
Extracts from Allium pseudojaponicum Makino Target STAT3 Signaling Pathway to Overcome Cisplatin Resistance in Lung Cancer

 , , , ,
, , , ,  , , and
, , and
Abstract

1. Introduction
2. Results
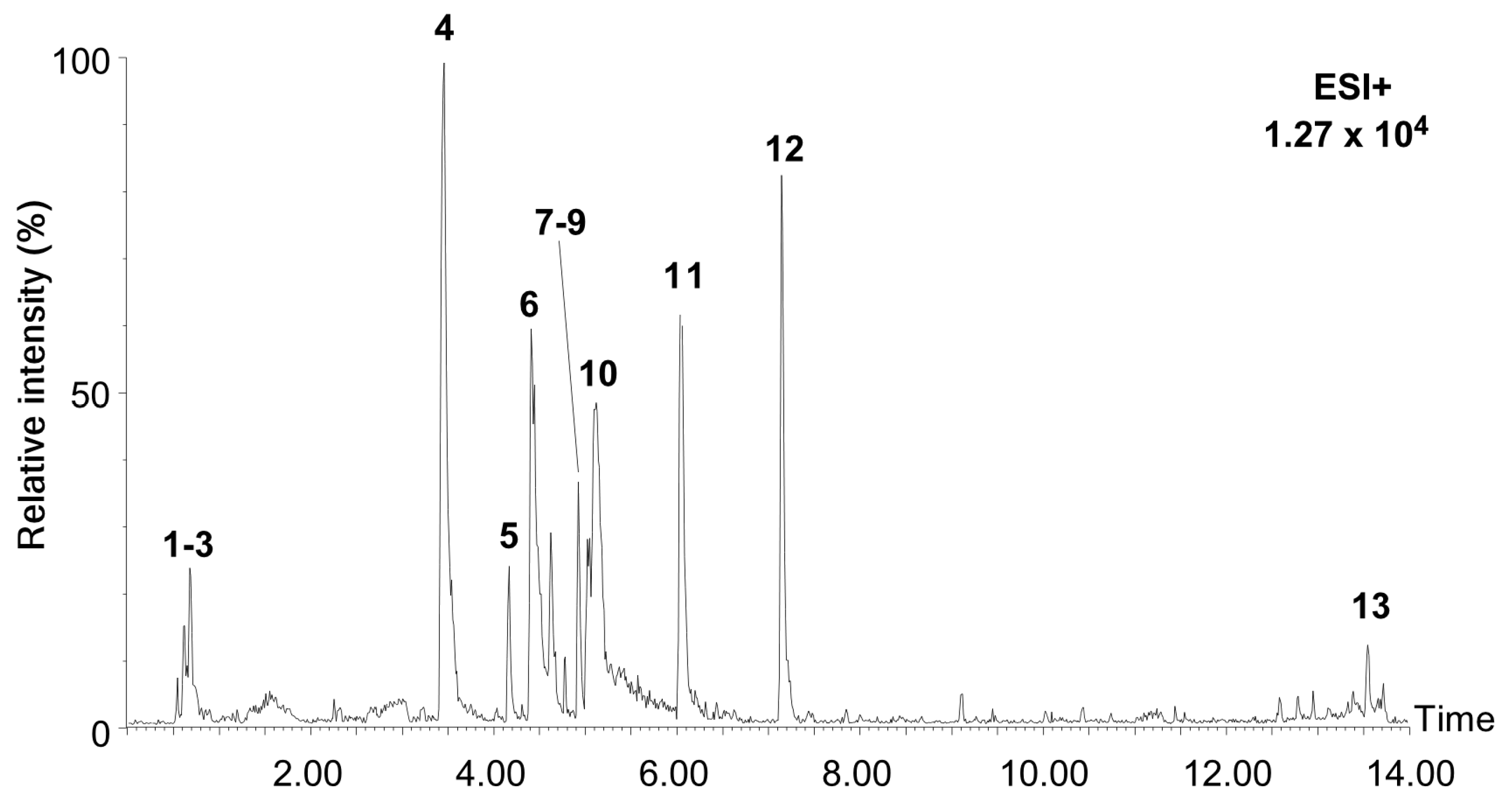
2.1. Characterization of Chemical Constituents in the Extract of Allium pseudojaponicum Makino by UPLC-MSE
2.2. APE Suppresses Proliferation and Colony Formation of Cisplatin-Resistant NSCLC Cells
2.3. APE Attenuates Cell Cycle Progression at the G1 Phase
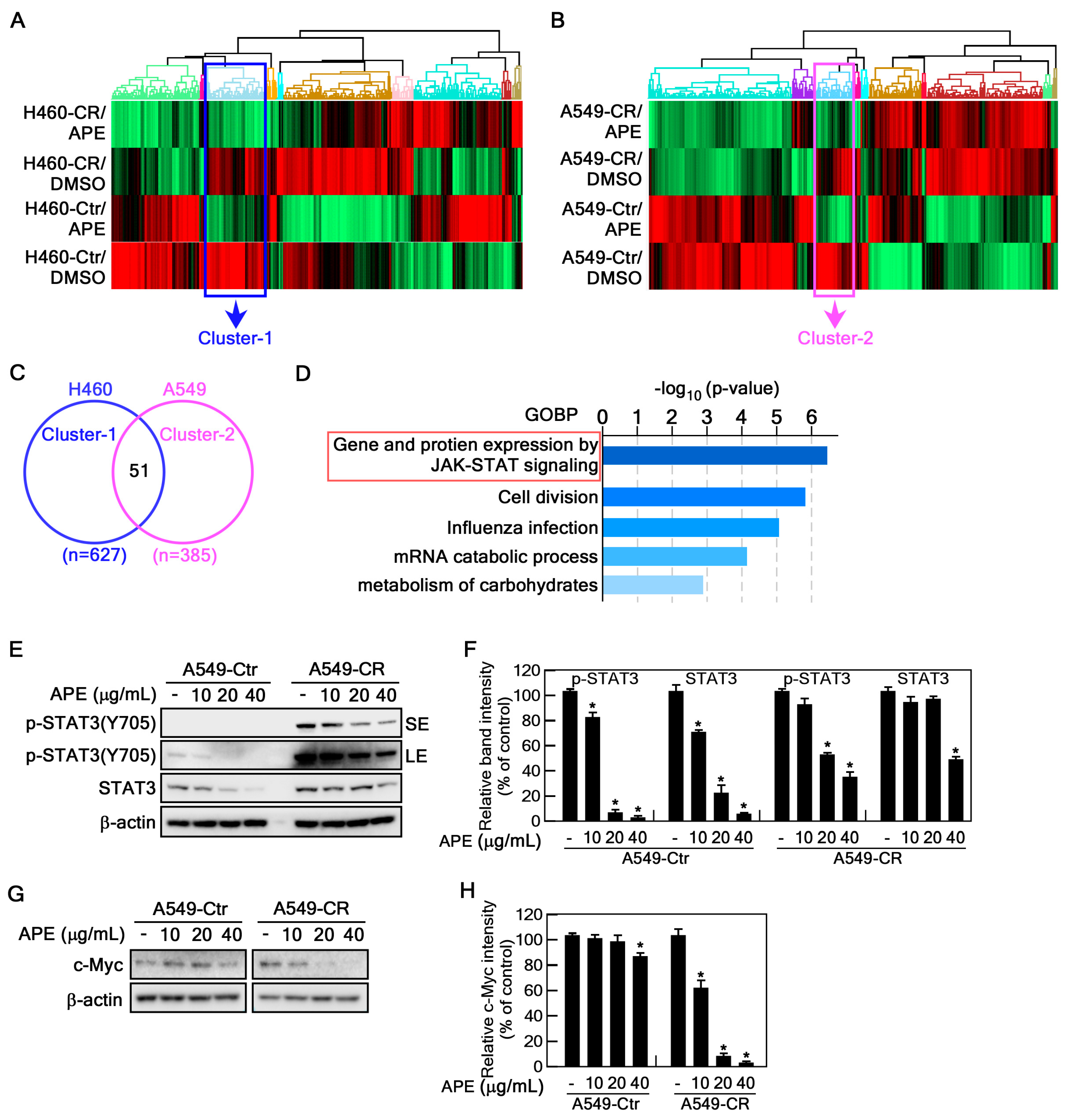
2.4. APE Targets STAT3 Signaling Pathway
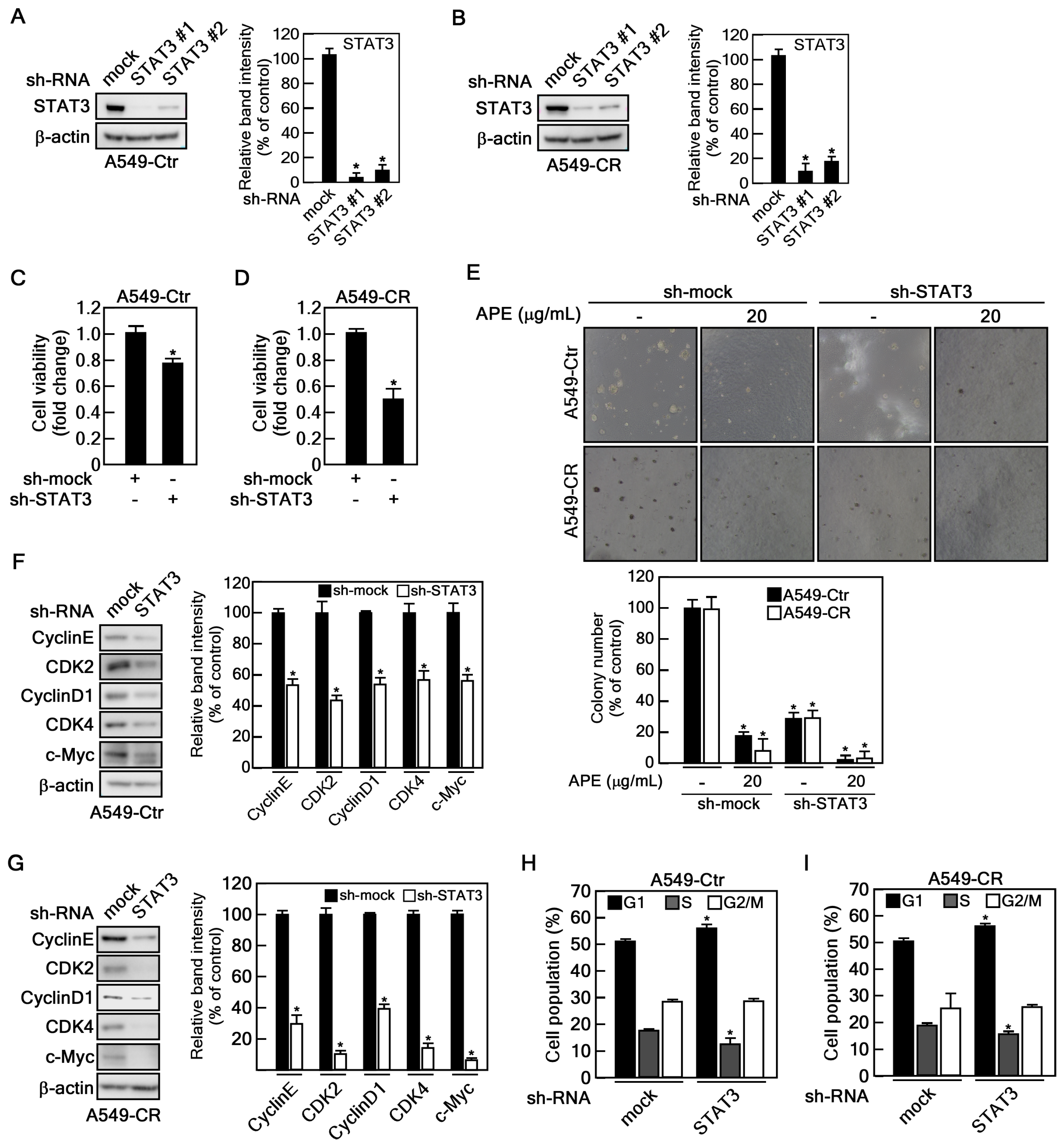
2.5. STAT3 Depletion Showed SIMILAR Anticancer Effect of APE
3. Discussion
4. Materials and Methods
4.1. Extract of Allium Pseudojaponicum Makino (APE)
4.2. Reagents
4.3. Cell Culture and Treatment
4.4. Cell Viability Assay
4.5. Western Blotting
4.6. Gene Silencing
4.7. Anchorage-Independent Colony Growth Assay (Soft Agar Assay)
4.8. Cell Cycle Analysis
4.9. S-Trap-Based Protein Digestion
4.10. TMT Labeling and Fractionation
4.11. LC-MS/MS Analysis
4.12. Protein Identification and Quantification
4.13. Bioinformatics Analysis
4.14. Metabolomic Analysis Using UPLC-QTOF-MSE
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APE | Extracts of Allium pseudojaponicum Makino |
| ATCC | American type culture collection |
| BME | Basal media eagle |
| BPI | Base peak intensity |
| BSA | Bovine serum albumin |
| CCK-8 | Cell counting kit-8 |
| CDK2 | Cyclin-dependent kinase 2 |
| CDK4 | Cyclin-dependent kinase 4 |
| DDA | Data-dependent acquisition |
| DMSO | Dimethyl sulfoxide |
| EBC | Eukaryotic buffer for cytoplasm |
| ECL | Enhanced chemiluminescence |
| FBS | Fetal bovine serum |
| FDR | False discovery rate |
| GOBP | Gene ontology biological process |
| HCD | High-energy collision dissociation |
| HR LC-MS | High-resolution liquid chromatograph-mass spectrometer |
| IAA | Iodoacetamide |
| JAK | Janus kinase |
| NSCLC | Non-small-cell lung cancer |
| PBS | Phosphate-buffered saline |
| PVDF | Polyvinylidene difluoride |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| STAT3 | Signal transducer and activator of transcription 3 |
| TBST | Tris-Buffered Saline with Tween-20 detergent |
| TCEP | Tris(2-carboxyethyl) phosphine |
| TEAB | Triethylammonium bicarbonate |
| TMT | Tandem mass tag |
| UPLC-QTOF-MS | Ultra-high performance liquid chromatography with quadrupole time-of-flight mass spectrometry |
References
- Mohammed, H.A.; Emwas, A.H.; Khan, R.A. Salt-Tolerant Plants, Halophytes, as Renewable Natural Resources for Cancer Prevention and Treatment: Roles of Phenolics and Flavonoids in Immunomodulation and Suppression of Oxidative Stress towards Cancer Management. Int. J. Mol. Sci. 2023, 24, 5171. [Google Scholar] [CrossRef]
- Ferreira, M.J.; Pinto, D.C.G.A.; Cunha, A.; Silva, H. Halophytes as Medicinal Plants against Human Infectious Diseases. Appl. Sci. 2022, 12, 7493. [Google Scholar] [CrossRef]
- Stankovic, M.; Stojanovic-Radic, Z.; Jakovljevic, D.; Zlatic, N.; Lukovic, M.; Dajic-Stevanovic, Z. Coastal Halophytes: Potent Source of Bioactive Molecules from Saline Environment. Plants 2023, 12, 1857. [Google Scholar] [CrossRef]
- Shim, J. Anti-aging effect of Allium pseudojaponicum in UVA-irradiated human epidermal keratinocytes. Cell Mol. Biol. 2023, 69, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Padinharayil, H.; Varghese, J.; John, M.C.; Rajanikant, G.K.; Wilson, C.M.; Al-Yozbaki, M.; Renu, K.; Dewanjee, S.; Sanyal, R.; Dey, A.; et al. Non-small cell lung carcinoma (NSCLC): Implications on molecular pathology and advances in early diagnostics and therapeutics. Genes Dis. 2023, 10, 960–989. [Google Scholar] [CrossRef] [PubMed]
- Araghi, M.; Mannani, R.; Heidarnejad Maleki, A.; Hamidi, A.; Rostami, S.; Safa, S.H.; Faramarzi, F.; Khorasani, S.; Alimohammadi, M.; Tahmasebi, S.; et al. Recent advances in non-small cell lung cancer targeted therapy; an update review. Cancer Cell Int. 2023, 23, 162. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef]
- Wang, H.Q.; Man, Q.W.; Huo, F.Y.; Gao, X.; Lin, H.; Li, S.R.; Wang, J.; Su, F.C.; Cai, L.; Shi, Y.; et al. STAT3 pathway in cancers: Past, present, and future. MedComm 2022, 3, e124. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Cascio, A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int. J. Mol. Sci. 2021, 22, 603. [Google Scholar] [CrossRef] [PubMed]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef]
- Schulze, A.; Zerfass, K.; Spitkovsky, D.; Henglein, B.; Jansen-Dürr, P. Activation of the E2F transcription factor by cyclin D1 is blocked by p16INK4, the product of the putative tumor suppressor gene MTS1. Oncogene 1994, 9, 3475–3482. [Google Scholar]
- Fukada, T.; Ohtani, T.; Yoshida, Y.; Shirogane, T.; Nishida, K.; Nakajima, K.; Hibi, M.; Hirano, T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998, 17, 6670–6677. [Google Scholar] [CrossRef]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal 2020, 18, 33. [Google Scholar] [CrossRef]
- Sepúlveda, P.; Encabo, A.; Carbonell-Uberos, F.; Miñana, M.D. BCL-2 expression is mainly regulated by JAK/STAT3 pathway in human CD34+ hematopoietic cells. Cell Death Differ. 2007, 14, 378–380. [Google Scholar] [CrossRef]
- Heiss, D.R.; Amoah, E.; Badu-Tawiah, A.K. Two-dimensional isomer differentiation using liquid chromatography-tandem mass spectrometry with in-source, droplet-based derivatization. Analyst 2023, 148, 5270–5278. [Google Scholar] [CrossRef]
- Mu, L.; Tong, Q.; Liu, Y.; Meng, X.; He, P.; Li, G.; Ye, L. Application of Gas-Liquid Microextraction (GLME)/GC-MS for Flavour and Fragrance in Ice Cream Detection and Composition Analysis. Molecules 2023, 28, 522. [Google Scholar] [CrossRef]
- Peixoto Araujo, N.M.; Arruda, H.S.; Dos Santos, F.N.; de Morais, D.R.; Pereira, G.A.; Pastore, G.M. LC-MS/MS screening and identification of bioactive compounds in leaves, pulp and seed from Eugenia calycina Cambess. Food Res. Int. 2020, 137, 109556. [Google Scholar] [CrossRef] [PubMed]
- Torras-Claveria, L.; Berkov, S.; Viladomat, F.; Bastida, J. QToF exact mass and ESI fragmentation of bioactive Amaryllidaceae alkaloids. S. Afr. J. Bot. 2021, 136, 81–90. [Google Scholar] [CrossRef]
- Rao, M.J.; Feng, B.; Ahmad, M.H.; Tahir Ul Qamar, M.; Aslam, M.Z.; Khalid, M.F.; Hussain, S.; Zhong, R.; Ali, Q.; Xu, Q.; et al. LC-MS/MS-based metabolomics approach identified novel antioxidant flavonoids associated with drought tolerance in citrus species. Front. Plant Sci. 2023, 14, 1150854. [Google Scholar] [CrossRef]
- Minh, T.N.; Khang, D.T.; Tuyen, P.T.; Minh, L.T.; Anh, L.H.; Quan, N.V.; Ha, P.T.; Quan, N.T.; Toan, N.P.; Elzaawely, A.A.; et al. Phenolic Compounds and Antioxidant Activity of Phalaenopsis Orchid Hybrids. Antioxidants 2016, 5, 31. [Google Scholar] [CrossRef]
- Xia, H.; Dai, Y.; Zhao, C.; Zhang, H.; Shi, Y.; Lou, H. Chromatographic and mass spectrometric technologies for chemical analysis of Euodiae fructus: A review. Phytochem. Anal. 2023, 34, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef]
- Barré, B.; Vigneron, A.; Coqueret, O. The STAT3 transcription factor is a target for the Myc and riboblastoma proteins on the Cdc25A promoter. J. Biol. Chem. 2005, 280, 15673–15681. [Google Scholar] [CrossRef]
- Liu, X.; Chen, L.; Wang, T. Overcoming cisplatin resistance of human lung cancer by sinomenine through targeting the miR-200a-3p-GLS axis. J. Chemother. 2023, 35, 357–366. [Google Scholar] [CrossRef]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J., Jr.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Dong, Z.; Liu, K. Unraveling the complexity of STAT3 in cancer: Molecular understanding and drug discovery. J. Exp. Clin. Cancer Res. 2024, 43, 23. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.P.; Tortelli, T.C., Jr.; Mancini, M.C.S.; Pavan, I.C.B.; Silva, L.G.S.; Severino, M.B.; Granato, D.C.; Pestana, N.F.; Ponte, L.G.S.; Peruca, G.F.; et al. STAT3 contributes to cisplatin resistance, modulating EMT markers, and the mTOR signaling in lung adenocarcinoma. Neoplasia 2021, 23, 1048–1058. [Google Scholar] [CrossRef]
- Parakh, S.; Ernst, M.; Poh, A.R. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef]
- Groot, J.; Ott, M.; Wei, J.; Kassab, C.; Fang, D.; Najem, H.; O’Brien, B.; Weathers, S.P.; Matsouka, C.K.; Majd, N.K.; et al. A first-in-human Phase I trial of the oral p-STAT3 inhibitor WP1066 in patients with recurrent malignant glioma. CNS Oncol. 2022, 11, CNS87. [Google Scholar] [CrossRef]
- Alexandrow, M.G.; Song, L.J.; Altiok, S.; Gray, J.; Haura, E.B.; Kumar, N.B. Curcumin: A novel Stat3 pathway inhibitor for chemoprevention of lung cancer. Eur. J. Cancer Prev. 2012, 21, 407–412. [Google Scholar] [CrossRef]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef]
- Wang, Y.; Ren, X.; Deng, C.; Yang, L.; Yan, E.; Guo, T.; Li, Y.; Xu, M.X. Mechanism of the inhibition of the STAT3 signaling pathway by EGCG. Oncol. Rep. 2013, 30, 2691–2696. [Google Scholar] [CrossRef]
- Gao, L.; Feng, Y.; Ge, C.; Xu, X.; Wang, S.; Li, X.; Zhang, K.; Wang, C.; Dai, F.; Xie, S. Identification of molecular anti-metastasis mechanisms of lycorine in colorectal cancer by RNA-seq analysis. Phytomedicine 2021, 85, 153530. [Google Scholar] [CrossRef]
- Rho, J.K.; Choi, Y.J.; Choi, Y.R.; Kim, S.Y.; Choi, S.J.; Choi, C.M.; Na, I.I.; Lee, J.C. The effect of acquired cisplatin resistance on sensitivity to EGFR tyrosine kinase inhibitors in EGFR mutant lung cancer cells. Oncol. Res. 2011, 19, 471–478. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | RT (min) | MS1 (m/z) | Δppm | Molecular Formula | Identification | Ontology | MS/MS (m/z) | Peak Area (%) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.66 | 365.1046 | 2.44 | C12H22O11 | Isomaltulose | O-glycosyl compounds | 85.03, 127.04, 145.05, 163.06 | 1.79 | [20] |
| 2 | 0.70 | 127.0383 | 5.35 | C6H6O3 | Triacetate lactone | Pynone derivatives | 69.03, 85.03, 97.03, 99.04, 109.03 | 1.52 | [21] |
| 3 | 0.70 | 522.2040 | 2.22 | C18H32O16 | Melezitose | Oligosaccharides | 97.03, 127.04, 145.05, 163.06, 289.09, 487.16 | 1.03 | [22] |
| 4 | 3.47 | 288.1241 | 1.74 | C16H17NO4 | Lycorine | Alkaloids | 119.05, 147.04, 177.05, 194.10, 222.09, 252.10, 270.11 | 29.81 | [23] |
| 5 | 4.19 | 348.1817 | 1.64 | C19H25NO5 | Ungvedine | Alkaloids | 206.93, 175.00, 141.97, 121.90 | 5.49 | [23] |
| 6 | 4.44 | 332.1486 | 3.55 | C18H21NO5 | Tazettine | Alkaloids | 181.06, 211.07, 282.11, 314.14 | 17.22 | [23] |
| 7 | 4.94 | 303.0497 | 0.53 | C15H10O7 | Tricetin | Flavonoids | 137.02, 149.02, 153.01, 229.05, 257.04 | 8.19 | [24] |
| 8 | 4.94 | 465.1018 | 2.09 | C21H20O12 | Hyperoside | Flavonoid-glycosides | 137.02, 149.02, 153.01, 229.05, 257.04, 303.05 | 1.63 | [24] |
| 9 | 4.94 | 611.1630 | 3.89 | C27H30O16 | Rutoside | Flavonoid-glycosides | 257.05, 273.03, 303.05, 465.10 | 1.36 | [24] |
| 10 | 5.34 | 317.0629 | 8.64 | C16H12O7 | 3-Methylquercetin | Flavonoids | 221.09, 272.13, 302.13, 314.15 | 0.62 | [24] |
| 11 | 6.07 | 362.1973 | 1.21 | C20H27NO5 | Phalaenopsine T | Alkaloids | 211.08, 266.08, 302.18, 330.17 | 2.99 | [25] |
| 12 | 7.17 | 244.1327 | 4.34 | C15H17NO2 | Atanine | Alkaloids | 78.03, 104.05, 156.08, 182.10, 184.07 | 12.59 | [26] |
| 13 | 13.55 | 609.2728 | 12.92 | C33H40N2O9 | Reserpine | Alkaloids | 182.10, 196.11, 330.17, 544.27, 593.28 | 3.55 | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, S.-B.; Choi, J.H.; Lee, G.-E.; Kim, J.Y.; Lee, M.-H.; Yang, G.; Cho, Y.-Y.; Jeong, H.G.; Bang, G.; Lee, C.-J. Extracts from Allium pseudojaponicum Makino Target STAT3 Signaling Pathway to Overcome Cisplatin Resistance in Lung Cancer. Mar. Drugs 2025, 23, 167. https://doi.org/10.3390/md23040167
Nam S-B, Choi JH, Lee G-E, Kim JY, Lee M-H, Yang G, Cho Y-Y, Jeong HG, Bang G, Lee C-J. Extracts from Allium pseudojaponicum Makino Target STAT3 Signaling Pathway to Overcome Cisplatin Resistance in Lung Cancer. Marine Drugs. 2025; 23(4):167. https://doi.org/10.3390/md23040167
Chicago/Turabian StyleNam, Soo-Bin, Jung Hoon Choi, Ga-Eun Lee, Jin Young Kim, Mee-Hyun Lee, Gabsik Yang, Yong-Yeon Cho, Hye Gwang Jeong, Geul Bang, and Cheol-Jung Lee. 2025. "Extracts from Allium pseudojaponicum Makino Target STAT3 Signaling Pathway to Overcome Cisplatin Resistance in Lung Cancer" Marine Drugs 23, no. 4: 167. https://doi.org/10.3390/md23040167
APA StyleNam, S.-B., Choi, J. H., Lee, G.-E., Kim, J. Y., Lee, M.-H., Yang, G., Cho, Y.-Y., Jeong, H. G., Bang, G., & Lee, C.-J. (2025). Extracts from Allium pseudojaponicum Makino Target STAT3 Signaling Pathway to Overcome Cisplatin Resistance in Lung Cancer. Marine Drugs, 23(4), 167. https://doi.org/10.3390/md23040167