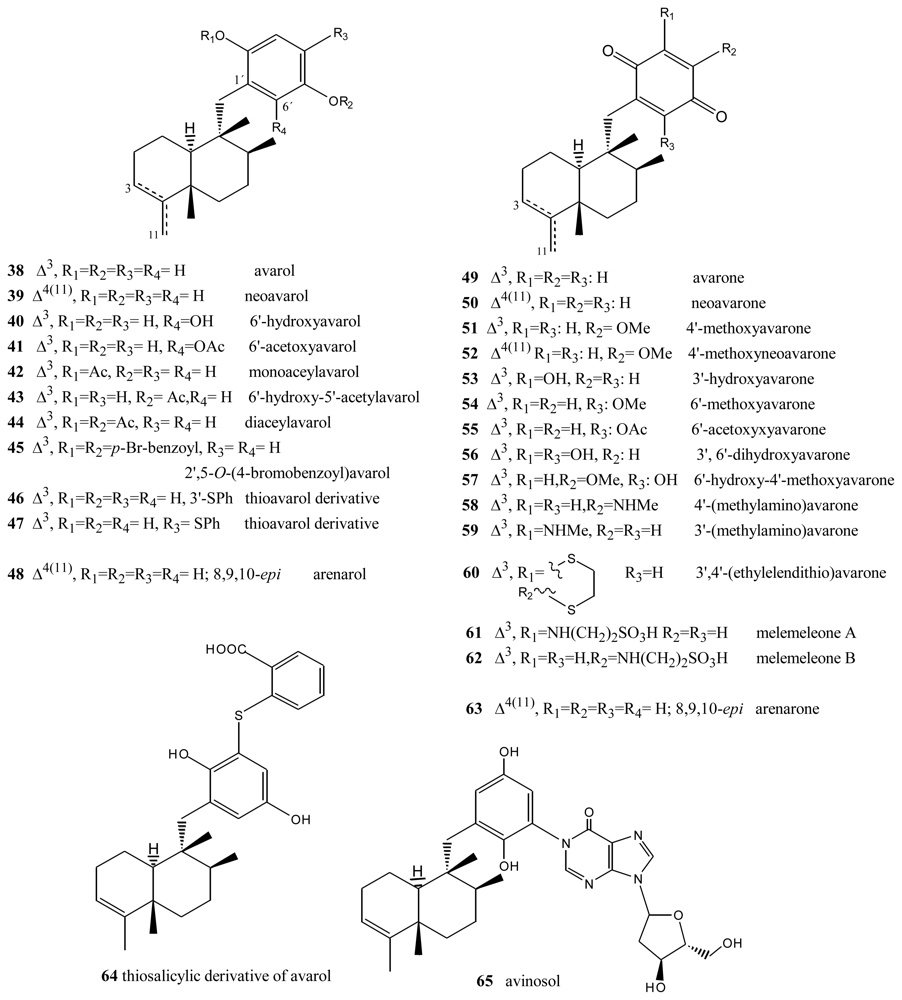
3. Terpenylquinones with a Labdane-Type Decalin Ring
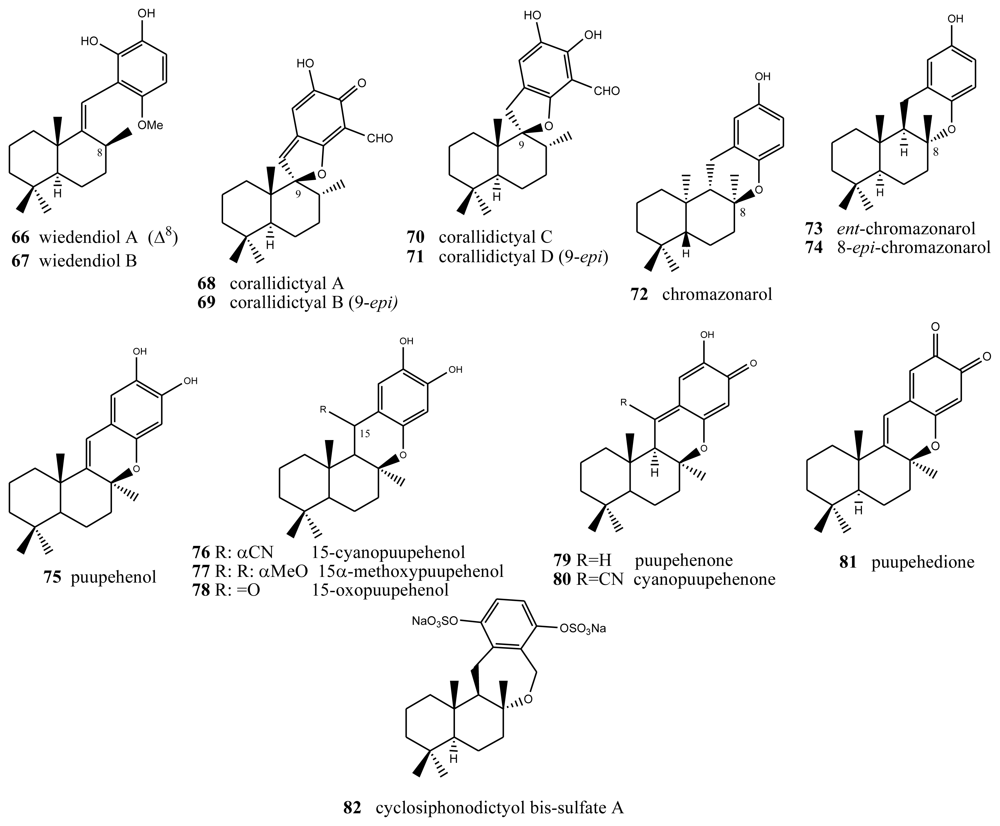
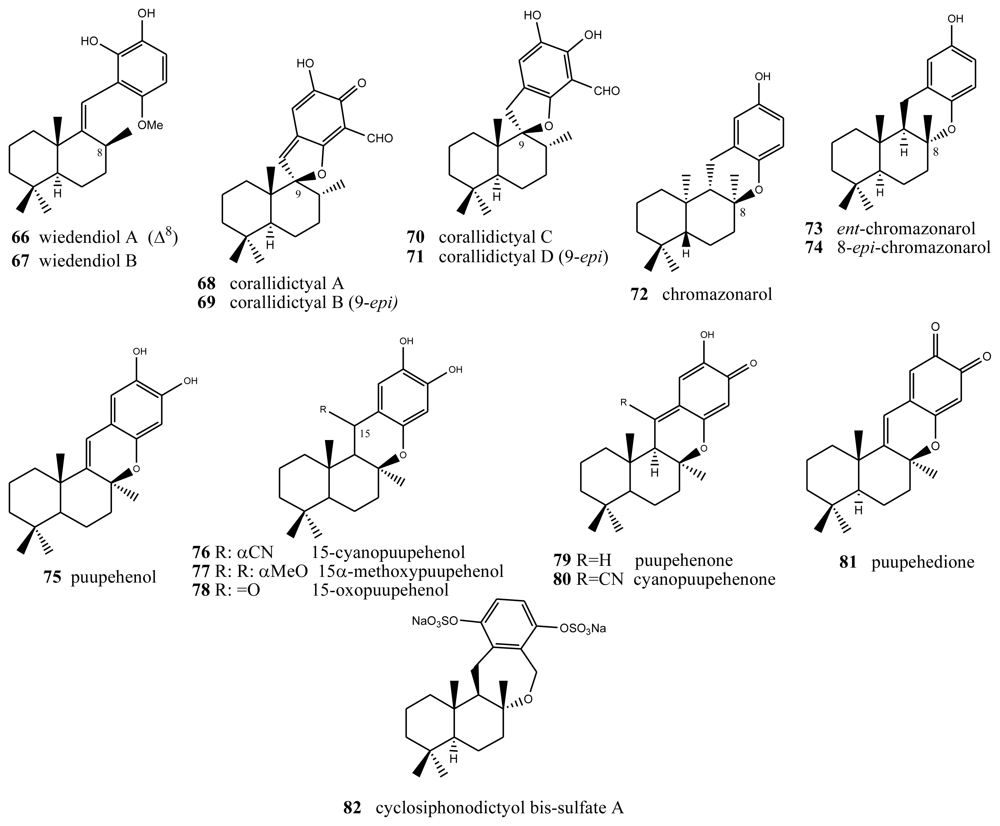
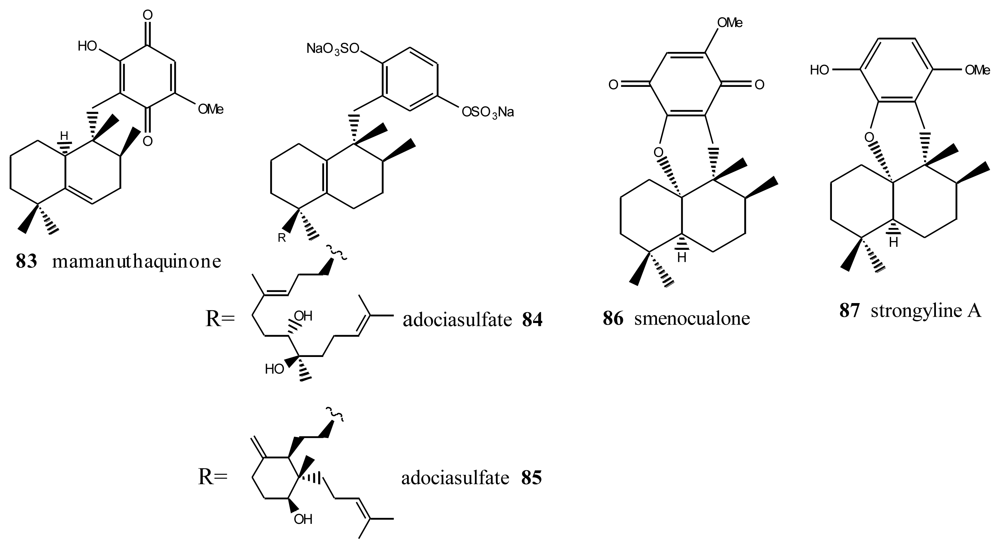
4. Terpenylquinones with a Halimane-Type Decalin Ring
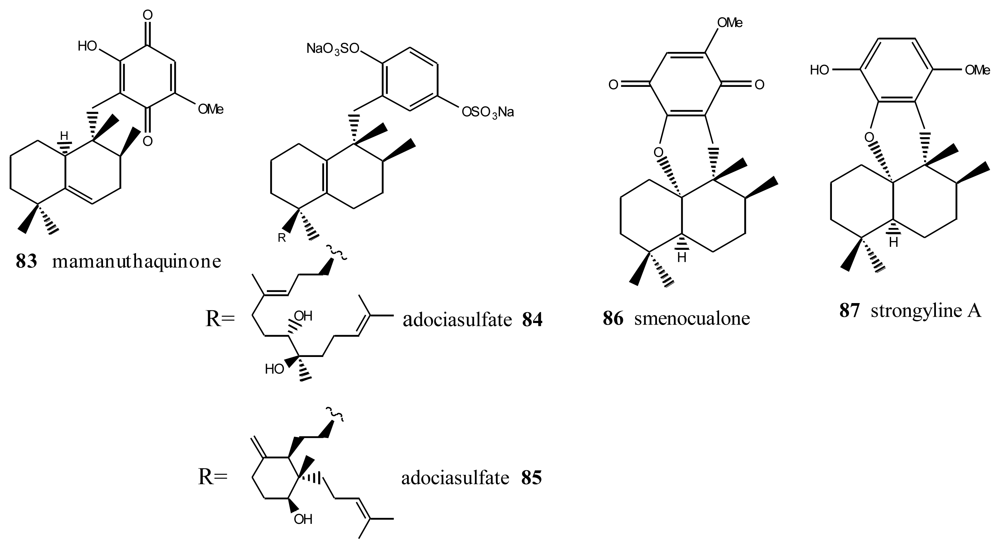
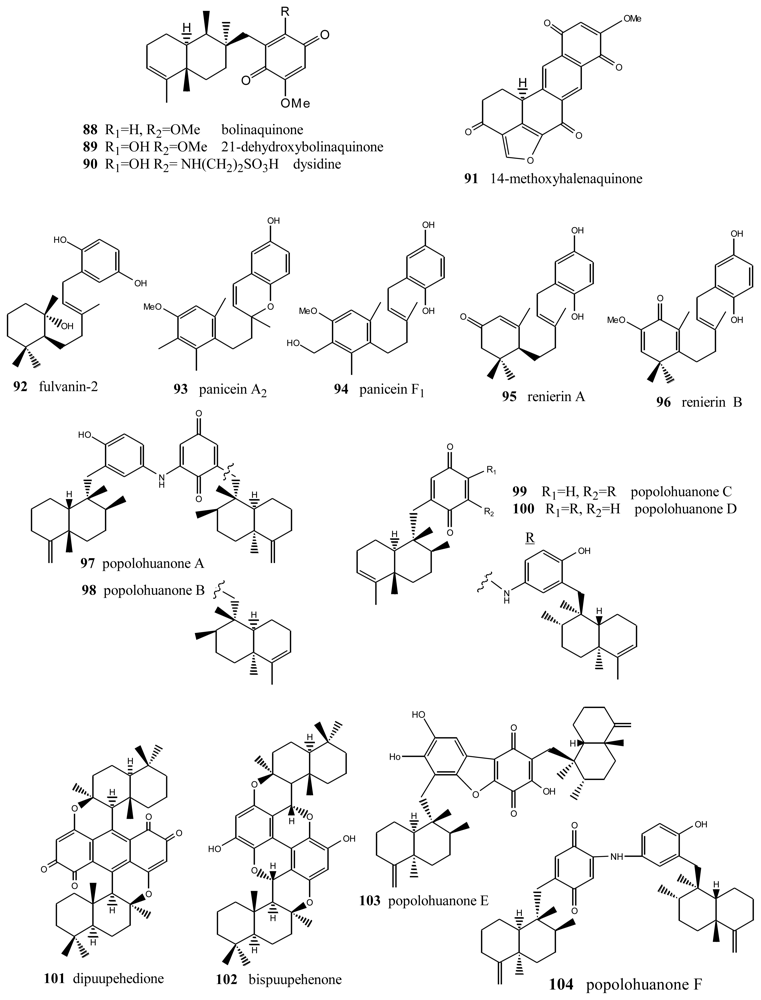
5. Other Related Compounds
6. SAR Studies and Mechanism of Action
7. Summary
Acknowledgements
- Samples Availability: Available from the authors.
References
- Skropeta, D. Deep-sea natural products. Nat Prod Rep 2008, 25, 1131–1166. [Google Scholar]
- Thornburg, C; Zabriskie, TM; McPhail, KL. Deep-sea hydrothermal: Potencial hot spot for natural products discovery. J Nat Prod 2010, 73, 489–499. [Google Scholar]
- Molinski, TT; Dalisay, DS; Lievens, SL; Saludes, JP. Drug development from marine natural products. Nat Rev Drug Discov 2009, 8, 69–85. [Google Scholar]
- Simmons, TL; Andrianasolo, E; McPhail, K; Flatt, P; Grewick, WH. Marine natural products as anticancer drugs. Mol Cancer Ther 2005, 4, 333–342. [Google Scholar]
- Villa, FA; Gerwick, L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharmacol Immunotoxicol 2010, 32, 228–237. [Google Scholar]
- Shi, Q; Huo, C; Li, L; Zhang, M. History retrospection on chemistry research of marine natural products. Zhongcaoyao 2009, 40, 1687–1695. [Google Scholar]
- Fusetani, N. Biotechnological potential of marine natural products. Pure Appl Chem 2010, 82, 17–26. [Google Scholar]
- Blunt, JW; Copp, BR; Munro, MHG; Northcote, PT; Prinsep, MR. Marine natural products. Nat Prod Rep 2010, 27, 165–237. [Google Scholar]
- Morris, JC; Phillips, AJ. Marine natural products: Synthetic aspects. Nat Prod Rep 2010, 27, 1186–1203. [Google Scholar]
- Hill, RA. Marine natural products. Annu Rep Prog Chem B Org Chem 2009, 105, 150–166. [Google Scholar]
- Nagle, DG; Zhou, Y-D. Marine natural products as inhibitors of hypoxic signaling in tumors. Phytochem Rev 2009, 8, 415–429. [Google Scholar]
- Arai, M; Kobayashi, M. Chemical biology of marine natural products. Kagaku to Seibutsu 2009, 47, 275–282. [Google Scholar]
- Glaser, KB; Mayer, AMS. A renaissance in marine pharmacology: From preclinical curiosity to clinical reality. Biochem Pharmacol 2009, 78, 440–448. [Google Scholar]
- Ravina, E. The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs; Wiley-WCH: Weinheim, Germany, 2010. [Google Scholar]
- Bailly, C. Ready for comeback of natural products in oncololoy. Biochem Pharmacol 2009, 77, 1447–1457. [Google Scholar]
- Cragg, GM; Grothaus, PG; Newman, DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev 2009, 109, 3012–3043. [Google Scholar]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin Transl Oncol 2007, 9, 767–776. [Google Scholar]
- Gordaliza, M. Terpeny-purines from the sea. Mar Drugs 2009, 7, 833–847. [Google Scholar]
- Lee, K-H. Discovery and development of natural product-derived chemotherapeutic agents based on a medicinal chemistry approach. J Nat Prod 2010, 73, 500–516. [Google Scholar]
- Miljanich, GP. Ziconotide: Neural calcium channel blocker for treating severe chronic pain. Curr Med Chem 2004, 11, 3029–3040. [Google Scholar]
- Cuevas, C; Francesch, A. Development of Yondelis® (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat Prod Rep 2009, 26, 322–333. [Google Scholar]
- Alday, PH; Correia, JJ. Macromolecular interaction of halichondrin B analogues eribulin (E7389) and E-076349 with tubulin by analitical ultacentrifugation. Biochemistry 2009, 48, 7927–7938. [Google Scholar]
- Smith, JA; Wilson, L; Azarenko, O; Zhu, X; Lewis, BM; Littlefield, BA; Jordan, MA. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 2010, 49, 1331–1337. [Google Scholar]
- Sladic, D; Gasic, MJ. Reactivity and biological activity of marine sesquiterpene hydroquinones avarol and related compound from sponges of Order Dictyoceratida. Molecules 2006, 11, 1–33. [Google Scholar]
- Motti, CA; Bourguet-Kondracki, M-L; Longeon, A; Doyle, JR; Llewellyn, LE; Tapiolas, DM; Yin, P. Comparison of biological properties of several marine sponge-derived sesquiterpenoid quinines. Molecules 2007, 12, 1376–1388. [Google Scholar]
- Cragg, GM; Newman, DJ. Industrial applications natural products for medicinal purposes. Drugs from nature: Present development and future prospects. In Natural Products in the New Millenium: Prospects and Industrial Applications; Rauter, AP, Palma, FB, Justino, J, Araújo, ME, Dos Santos, SP, Eds.; Kluwer: Dordrecht, The Netherlands, 2002; pp. 441–461. [Google Scholar]
- Gordaliza, M; Miguel del Corral, JM; Mahiques, MM; San Feliciano, A; García-Grávalos, MD. Terpenequinone with antitumor activity. PCT Int Appl EP 0731078 A1 1994. [Google Scholar]
- Miguel del Corral, JM; Castro, MA; Gordaliza, M; Martín, ML; Gamito, AM; Cuevas, C; San Feliciano, A. Synthesis and citotoxicity of new heterocyclic terpenylnaphthoquinones. Bioorg Med Chem 2006, 14, 2816–2827. [Google Scholar]
- Alegría, A; Sánchez, S; Sánchez-Cruz, P; Nieves, I; Cruz, NG; Gordaliza, M; Martín-Martín, ML. Terpenylnaphthoquinones are reductively activated by NADH/NADH dehydrogenase. Toxicol Environ Chem 2005, 87, 237–245. [Google Scholar]
- Alegría, A; Cordones, E; Marcano, Y; Sanchez, S; Gordaliza, M; Martín-Martín, ML. Reductive activation of terpenylnaphtoquinones. Toxicology 2002, 175, 167–175. [Google Scholar]
- Bozic, T; Novakovic, I; Gasic, MJ; Juranic, Z; Stanojkovic, T; Tufegdzic, S; Kljajic, Z; Sladic, D. Synthesis and biological activity of derivatives of the marine quinone avarone. Eur J Med Chem 2010, 45, 923–929. [Google Scholar]
- Lu, P-H; Chueh, S-C; Kung, F-L; Pan, S-L; Shen, Y-C; Guh, J-H. Illimaquinone, a marine sponge metabolite, displays anticancer activity via GADD153-mediated pathway. Eur J Pharmacol 2007, 556, 45–54. [Google Scholar]
- Parks, J; Gyeltshen, T; Prachyawarakorn, V; Mahidol, C; Ruchirawat, S; Kittakoop, P. Glutarimide alkaloids and a terpenoid benzoquinone from Cordia globifera. J Nat Prod 2010, 73, 992–994. [Google Scholar]
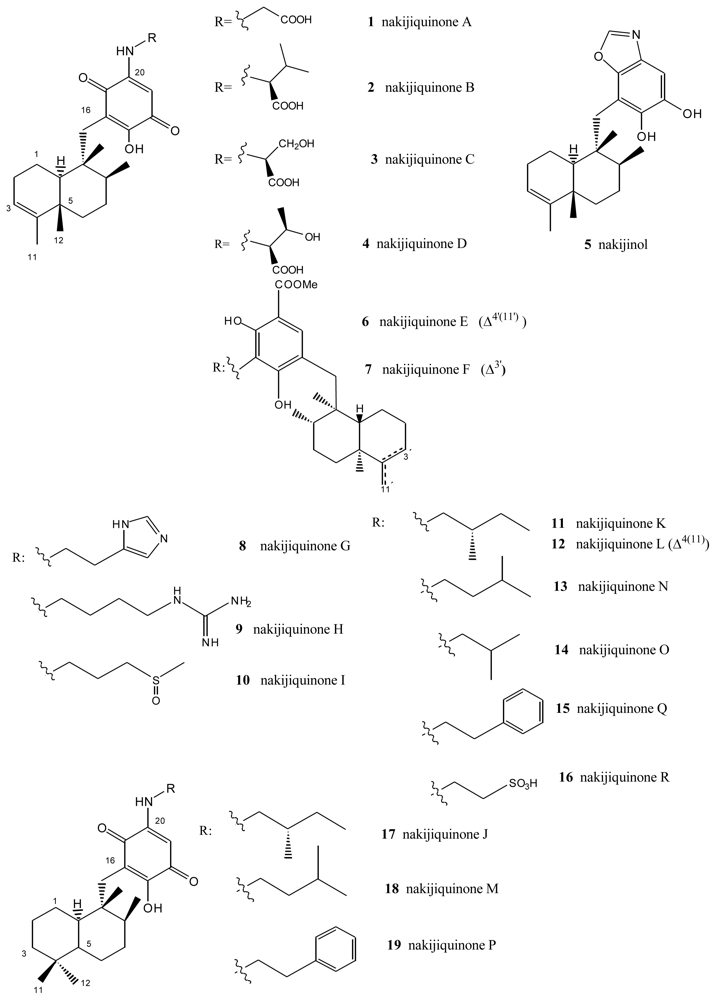
- Kobayashi, J. Chemistry and biology of Okinawan marine natural products. Pure Appl Chem 2009, 81, 1009–1018. [Google Scholar]
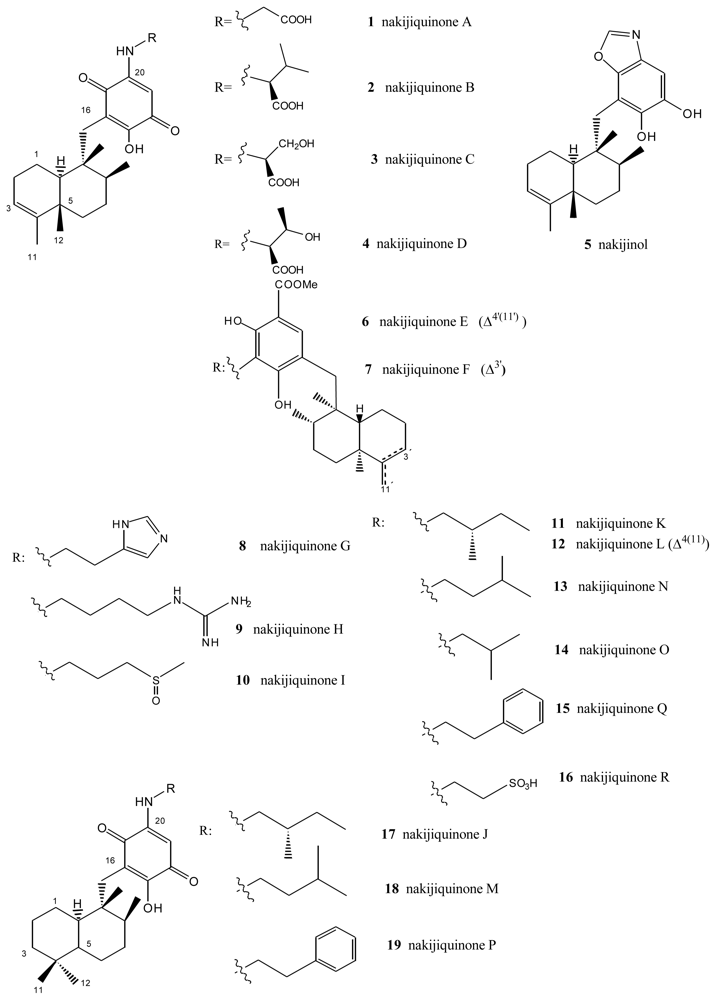
- Shigemori, H; Madono, T; Sasaki, T; Mikami, Y; Kobayashi, J. Nakijiquinones A and B, new antifungal sesquiterpenoid quinones with an amino acid residue from an Okinawan marine sponge. Tetrahedron 1994, 50, 8347–8354. [Google Scholar]
- Kobayashi, J; Madono, T; Shigemori, H. Nakijiquinones C and D, new sesquiterpenoid quinones with a hydroxy amino acid residue from a marine sponge inhibiting c-erbB-2 kinase. Tetrahedron 1995, 51, 10867–10874. [Google Scholar]
- Takahashi, Y; Kubota, T; Kobayashi, J. Nakijiquinones E and F, new dimeric sesquiterpenoid quinones from marine sponge. Bioorg Med Chem 2009, 17, 2185–2188. [Google Scholar]
- Takahashi, Y; Kubota, T; Ito, J; Mikami, Y; Fromont, J; Kobayashi, J. Nakijiquinones G–I, new sesquiterpenoid quinones from marine sponge. Bioorg Med Chem 2008, 16, 7561–7564. [Google Scholar]
- Takahashi, Y; Ushio, M; Kubota, T; Yamamoto, S; Fromont, J; Kobayashi, J. Nakijiquinones J–R, Sesquiterpenoid quinones with an qmine residue from Okinawan marine sponges. J Nat Prod 2010, 73, 467–471. [Google Scholar]
- Stahl, P; Kissau, L; Mazitschek, R; Huwe, A; Furet, P; Giannis, A; Waldmann, H. Total synthesis and biological evaluation of the nakijiquinones. J Am Chem Soc 2001, 123, 11586–11593. [Google Scholar]
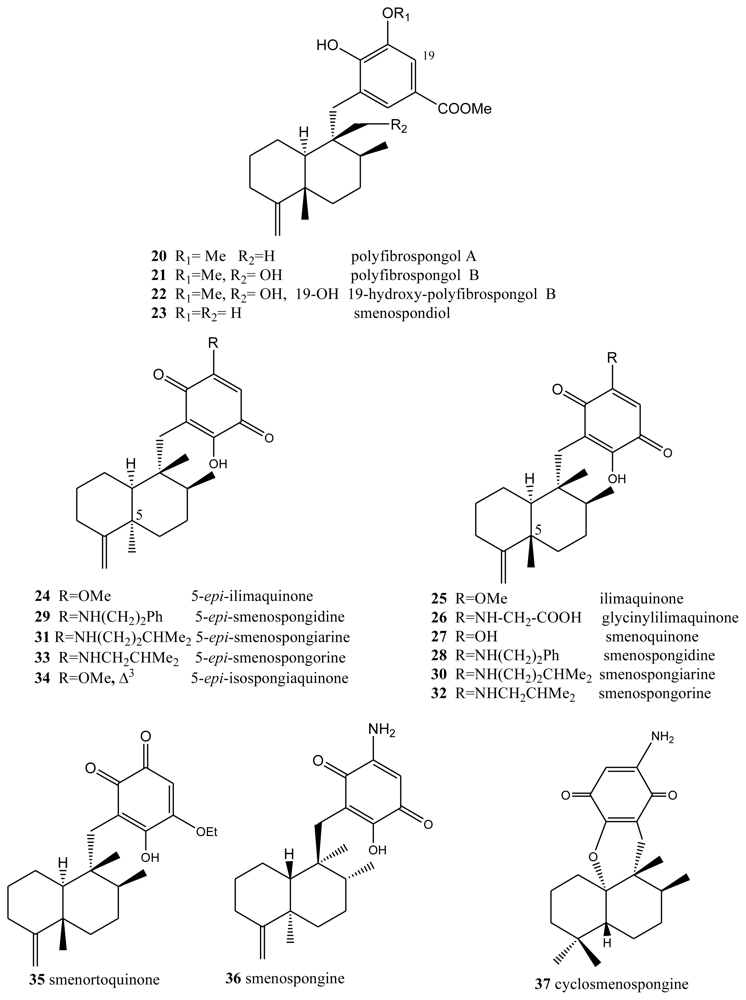
- Shen, Y-C; Hsieh, P-W. New sesquiterpene hydroquinones from a Taiwanese marine sponge Polyfibrospongia australis. J Nat Prod 1997, 60, 93–97. [Google Scholar]
- Qiu, Y; Wang, XM. A new sesquiterpenoid hydroquinone from the marine sponge Dysidea arenaria. Molecules 2008, 13, 1275–1281. [Google Scholar]
- Haruo, Y; Hasegawa, T; Tanaka, H; Takahashi, T. Total Synthesis of (±)-smenospondiol by titanium(III)-mediated tandem radical cyclization. Synlett 2001, 2001, 1935–1937. [Google Scholar]
- Kondracki, M-L; Davoust, D; Guyot, M. Smenospondiol, a biologically active hydroquinone from the sponge Smenospongia sp. J Chem Res (Synop) 1989, 3, 74–75. [Google Scholar]
- Nakamura, H; Deng, S; Kobayashi, J; Ohizumi, Y. Dictyioceratin-A and B, novel antimicrobial terpenoids from Okinawan marine sponge Hippospongia sp. Tetrahedron 1986, 42, 4197–4201. [Google Scholar]
- Rodriguez, J; Quinoá, E; Riguera, R; Peters, BM; Abrell, LM; Crews, P. The structures and stereochemistry of cytotoxic sesquiterpene quinones from Dactylospongia elegans. Tetrahedron 1992, 48, 6667–6680. [Google Scholar]
- Liu, H; Wang, G; Namikoshi, M; Kobayashi, H; Yao, X; Cai, G. Sesquiterpene quinones from a marine sponge Hippospongia sp. that inhibit maturation of starfish oocytes and induce cell cycle arrest with HepG2 cells. Pharm Biol 2006, 44, 522–527. [Google Scholar]
- Kondracki, ML; Guyot, M. Biologically active quinine and hydroquinones sesquiterpenoids from the sponge Smenospongia sp. Tetrahedron 1989, 45, 1995–2004. [Google Scholar]
- Evans, TP; Cornell, L; Peterson, RW; Faulkner, DJ. Isolation and synthesis of a glycine derivative of ilimaquinone from Fasciospongia sp. Nat Prod Lett 1994, 4, 287. [Google Scholar]
- Kondracki, ML; Guyot, M. Smenospongine: A cytotoxic and antimicrobial aminoquinone isolated from Smenospongia sp. Tetrahedron Lett 1987, 28, 5815–5818. [Google Scholar]
- Kong, D; Aoki, S; Sowa, Y; Sakai, T; Kobayashi, M. Smenospongine, a sesquiterpene aminoquinone from a marine sponge, induces G1 arrest or apoptosis in different leukemia cells. Mar Drugs 2008, 6, 480–488. [Google Scholar]
- Aoki, S; Kong, D; Matsui, K; Rachmat, R; Kobayashi, M. Sesquiterpene aminoquinoes from a marine sponge induce erytnroid differentiation in human chronic myelogenous leukemia K562 cells. Chem Pharm Bull 2004, 52, 935–937. [Google Scholar]
- Utkina, NK; Denisenko, VA; Scholokova, OV; Virovaya, MV; Prokof’eva, NG. Cyclosmenospongine, a new sequiterpenoid aminoquinone from an Australian marine sponge Spongia sp. Tetrahedron Lett 2003, 44, 101–102. [Google Scholar]
- Ling, T; Poupon, E; Rueden, EJ; Kim, SH; Theodorakis, EA. Unified synthesis of quinone sesquiterpenes based on a radical decarboxilation and quinone addition reaction. J Am Chem Soc 2002, 124, 12261–12267. [Google Scholar]
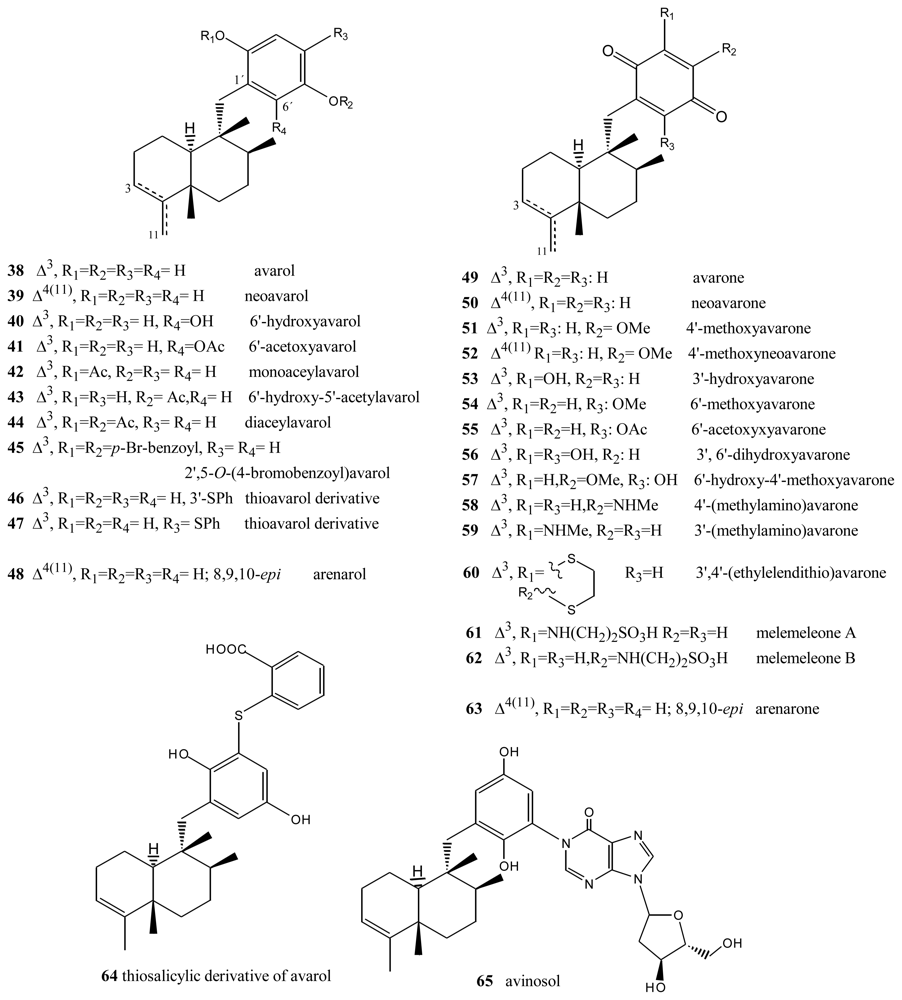
- Cimino, G; De Rosa, S; De Stefano, S; Cariello, L; Zanetti, L. Structure of two biologically active sesquiterpenoid amino-quinones from de marine sponge Dysidea avara. Experientia 1982, 38, 896. [Google Scholar]
- Cozzolino, R; De Giulio, A; De Rosa, S; Strazzullo, G; Gašič, MJ; Sladić, D; Zlatović, M. Biological activities of avarol derivatives, 1. Amino derivatives. J Nat Prod 1990, 53, 699–702. [Google Scholar]
- Müller, WEG; Maidhof, A; Zahn, RK; Schröder, HCM; Gasic, MJ; Heidemann, D; Bernd, A; Kurelec, B; Eich, E; Seibert, G. Potent antileukemic activity of the novel cytostatic agent avarone and its analogues in vitro and in vivo. Cancer Res 1985, 45, 4822–4826. [Google Scholar]
- Müller, WEG; Sobel, C; Sachsse, W; Diehl-Seifert, B; Zahn, RK; Eich, E; Kljajić, Z; Schröder, HC. Biphasic and differential effects of the cytostatic agents avarone and avarol on DNA metabolism of human and murine T and B lymphocytes. Eur J Cancer Clin Oncol 1986, 22, 473–476. [Google Scholar]
- Müller, WEG; Sobel, C; Diehl-Seifert, B; Maidhof, A; Schöder, HC. Influence of the antileukemic and anti-human immunodeficiency virus agent avarol on selected immune responses in vitro and in vivo. Biochem Pharmacol 1987, 36, 1489–1494. [Google Scholar]
- Sarin, PS; Sun, D; Thornton, A; Müller, WEG. Inhibition of replication of the etiologic agent of acquired immune deficiency syndrome (human T-lymphotropic retrovirus/lymphadenopathyassociated virus) by avarol and avarone. J Natl Cancer Inst 1987, 78, 663–666. [Google Scholar]
- De Giulio, A; De Rosa, S; Strazzullo, G; Diliberto, L; Obino, P; Marongiu, ME; Pani, A; La Colle, P. Synthesis and evaluation of cytostatic and antiviral activities of 3′- and 4′-avarone derivatives. Antivir Chem Chemother 1991, 4, 223–227. [Google Scholar]
- Iguchi, K; Sahashi, A; Khono, J; Yamada, Y. New sesquiterpenoid hydroquinone and quinones from the Okinawan marine sponge (Dysidea sp.). Chem Pharm Bull 1990, 38, 1121–1123. [Google Scholar]
- Hirsch, S; Rudi, A; Kashman, Y; Loya, Y. New avarone and avarol derivatives from the marine sponge Dysidea cinerea. J Nat Prod 1991, 54, 92–97. [Google Scholar]
- Alvi, KA; Diaz, MC; Crews, P; Slate, DL; Lee, RH; Moretti, R. Evaluation of new sesquiterpene quinones from two Dysidea sponge species as inhibitors of protein tyrosine kinase. J Org Chem 1992, 57, 6604–6607. [Google Scholar]
- Crispino, A; De Giulio, A; De Rosa, S; Strazzullo, G. A New bioactive derivative of avarol from the marine Sponge Dysidea avara. J Nat Prod 1989, 52, 646–648. [Google Scholar]
- De Giulio, A; De Rosa, S; Di Vincenzo, G; Strazzullo, G. Further bioactive derivative of avarol from Dysidea avara. Tetrahedron 1990, 46, 7971–7976. [Google Scholar]
- Sakurai, J; Oguchi, T; Watanabe, K; Abe, H; Kanno, S-I; Ishikawa, M; Katoh, T. Highly efficient total synthesis of the marine natural products. (+)-avarone, (+)-avarol, (−)-neoavarone, (−)-neoavarol and (+)-aureol. Chem Eur J 2008, 14, 829–837. [Google Scholar]
- Shen, Y-C; Lu, C-H; Chakraborty, R; Kuo, Y-H. Isolation of sesquiterpenoids from sponge Dysidea avara and chemical modification of avarol as potencial antitumor agents. Nat Prod Rep 2003, 17, 83–89. [Google Scholar]
- Amigo, M; Terencio, MC; Paya, M; Iodice, C; De Rosa, S. Synthesis and evaluation of diverse thio avarol derivatives as potential UVB photoprotectives candidates. Bioorg Med Chem Lett 2007, 17, 2561–2565. [Google Scholar]
- Amigo, M; Terencio, MC; Mitova, M; Iodice, C; Paya, M; De Rosa, S. Potential antipsoriatic avarol derivatives as antioxidants and inhibitors of PGE2 generation and proliferation in HaCaT cell line. J Nat Prod 2004, 67, 1459–1463. [Google Scholar]
- Diaz-Marrero, AR; Austin, P; van Soest, R; Matainaho, T; Roskelley, CD; Roberge, M; Andersen, RJ. Avinosol, a meroterpenoid-nucleoside conjugate with antiinvasion activity isolated from the marine sponge Dysidea sp. Org Lett 2006, 8, 3749–3752. [Google Scholar]
- Schmitz, FJ; Lakshmi, V; Powell, DR; Van der Helm, D. Arenarol and arenarona: Sesquiterpenoids with rearranged drimane skeletons from marine sponge Dysidea arenaria. J Org Chem 1984, 49, 241–244. [Google Scholar]
- Utkina, NK; Denisenko, VA; Krasokhin, VB. Sesquiterpenoidd aminoquinones from marine sponge Dysidea sp. J Nat Prod 2010, 73, 788–791. [Google Scholar]
- Coval, SJ; Conover, MA; Mierzwa, R; King, A; Puar, MS; Phife, DW; Pai, JK; Burrier, RE; Ahn, HS. Widendiol A and B, cholesteryl esther transfer protein inhibitors from marine sponge Xetospongia widenmayeri. Bioorg Med Chem Lett 1995, 5, 605–610. [Google Scholar]
- Chackalamannil, S; Wang, Y; Xia, Y; Czarniecki, M. An efficient synthesis of wiedendiol A from (+)-sclareolide. Tetrahedron Lett 1995, 36, 5315–5318. [Google Scholar]
- Laube, T; Bernet, A; Dahse, H-M; Jacobsen, ID; Seifert, K. Synthesis and pharmacological activities of some sesquiterpene quinines and hydroquinones. Bioorg Med Chem 2009, 17, 1422–1427. [Google Scholar]
- Chan, JA; Freyer, AJ; Carté, BK; Hemling, ME; Hofmann, GH; Mattern, MR; Mentzer, MA; Westley, JW. Protein kinase C inhibitors: novel spitosequiterpene aldehydes from a marine sponge Aka (=Siphonodictyon). coralliphagum. J Nat Prod 1994, 57, 1543–1548. [Google Scholar]
- Grube, A; Assman, M; Lichte, E; Sasse, F; Pawlik, JR; Loeck, M. Bioactive metabolites from the Caribbean sponge Aka coralliphagun. J Nat Prod 2007, 70, 504–509. [Google Scholar]
- Cimino, G; De Stefano, S; Minale, L. Chromazonarol, a chroman-sesquiterpenoid from sponge Dysidea pallenscens. Experientia 1975, 31, 1117. [Google Scholar]
- Barrero, AJ; Alvarez-Manzaneda, EJ; Herrador, MM; Chahboun, R; Galera, P. Synthesis and antitumoral activities of marine ent-chromazonarol and related compounds. Bioorg Med Chem Lett 1999, 9, 2325–2328. [Google Scholar]
- Djura, P; Stierle, DB; Sullivan, B; Faulkner, DJ. Some metabolites of the marine sponges Smenospongia aurea and Smenospongia (Polyfibrospongia) echina. J Org Chem 1980, 45, 1435–1441. [Google Scholar]
- Song, F; Fan, X; Xu, X; Wang, S; Li, S; Yang, Y; Shi, J. Studies on chemical constituents of the brown alga Dictyopteris divaricata. Zhongguo Zhong Yao Za Zhi 2006, 31, 125–128. [Google Scholar]
- Hideaki, I; Kazuaki, I; Hisashi, Y. A new artificial cyclase for poliprenoids: Enantioselective total synthesis of (−)-chromazonarol, (+)-8-epi-puupehedione and (−)-11′-deoxytaondiol methyl ether. J Am Chem Soc 2004, 126, 11133–11123. [Google Scholar]
- Bourguet-Kondracki, M-L; Lacombe, F; Guyot, M. Methanol adduct of puupehenone, a biologically active derivative from the marine sponge Hyrtios species. J Nat Prod 1999, 62, 1304–1305. [Google Scholar]
- Hamann, MT; Scheuer, PJ; Kelly-Borges, M. Biogenetically diverse, bioactive constituents of a sponge, order Verongida: Bromotyramines and sesquiterpene-shikimate derived metabolites. J Org Chem 1993, 58, 6565–6569. [Google Scholar]
- Kohmoto, S; McConnell, OJ; Wright, A; Koehn, F; Thompson, W; Lui, M; Snader, KM. Puupehenone, a cytotoxic metabolite from a deep water marine sponge Stronglyophora hartmani. J Nat Prod 1987, 50, 336. [Google Scholar]
- Nasu, SS; Yeung, BKS; Hamann, T; Scheuer, PJ; Kelly-Borges, M; Goins, K. Puupehenone-related metabolites from two Hawaiian sponges, Hyrtios sp. J Org Chem 1995, 60, 7290–7292. [Google Scholar]
- Arjona, O; Garranzo, M; Mahugo, J; Maroto, E; Plumet, J; Saez, B. Total synthesis of both enantiomers of 15-oxopuupehenol methylendioxy derivatives. Tetrahedron Lett 1997, 38, 7249–7252. [Google Scholar]
- Barrero, AJ; Alvarez-Manzaneda, EJ; Chahboun, R; Cortes, M; Armstrong, V. Synthesis and antitumoral activities of puupehedione and related compounds. Tetrahedron 1999, 55, 15181–15208. [Google Scholar]
- Alvarez-Manzaneda, E; Chhboun, R; Cabrera, E; Alvarez, E; Haidour, A; Ramos, JM; Alvarez-Manzaneda, R; Tapia, R; Es-Samti, H; Fernandez, A; Barranco, I. A convenient enantiospecific route towards bioactive merosesquiterpenes by cationic-resin-promoted Friedel-Crafts alkylation with α,β-enones. Eur J Org Chem 2009, 2009, 1139–1143. [Google Scholar]
- Castro, ME; Gonzales-Iriarte, M; Barrero, AF; Salvador-Tormo, N; Munoz-Chapuli, R; Medina, MA; Quesada, AR. Study of puupehenone and related compounds as inhibitors of angiogenesis. Int J Cancer 2004, 110, 31–38. [Google Scholar]
- Pina, I; Sanders, ML; Crews, P. Puupehenones congeners from an Indo-pacific Hyrtios sponge. J Nat Prod 2003, 66, 2–6. [Google Scholar]
- Killday, KB; Wright, AE. Bis(Sulfato)-Cyclosiphonodictyol A, a new disulfated sesquiterpenehydroquinone from a deep water collection of the marine sponge Siphonodictyon coralliphagum. J Nat Prod 1995, 58, 958–960. [Google Scholar]
- Swersey, JC; Barrows, LR; Ireland, CM. Mamanuthaquinone: An antimicrobial and cytotoxic metabolite of Fasciospongia sp. Tetrahedron Lett 1991, 32, 6687–6690. [Google Scholar]
- Blackburn, CL; Hopmann, C; Sakowicz, R; Berdelis, MS; Goldstein, LSB; Faulkner, DJ. Adociasulfates 1–6 inhibitors of kinesin motor proteins from the sponge Haliclona (aka Adocia) sp. J Org Chem 1999, 64, 5565–5570. [Google Scholar]
- Bourghet-Kondracki, M-L; Martin, M-T; Guyot, M. Smenoqualone a novel sesquiterpenoid from the marine sponge Smenospongia sp. Tetrahedron Lett 1992, 33, 8079–8080. [Google Scholar]
- Wright, AE; Rueth, SA; Cross, SS. An antiviral sesquiterpene hydroquinone from the marine sponge Strongylophora hartmani. J Nat Prod 1991, 54, 1108–1111. [Google Scholar]
- Guzmán, FS; Copp, BR; Mayne, CL; Concepcion, GP; Mangalindan, GC; Barrows, LR; Ireland, CM. Bolinaquinone: A novel cytotoxic sesquiterpene hydroquinone from Philippine Dysidea sponge. J Org Chem 1998, 63, 8042–8044. [Google Scholar]
- Li, Y; Zhang, Y; Shen, X; Guo, Y-W. A novel sesquiterpene quinine from Haian sponge Dysidea villosa. Bioorg Med Chem Lett 2009, 19, 390–392. [Google Scholar]
- Giannini, C; Debitus, C; Lucas, R; Ubeda, A; Paya, M; Hooper, JN; D’Auria, MV. New sesquiterpene derivatives from sponge Dysidea species with a selective inhibitor profile against hyman phosfopholipase A2 and other leukocyte functions. J Nat Prod 2001, 64, 612–615. [Google Scholar]
- Alvi, KA; Rodríguez, J; Diaz, MC; Moretti, R; Lee, RH; Slate, DL; Crews, P. Protein tyrosine kinase inhibitory properties of planar polycyclics obtained from the marine sponge Xestospongia cf. carbonaria and from total synthesis. J Org Chem 1993, 58, 4871–4880. [Google Scholar]
- Casapullo, A; Minale, L; Zollo, F. Paniceins and related sesquiterpenoids from the Mediterranean sponge Reniera fulva. J Nat Prod 1993, 56, 527–533. [Google Scholar]
- Zubia, E; Ortega, MJ; Carballo, JL; Salva, J. Sesquiterpene hydroquinone from the sponge Reniera mucosa. Tetrahedron 1994, 50, 8153–8160. [Google Scholar]
- Rodriguez, AD; Yoshida, WY; Scheuer, PJ. Popolohuanone A and B. Two new sesquiterpenoid aminoquinones from Pacific sponge Dysidea sp. Tetrahedron 1990, 46, 8025–8030. [Google Scholar]
- Carney, JR; Scheuer, PJ. Popolohuanone E, a topoisomerase II inhibitor eith selective lung citotoxicity from Pohnpei sponge Dysidea sp. Tetrahedron Lett 1993, 34, 3727–3730. [Google Scholar]
- Munday, RH; Denton, RM; Anderson, JC. Asymmetric synthesis of 6′-hydroxyarenarol: The proposed biosynthetic precursor to popolohuanone E. J Org Chem 2008, 73, 8033–8038. [Google Scholar]
- Yong, KWL; Jankam, A; Hooper, JNA; Suksamrarn, A; Garson, MJ. Stereochemical evaluation of sesquiterpene quinones from two sponges of the genus Dactylospongia and the implication for enantioselective processes in marine terpene biosynthesis. Tetrahedron 2008, 64, 6341–6348. [Google Scholar]
- Prokof’eva, NG; Utkina, NK; Chaikina, EL; Makarchenko, AE. Biological activities of marine sesquiterpenoid quinones: Structure-activity relationships in cytotoxic and hemolytic assays. Comp Biochem Physiol B: Biochem Mol Biol 2004, 139, 169–173. [Google Scholar]
- Schirmer, RH; Müller, JG; Krauth-Siegel, RL. Disulfide-Reductase Inhibitors as Chemotherapeutic Agents: The Design of Drugs for Trypanosomiasis and Malaria. Angew Chem Int Ed Engl 1995, 34, 141–154. [Google Scholar]
- Monks, TJ; Hanzlik, RP; Cohen, GM; Ross, D; Graham, DG. Quinone chemistry and toxicity. Toxicol Appl Pharmacol 1992, 112, 2–16. [Google Scholar]
- O’Brien, PJ. Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact 1991, 80, 1–41. [Google Scholar]
- Schröder, HC; Wenger, R; Gerner, H; Reuter, P; Kuchino, Y; Müller, WEG. Suppression of the Modulatory Effects of the Antileukemic and Anti-Human Immunodeficiency Virus Compound Avarol on Gene Expression by Tryptophan. Cancer Res 1989, 49, 2069–2076. [Google Scholar]
- Sladić, D; Gašić, MJ. Effects of iron(II) compounds on the amount of DNA damage in Friend erythroleukemia cells induced by avarol. Role of hydroxyl radicals. J Serb Chem Soc 1994, 59, 915–920. [Google Scholar]
- Novaković, I; Vujčić, Z; Božić, T; Božić, N; Milosavić, N; Sladić, D. Chemical modification of β-lactoglobulin by quinones. J Serb Chem Soc 2003, 68, 243–248. [Google Scholar]
- Sladić, D; Novaković, I; Vujčić, Z; Božić, T; Božić, N; Milić, D; Šolaja, B; Gašić, MJ. Protein covalent modification by biologically active quinones. J Serb Chem Soc 2004, 69, 901–907. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gordaliza, M. Cytotoxic Terpene Quinones from Marine Sponges. Mar. Drugs 2010, 8, 2849-2870. https://doi.org/10.3390/md8122849
Gordaliza M. Cytotoxic Terpene Quinones from Marine Sponges. Marine Drugs. 2010; 8(12):2849-2870. https://doi.org/10.3390/md8122849
Chicago/Turabian StyleGordaliza, Marina. 2010. "Cytotoxic Terpene Quinones from Marine Sponges" Marine Drugs 8, no. 12: 2849-2870. https://doi.org/10.3390/md8122849
APA StyleGordaliza, M. (2010). Cytotoxic Terpene Quinones from Marine Sponges. Marine Drugs, 8(12), 2849-2870. https://doi.org/10.3390/md8122849