
Effects of Amphidinium carterae Phytocompounds on Proliferation and the Epithelial–Mesenchymal Transition Process in T98G Glioblastoma Cells
 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Results
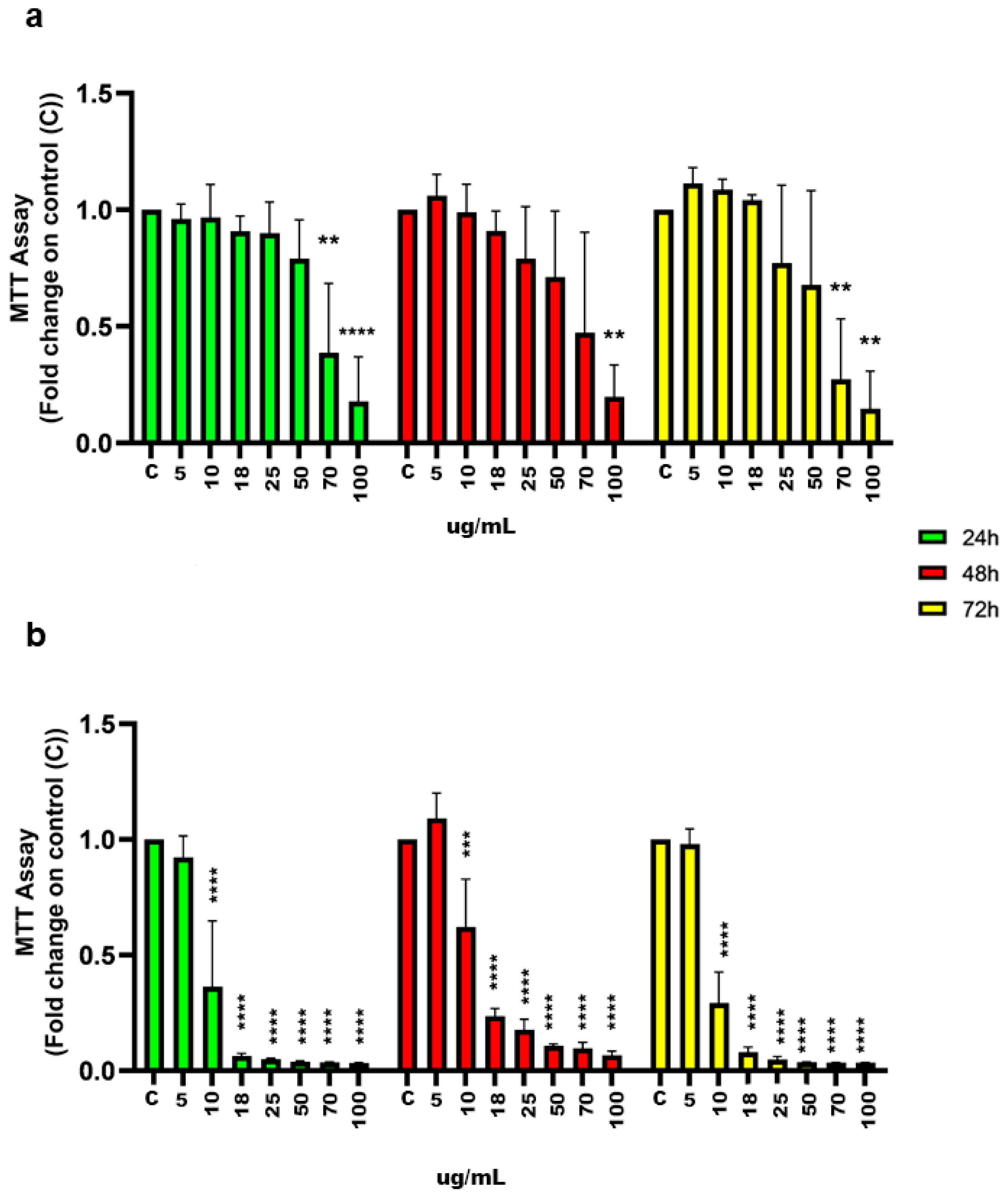
2.1. Inhibition of Cell Proliferation
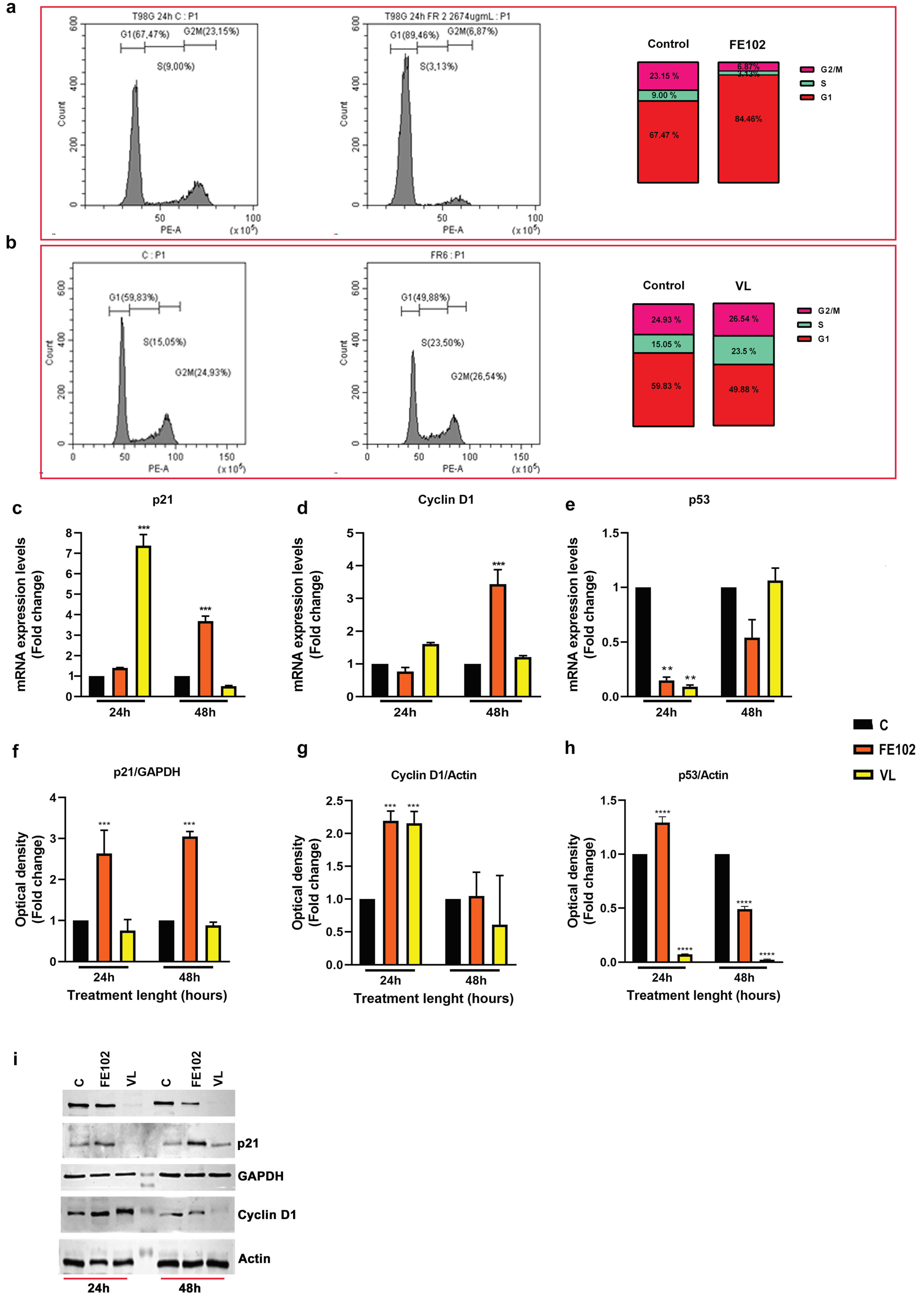
2.2. Cell Cycle Impairment
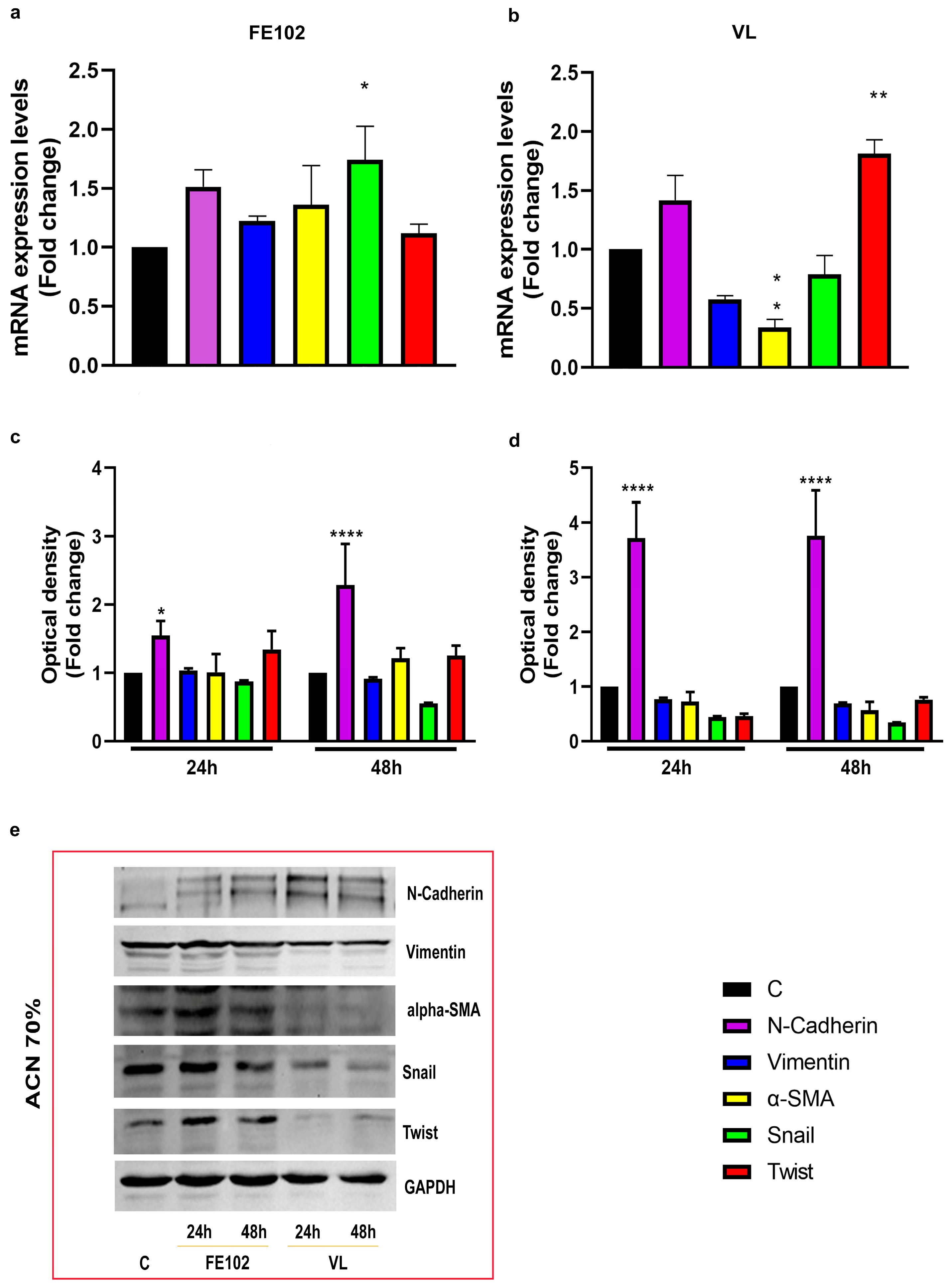
2.3. Epythelial Mesenchimal Transition Modulation
2.4. Modulation of Cell Migration
3. Discussion
4. Materials and Methods
4.1. Cells and Strains
4.2. Cell Culture and Harvesting
4.2.1. A. carterae
4.2.2. T98G
4.3. Chemical Extraction
4.3.1. Total Extract
4.3.2. Total Extract Fractionation
4.4. In Vitro Tests on Glioblastoma Cell Lines
4.4.1. Reagents
4.4.2. MTT Assay
4.4.3. Cell Cycle Analysis
4.4.4. Gene Expression Studies by Real Time-Quantitative Polymerase Chain Reaction (rt-qPCR)
4.4.5. Immunoblotting Analysis
4.4.6. Scratch Assay
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| T98G | Human glioblastoma multiforme fibroblast cells |
| A. carterae | Amphidinium carterae |
| FE102 | FE (Francesco Esposito) 102 |
| VL | A. carterae strain VL (Villa Lauro) |
| GBM | Glioblastoma |
| EMT | Epithelial–mesenchymal transition |
| MET | Mesenchymal–epithelial transition |
| HAB | Harmful algal bloom |
| CDH1, E-cadherin | Epithelial cadherin |
| CDH2, N-cadherin | Neuronal cadherin |
| α-SMA | Alpha smooth muscle actin |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide |
| PCNA | Proliferating cell nuclear antigen |
| DNA | DeoxyriboNucleic acid |
| IF | Intermediate filament |
| K medium | Keller medium |
| PBS | Phosphate buffered saline |
| CO2 | Carbon dioxide |
| FBS | Fetal bovine serum |
| A | Absorbance |
| cDNA | Coding DNA |
| Atm | Atmosphere |
| h | Hour |
| V | Volume |
| g | Microgram |
| mL | Milliliters |
| L | Liter |
| Mm | Millimeter |
| Mg | Milligram |
| cm | Centimeter |
| U | Unit |
| min | Minute |
| °C | Centigrade |
| TP | Time point |
| FC | Fold change |
| ACN | Acetonittile |
| MeOH | Methanol |
| CHCl3 | Chloroform |
| DCM | DiChloroMethane |
| IC50 | Inhibitory concentration |
| C | Control |
| MEM | Minimum essential medium |
| DMSO | Dimethyl sulfoxide |
| Tris–HCl | Tris(hydroxymethyl)aminomethane hydrochloride |
| NaCl | Sodium chloride |
| EDTA | (Ethylenedinitrilo) tetraacetic acid |
| SDS | Sodium dodecyl sulfate |
| RIPA | Radio-immunoprecipitation assay |
| HRP | Horseradish peroxidase |
| ECL | Enhanced chemiluminescence |
| PCR | Polymerase chain reaction |
| qPCR | Quantitative polymerase chain reaction |
| ANOVA | Analysis of variance |
| J.O.D. | Julia Oyón Díaz de Cerio |
| G.V. | Giulia Venneri |
| I.O. | Ida Orefice |
| M.F. | Martina Forestiero |
| C.R.B. | Carlos Roman Baena |
| G.B.T. | Gianluca Bruno Tassone |
| I.P. | Isabella Percopo |
| A.S. | Angela Sardo |
| M.L.P. | Maria Luisa Panno |
| F.G. | Francesca Giordano |
| V.D.D. | Valeria Di Dato |
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Smoll, N.R.; Schaller, K.; Gautschi, O.P. Long-Term Survival of Patients with Glioblastoma Multiforme (GBM). J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2013, 20, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.-G.; Oh, S.-J.; Ahn, E.-J.; Kim, Y.-J.; Jung, T.-Y.; Jung, S.; Kim, K.-K.; Lee, J.-H.; Lee, K.-H.; Moon, K.-S. Prognostic Significance of E-Cadherin and N-Cadherin Expression in Gliomas. BMC Cancer 2017, 17, 583. [Google Scholar] [CrossRef]
- Brabletz, T. To Differentiate or Not--Routes towards Metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Leung, D.H.L.; Phon, B.W.S.; Sivalingam, M.; Radhakrishnan, A.K.; Kamarudin, M.N.A. Regulation of EMT Markers, Extracellular Matrix, and Associated Signalling Pathways by Long Non-Coding RNAs in Glioblastoma Mesenchymal Transition: A Scoping Review. Biology 2023, 12, 818. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-Mesenchymal Transition in Glioblastoma Progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef]
- Ansieau, S.; Collin, G.; Hill, L. EMT or EMT-Promoting Transcription Factors, Where to Focus the Light? Front. Oncol. 2014, 4, 353. [Google Scholar] [CrossRef]
- Lee, H.-M.; Hwang, K.-A.; Choi, K.-C. Diverse Pathways of Epithelial Mesenchymal Transition Related with Cancer Progression and Metastasis and Potential Effects of Endocrine Disrupting Chemicals on Epithelial Mesenchymal Transition Process. Mol. Cell. Endocrinol. 2017, 457, 103–113. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Noronha, C.; Ribeiro, A.S.; Taipa, R.; Castro, D.S.; Reis, J.; Faria, C.; Paredes, J. Cadherin Expression and EMT: A Focus on Gliomas. Biomedicines 2021, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; van Roy, F. Involvement of Members of the Cadherin Superfamily in Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Nikkhah, G.; Maciaczyk, J. Epithelial-to-Mesenchymal(-like) Transition as a Relevant Molecular Event in Malignant Gliomas. Cancer Lett. 2013, 331, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.-P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 Promotes Invasion through Mesenchymal Change in Human Glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef]
- Han, S.-P.; Kim, J.-H.; Han, M.-E.; Sim, H.-E.; Kim, K.-S.; Yoon, S.; Baek, S.-Y.; Kim, B.-S.; Oh, S.-O. SNAI1 Is Involved in the Proliferation and Migration of Glioblastoma Cells. Cell. Mol. Neurobiol. 2011, 31, 489–496. [Google Scholar] [CrossRef]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kögel, D. Dietary Curcumin Attenuates Glioma Growth in a Syngeneic Mouse Model by Inhibition of the JAK1,2/STAT3 Signaling Pathway. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-Cadherin Expression Associated with an Aggressive Brain Tumor Phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef]
- Trendowski, M. Exploiting the Cytoskeletal Filaments of Neoplastic Cells to Potentiate a Novel Therapeutic Approach. Biochim. Biophys. Acta 2014, 1846, 599–616. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Dong, X.; Liu, L.; Huo, L.; Chen, H. High Expression of Vimentin Is Associated with Progression and a Poor Outcome in Glioblastoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2018, 26, 337–344. [Google Scholar] [CrossRef]
- Noh, H.; Yan, J.; Hong, S.; Kong, L.-Y.; Gabrusiewicz, K.; Xia, X.; Heimberger, A.B.; Li, S. Discovery of Cell Surface Vimentin Targeting mAb for Direct Disruption of GBM Tumor Initiating Cells. Oncotarget 2016, 7, 72021–72032. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wang, G.; Ming, J.; Meng, X.; Han, B.; Sun, B.; Cai, J.; Jiang, C. Analysis of Expression and Prognostic Significance of Vimentin and the Response to Temozolomide in Glioma Patients. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 15333–15339. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef]
- Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Marine Microorganisms as a Promising and Sustainable Source of Bioactive Molecules. Mar. Environ. Res. 2017, 128, 58–69. [Google Scholar] [CrossRef]
- Munro, M.H.G.; Blunt, J.W.; Dumdei, E.J.; Hickford, S.J.H.; Lill, R.E.; Li, S.; Battershill, C.N.; Duckworth, A.R. The Discovery and Development of Marine Compounds with Pharmaceutical Potential. In Progress in Industrial Microbiology; Osinga, R., Tramper, J., Burgess, J.G., Wijffels, R.H., Eds.; Marine Bioprocess Engineering; Elsevier: Amsterdam, The Netherlands, 1999; Volume 35, pp. 15–25. [Google Scholar]
- Gallardo-Rodríguez, J.; Sánchez-Mirón, A.; García-Camacho, F.; López-Rosales, L.; Chisti, Y.; Molina-Grima, E. Bioactives from Microalgal Dinoflagellates. Biotechnol. Adv. 2012, 30, 1673–1684. [Google Scholar] [CrossRef]
- Ebrahimi Nigjeh, S.; Yusoff, F.M.; Mohamed Alitheen, N.B.; Rasoli, M.; Keong, Y.S.; Omar, A.R. bin Cytotoxic Effect of Ethanol Extract of Microalga, Chaetoceros calcitrans, and Its Mechanisms in Inducing Apoptosis in Human Breast Cancer Cell Line. BioMed Res. Int. 2012, 2013, 783690. [Google Scholar] [CrossRef]
- Samarakoon, K.W.; Ko, J.Y.; Rahman, S.M.; Lee, J.H.; Kang, M.C.; Kwon, O.N.; Lee, J.B.; Jeon, Y.J. In Vitro Studies of Anti-Inflammatory and Anticancer Activities of Organic Solvent Extracts from Cultured Marine Microalgae. ALGAE 2013, 28, 111–119. [Google Scholar] [CrossRef]
- Sanmukh, S. Bioactive Compounds Derived from Microalgae Showing Antimicrobial Activities. J. Aquac. Res. Dev. 2014, 5, 1000224. [Google Scholar] [CrossRef]
- Lauritano, C.; Andersen, J.H.; Hansen, E.; Albrigtsen, M.; Escalera, L.; Esposito, F.; Helland, K.; Hanssen, K.Ø.; Romano, G.; Ianora, A. Bioactivity Screening of Microalgae for Antioxidant, Anti-Inflammatory, Anticancer, Anti-Diabetes, and Antibacterial Activities. Front. Mar. Sci. 2016, 3, 68. [Google Scholar] [CrossRef]
- Bule, M.H.; Ahmed, I.; Maqbool, F.; Bilal, M.; Iqbal, H.M.N. Microalgae as a Source of High-Value Bioactive Compounds. Front. Biosci. Sch. Ed. 2018, 10, 197–216. [Google Scholar] [CrossRef]
- Hallegraeff, G.M.; Anderson, D.M.; Cembella, A. IOC Manuals and Guides No.33. Manual on Harmful Marine Microalgae; UNESCO: Paris, France, 1995. [Google Scholar]
- Pradhan, B.; Ki, J.-S. Phytoplankton Toxins and Their Potential Therapeutic Applications: A Journey toward the Quest for Potent Pharmaceuticals. Mar. Drugs 2022, 20, 271. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, W.; Wang, J.; He, Y.; Yang, S.; Sun, H. A Comprehensive Review on the Heterotrophic Production of Bioactive Compounds by Microalgae. World J. Microbiol. Biotechnol. 2024, 40, 210. [Google Scholar] [CrossRef] [PubMed]
- Di Dato, V.; Ianora, A.; Romano, G. Identification of Prostaglandin Pathway in Dinoflagellates by Transcriptome Data Mining. Mar. Drugs 2020, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Taipale, S.; Peltomaa, E.; Salmi, P. Variation in ω-3 and ω-6 Polyunsaturated Fatty Acids Produced by Different Phytoplankton Taxa at Early and Late Growth Phase. Biomolecules 2020, 10, 559. [Google Scholar] [CrossRef] [PubMed]
- Galasso, C.; Nuzzo, G.; Brunet, C.; Ianora, A.; Sardo, A.; Fontana, A.; Sansone, C. The Marine Dinoflagellate Alexandrium Minutum Activates a Mitophagic Pathway in Human Lung Cancer Cells. Mar. Drugs 2018, 16, 502. [Google Scholar] [CrossRef]
- Cappello, E.; Nieri, P. From Life in the Sea to the Clinic: The Marine Drugs Approved and under Clinical Trial. Life 2021, 11, 1390. [Google Scholar] [CrossRef]
- Martínez, K.A.; Lauritano, C.; Druka, D.; Romano, G.; Grohmann, T.; Jaspars, M.; Martín, J.; Díaz, C.; Cautain, B.; de la Cruz, M.; et al. Amphidinol 22, a New Cytotoxic and Antifungal Amphidinol from the Dinoflagellate Amphidinium Carterae. Mar. Drugs 2019, 17, 385. [Google Scholar] [CrossRef]
- Orefice, I.; Balzano, S.; Romano, G.; Sardo, A. Amphidinium Spp. as a Source of Antimicrobial, Antifungal, and Anticancer Compounds. Life 2023, 13, 2164. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition in Cancer: Complexity and Opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial-Mesenchymal Plasticity in Carcinoma Metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for Epithelial-Mesenchymal Transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Busseni, G.; Rocha Jimenez Vieira, F.; Amato, A.; Pelletier, E.; Pierella Karlusich, J.J.; Ferrante, M.I.; Wincker, P.; Rogato, A.; Bowler, C.; Sanges, R.; et al. Meta-Omics Reveals Genetic Flexibility of Diatom Nitrogen Transporters in Response to Environmental Changes. Mol. Biol. Evol. 2019, 36, 2522–2535. [Google Scholar] [CrossRef]
- Stengel, D.B.; Connan, S. Marine Algae: A Source of Biomass for Biotechnological Applications. In Natural Products from Marine Algae: Methods and Protocols; Stengel, D.B., Connan, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; pp. 1–37. ISBN 978-1-4939-2684-8. [Google Scholar]
- Mimouni, V.; Ulmann, L.; Pasquet, V.; Mathieu, M.; Picot, L.; Bougaran, G.; Cadoret, J.-P.; Morant-Manceau, A.; Schoefs, B. The Potential of Microalgae for the Production of Bioactive Molecules of Pharmaceutical Interest. Curr. Pharm. Biotechnol. 2012, 13, 2733–2750. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.R.; Samarakoon, K.W.; Ko, J.; Lakmal, H.H.C.; Lee, J.; An, S.; Jeon, Y.; Lee, J. Potentiality of Benthic Dinoflagellate Cultures and Screening of Their Bioactivities in Jeju Island, Korea. Afr. J. Biotechnol. 2014, 13, 792–805. [Google Scholar] [CrossRef]
- Bui, Q.T.N.; Pradhan, B.; Kim, H.-S.; Ki, J.-S. Environmental Factors Modulate Saxitoxins (STXs) Production in Toxic Dinoflagellate Alexandrium: An Updated Review of STXs and Synthesis Gene Aspects. Toxins 2024, 16, 210. [Google Scholar] [CrossRef]
- Gerecht, A.; Romano, G.; Ianora, A.; d’Ippolito, G.; Cutignano, A.; Fontana, A. Plasticity of Oxylipin Metabolism Among Clones of the Marine Diatom Skeletonema marinoi (Bacillariophyceae)(1). J. Phycol. 2011, 47, 1050–1056. [Google Scholar] [CrossRef]
- Di Dato, V.; Orefice, I.; Amato, A.; Fontanarosa, C.; Amoresano, A.; Cutignano, A.; Ianora, A.; Romano, G. Animal-like Prostaglandins in Marine Microalgae. ISME J. 2017, 11, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Schafer, K.A. The Cell Cycle: A Review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Jamasbi, E.; Hamelian, M.; Hossain, M.A.; Varmira, K. The Cell Cycle, Cancer Development and Therapy. Mol. Biol. Rep. 2022, 49, 10875–10883. [Google Scholar] [CrossRef]
- Ho, C.-J.; Tsai, C.-Y.; Zhu, W.-H.; Pao, Y.-H.; Chen, H.-W.; Hu, C.-J.; Lee, Y.-L.; Huang, T.-S.; Chen, C.-H.; Loh, J.-K.; et al. Compound Cellular Stress Maximizes Apoptosis Independently of P53 in Glioblastoma. Cell Cycle Georget. Tex 2022, 21, 1153–1165. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the P53 Gene and Its Expression in Human Glioblastoma Cells. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. P53-Dependent and Independent Expression of P21 during Cell Growth, Differentiation, and DNA Damage. Genes Dev. 1995, 9, 935–944. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in Cancer and Its Potential as a Molecular Target for Cancer Therapy. Cell. Mol. Life Sci. CMLS 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Sakane, F.; Hashimoto, K. N-Cadherin-Based Adherens Junction Regulates the Maintenance, Proliferation, and Differentiation of Neural Progenitor Cells during Development. Cell Adhes. Migr. 2015, 9, 183–192. [Google Scholar] [CrossRef]
- Van Roy, F. Beyond E-Cadherin: Roles of Other Cadherin Superfamily Members in Cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-Cadherin in Cancer Metastasis, Its Emerging Role in Haematological Malignancies and Potential as a Therapeutic Target in Cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Motta, F.J.N.; Valera, E.T.; Lucio-Eterovic, A.K.B.; Queiroz, R.G.P.; Neder, L.; Scrideli, C.A.; Machado, H.R.; Carlotti-Junior, C.G.; Marie, S.K.N.; Tone, L.G. Differential Expression of E-Cadherin Gene in Human Neuroepithelial Tumors. Genet. Mol. Res. GMR 2008, 7, 295–304. [Google Scholar] [CrossRef]
- Bar, J.K.; Zub, L.; Lis-Nawara, A.; Noga, L.; Jeleń, M.; Paradowski, B. Expression and Interactions Between Cell Adhesion Molecules CD44v6 and E-Cadherin in Human Gliomas. Adv. Clin. Exp. Med. 2014, 23, 827–834. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Méresse, S.; Fodil, M.; Fleury, F.; Chénais, B. Fucoxanthin, a Marine-Derived Carotenoid from Brown Seaweeds and Microalgae: A Promising Bioactive Compound for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 9273. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Chiang, Y.-F.; Huang, C.-Y.; Shieh, T.-M.; Kao, C.; Chang, F.-K.; Huang, T.-C.; Ali, M.; Chang, H.-Y.; Hong, Y.-H.; et al. Spirulina Phycocyanin Extract and Its Active Components Suppress Epithelial-Mesenchymal Transition Process in Endometrial Cancer via Targeting TGF-Beta1/SMAD4 Signaling Pathway. Biomed. Pharmacother. 2022, 152, 113219. [Google Scholar] [CrossRef]
- Smith, D.; Shimamura, T.; Barbera, S.; Bejcek, B.E. NF-kappaB Controls Growth of Glioblastomas/Astrocytomas. Mol. Cell. Biochem. 2008, 307, 141–147. [Google Scholar] [CrossRef]
- Velásquez, C.; Mansouri, S.; Mora, C.; Nassiri, F.; Suppiah, S.; Martino, J.; Zadeh, G.; Fernández-Luna, J.L. Molecular and Clinical Insights into the Invasive Capacity of Glioblastoma Cells. J. Oncol. 2019, 2019, 1740763. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yuan, X.; Mu, J.; Zou, Y.; Xu, L.; Chen, J.; Zhu, X.; Li, B.; Zeng, Z.; Wu, X.; et al. Quercetin Induces MGMT + Glioblastoma Cells Apoptosis via Dual Inhibition of Wnt3a/β-Catenin and Akt/NF-κB Signaling Pathways. Phytomed. Int. J. Phytother. Phytopharm. 2023, 118, 154933. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R.L. Media for the Culture of Oceanic Ultraphytoplankton1,2. J. Phycol. 1987, 23, 633–638. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Type of Extract | Weight | Stock Solution | Working Stock |
|---|---|---|---|---|---|
| Amphidinium carterae | FE102 | Total extract Replicate 1 | 100 mg | 45,454 μg/mL | 10,000 μg/mL |
| Total extract Replicate 2 | 120 mg | 54,545 μg/mL | 10,000 μg/mL | ||
| Total extract Replicate 3 | 80 mg | 47,059 μg/mL | 10,000 μg/mL | ||
| MeOH 50% | 3.1 mg | 31,000 μg/mL | 1000 μg/mL | ||
| ACN 70% | 3.5 mg | 70,000 μg/mL | 1000 μg/mL | ||
| ACN 100% | 1.3 mg | - | - | ||
| DCM:MeOH 9:1 | 53.3 mg | - | - | ||
| VL | Total extract Replicate 1 | 50 mg | 50,000 μg/mL | 10,000 μg/mL | |
| Total extract Replicate 2 | 20 mg | 33,334 μg/mL | 10,000 μg/mL | ||
| Total extract Replicate 3 | 200 mg | 117,647 μg/mL | 10,000 μg/mL | ||
| MeOH 50% | 6.6 mg | 29,000 μg/mL | 1000 μg/mL | ||
| ACN 70% | 12.4 mg | 62,000 μg/mL | 1000 μg/mL | ||
| ACN 100% | 2.9 mg | 58,000 μg/mL | 1000 μg/mL | ||
| DCM:MeOH 9:1 | 19.3 mg | - | - |
| A. carterae Strain | Type of Extract | Concentration |
|---|---|---|
| FE102 | Total extract | 5 μg/mL |
| MeOH 50% | 10 μg/mL | |
| ACN 70% | 25.74 μg/mL | |
| VL | Total extract | 5 μg/mL |
| MeOH 50% | 18 μg/mL | |
| ACN 70% | 18 μg/mL | |
| ACN 100% | 18 μg/mL |
| Oligo Name | Forward | Reverse |
|---|---|---|
| 18S | 5′-CGGCGACGACCCATTCGAAC-3′ | 5′GAATCGAACCCTGATTCCCCGTC-3′ |
| P21F | 5′ GACACAGGTGTTGTGACACAT-3′ | 5′CTCATTCAGCCTGACTTGGGA-3′ |
| CYCLIN D1 | 5′-GATGCCAACCTCCTCAACGAC-3′ | 5′-CTCCTCGCACTTCTGTTCCTC-3′ |
| N-CADHERIN | 5′-ACAGTGGCCACCTACAAAGG-3′ | 5′-CCGAGATGGGGTTGATAATG-3′ |
| VIMENTIN | 5′-GAGAACTTTGCCGTTGAAGC-3′ | 5′-GCTTCCTGTAGGTGGCAATC-3′ |
| TWIST1 | 5′-TCCAAATTCAAAGAAACAGGCG-3′ | 5′-CAGAATGCAGAGGTGTGAGGA-3′ |
| SNAIL | 5′-CGAGTGGTTCTTCTGCGCTA-3′ | 5′-GGGCTGCTGGAAGGTAAACT-3′ |
| α-SMA | 5′-AGACATCAGGGGGTGATGGT-3′ | 5′-CATGGCTGGGACATTGAAAG-3′ |
| A. carterae Strain | Extract | Concentration |
|---|---|---|
| FE102 | Total extract | 5 μg/mL |
| ACN 70% | 25.74 μg/mL | |
| VL | Total extract | 5 μg/mL |
| MeOH 50% | 18 μg/mL | |
| ACN 70% | 18 μg/mL | |
| ACN 100% | 18 μg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyón Díaz de Cerio, J.; Venneri, G.; Orefice, I.; Forestiero, M.; Baena, C.R.; Tassone, G.B.; Percopo, I.; Sardo, A.; Panno, M.L.; Giordano, F.; et al. Effects of Amphidinium carterae Phytocompounds on Proliferation and the Epithelial–Mesenchymal Transition Process in T98G Glioblastoma Cells. Mar. Drugs 2025, 23, 173. https://doi.org/10.3390/md23040173
Oyón Díaz de Cerio J, Venneri G, Orefice I, Forestiero M, Baena CR, Tassone GB, Percopo I, Sardo A, Panno ML, Giordano F, et al. Effects of Amphidinium carterae Phytocompounds on Proliferation and the Epithelial–Mesenchymal Transition Process in T98G Glioblastoma Cells. Marine Drugs. 2025; 23(4):173. https://doi.org/10.3390/md23040173
Chicago/Turabian StyleOyón Díaz de Cerio, Julia, Giulia Venneri, Ida Orefice, Martina Forestiero, Carlos Roman Baena, Gianluca Bruno Tassone, Isabella Percopo, Angela Sardo, Maria Luisa Panno, Francesca Giordano, and et al. 2025. "Effects of Amphidinium carterae Phytocompounds on Proliferation and the Epithelial–Mesenchymal Transition Process in T98G Glioblastoma Cells" Marine Drugs 23, no. 4: 173. https://doi.org/10.3390/md23040173
APA StyleOyón Díaz de Cerio, J., Venneri, G., Orefice, I., Forestiero, M., Baena, C. R., Tassone, G. B., Percopo, I., Sardo, A., Panno, M. L., Giordano, F., & Di Dato, V. (2025). Effects of Amphidinium carterae Phytocompounds on Proliferation and the Epithelial–Mesenchymal Transition Process in T98G Glioblastoma Cells. Marine Drugs, 23(4), 173. https://doi.org/10.3390/md23040173