2.2. Acetophenones
A natural compound, 2,4-dihydroxyacetophenone (
6,
Figure 8), showed robust inhibition of
Ulva pertursa and a relevant decrease in fouling biomass when incorporated in a controlled depletion paint [
38]. On the other hand, triazole derivatives are remarkable compounds in medicinal chemistry, showing a wide range of bioactivities, such as antimicrobial. Considering the relevance of both 1,2,3-triazole and benzo/acetophenone moieties, a library of 14 new acetophenone-1,2,3-triazole hybrids (
7–
20,
Figure 8) containing different substitution patterns, such as aromatic nitriles (
7 and
8), aromatic halogens (
9–
14), aliphatic alcohols (
15 and
16), and N-acetylglucosamines (
19 and
20), were synthesized through the copper(I)-catalyzed alkyne-azide Huisgen cycloaddition, a “click” reaction [
38].
The most promising compounds against the settlement of a heavy macrofouler, mussel
Mytilus galloprovincialis larvae, were three compounds containing methoxy groups in the phenyl ketone core with different substituents linked to the heterocyclic ring. In particular, compounds
13 (EC
50 = 11.20 µg·mL
−1/28.87 µM) with an aromatic chlorine,
15 (EC
50 = 13.46 µg·mL
−1/40.14 µM) with an aliphatic alcohol, and compound
19 (EC
50 = 9.94 µg·mL
−1/20.68 µM), an acetophenone derivative with an acetamide glucose moiety. It is crucial to observe that none of these three compounds caused mortality to the target species
M. galloprovincialis plantigrades at the highest concentration tested (200 µM), and revealed an EC
50 < 25 µg·mL
−1, a recommended value by the U.S. Navy program for antifoulants. Regarding SAR, the presence of two methoxy groups at C-3′ and C-5′ on the phenyl ketone core proved to be more favorable than the presence of hydroxyl groups at C-2′ for the mussel larvae anti-settlement activity [
38].
These acetophenones were also evaluated for their ability to inhibit the growth of marine biofilm-forming bacteria of five strains (
Vibrio harveyi,
Cobetia marina,
Halomonas aquamarina,
Pseudoalteromonas atlantica, and
Roseobacter litoralis), fungi (
Candida albicans,
Aspergillus fumigatus, and
Trichophyton rubrum), and microalgae (
Navicula sp.) [
38]. Compounds
8,
10, and
16 were shown to robustly inhibit
R. litoralis growth. Regarding SAR, the presence of hydroxyl groups at C-2′ on the phenyl ketone core appears to increase the antibacterial activity. None of the compounds tested revealed activity against the fungal strains, with MICs higher than the maximum tested concentration (128 µg·mL
−1). The most promising compounds with antifouling activity (
13,
15, and
19) were evaluated for their ability to inhibit the growth of the biofilm-forming marine diatom
Navicula sp. Acetophenone
15 showed inhibitory activity against
Navicula sp. with a EC
50 of 26.73 µM, which suggests a complementary action of this compound against macro and microfouling species and also reinforces the potential of this compound as an AF agent [
38]. Some insights of the SAR for acetophenones are evidenced on
Scheme 1.
Congruent to find eco-friendly compounds, the ecotoxicity and bioaccumulative potential of compounds
13,
15, and
19 were also assessed. The compounds were found to be less toxic to crustacean
Artemia salina at both concentrations tested (25 and 50 µM) than the commercial biocide econea
® (100% lethality), with the mortality rates of acetophenones
15 and
19 not significantly different from the negative control. Acetophenones
15 and
19 showed a LogKow value lower than 3 (in silico prediction), the threshold value from which compounds are considered bioaccumulative, which indicates their low bioaccumulative potential. In this line of thinking, compounds
15 and
19 could be considered hits for the development of effective and eco-friendly AF compounds [
38].
2.3. Anthraquinone Derivatives
Anthraquinones (AQs) are chemical scaffolds characterized by a 9,10-dioxoanthracene core structure substituted by three fused benzene rings with two ketone functional groups on the central ring. Until now, about 700 molecules with AQ skeletons were characterized, of which about a third were isolated from plants, while most were isolated from bacteria, lichens, fungi, and sponges or other marine invertebrates [
39]. Due to the diversity of biological properties, AQs are a relevant class of bioactive compounds and there is strong evidence of their potential interest for use as additives in AF coatings to prevent marine biofouling. Specifically, citreorosein and emodin were described to reveal robust AF activity against the settlement of
A. amphitrite larvae [
39]. On this wise, Preet and colleagues selected 19 structurally distinct AQ compounds (
21–
39,
Figure 9), based on the same anthraquinone core structure, and studied them regarding their microbial growth and biofilm adhesion inhibition activity against three marine bacterial species which are key players in the marine biofilm process:
Vibrio carchariae,
Pseudoalteromonas elyakovii, and
Shewanella putrefaciens [
39].
All AQ analogs were effective at inhibiting the biofilm growth of
P. elyakovii at a very low concentration (20% to 56% at 0.001 μg·mL
−1). Furthermore, 60% of the compounds reveal a minimal inhibitory concentration (MIC) above 10 μg·mL
−1, which shows that although the overall growth of the microbes is not affected between 10 μg·mL
−1 and 0.001 μg·mL
−1, the biofilm adhesion is affected by these compounds. A biofilm adhesion MIC of 0.01 μg·mL
−1 was observed for
26,
27, and
35 against
V. carchariae, while in general, the compounds tested exhibited a larger degree of variation in their activity against this bacterium. Curiously, the commercially applied compound
30 showed one of the highest microbial growth and biofilm adhesion MICs against
V. carchariae of 10 μg·mL
−1 [
39].
Additionally, SAR of the best-performing compounds were evaluated to conclude the structural properties which are crucial in contributing to the biofilm-associated AF activity. The presence of phenolic hydroxyl (OH) groups at positions 2 and/or 4 of the AQ skeleton provides the lowest MIC of 0.01 μg·mL
−1 (compound
26). In a structural vision, compared with the other compounds investigated in this study, compounds
35 and
38 were the most distinct. In this way, the pronounced activity of compound
35 (0.01 μg·mL
−1 against
V. carchariae) can be attributed to the presence of additional aromatic rings within the structure, while the presence of an additional sterically demanding heterocyclic ring system in compound
38 was conducted to decreased activity (0.1 μg·mL
−1) [
39]. Some insights of SAR for anthraquinones are summarized in
Scheme 2.
Because of the absence of molecular targets identification and the unknown mechanism of action of AF compounds, the research of potential mechanisms that explain how compounds are inhibiting biofim adhesion becomes a challenge. In this point of view, the authors proposed the hypothesis that the interruption of the quorum sensing signaling system associated with the production of biofilms in
V. carcharie could be involved in the mechanism of action for the AF activity observed [
39]. In this way, a molecular docking study was performed to evaluate the differences in binding between the compounds and a crucial protein involved in the transportation of autoinducers which are signaling compounds within the quorum sensing system, the LuxP protein. Compounds
21,
27,
30, and
39 revealed the best docking to the receptor site with binding energies ranging between −8.4 kcal/mol and −7.7 kcal/mol [
39]. It deserves to be highlighted that one of the most active AQs in the MIC study was compound
27, so the authors speculated that the compound’s activity may be based on binding to LuxP protein. A pharmacophore model was created based on the four best docked AQs:
21,
27,
30, and
39. The generated pharmacophore revealed three main features: hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), and aromatic rings. The common feature pharmacophore model with a score of 0.9242 showed certain features: two HBDs, four HBAs, and two aromatic rings [
39]. In a subsequent work, Preet and colleagues aimed to identify the pharmacophore of anthraquinones with antifouling activity by targeting LuxP protein [
40]. For that, the authors performed a virtual screening using a dataset of naturally occurring anthraquinones-related compounds against LuxP protein of
V. carchariae and found that there are six possible pharmacophoric features important for AF activity, particularly hydrophobic interactions, HBAs, HBDs, aromatic interaction, negative ionisable area interaction, and positive ionisable area interaction, that may guide the selection, design, and synthesis of anthraquinone derivatives [
40].
2.6. Bile Acid Derivatives
Among the plethora of structurally diverse steroids isolated from marine invertebrates [
43,
44], there are only a few reports of bile acids and derivatives isolated mainly from octocorals and sponges [
45]. These rare findings of bile acid derivatives in marine invertebrates show an interesting structural feature, the increased lipophilicity, for example, by acetylation of their polar compounds, which decreases their water solubility and allows a high concentration of the bioactive compound on the surface of the organism. In this way, this structural feature may constitute a requirement for its ecological function, which is especially interesting in the case of interactions that take place at the surface of the invertebrate, such as acting as a natural shield against epibiosis [
45]. Specifically, peracetylated cholic acid, a natural biodegradable bile acid derivative isolated from the Patagonian sponge
Siphonochalina fortis, was evaluated in laboratory and field AF trials. The results reveal that peracetylated cholic acid exhibited AF activity and low toxicity against the mussel
Mytilus edulis platensis. Moreover, the experimental soluble matrix paints additivated with 0.6% (
w/
w) showed promissory performances in the in situ sea field trials [
45].
To explore the interest of bile acid scaffolds in the development of new antifouling compounds, a series of derivatives of three bile acids (deoxycholic acid, chenodeoxycholic acid, and cholic acid) with diverse polarities and with the ability to form both hydrophobic and electrostatic interactions was planned and synthesized (
Figure 12) [
46]. The AF effects of the synthesized derivatives, as well as of the three parent bile acids, were assessed through a set of AF bioassays, including antimacrofouling tests against the mussel
M. galloprovincialis and antimicrofouling tests against five biofilm-forming marine bacteria and four representative biofouling microalgae species. The bile acid
56 and derivatives
60–
64 showed robust bacterial growth inhibition, revealing inhibitory values around 40% at concentrations of 12.5 μM and
R. litoralis was the most sensitive species, specially to compounds
60 and
62. Concerning to microalgal, bile acids
56 and
57 and derivatives
60,
63, and
64 inhibited the growth of all the diatom species. It is important to note that compounds
56,
60, and
62–
64 presented EC
50 values between 3 μM and 10 μM for all the tested species, except for
Navicula sp. The most potent bile acid against the settlement of
M. galloprovincialis larvae was the methyl ester derivative of deoxycholic acid (
65) (EC
50 = 3.7 μM; LC
50 > 200 μM; and LC
50/EC
50 > 50) followed by methyl ester derivatives of chenodeoxycholic acid (
64) and cholic acid (
60). The lengthening the side chain in ester derivative of deoxycholic acid (
61) did not lead to an increase in the AF activity, but it appears to affect the broad spectrum to a species-specific profile considering that compounds
60 and
63–
65 showed AF activity against micro and macrofouling species, and that compound
61 only inhibited the bacteria
R. litoralis and diatom
Halamphora sp. Moreover, sulfation at position 3 did not affect pronouncedly the AF activity, but increased the solubility of compound
60 in water. It is also relevant to analyze that compound
59, without free hydroxyl groups, did not show any AF activity, which suggests that free hydroxyl groups in the bile acid scaffold may be important to AF activity. In contrast, structural modifications such as esterification (compounds
60,
61 and
63–
65) and the presence of a primary amine (compound
62) increased the inhibitory activity against the growth of diatoms and marine biofilm-forming bacteria (
Figure 12) [
46]. A resuming of using the SAR for bile acid derivatives is evidenced in
Scheme 3. Furthermore, compounds
63 and
65 demonstrated better inhibition activity than econea
® against
Halamphora sp., and
Cylindrotheca sp., respectively. With regard to eco-toxicological aspects, compounds
60 and
63–
65 were not toxic to
A. salina and compound
63 showed the lowest bioaccumulation potential [
46]. Bile acid derivative
60 was selected for direct incorporation in two polymeric coatings, polydimethylsiloxane (PDMS) and polyurethane (PU), and showed good compatibility with both systems. The anti-settlement activity on
M. galloprovincialis larvae of this bile acid derivative was maintained even after incorporation. These results indicate that
60 is a good candidate for further in situ testing [
46].
In the direction to elucidate de mechanism of action of bile acid derivatives, the in vitro inhibition of acetylcholinesterase (AChE), which is known to play an important role in the settlement of macrofouling organisms, was evaluated for the compounds that revealed an EC
50 < 10 μM in the settlement of
M. galloprovincialis (compounds
60 and
63–
65), and only in the presence of derivative
64, the activity of this enzyme decreased slightly [
46].
2.7. Bromotyramines
Bromotyrosine-derived MNPs are frequently isolated from sponges and ascidians [
47]. Moloka’iamine (
66), a compound first isolated in 1993, is based on an
O-alkylated dibromotyramine core and displays cytotoxic and AF activities [
48]. Particularly, compound
66 is well-known due its robust AF activity against barnacle crypids of
A. amphitrite. Nearly 30 natural bromotyramines and their synthetic derivatives were reported in the literature as follows.
Analogs of hydrochlorides of natural bromotyramines
66 and 3,5-dibromo-4-methoxy-b-phenethylamine (
67) were synthesized (
68–
73,
Figure 13) to establish SAR for this relatively simple core structure [
49].
The substitution of the bromines with hydrogens and chlorines in
70 (EC
50 > 50 µg·mL
−1) and
71 (EC
50 = 33 µg·mL
−1), respectively, led to a decrease in AF activity against barnacle
A. amphitrite, indicating that bromines were strictly required for AF performance. Compound
68 (EC
50 = 0.8 µg·mL
−1) was the most active compound tested among the analogs of bromotyramine
66–
73. The authors also investigated the AF activity of compound
67 (EC
50 = 0.07 µg·mL
−1) and found that this natural compound is the most potent AF bromotyramine reported to date. The structure of bromotyramines
72 and
73 was designed to hybridize the aromatic portion of
67 with an aliphatic portion to obtain a more lipophilic derivative of
66 that would be more soluble in marine coatings (lipophilic) with a solvent base, and therefore more practical as an AF marine paint additive. Compounds
72 (EC
50 = 0.2 µg·mL
−1) and
73 (EC
50 = 0.008 µg·mL
−1) potently inhibited the settlement of barnacle cyprids, and the AF performance of compound
73 was far superior to the AF performance of the natural compound
67. Regarding cyprid toxicity, bromotyramines
67,
72, and
73 exhibited different levels of toxicity which may be attributed to the observed AF activity [
49].
In another study, 23 analogs based on the
O-alkylated dibromotyramine core were synthesized through the application of click chemistry (
74–
96,
Figure 13) on an appropriately large scale [
50]. Compounds
74–
76 comprise the bromotyramine core bound to the methyl ether group through a 1,2,3-triazole. The antibiofilm activities in three Gram-negative marine bacteria,
Pseudoalteromonas ulvae,
Pseudoalteromonas lipolytica, and
Paracoccus sp. were compared to compounds
66 and
67, the natural bromotyramines, to understand the effect of the triazole ring. It was possible to infer that most of the bromotyramine derivatives were more active than
66 and
67 against biofilm formation by the three bacterial strains. Regarding to SAR, the antibiofilm activity of compound
74 suggests that the introduction of a triazole ring increases the inhibition of biofilm formation. The absence of the bromine atom at the 5-position on the aromatic ring in
75 slightly increased the activity against all strains. Monobrominated derivative
75 was selected as a starting point to study the impact of the length of the chain between the triazole and aromatic rings on the antibiofilm activity. Decreasing the methylene spacer from two carbons (
75) to one carbon atom (
77) resulted in a decrease in antibiofilm activity. The addition of one methylene group between the aromatic and triazole rings (
78) enhanced activity against
Paracoccus sp., but surprisingly led to a decrease in activity against
P. ulvae and
P. lipolytica. Following, an evaluation of the impact of the nature of substituents on the 4-position of the triazole was conducted. The replacement of the ether group of compounds
75,
77, and
78 with a methyl ester or phenyl in compounds
79–
81 and
82–
84, respectively, led to a substantial decrease in the antibiofilm activity against all strains. From this point, bromotyramines
75 and
78 were the most promising compounds and more analogs were synthesized (
87–
96) to understand how modifications of the aromatic core would change the antibiofilm activity. Replacing a methyl group by a
N,
N-dimethylethanamine chain (
87) increased the antibiofilm activity and lowered the values of the EC
50 to less than 100 μM. Analogs with a longer chain were synthesized (
88–
92), including primary amine derivatives (
93–
96), but the extension of the chain did not bring any benefits for the antibiofilm activity. A primary amine was introduced instead of a tertiary amine in compounds
87,
88,
90, and
91 to generate compounds
93–
96. Replacement of dimethylamine by a primary amine was prejudicial to the antibiofilm activity. The information obtained with the SAR for this class of compounds, such as the essential presence of bromine and the presence of a triazole ring, might help researchers to develop more potent antibiofilm compounds. Toxicity assays confirmed that the antibiofilm activity of all compounds, except for compound
89, was not a result of a bactericidal effect [
50]. Some insights of the SAR for bromotyramine derivatives are evidenced in
Scheme 4.
Hemibastadins are typical specialized metabolites of the Pacific elephant ear sponge
lanthella basta and consist of a brominated tyrosine moiety featuring an oxime function instead of the amino group and a likewise brominated tyramine unit linked to tyrosine through an amide bond [
51]. In 2007, Ortlepp and colleagues reported the AF activity of 15 brominated NPs isolated from several marine sponges and three synthetic analogs [
52]. These compounds were tested against the inhibition of
Amphibalanus improvisus cyprid settlement. Among the NPs present in the study, hemibastadin-1 (compound
97) was selected as a lead compound for the preparation of synthetic analogs
98–
100 (
Figure 14). Through these analogs, the importance of the oxime substituent and the bromide atoms for the AF activity was studied.
The removal of the oxime function (compound
100) and bromine (compound
99) resulted in a decrease in the activity, which indicates that the presence of the oxime function and bromine modulates the activity of the respective derivatives. Interestingly, while the natural compound
97 caused significant larval mortality at low concentrations, no toxicity was observed for its synthetic analogs, especially compound
98, which exhibited the same potency as the natural compound
97. Hemibastadin-1 (
97) and synthetic products
98–
100 showed no general toxicity when tested against brine shrimp (
A. salina) larvae. Synthetically derived 5,5′-dibromohemibastadin-1 (
98) was later found to be a potent inhibitor of blue mussel
M. edulis phenoloxidase (IC
50 = 0.8 µM), an enzyme involved in the firm attachment of this invertebrate to substrates [
53]. The oxime function of hemibastadins, which was shown to be important for the anti-settlement displayed by compound
98, is responsible for the strong copper-chelating properties, thereby presumably causing enzyme inhibition. Furthermore, the presence of bromine substituents at the phenolic rings of compound
98 increased the enzyme inhibitory properties [
53].
Later, to investigate the enzyme inhibitory activity of hemibastadin derivatives, nine new compounds (
101–
108,
Figure 14) were synthesized with structural variations regarding compound
98’s core structure, namely, different halogen substituents present at the aromatic rings (
101 and
102) and different amine moieties linked to the (
E)-2-(hydroxyimino)-3-(4-hydroxyphenyl)propionic acid (
103–
108) [
51]. According to the results obtained, it was possible to establish that the presence of bulky halogen atoms ortho to the phenolic hydroxy groups increases the inhibitory activity [
51]. Overall, derivatives structurally close to sponge-derived hemibastadins revealed superior enzyme inhibitory properties vs. derivatives featuring structural moieties that are absent in the respective MNPS. The antibacterial activity of compound
98 was explored, and it was discovered that this molecule has antibiofilm activity without mortality for marine and terrestrial bacteria [
54].
Important molecular features of hemibastadin derivatives are highlighted in
Scheme 5.
Compound
98 (
Figure 14) was incorporated in a biodegradable polymer poly(
ε-caprolactone-co-
δ-valerolactone), and demonstrated an ability to reduce the biofilm development of a mixture of bacteria and diatoms species both in situ and in vivo assays, realized during 28 and 21 days, respectively [
55].
In what concerns the mechanism of action, synthetic hemibastadin derivatives were shown to modify the intracellular Ca
2+ levels inhibiting the attachment of cyprid barnacles and the catalytic activity of blue mussel phenoloxidase, explaining their ability to disturb the settlement of this invertebrate [
51,
55,
56].
2.8. Chalcones and Flavonoids
Chalcones are privileged scaffolds in medicinal chemistry, being a central core of many bioactive compounds and are biogenetic precursors of important molecules such as flavonoids. The carbonyl-conjugated system with two electrophilic centers make it susceptible to a wide range of reactions such as nuchleophilic additions and Dies-alder cycloadditions [
57]. Chalcones represent one of the major subclasses of flavonoids and their importance in medicinal chemistry is undeniable [
58,
59]. They can be isolated from natural sources or easily obtained from simple synthetic methods. Several biological activities of natural and synthetic chalcones were reported. Moreover, they have slimicidal and anticorrosive properties, which make them ideal candidates for use in AF paints [
60,
61]. Since 2010, more than 70 synthetic chalcone derivatives were reported to have AF activities against micro and macrofoulers [
60,
62,
63].
A series of chalcone derivatives was synthesized (
109–
155,
Figure 15) and their AF activities were evaluated against three marine bacteria:
Bacillus flexus,
Pseudomonas fluorescens, and
Vibrio natriegens isolated from biofilms formed on polymer and metal surfaces immersed in ocean water [
60]. Regarding
B. flexus, compounds
113,
115,
124,
131, and
144 were the most active compounds (MIC = 0.002–0.116 µM) and compounds
114,
116,
129, and
134 (MIC = 0.014–0.466 µM) were the least active. Most of the compounds showed high activity against
P. fluorescens, except compounds
109,
148, which were poorly active. Compounds
113,
131,
139,
140, and
144 were the most active compounds against
V. natriegens. Overall, compounds
113,
124,
131,
140,
141, and
144 were found to be the most promising due to the broad activity against the three marine bacteria species. Interestingly, most of them had a hydroxy pattern of substitution.
Quantitative structure–activity relationship (QSAR) studies were performed and showed the contribution of the spatial, structural, and electronic descriptors towards the biological activity. Steric factors were found to contribute negatively to the activity and consequently, small molecules were more active. Hydrophobicity was found to be positively correlated to the activity, which indicates that hydrophobic molecules have better activity. An increase in the number of free rotatable bonds in these compounds showed a negative contribution to the activity, while an increase in the relative negative charged surface area of the molecules led to an increase in the activity [
60].
A series of chalcone derivatives displaying AF properties (
156–
171,
Figure 15) was also investigated [
62]. Firstly, compounds were evaluated against the adhesive larvae of the macrofouling mussel
M. galloprovincialis. This was the first report of chalcones with anti-macrofouling activity: compounds
166,
167, and
171 (EC
50 = 34.63, 7.24, and 16.48 µM, respectively) were active, with no toxicity against
M. galloprovincialis, for the maximal concentration tested (200 µM). These compounds were then evaluated concerning their inhibitory activity against the growth of five biofilm-forming marine bacteria (
C. marina,
V. harveyi,
P. atlantica,
H. aquamarina, and
R. litoralis) and four marine diatom strains (
Cylindrotheca sp.,
Halamphora sp.,
Nitzschia sp., and
Navicula sp.). Compounds
167 and
171 showed activity against
H. aquamarina (EC
50 = 18.67 and 18.78 µM, respectively) and
R. litoralis (EC
50 = 4.09 and 12.34 µM, respectively) and compound
171 also showed activity against the four diatoms strains (EC
50 = 6.75–20.31 µM). Ecotoxicity for the most promising chalcones (
166,
167, and
171) was assessed using
A. salina standard ecotoxicity assay, and no toxicity was observed (50 µM). QSAR studies were also performed in this work and showed the contribution of geometric and relative charged surface area descriptors towards the AF activity against the settlement of mussel larvae of
M. galloprovincialis [
62].
Following the report of several sulfated compounds with AF activity (
Section 2.26) [
65], including glycosylated flavones, new potential AF polymethoxylated chalcone and flavone derivatives with glycosyl groups incorporating a 1,2,3-triazole moiety (
189,
Figure 15) were synthesized and their AF potential was studied, namely anti-settlement activity towards
M. galloprovincialis larvae and antibacterial activity of five biofilm-forming marine bacteria species (
C. marina,
V. harveyi,
P. atlantica,
H. aquamarina, and
R. litoralis) [
64]. Chalcones
183,
188, and
189 and flavone
173 effectively inhibit the settlement of
M. galloprovincialis larvae (EC
50 < 25 µg·mL
−1). Among these compounds, chalcone
183 was the most potent (EC
50 = 3.28 µM; 2.43 µg·mL
−1) and showed a high therapeutic ratio (LC
50/EC
50 > 60) [
64]. The hydrogen bonding acceptor ability of the compounds, the average complementary information content of order 2, and the presence of triple bonds are the three descriptors that were found to have an impact on the AF activity displayed by this series of compounds in the QSAR model obtained. The first descriptor increased the AF activity (chalcone
183), while the other two descriptors negatively affected the AF activity (
175,
180, and
181) [
64]. Regarding the inhibition of biofilm-forming marine bacteria, only chalcones
180 and
181 were able to inhibit the growth of one of the species tested,
R. litoralis (EC
30 = 135 and 83.5 µM). Compounds
173,
183,
190, and
189 were also tested against the inhibition of the growth of
Navicula sp., a marine diatom species, but only compound
183 showed significant activity (EC
50 = 41.76 µM) [
64]. Ecotoxicity results for the most promising compounds (
173,
183,
188, and
189) showed that these compounds were not toxic against the non-target species
A. salina at 25 and 50 µM [
64]. Overall, compound
183 was the most promising, showing both anti-macrofouling and anti-microfouling effects and no toxicity against a non-target organism.
More recently, the AF activity of six synthetic furylchalcones was verified in field tests and the results are described in the furan and furanones section (
Section 2.11) [
63].
2.10. Diterpene Derivatives
Diterpenes are a class of natural products originating from C20 precursor geranylgeranyl diphosphate [
69]. Pimarane diterpenes are a kind of tricyclic diterpene and could be isolated mainly from plants and fungi [
70]. Due to the stereochemistry diversity, pimarane diterpenes are distinguished into pimarane, isopimarane, and
ent-pimarane. This group of compounds became a direction in the research of novel active compounds because of its wide applications in medicine and agriculture [
71]. During the investigations on environmentally friendly AF compounds, it was observed that pimarane diterpenoids can exhibit non-toxic AF activities against the larval settlement of the barnacle
B. albicostatus [
72]. To investigate the SAR of these pimarane diterpenoids, they synthesized five new pimarane diterpenoids (
199–
203,
Figure 17) and tested their ability to inhibit the settlement of the larvae of barnacle
B. albicostatus [
73]. It was observed that substitution patterns at C-15 and C-16 positions of pimarane diterpenoid significantly affected the AF activity. For example, substituents, such as the acetoxyl, methoxyl, or methanesulfonyloxyl group at C-15 and C-16 positions of the pimarane diterpenoid led to less potent compounds. Additionally, the AF activity of acyl compounds
199 and
200 (EC
50 = 0.30 and 0.14 µg·cm
−2, respectively) was higher than for the methylated compound
201 (EC
50 = 0.57 µg·cm
−2), which suggests that an acetoxyl group at a suitable position of the pimarane diterpenoid scaffold might result in increased AF activity compared to the methoxyl group. The methanesulfonyloxy group showed no significant impact on the AF activity of pimarane diterpenoids, as compound
202 showed weak AF activity (EC
50 = 7.47 µg·cm
−2) [
73]. Additionally, an epoxy group replacing hydroxy groups at the C-15 and C-16 positions in pimarane diterpenoid greatly increased AF activity as seen by compound
203 (EC
50 = 0.05 µg·cm
−2), which suggests that the epoxy group might be an important functional group for potent AF activity in pimarane diterpenoids [
73]. No toxicity was observed for all the compounds (LC
50 > 10 µg·cm
−2), indicating that the substituents in the side chain of pimarane diterpenoids did not influence the toxicity [
73]. Some insights of the SAR for pimarane diterpenes are evidenced in
Scheme 6.
Bromosphaerol (
204,
Figure 18) is a brominated diterpene isolated from the red alga
Sphaerococcus coronopifolius [
74,
75]. This molecule showed robust anti-settlement activity against larvae of
A. amphitrite with very low toxicity. From this perspective, 15 structural analogs involving transformations at ∆
1 double bond and positions C-11, C-16, and C-17 (
205–
219,
Figure 18) were designed and synthesized to improve bromosphaerol AF activity [
76]. Using cyprids and nauplii of
A. amphitrite as a model organism, the anti-settlement activity (EC
50) and the degree of toxicity (LC
50) of the bromosphaerol derivatives were evaluated. The design thinking to achieve SAR embraced introducing polar groups at C-1 and/or C-2 (
205–
209 and
211), removing the C-11 hydroxyl group (
212 and
213) and substituting C-2 with functional groups, in specific, an ester and an oxime, while the ∆
1 double was repositioned to C-1-C-10, providing the generation of an extended conjugated system (
210 and
214–
219) [
76].
Analogs
212,
216,
218, and
219 revealed promising AF efficacy (EC
50 < 0.5 mg·L
−1). Compounds that bear an oxygen moiety at C-1 and C-2 (
205,
207,
209, and
210) exhibited similar EC
50 values ranging from 10.44 to 8.75 mg·L
−1. The introduction of bromine at C-2 abolished the activity (compound
211). α,β-unsaturated ester analog
214 did not reveal a promising AF compound (EC
50 > 50 mg·L
−1). The elimination of the hydroxyl group at C-11 to form the exocyclic double bound showed robust AF activity with na EC
50 < 0.5 mg·L
−1 (
212). The exocyclic elimination congener was less potent (EC
50 = 3.87 mg·L
−1). It was possible to conclude that the nature of oxime functionality impacts AF activity. Supporting that, the unsubstituted oxime compound
215 and the carboxy oxime were revealed to be inactive, contrasting with the methoxy oxime analog
216 and the methyl ester congener of
217, analog
218, which presents EC
50 < 0.5 mg·L
−1. In the same point of view, the dimethylaminoethyl oxime derivative
219 showed EC
50 < 0.5 mg·L
−1 [
76].
Concerning the toxicity assessment, structural analogs
205,
207, and
209–
219 showed diverse levels of AF activity. While the LC
50 values for derivatives
212 and
216 were >50 mg·L
−1, compounds
218 and
219 revealed high toxicity towards the barnacle naupliar stage with LC
50 values of 2.7 and 12.5 mg·L
−1, respectively. Epoxy derivative
205 and the tetrahydrofuranyl analog
209 possess LC
50 values of 25.2 and 10.2 mg·L
−1, respectively. All the other compounds were not toxic against cypris larvae with LC
50 values > 50 mg·L
−1. Compounds
212 and
216 were demonstrated as well-performing antifoulants, acting through a non-toxic mechanism with therapeutic ratio values > 100 on
A. amphitrite cypris [
76]. Some insights of the SAR for brominated diterpenes are evidenced in
Scheme 7.
2.12. Indole Derivatives
The indole heterocycle is found in natural products. Indoles show prominent biological activities including anticancer, antioxidant, anti-inflammatory, antifungal, anticholinesterase, and antibacterial properties [
79,
80]. Isatin (
229), an antifungal and ecologically defensive marine natural product isolated from marine bacterium isolated from healthy embryos of the caridean shrimp Palaemon macrodectylus (
altermonas sp.), was proposed as an alternative molecular AF scaffold and as a source for structural insight to guide the molecular design of AF compounds [
81]. Nearly 30 natural and synthetic indole derivatives were reported in the literature as follows.
Derivatives of isatin (
230–
244,
Figure 20) were obtained to understand the structural requirements for AF activity and tested against the growth of bacteria isolated from natural biofilms [
81].
Briefly, the use of bromine substituent at the C-5 carbon atom in the derivatives of isatin led to a decrease in antibacterial activity as compared with isatin (
229). A free NH is necessary for good inhibitory activity of isatin (
229) against fouling bacteria, as
N-methyl and
N-butyl isatin derivatives showed decreased activity. The presence of the 3-acetonylidene group and a free NH moiety increased antibacterial activity as observed for compound
233, which was the most potent compound. The effects of synthetic indole derivatives (
245–
253,
Figure 20) were evaluated against two marine benthic diatoms (
Nitzschia closterium f. minutissima and
Navicula climacospheniae) [
82]. Indole derivatives containing a simple indole ring and halogenated substituents significantly inhibited the growth of the two types of diatoms. In general, the presence of a halogen increased anti-algal activity, but it was dependent on the position of the halogen in the indole ring. Indole derivatives with chlorine at position 7 (
248) showed increased activity compared with derivatives with chlorine at positions 5 and 6 (
246 and
247). On the other hand, bromine at position 6 (
250) was more favorable for antialgal activity compared with positions 5 and 7 (
251 and
252). Diatom species determined the sensitivity of various indole derivatives. Derivatives with chlorine or bromide (
246–
252) were more potent against
N. closterium f. minutissima than against
N. climacospheniae. Recently, indole derivatives were tested in a marine environment after incorporating them in marine AF coatings (
254–
260,
Figure 20), after performing antibacterial and anti-algal experiments [
83]. The antibacterial activity of indole derivative
256 was significantly higher than those of indole derivative
254, which suggests that phenyl substituents increase the antibacterial activity. Indole derivative substituted with a bromine group (
255) showed slightly higher antibacterial activity compared to indole derivative
254. Substituents of the indole ring resulted in great differences in algal inhibition. An acrylamide methylene group (
260) was more favorable than a benzamide methylene group (
257) in the inhibition of
N. closterium and
Platymonas subcordiformis. A bromine atom seems to increase anti-algal activity, as observed for the activity compound
255 against
N. closterium when compared to compound
254, and the activity of compound
255 against three algae species when compared to compound
257 [
83]. Some insights of SAR for indole derivatives are evidenced in
Scheme 8. Compound
254 was selected for the net Ca
2+ flux test due to the higher inhibition rate of
P. subcordiformis and the results might indicate that indole derivatives can act as inhibitors of transmembrane transport and trigger algal cellular Ca
2+ efflux. AF coatings containing indole derivatives
254–
260 were prepared and tested in a marine environment (Qingdao Harbor) for five months. Polyvinyl chloride (PVC) panels brushed with coating containing Cu
2O and the indole derivatives (compounds
254–
256 and
260) were adhered by fewer fouling organisms, which is consistent with the results obtained in the laboratory experiments [
83].
A novel indole derivative, compound
261 (
Figure 20), with an acrylamide group and structurally similar to compounds
254–
260, was obtained by synthesis, and its AF properties were studied concerning their ability to inhibit the growth of bacteria (
Escherichia coli and
Staphylococcus aureus) and marine algae (
Phaeodactylum tricornutum,
N. closterium, and
Skeletonema costatum) [
84]. Results show that compound
261 has a bacteriostasis rate of 94.91% and 94.91% against
E. coli and
S. aureus, respectively. The inhibition rates of compound
261 against marine algae were between 92% and 95%, showing also evident anti-algal properties. Further studies regarding the performance of compound
261 immobilized in an acrylate resin were performed. Inhibition rates of acrylate resin containing compound
261 against
P. tricornutum,
N. closterium were improved by one-fold, compared with acrylate resins alone, in static conditions. In dynamic conditions, acrylate resins with compound
261 were immersed in the sea for three months and distinct differences between different contents of indole derivative
261 were observed, as the control acrylate resin was completely covered by algae. In contrast, resins containing compound
261 showed less fouling and the observed fouling was directly proportional to the indole content in the resin [
84]. Overall, compound
261 displayed antimicrobial and antialgal properties, even after being incorporated in acrylate resin, highlighting its potential to be used in marine paints and have a real application [
84]. These results are the proof of concept that these indole derivatives have AF properties and can be further studied to be used as AF ingredients in marine paints.
Scheme 8 summarizes the moyeties important for AF activity based on the described studies.
2.13. Isocyanide-Containing Compounds
The first isocyanide was synthesized in 1859 and the first natural isocyanide was identified in 1950. The isocyanide concerns an interesting functional group in organic chemistry, as its carbon can act as (i) a nucleophile attacking activated electrophiles, (ii) as an electrophile being intercepted by different nucleophiles, (iii) as a carbene involved in formal [4 + 1] cycloaddition, and (iv) as a radical acceptor to form imidoyl radical reaction intermediates. Additionally, lone pair interactions enable the preparation of various coordination complexes [
85]. Isocyanides-containing molecules reveal a powerful diversity of biological activities.
Although compounds containing an isocyano group are a rare sort of MNPs, some were isolated from certain marine sponges [
86]. Most natural isocyanides are toxic toward fish and crustaceans, which indicates a role in chemical defense and perhaps a role as repellants of epibiotic organisms [
86]. Natural isocyanides have complex structures and the synthesis of simpler analogs with feasible chemical reactions seem to be a suitable solution to obtain these compounds in higher quantities. Isocyanides were the most explored compounds as AF agents, with 110 synthetic compounds reported from 2002 to 2017.
3-Isocyanotheonellin (compound
262,
Figure 21), a marine sesquiterpene from a nudibranch species, with an isocyano group at the C-3 position, exhibited potent AF activity against the larvae of the barnacle
A. amphitrite (EC
50 = 0.13 μg·mL
−1). The SAR for compound
262 analog was first explored through the synthesis of seven analogs (compounds
263–
269,
Figure 21) and the AF activity evaluation against the settlement of
A. amphitrite larvae [
87,
88]. Modifications were based on the stereochemistry of the isocyano group and the double bonds. The stereochemistry of the double bond did not significantly influence the AF activity, as compound
263 (EC
50 = 0.29 µg·mL
−1) only displayed a slight activity difference compared to compound
262 (EC
50 = 0.19 µg·mL
−1) [
87]. Concerning the stereochemistry of the isocyano group, the AF performance of compounds
268 and
269 (EC
50 = 0.18 and 0.41 µg·mL
−1) was not significantly different from the performance of compounds
262 and
263 [
88]. The LC
50 of cyprids was calculated for all the compounds (
262–
269,
Figure 21), and all of them were not toxic at high concentrations to the cyprid larvae (LC
50 > 100 µg·mL
−1) [
88].
Another detailed SAR for compound
262 was attempted with the synthesis of four analogs (
266–
269,
Figure 21) [
88]. Analogs
266–
269 were less potent than analogs
263–
265, showing only moderate AF activity (EC
50 = 1.80–7.20 µg·mL
−1). The methyl group at C-14 seems to contribute to the potent AF activity displayed by compounds
263–
265, probably by influencing the conformation of the molecules [
88]. This work was the inspiration for the synthesis of twelve simple linear isocyanides (
270–
281,
Figure 21) [
89]. All the compounds displayed AF activity, suggesting that the isocyano group is important for the AF activity. Compound
270, together with phenylthiol
278, showed the most potent AF activity (EC
50 = 0.046 and 0.056 µg·mL
−1, respectively) without toxicity to the cyprid larvae (LD
50 > 30 µg·mL
−1). Compounds
270,
280, and
281, which differ only in the number of methyl groups in the isocyano moiety, showed differences in AF activity, in which compound
270 with two methyl groups was the most active and compound
281 with no methyl groups was the less active (EC
50 = 0.14 µg·mL
−1). The differences in the AF activity in these compounds could indicate that the lipophilicity near the isocyano group influences AF activity [
89]. The most hydrophobic compounds (
275–
277) showed only moderate AF activity, which might be associated with their poor solubility. Compounds
271–
273, possessing a hydrogen bonding donor group, showed some toxicity against the cyprid larvae (LD
50 = 21.28–22.25 µg·mL
−1), which puts forward the possible association of the hydrogen bonding donor with toxicity [
89].
Scheme 9 summarizes the SAR based on compound
270 [
85].
Studies concerning the mechanism of action of compound
270 in two target organisms, the bryozoan
B. neritina and the barnacle
A. amphitrite, and one non-target organism, the species zebrafish
Danio rerio, were performed [
95]. Compound
270 bound to mitochondrial proteins both in
B. neritina and
A. amphitrite, suggesting that it may influence mitochondrial functions, which can lead to changes in behaviors, such as the selection of an attachment site and/or inhibition of attachment and metamorphosis [
95]. Specifically, compound
270 interacts with three proteins of
B. neritina, in which two are identical to voltage dependent anion channels (VDAC), located on the outer part of the mitochondrial membrane involved in cellular mechanisms, such as cell metabolism and cell survival. In
A. amphitrite, compound
270 targeted a cytochrome P450 complex enzyme, similar to a NADH-ubiquinone oxidoreductase-like protein, located in the inner part of the mitochondrial membrane, involved in the oxidative phosphorylation, where it catalyzes the electron transfer from NADH to coenzyme Q. Compound
270 in the zebrafrish embryo caused a typical signature due to copper deficiency (e.g., pericardial edema, poor blood circulation, pigmentation defects, and defect on hematopoiesis) [
95].
Previous results suggest that an isocyano group and a hydrophobic site at a suitable position are important in the expression of potent AF activity [
87,
88]. To cover more, novel isocyano cyclohexane derivatives (
282–
293,
Figure 21) with an ester or an ether functional group were synthesized [
90]. The results obtained with compounds
282–
285 indicate that the ester group might be important for the expression of potent AF activity against the cyprid larvae of
A. amphitrite (EC
50 = 0.0096–0.98 μg·mL
−1). Following this, other ester derivatives were obtained, and a very potent AF activity was observed for compounds
286–
289 (EC
50 = 0.048–0.54 μg·mL
−1). To explore the role of the ester group in detail, the synthesis of ether compounds lacking the carbonyl function was undertaken (compounds
290 and
291), and their AF activity showed that the carbonyl moiety of the ester group was more important than the oxygen–carbon moiety (
290: EC
50 = 0.176 μg·mL
−1 and
291: EC
50 = 17.0 μg·mL
−1). The synthesis of compounds lacking the isocyano group was performed to verify the role of this functional group in this series of compounds. Compounds
292 and
293 did not show a significant settlement inhibition activity (EC
50 > 30 μg·mL
−1 for both compounds), which suggests that the isocyano group is essential for the expression of potent AF activity. On the other hand, the results of this study do not make clear the difference in AF activity that exists between the stereoisomers. For all the synthesized compounds, no significant mortality at high concentrations was observed [
90].
Although isocyano cyclohexanes showed promising AF activity without showing toxicity, the yields on the synthesis of these compounds were poor. Therefore, simpler isocyano benzenes were prepared (
294–
298,
Figure 21) [
91]. Compound
294, a phenyl version of 3-isocyanotheonellin (
262), was found to be potent against the settlement of
A. amphitrite, without significant toxicity (EC
50 = 0.078 μg·mL
−1; LD
50 > 100 µg·mL
−1). The increase in chain length led to an increase in AF activity, as observed for compounds
295–
297, but only compound
297 did not show significant toxicity (LD
50 > 3.0 µg·mL
−1). Compound
298 was the most potent compound; however, it was very toxic against the larvae of
A. amphitrite (EC
50 = 0.054 µg·mL
−1; LC
50 = 3.0 µg·mL
−1). Previous SAR studies indicated that an isocyanide with a tertiary alkyl isocyano group was more active than the congeners containing primary or specialized alkyl isocyano groups [
91]. Twenty novel simple isocyanides were synthesized (
299–
318,
Figure 21) containing a tertiary alkyl isocyano group derived from citronellol without using a Grignard reaction [
96]. All the isocyano compounds exhibited potent AF activity against cypris larvae of the barnacle
A. amphitrite, without significant toxicity. Benzoate
301 and chloride
303 were the most potent AF compounds in this study and did not show significant toxicity (EC
50 = 0.08 μg·mL
−1 and LC
50 > 100 µg·mL
−1 for both compounds). Both compounds with a tertiary and with a primary alkyl (compounds
313 and
314) isocyanide showed potent AF activity (EC
50 < 1.0 µg·mL
−1). Compounds
299–
302 (EC
50 = 0.08–0.38 µg·mL
−1) and
307 (EC
50 = 0.32 µg·mL
−1) with hydroxy, ether, ester, or the sulfonate ester functional group, and halides
303–
306 (EC
50 = 0.08–0.057 µg·mL
−1) showed good AF activity. Additionally, compounds
308,
309,
311, and
312, which possess an imide, amide, or amine group, also exhibited good AF activity. Diisocyano sulfide
316 and diisocyano sulfone
317 showed only moderate AF activity (EC
50 = 1.49 and 1.32 µg·mL
−1, respectively), suggesting that an increase in the number of isocyano groups does not translate to an increase in the AF activity [
96]. The results clearly show that in comparison with a linear alkyl chain, the isoprene framework derived from citronellol was a better spacer to achieve AF activity without toxicity.
Isocyanides derived from amino acids as suitable candidates to be environmentally friendly AF agents were also explored [
92]. Some factors contribute to the suitability of isocyanides derived from amino acids, specifically, (i) amino acids can be converted to the corresponding isocyanides (amino acid-isocyanides) in only a few reactional steps; (ii) amino acid-isocyanides are expected to show potent AF activity without significant toxicity due to the presence of the isocyano group, and (iii) are expected to biodegrade to the original non-toxic amino acids. All the 15 synthesized isocyano compounds (
319–
333,
Figure 21) showed effective AF activity against cyprid larvae of
A. amphitrite. Isocyanides
319–
323 (EC
50 = 0.97–2.67 µg·mL
−1) and
333 (EC
50 = 2.63 µg·mL
−1) exhibited relatively potent AF activity, whereas isocyanides
324 (EC
50 = 7.99 µg·mL
−1),
326 (EC
50 = 3.97 µg·mL
−1),
327 (EC
50 = 10.34 µg·mL
−1),
331 (EC
50 = 5.49 µg·mL
−1), and
332 (EC
50 = 3.96 µg·mL
−1) showed moderate AF activity, which indicates that the aromatic ring of the α-side chain might be effective in improving AF activity [
92]. Secondary alkyl isocyanides derived from α-amino acids had potent AF activity, as well as the tertiary alkyl isocyanides [
92]. All the synthesized amino acid-isocyanides showed low mortality rates at high concentrations, after 120 h of exposure [
92].
Umezawa et al. performed the total synthesis of 10-isocyano-4-cadinene (
334,
Figure 21), a MNP obtained from nudibranchs of the family
Phyllidiidae [
93]. The synthetic intermediates without the isocyanate group were also investigated and showed weak AF activity against cypris larvae of the barnacle
A. amphitrite [
93]. After these results, glucosamine-based isocyanides (
335–
345,
Figure 21) were synthesized and the effect of substituents at C-1, C-3, C-4, and C-6 positions on the AF activity was evaluated [
93]. Compounds with an ether group at C-1 (compounds
337–
341; EC
50 = 0.23–0.71 µg·mL
−1) were more active than compounds with an ester group (compounds
335 and
336; EC
50 = 5.54 and 6.36 µg·mL
−1). Changes at C-3, 4, and 6-positions (compounds
342–
345; EC
50 = 0.25–0.81 µg·mL
−1) did not significantly affect the AF activity [
93].
Although almost all the amino acid isocyanides described before (
319–
333) effectively inhibited the settlement of barnacle larvae without exhibiting significant toxicity, they were obtained as mixtures of enantiomers. To circumvent this problem, the structure of amino acid isocyanides was modified by introducing an identical side chain at the α-position to afford achiral α,α-disubstituted amino acid isocyanides (
346–
363,
Figure 21) [
94]. Overall, the most potent AF compounds in this series were hydrophobic amino acid-derived isocyanides
347 (EC
50 = 0.07 µg·mL
−1),
348 (EC
50 = 0.30 µg·mL
−1),
350 (EC
50 = 0.07 µg·mL
−1), and
352 (EC
50 = 0.14 µg·mL
−1), which suggested that hydrophobicity might be associated with AF activity. The AF activity of compounds
347–
352 was higher than the AF activity displayed by the corresponding monosubstituted amino acid isocyanides, suggesting that the introduction of the second side chain increases AF activity [
94]. Additionally, the mortalities observed for all disubstituted amino acid isocyanides were lower than those of monosubstituted ones, indicating that the introduction of the second side chain also effectively reduces toxicity, as observed in a previous work of the same authors [
89]. Previously, the authors showed that aromatic amino acid (Phe and Tyr)-derived isocyanides exhibited high AF activity (EC
50 = 0.14 and 0.17 µg·mL
−1, respectively) and compound
352 (EC
50 = 0.14 µg·mL
−1) also showed potent AF activity [
92]. Therefore, the presence of aromatic rings in the α-side chain was suggested to effectively improve AF activity, inspiring the authors to prepare isocyanides
357–
363 (
Figure 21) [
94]. However, these aromatic compounds only showed moderate to low AF activity except for compound
362 (EC
50 = 0.32 µg·mL
−1), which was attributed to sterically hindrance effects [
94]. The safety of the synthesized AF-active compounds was evaluated based on their therapeutic ratios, and notably, isocyanides
347,
348,
350, and
352, which showed high AF activity, also exhibited high therapeutic ratios [
94].
From the results obtained from all the isocyanide derivatives, it was possible to conclude that the presence of the isocyano group was crucial to the potent AF activity of these compounds. Additionally, lipophilicity near the cyano group was found to increase AF activity, although overall hydrophobicity was associated only with moderate AF activity due to poor solubility. Another conclusion for isocyano compounds reported was that the increase in the number of isocyano groups did not translate into an increase in AF activity. All isocyanide derivatives were tested against the settlement of the cyprid larvae of A. amphitrite, a macrofouling organism, although it would be interesting to test this class of compounds against microfouling species, such as marine bacteria or diatoms.
2.14. Lactones
Butenolides, or butyrolactones, are isolated from fungi, especially the
Aspergillus sp., and possess the skeleton of α,β-unsaturated
γ-butyrolactone scaffold [
97]. These molecules were demonstrated to reveal distinct biological activities, including anti-inflammatory, cytotoxic, antiviral, antioxidant, antimicrobial, antidiabetic, protein kinase-inhibitory, and α-glucosidase inhibitory activities [
98]. Proteomic profiling studies revealed that butenolide can disrupt the neurotransmission in the nervous system by disorganizing the cytoskeletal structure in the brains of medaka
Oryzias melastigm, a fish model species, specifically valuable due to the relevance of ecosystem risk assessment of marine pollution [
99]. Nevertheless, butanolide also induces the detoxification system in their livers, ensuring lower non-target toxicity and higher biosafety [
100]. From 2010 to 2014, 61 natural butenolides and derivatives were reported in the literature.
The AF activity against the cyprids of the barnacle
A. amphitrite of nine NPs (
364–
372,
Figure 22) isolated from different marine
Streptomyces sp. was investigated [
77]. Compounds
367 and
368 were found to be less potent than compounds
364,
365, and
366, which suggested that the side chain may affect AF activity. Compound
369 is similar to compound
367 except that its side chain does not contain a hydroxy group and is around 20 times more potent, supporting the hypothesis that lipophilicity increases AF activity. The presence of a 2-furanone ring is also essential for AF activity since compounds
370,
371, and
372, which do not possess this structural feature, did not present AF activity. Based on this information, the authors synthesized a simplified molecule, compound
373 (
Figure 22), containing a 2-furanone ring and a linear carbon chain. This optimized compound presented excellent AF activity (EC
50 = 0.518 µg·mL
−1) and did not exhibit any toxicity against barnacle larvae [
77]. The AF activity of butenolide
373 was evaluated also against the polychaete
Hydroids elegans and the bryozoan
B. neritina. Again, a potent AF activity was observed, as well as low toxicity against these fouling organisms [
77]. To investigate the mechanism of the action of compound
373, an analysis of the proteome and phosphoproteome alterations during cyprid development/aging of the barnacle upon treatment with butenolide
373 was performed [
101]. This analysis revealed that the expression of two groups of proteins, stress-associated and energy metabolism-related proteins, which are differentially expressed during cyprid development, was sustained by butenolide
373 [
101]. Following this, the effect of butenolide on behavioral and morphological changes in the barnacle
A. amphitrite and the bryozoan
B. neritina was studied and butanolide was showed to decrease their phototactic behavior and attachment and inhibited secretory granules in the cement gland of
A. amphitrite cyprids [
102]. Investigations concerning the molecular mechanism of action of butenolide
373 in three fouling species,
A. amphitrite,
B. neritina, and
Vibrio sp. UST020129-010 were conducted [
103]. Butenolide
373, in a pull-down assay with lysates, was found to bound acetyl-CoA acetyltransferase 1 in the barnacle
A. amphitrite, while in the bryozoan
B. neritina, it binds to acyl-CoA dehydrogenase, actin, and glutathione S-transferases. Finally, in the marine bacterium
Vibrio sp. UST020129-010, butenolide
373 was found to bind succinyl-CoA synthetase β subunit and to inhibit bacterial growth [
103]. Further, the authors predicted by molecular docking with models of acetyl-CoA acetyltransferase 1 of
A. amphitrite and acyl-CoA dehydrogenase of
B. neritina, that butanolide
373 binds to the acyl-CoA pocket or to the flavin adenine dinucleotide coenzyme pocket, respectively, influencing the binding of the substrate or the coenzyme [
103]. Overall, these results suggest that butenolide
373 inhibits the fouling of these organisms by influencing their primary metabolism. A previous work also reported the differential expression of proteome and phosphoproteome in
B. neritina larvae exposed to butanolide
373 [
104]. The acute toxicity of butenolide
373 was also assessed in several non-target organisms, including microalgae (
S. costatum), crustaceans (
Melita longidactyla,
Tigriopus japonicus, and
Daphnia magna), and fish (
Lutjanus erythropterus and
D. rerio) [
105]. Results suggest that butenolide
373 induced cell apoptosis and pericardial edema in zebrafish
D. rerio embryos [
105]. It was possible to calculate the predicted no-effect concentration (PNEC), which was among one of the highest in representative new biocides (PNEC = 0.168 µg·mL
−1), although this value should be lower than the predicted environmental concentration (PEC), which was not possible to calculate in this study [
105]. Later, chronic effects of compound
373 on oxidative stress, neurotransmission, endocrine homeostasis, and reproductive success in adult
O. melastigma (marine medaka) were evaluated and compared to the effects of sea nine 211
® (
Figure 1) [
106]. Butenolide
373 induced at a lower extent the oxidative stress in the liver of both male and female medaka, compared to sea nine 211
®. Moderate effects were observed on sex hormone levels in males exposed to butenolide
373, which were gradually recovered during depuration, in contrast to sea nine 211
®. These results show that butenolide
373 exerted transient, reversible biological effects on marine medaka [
106]. The degradation kinetics of butenolide
373 under various environmental conditions were studied, and it was found that the half-life of the compound was 0.5 days in natural seawater [
107]. It was concluded that the main contributor to degradation in natural seawater was caused by marine bacteria [
107].
SAR studies revealed that both furan and furanone were important pharmacophores for anti-settlement activity against barnacle larvae (
364–
366,
Figure 22) [
77]. Following this, modification in the structure of AF compounds based on SAR information was performed to tune the physicochemical properties by modifying the side chain of alkyl butenolide
373 [
108]. A series of butenolides (
374–
401,
Figure 22) was obtained and the anti-settlement activity was assessed against the larvae of the barnacle
A. amphitrite [
108]. First, the impact of the nature of the side chain on anti-settlement activity was studied and 2-furanone derivatives with a five or six-carbon alkyl amine substituent at the 5-position were obtained. The results indicate that the Boc group was the most favorable terminal group as a substituent at the
N-terminal since Boc carbamate
374 (EC
50 = 5.43 µM) and
375 (EC
50 = 4.00 µM) exhibited the best anti-settlement efficacy. Variation in the length of the alkyl side chain at the 5-position improved lipophilicity, which appeared to increase the AF activity and led to the development of more potent analogs (
376 and
377; EC
50 = 2.13 and 2.22 µM, respectively). The fact that analogs
380 and
379 (EC
50 = 444 and 255 µM, respectively), with a high degree of hydrophilic substitution in the side chain, exhibited poor anti-settlement activity further corroborates the concept that lipophilicity of the side chain might be a key aspect for AF activity. An increase in the number of carbons in the side chain of an amine analog from five to ten carbon atoms (
380–
385) significantly increased the anti-settlement activity (EC
50 = 16.7–444 µM). For amide analogs,
386–
391 (EC
50 = 5.48–40.3 µM), an increase in the number of carbons in the alkyl side chain also increases the anti-settlement activity. However, no enhancement in anti-settlement activity was observed when the number of carbons in the alkyl chain side was increased beyond seven carbons [
108]. Using a Pearson correlation analysis, a positive association between lipophilicity and bioactivity was also demonstrated. Butenolides
376 and
377 were found to be the most potent antifoulants with desirable physicochemical properties [
108].
Twenty-one
γ-hydroxybutenolides (
404–
421,
Figure 22) inspired by two natural AF sesterterpene
γ-hydroxybutenolides, previously identified from a New Zealand marine sponge, cavernosolide (
402) and lintenolide A (
403) (
Figure 22), were synthesized and tested for AF activity against the marine bryozoan larvae of
B. neritina and the marine algae
Isochrysis galbana [
109].
γ-Hydroxybutenolides
404,
405,
414,
416,
419,
420, and
423 were inactive against both marine organisms, while
γ-hydroxybutenolides
406,
407,
408,
413, and
422 were only inactive against
I. galbana. Concerning the AF activity against the marine bryozoan, efficacy decreased (higher EC
50) with lower lipophilicity and the attachment of an aromatic group appeared to increase activity [
109].
Scheme 10 summarizes SAR for butenolide derivatives.
Compounds
407,
411,
417, and
418 were further selected for in situ field trials after incorporation into a rosin-modified acrylic base at a loading of ~10% (
w/
w). After four months, coatings containing
407 and
410 did not present any substantial fouling, as did the commercial AF reference containing Cu
2O [
109]. This work showed, once again, that natural AF compounds can be used as inspiration for the synthesis of structurally simpler analogs.
Synthetic butenolide (
373) (
Figure 22) was incorporated into a marine paint and 5% (
w/
w) of the compound was required to see differences between the treated surface and the control after three months submerged in seawater [
77]. Following this, an optimization of the formulation containing butenolide
373 was performed [
25] regarding concentrations (5, 10, and 15% (
w/
w)), different pigment choices, and binder compositions. After six months of submersion in seawater, coatings with 1:2.5 (acrylic copolymer and rosin) paints containing 10% butenolide had the best AF performance [
25]. In the same year, a novel AF coating incorporating butanolide
373 into biodegradable poly(
ε-caprolactone)-based polyurethane was reported [
112]. Butenolide
373 can be released from the biodegradable polymer during at least three months and the rate of release depends on the concentration of the compound and the temperature. The addition of rosin into the formulation improved the late release of butenolide
373 [
112]. Moreover, in another study, the modified Boc-butenolide
374 was formulated into an antifouling paint using polymer matrix poly(
ε-caprolactone)-based polyurethane (PCL-PU), which is environmentally friendly and biodegradable [
113]. Interestingly, PCL-PU/Boc-butenolide revealed a large decrease in release rate related to PCL-PU/butenolide, which can be attributed relatively to the compatibility of Boc-butenolide in PCL-PU. In the marine field test, these Boc-butenolide coatings showed good performance with low coverage of biofouler after two months [
113]. Furthermore, anti-settlement bioassays against
A. amphitrite and
H. elagans larval suggested that Boc-butenolide has lower toxicity at high concentrations and similar AF activity than butanolide against macrofoulers, which indicates that Boc-butenolide could be a substitute in AF paints considering both antifouling effect and environmental impact [
113].
Combining different bioactive ligands/pharmacophores into a single molecule is a strategy currently employed in medical research where such multi-target-directed ligands are investigated as improved drug leads. The structural motifs of butenolide
371 were fused with geraniol
425 (
Figure 22) (two NPs with AF activity against the settlement of
A. amphitrite [
77]) to generate a library of AF hybrid molecules with potentially higher potency (
426–
433,
Figure 22) [
110]. This work represented an attempt to extrapolate the multi-target-directed ligands strategy into a marine setting. The major structural differences in these compounds are at the
A. amphitrite moiety and the oxidation degrees C-5 and C-12. All hybrid molecules
427–
433 showed AF activity and most of them with no toxicity, which suggested that the hybridization of the geraniol (
425) and butanolide (
426) structures led to the enhancement of the AF activity [
110].
A natural polyketide, 6-pentyl-2
H-pyrone-2-one (
434,
Figure 22), isolated from a marine-derived fungus,
Trichoderma atroviride, and its synthetic analogs (
435–
440,
Figure 22) were tested for potential antimicrobial activity, antibiofilm formation, and anti-settlement activity against barnacles [
111]. Compounds
434,
436,
439, and
440 were active against the settlement of
A. amphitrite (EC
50 = 8.82, 3.83, 4.32, and 4.48 µg·mL
−1, respectively) [
111]. Regarding antibacterial activity, all the tested compounds, except compound
438, were active against Gram-positive
Loktanella hongkongensis, while all except compound
440 were active against Gram-negative
Photobacterium angustum. Compound
439 was the most potent compound against
Staphylococcus cohnii and
L. hongkongensis (MIC
50 = 12.5 and 25.6 µg·mL
−1, respectively) [
111]. These results reveal that bulky groups, especially plane aromatic benzyl groups on both sides of the α-pyrone moiety, appeared to increase the antibacterial nature of these derivatives. The antibiofilm effects of compounds
434 and
435–
440 (25.6 and 6.4 µg·mL
−1) were studied using the same bacterial species. Compounds
435,
437, and
438 did not show any antibiofilm effects at both concentrations tested either against
L. hongkongensis and
S. cohnii. At 25.6 µg·mL
−1, natural compound
434 and compound
437 exhibited only weak antibiofilm activity, while compounds
439 and
440 showed a significant antibiofilm activity towards Gram-positive
L. hongkongensis [
111]. The benzyl group at the C-3-position of the α-pyrone moiety seemed to increase the antibiofilm activity of compounds
439 and
440 compared to
437 and
435, respectively, against
L. hongkongensis. Moreover, the presence of a methyl group at the C-3-position of the pyrone ring was detrimental to the antibiofilm activity against the same bacteria [
111]. Compound
439 was the only compound to show antibiofilm effects against
S. cohnii at both concentrations. The presence of a benzyl group at the C-3 and C-5-positions of the α-pyrone moiety appeared to improve the antibiofilm activity (compounds
434 and
439) against Gram-positive bacteria. Compound
439 had both macro and micro-AF-superior effects when compared with natural compound
434, and it is a good candidate for further field testing and marine industrial application [
111]. More recently, the same group confirmed the importance of a bulky or a suitable aliphatic chain (approximately 5 carbons) in C-5 and a bulky group in C-3, for a strong anti-settlement activity against
A. amphitrite barnacles [
114].
Scheme 11 evidences SAR for natural poliketide
434 and derivatives.
2.17. Peptides
Until 2015, innate immune peptides represented a class of bioactive compounds that was under evaluated in the marine environment. A strong effect on biofilm-forming bacteria such as
Staphylococcus epidermidis and
S. aureus was the inspiration for evaluating the AF effect of a series of short amphiphilic micropeptides (
478–
490,
Figure 25) [
118].
Most of the investigated peptides showed a high anti-settlement effect on
A. improvisus cyprids, with peptides
483 and
490 being the most potent compounds (IC
50 = 1.0 and 0.5 µg·mL
−1, respectively) [
118]. A clear link between the antibacterial activity, the settlement inhibition, and the hydrophobicity of the most active peptides was observed [
118]. Peptides
481,
483, and
487–
490 containing one or two artificial phenylalanine derivatives, mimicking arginine and lysine, were also the most active ones, suggesting that these two structurally similar synthetic amino acids have a significant role in the AF activity. A balance between charge and hydrophobicity is needed for AF activity and a high hydrophobicity is not enough, as depicted by dipeptide
478 (IC
50 > 5 µg·mL
−1), lacking a cationic residue [
118]. The AF activity observed for peptides
480,
487–
490 was reversible and cyprids displayed normal behavior after being placed in fresh seawater [
118]. Additionally, none of the peptides appeared to display any toxic effects on the cyprids within the tested concentration range, suggesting a reversible nontoxic AF mechanism. Peptides
480,
483, and
490 were the most active compounds towards the growth and adhesion of both marine bacteria (
H. aquamarine,
Polaribacter irgensii,
P. elyakovii,
R. litoralis,
S. putrefaciens,
Vibrio aestuarianus,
V. carchariae,
V. harveyi,
V. natriegens and
Vibrio proteolyticus) and microalgae (
Cylindrotheca closterium,
Exanthemachrysis gayraliae,
Halamphora coffeaeformis,
Pleurochrysis roscoffensis,
Porphyridium purpureum,
Hymenomonas coronate,
Rhodosorus marinus, and
Pleurochrysis carterae) [
118]. Overall, peptides
483 and
490 were the most promising displaying AF against both micro and macrofouling species.
Synoxazolidinones are a type of heterocyclic organic compound that contain an oxazolidinone ring, which is a five-membered ring with a nitrogen atom in the 3-position and a carbonyl group in the 4-position. Synoxazolidinones A (
491,
Figure 26) and C (
492,
Figure 26), isolated from the sub-artic ascidian
Synoicum pulmonaria, are novel bromotyrosine scaffolds with powerful compounds described in the literature as potent anti-microfoulers and anti-macrofoulers [
119]. These natural compounds were able to inhibit the settlement of the barnacle
A. improvisus (IC
50 = 15 and 2 µM, respectively) and the adhesion and the growth of marine bacteria and diatoms (MIC = 0.02–20 µM) [
119]. Remarkably, while antibarnacle activity of synoxazolidinones is similar to other promising marine antifouling agents, such as barettin (
534) and iantheline (
493,
Figure 26), the antibacterial and antialgal effects are superior [
120]. Based on these data, analogues of synoxazolidinone A (
491) were synthesized (
216–
219), employing the 2,5-diketopiperazine central core (
Section 2.19). Dipeptic derivatives of brominated synoxazolidinones represent a particular group of MNPs with broad AF activities [
120]. Ianthelline (
493) is a potent natural antifouling compound, which inhibits the settlement and metamorphosis of barnacle crypids (IC
50 = 6 μM) and shows antifouling effects in marine bacteria (MIC values of 0.1–10 μg·mL
−1) [
119]. Phidianidine A (
494) was isolated from the aeolid opisthobranch mollusk
Phidiana militaris and it is a structural analogue of ianthelline (
493), barettin (
534,
Figure 26), and the synoxazolidinones
491 and
492. The phidianidine A (
494) structure comprises an 1,2,4-oxodiazole ring linked to the brominated indole system and a guanidine moiety. Remarkably, many AF bromotyrosine derivatives have a cationic guanidine or guanidine-like group [
120]. A library of 10 synthetic analogs of phidianidine A (
494) was studied for the antifouling activity against
A. improvises cyprid and the inhibition results were compared with the positive control sea nine 211
® and potent MNPs (ianthelline, barettin, and synoxazolidinone A) [
119]. Compound
494 presented an IC
50 value of 4.0 μg·mL
−1, which is comparable to the reference molecules and it caused a 3% mortality of cyprids at a concentration of 5 μg·mL
−1, so this compound exerts its antifouling effect through a non-toxic mechanism at the employed concentration. Interestingly, synthetic analogs
502 and
503 demonstrated promising antifouling activity, with lower IC
50 values related to compound
494 at 2.2 and 0.7 μg·mL
−1, respectively [
119]. In this way, compound
503 was revealed to be the most effective inhibitor, with better antifouling performance than several robust antifouling MNPs. Additionally, compound
503 displayed low toxicity against the target organism [
119]. Compounds
494,
502, and
503, with longer alkane linkers, were more potent, the presence of a 1,2,4-oxadiazole ring was not essential (as compounds
502 and
503, without 1,2,4-oxadiazole ring, were more potent than compound
494), and the basicity of the cationic group seems to increase the AF activity. Important molecular features of synoxazolidinones and derivatives are highlighted in
Scheme 12. Compound
503 (
Figure 26) was selected to be incorporated in a biodegradable poly(
ε-caprolactone-co-
δ-valerolactone) polymer for AF applications [
119]. The anti-macrofouling activity of compound
503 was not possible to observe since none of the panels, including the control, had macroorganisms. Nonetheless, the biofilm formation was analyzed by confocal laser scanning microscopy and it was possible to observe a reduction in the biofilm mass in the panel containing compound
503 [
119]. Therefore, compound
503 was found to be the most potent anti-macrofouling compound of this series of synthetic derivatives of compound
494 and also showed anti-microfouling activity in the field tests [
119].
Dolastatin 16 (
504,
Figure 27) is a natural depsipeptide product obtained from the sea hare
Dolabella auricularia and contains two rare amino acids, dolafenvaline and dolamethylleuin, and was first reported by Pettit and his colleagues in 1997 [
121]. In 2010, Tan and co-workers demonstrated robust activity of dolastatin 16 (
504) toward the larval settlement and metamorphosis of barnacle
A. amphitrite with EC
50 and LC
50 values of 0.003 and 20 µg·mL
−1, respectively, being a promising lead compound for the development of novel AF materials [
122]. In 2017, it was reported that the AF activity of dolastatin 16 (
504) and two intermediates, northern carboxylic acid fragment
511 and southern amine fragment
506, showed the highly potent activity of
504 (EC
50 < 0.03 µg·mL
−1) and moderate to low activities of
511 and
506 (EC
50 > 10 and 1.17 µg·mL
−1, respectively) against barnacle
A. amphitrite [
121]. In 2022, derivatives of dolaphenvaline and dolamethylleuine, as well as some derivatives of compound
504 were synthesized and investigated to analyze their potential activity toward the development of a green AF material [
121]. The compounds evaluated (
504–
512) are shown in
Figure 27 [
121].
All samples showed AF profiles with low toxicity against cypris larvae of the barnacle
A. amphitrite [
121]. At the level of SAR, the introduction of a functional group on the aromatic ring of compound
504 reduced the AF activity to moderate (
505, EC
50 = 1.74 µg·mL
−1). Compounds
507,
508, and
512 were revealed to be more active with EC
50 values below 1 µg·mL
−1. Protection of the southern fragment with a Boc group enhanced the EC
50 value (
507, EC
50 = 0.79 µg·mL
−1), which can be attributed to its higher hydrophobicity than
506 (EC
50 = 1.17 µg·mL
−1) by the protection of the amino group. The functional groups at the
p-position of the aromatic ring affected the AF activity of the southern fragment due to steric bulkiness: a hydroxy group (
508, EC
50 = 0.60 µg·mL
−1) had a slightly decreased EC
50 value compared to
507, but a benzyloxy group (
509, EC
50 = 4.62 µg·mL
−1) abruptly reduced the AF activities to 4.62 µg·mL
−1. A benzyl ester of the northern fragment (
512, EC
50 = 0.90 µg·mL
−1) revealed much higher potency than
511 (EC
50 > 10 µg·mL
−1), suggesting the higher hydrophobic fragment was more active than the corresponding more polar one. A benzyl ether (
510, EC
50 = 3.27 µg·mL
−1) decreased the AF activity, which suggests the lactate moiety or the carbonyl group for the northern fragment are crucial factors [
121].
2.18. Phenyl Ether Derivatives
Natural-occurring biphenyl ethers were found to possess AF activity along with four synthetic biphenyl ethers (
513–
516,
Figure 28) against a panel of several fouling organisms, such as mussel, barnacle, diatom, and bacteria isolated from marine biofilms [
123]. All four synthetic compounds inhibited the attachment of the mussel
M. edulis, with derivative
514 being the most potent compound (EC
50 = 110 nM), and the growth and attachment of the benthic diatom
H. coffeaeformis, with compound
513 being the most potent (EC
50 = 7.74 µM). Regarding antibacterial activity, only diphenyl ether
514 did not inhibit the growth of
Bacillus sp. or
Zooshikella sp. Compound
513 demonstrated the strongest antibacterial activity (MIC = 0.76–1.90 µM). The presence of hydroxy in compound
513 may be influencing the marked AF activity. Nevertheless, compounds
513–
515 were found to be toxic to cyprids of
A. amphitrite [
123].
Phenyl ether derivatives
517–
522 (
Figure 28) were isolated from the fungus
Aspergillus sp. XS-20090066. The phenyl ether derivatives
523–
531 (
Figure 28) were synthesized by structure modification from diorcinol (
517) and 4-methoxycarbonyl-diorcinol (
519). All these compounds were tested for their AF activities against the cyprids of barnacle
A. amphitrite [
124]. Comparing the natural phenyl ethers
517–
522, 4-methoxyacyl-diorcinol (
519) showed the most robust AF activity (EC
50 = 7.43 μM), while others showed moderate activity (EC
50 = 18.2–57.3 μM). It is important to mention that compound
519 exhibited four times stronger activity than diorcinol (
517) (EC
50 = 32.6 μM). In a structural point of view, the ester group substitution at C-4 increases the activity. A hydroxy substitution at C-2, as in compound
518 (EC
50 = 57.3 μM), decreased the activity. Methoxy substituted at C-3 in compound
520 (EC
50 = 31.0 μM) did not show an impact on activity [
124]. The alkylated (compounds
523,
524, and
528) and acylated (compounds
525–
527,
531) synthetic phenyl ether derivatives revealed stronger AF activity (EC
50 = 2.23–27.9 μM) than the original compound
517. Concerning to the alkylated derivatives, compound
523 with a propionyloxy group substitution at C-3 had the most promising activity (EC
50 = 9.82 μM). Relative to the acylated products, a benzoyloxy substitution at C-3 (compound
526; EC
50 = 12.6 μM) increased the activity, but an acetoxy substitution (compound
525) (EC
50 = 3.05 μM) and a
p-bromobenzoyl substitution (compound
531; EC
50 = 2.42 μM) caused a significant increase in the activity. In this line, smaller acetoxy substitutions demonstrated to be better than larger benzoyloxy substitution at C-3 to improve the AF activity profile. Moreover, the introduction of a bromine atom could improve the activity (compound
531). Corroborating that, compounds
529 and
530 (EC
50 values of 0.71 μM and 1.17 μM, respectively) also showed robust anti-larval
A. amphitrite settlement [
124].
On one hand, the hydroxyl groups at C-3/C-3′ were not shown to be essential for the AF activity, and hydroxy substitution at C-2 might decrease AF activity. On the other hand, acylation at 3-OH and acetate substitution at C-4 could improve AF activity. Remarkably, bromine substituents could increase the AF activity [
124].
Scheme 13 summarizes the SAR for biphenyl ether derivatives.
From a toxicological view, derivative
523 showed toxicity with a therapeutic ratio of 11.4. In contrast, the phenyl ether derivatives
525,
527, and
529–
531, demonstrated low toxicity with LC
50/EC
50 ≥ 21.1. Interestingly, the polybrominated diphenyl ether derivative
529 showed the most favorable therapeutic ratio, with a value higher than 31.0. In this way, compound
529 was considered a promising candidate for environmentally friendly AF agents [
124].
In another study, phenyl ether derivatives of capsaicin were (
532 and
533,
Figure 28) shown to be able to inhibit algal growth of
P. tricotornum,
S. costatum, and
Chaetoceros curvisetus [
125]. It was demonstrated that the capsaicin synthetic derivatives were excellent algaecides and AF agents. Specifically, compound
533 revealed better AF performance compared to compound
532. Regarding the mechanism of action, the derivatives interfere with the permeability and structure of the algal cell membrane. Moreover, the marine field tests showed that the addition of compounds
532 and
533 as environmentally friendly auxiliaries to coatings could be a promising strategy to achieve long-term marine fouling resistance [
125].
2.19. Piperazines
2,5-Diketopiperazines derivatives are a class of naturally occurring privileged structures from fungi, bacteria, the plant kingdom, and mammals [
126]. Barettin (
534,
Figure 29), a diketopiperazine isolated from the marine sponge
Geodia barretti, is a potent AF agent, and its effect on barnacles can be attributed to the 2,5-diketopiperazine core and an exocyclic double bond to the brominated indole [
120]. To increase the knowledge into the SAR of barettin (
534) a series of 14 analogs (
535–
548,
Figure 29) was obtained, and their activity was evaluated against the larval settlement of
A. improvises [
127].
Slight modifications in the structure of these analogs had a high impact on their ability to inhibit the settlement of the larvae. The authors focused on the modifications of the position of bromine in the indole residue of tryptophan. When the bromine was changed from the 6-position to the 5-position (
535) stimulation of the settlement of
A. improvisus was observed (EC
50 = 4.1 µM), while the removal of the bromine at the 6-position resulted in a complete loss of activity (
536) [
127]. Regarding dipodazine derivatives, once again, the presence of bromine in the 6-position (
545 and
547) resulted in significant inhibition of the larval settlement of
A. improvisus (EC
50 = 2.4 and 6.7 µM, respectively). However, in dipodazine (
537) the insertion of methyl in the 5-position (
542) did not lead to the stimulatory effect of 5-bromobarettin (
535). Compounds with methoxyl and nitro groups in the dipodazine scaffold (
539 and
540) did not show any settlement inhibition. Substitution of the bromine for chlorine resulted in the loss of activity (
541), suggesting that the atom size in that position is critical for the activity. Compound
548 was the most potent compound against the settlement of barnacle larvae (EC
50 = 0.034 µM) [
127]. This result could indicate that the highly non-polar phenyl in the 6,7-position of benzo[
g]dipodazine present in compound
548 is electronically similar to the bromine in the 6-position present in barettin (
534). The position of the phenyl ring in compounds
543 and
548 influenced the AF activity displayed by these compounds, being that compound
548 is more active than compound
543. When the phenyl was substituted for a methyl group (
542), no activity was observed. The stereochemistry of the double bond of compounds
545 and
546 seems to play an important role in the inhibition of the settlement of barnacle larvae since the isomer
E (
545, EC
50 = 2.4 µM) was active, while the isomer
Z (
546) was inactive [
127].
A small library of synthetic analogues of synoxazolidinone A (compound
491,
Section 2.17) employing the 2,5-diketopiperazine central core found in barettin (
534), as a replacement for the synthetically more challenging 4-oxazolidione core, was prepared based on the high activities of synoxazolidinone A (compound
491) and synoxazolidinone C (compound
492) [
120]. Comparing the natural compounds (
491 and
492) with the synthetic analogues (
549–
552), it is possible to infer that the effect on microfouling can be maintained using a 2,5-diketopiperazine scaffold [
120]. However, the effects on microalgae are lower for the derivatives and the elimination of bromine led to the almost inactive compounds (
551 and
552) against microalgal growth and adhesion [
120]. Interestingly, none of the synthetic compounds were able to interfere with
A. improvisus larvae, suggesting that a certain degree of structural integrity is required for macro AF activity. In this way, the use of a saturated 2,5-diketopiperazine scaffold is not a crucial requirement for active macro AF activity, and the presence of a double bond is essential for high inhibitory activity toward barnacles and microfouling species [
120].
A small library of soluble 2,5-diketopiperazine derivatives was synthesized and evaluated against
A. amphitrite larval attachment [
128]. From the first series of compounds (
553–
560,
Figure 29), only compound
560 (EC
50 = 20.5 µg·mL
−1) showed AF effects, which indicate that 2-OCH
3 might be contributing to the AF effects. For this reason, compound
560 was selected as a template molecule for the synthesis of other 2,5-diketopiperazine derivatives (
561–
573). The new series showed better AF activity, with compound
564 exhibiting the most potent activity against
A. amphitrite (IC
50 = 1.6 µg·mL
−1) with low or no toxicity (LC
50 = 25 µg·mL
−1) and could be a new template molecule for further development as environmentally friendly AF agent [
128].
Piperamide is a substituted piperazine with a tertiary amine group and was used as an anthelmintic agent due to its effectiveness against
Trypanossoma [
129]. The AF activity of an acetone extract of the terrestrial plant
Piper betle was evaluated and four structurally similar AF piperamides were isolated (
574–
577,
Figure 30) [
130]. Following this, 15 piperamide analogs (
578–
592,
Figure 30) were synthesized and their AF activity was compared to determine SAR among this class of compounds [
130]. The effects of natural piperamides
574–
577 in the larval settlement and toxicity were assessed using cyprids of the barnacle
A. amphitrite, larvae of the polychaete
H. elegans, and larvae of the bryozoan
B. neritina. The AF activity of piperamide analogs containing modified side chains was evaluated against the settlement of the barnacle
A. amphitrite. Piperidine amides
574,
579,
584, and
589 exhibited the highest anti-settlement activity (EC
50 = 2.3, 1.3, 0.5, and 1.5 µg·mL
−1, respectively), indicating that the piperidine group was the optimal group at the
N-terminal [
130]. Increased AF activity might be related to the lipophilicity of the amine moieties, as compounds with the piperazine, morpholine, or the
N-isobutyl groups had significantly weaker AF activity compared to compounds containing the piperidine or the pyrrolidine groups. Among piperamide analogs with the piperidine group, compound
584, containing a C
7 alkyl chain, revealed the strongest AF activity, showing that the length of the alkyl chain influences AF activity. The introduction of the double bonds at the C-2 and C-4-positions and reduction in the double bonds at the C-6-positions decreased AF activity, as seen in compound
576 (EC
50 = 4.2 µg·mL
−1) [
130].
Scheme 14 summarizes the SAR for piperamides. Toxicity against zebrafish embryos was investigated for the two most active compounds, piperamides
574 and
584, and no effect was observed [
130].
2.20. Polyphenol Derivatives
Capsaicin (
593,
Figure 31), 8-methyl-
N-vanillyl-6-nonenamide, is isolated from
Capsicum sp., and it exhibits promising AF activity [
131]. Capsaicin became an important lead compound in the research of novel AF compounds. Capsaicin (
593) and two other commercially available compounds with one or more capsaicin-like structural features (
593–
595,
Figure 31) were tested against the settlement of zebra mussel (
D. polymorpha). Lethal effects were investigated for the most promising compounds against the non-target organism
D. magna [
131]. Significant AF activity was observed for compounds
593,
594, and
595 (EC
50 = 13.7, 17.7, and 25.7 µM, respectively). From the three active compounds, compound
594 was the most promising, as it was able to inhibit zebra mussel byssal attachment at concentrations that had no significant lethal effects on this species or the non-target species
D. magna [
131].
In another study, six capsaicin derivatives were synthesized to obtain environmentally friendly marine AF agents, two of which are diphenyl ethers (
532 and
533—
Figure 28,
Section 2.18 and
596–
599—
Figure 31). Structural analogs of capsaicin were decorated with distinct functional groups, particularly, phenyl, phenolic hydroxyl, amide, and alkeny groups [
125]. Capsaicin derivatives were shown to be able to inhibit algal growth of
P. tricotornum,
S.costatum and
C. curvisetus [
125]. The inhibition order of the six compounds on algae was compounds
599 >
598 >
596 >
597. Compound
599 revealed the best inhibition effect, with an inhibition rate of approximately 95% at 3 mg·L
−1 [
125].
Regarding toxicity evaluation, most compounds showed low toxicity to algae and EC
50 values (72 h) were lower than 250 mg·L
−1. The toxicity of compound
599 to
S. costatum was intermediate (0.3–3 mg·L
−1, and all other compounds demonstrated low toxicities (more than 3 mg·L
−1). Remarkably, compounds
596 and
597 have no significant toxicity to
P. triconurtum and
S. costatum (150 mg·L
−1). In this way, the toxicity of compounds
598 and
599 was greater than compounds
596 and
597 [
125].
In another study eight capsaicin derivatives (
600–
607,
Figure 31), decorated with distinct amide groups on the benzene ring, were evaluated for the antibacterial, anti-algal, and AF activities [
132]. To evaluate the AF performance of capsaicin derivatives, its antibacterial activity against
E.coli and
S. aureus, as well as the anti-algal effect and toxicity against
N. closterium and
Chlorella vulgaris, were determined [
132]. The capsaicin derivatives showed robust antibacterial effects against
E.coli and
S. aureus, with a small number of colonies on the test board compared to the control board. From a structural point of view, amide groups can impact the electron transport system of the bacterial respiratory chain, leading to their death. Moreover, compounds with phenol groups,
600,
601,
602, and
603 showed better antibacterial activity than compounds
604,
605,
606, and
607, which revealed that phenol groups are a key to antibacterial activity. Compounds
604,
606, and
607 showed better antibacterial activity compared to compound
605, which suggests that antibacterial activity can be directly related with the length of the side chain [
132]. Overall, the benzene ring and amide group are crucial for antibacterial activity and the phenolic hydroxy group is the preferred group. The similar inhibitory effects of capsaicin derivatives are probably related to damaging effects on the cell membranes of
E. coli and
S. aureus. The anti-algal activity of capsaicin derivatives increased with the enhancement of time and concentration. The compounds were shown to be environmentally friendly anti-algal agents, revealing much less toxicity compared with the currently used antifoulants, such as TBT and sea nine 211
® (
Figure 1) [
132]. Compounds
603 and
602 exhibited excellent anti-algal activity (I > 77.30% against
N. closterium and I > 82.50% against
C. vulgaris), compared with compounds
600 and
601. Compounds
607 and
606 revealed more promising anti-algal activity compared with compounds
604 and
605. Moreover, compounds
600,
601,
602, and
603 showed higher anti-algal activity than
604,
605,
606, and
607 [
132]. From a structural point of view, these results allow us to conclude that the presence of benzene rings and amide groups are crucial for anti-algal activity and phenolic groups and chlorine atoms are important to improve the anti-algal activity. In addition, the increase in the number of benzene groups improves the anti-algal activity. In situ AF assay was carried out for six months to evaluate the AF effects of the eight capsaicin derivatives (
600–
607) as AF adjuvants by incorporating them in coatings containing zinc acrylic resin, Cu
2O, rosin resin, and other auxiliaries, and is then applicated on PVC panels. The control plate had many microorganisms and algae attached, while the test plates only presented a small number of microorganisms adhered [
132]. Remarkably, capsaicin derivatives reveal excellent AF performance in the marine environment, eco-compatibility, and slow release rates as AF agents compared with the natural molecule capsaicin, which is easily soluble in water and shows a less promising AF effect and durability in seawater [
132]. In a following work, benzamide derivatives containing capsaicin (BDCC;
608–
612;
Figure 31) were synthesized, being the aromatic moieties the methyl gallate (
608), ethyl gallate (
609), propyl gallate (
610), 1,5-dihydroxynaphthalene (
611), and α-naphthol (
612) [
133]. BDCC showed antimicrobial activity against
S. aureus and
E. coli. Remarkably, compounds
608,
609, and
610 revealed better antimicrobial effects than compounds
611 and
612. Interestingly, all the compounds revealed better inhibitory effects on
E. coli than those on
S. aureus. This can be attributed to the abundance of peptidoglycan layers in cell walls, which is higher in
S. aureus, which makes the permeation of benzamide derivatives difficult. From a structural point of view, the presence of benzene rings, phenolic hydroxyl groups, ester groups and amide groups was demonstrated to be related to good antimicrobial activity. Moreover, the substitution of benzamide groups on aromatic compounds increased the antimicrobial activities of aromatic compounds. In addition, phenolic hydroxyl groups were shown to be associated with the antimicrobial activities of benzamide derivatives. In this line of thinking, the charge differences between H and O atoms in the phenolic hydroxyl groups of compounds
609,
610,
608,
611, and
612 decreased in turn, which is closely related to antimicrobial activities of compounds, explained by the reaction with protein through hydrogen bonds and the formation of precipitates [
133]. BDCC compounds (
608–
612) were incorporated in the same previously used coating formulation [
132,
133]. The AF assay was carried out for 180 days and it was verified that only a small amount of microorganisms and a small number of barnacle larvae were attached to test plates containing capsaicin derivatives, compared to control plate [
133]. Thus, the results suggest that the AF coating formulation shows a good repellent effect on microorganisms and the good hardness and adhesion of AF coating [
132,
133]. Specifically, the more stable compounds
608–
610 showed the most promising AF activities after 6 months [
133]. The AF effect of
611 was better than
612, which can be attributed to its greater polarity and, consequently, its higher solubility, which allowed a more uniform and effective AF coating [
133]. Due to the combination of BDCC with Cu
2O, and zinc acrylic resin, benzamide derivatives performed optimal AF properties. Moreover, these compounds presented uniform distribution, good thermal stability, and high surface tension [
133].
Concerning the mechanism of action, it was shown that the antibacterial activity of capsaicin can be attributed to its ability to change the cytomembrane fluidity, which led to the disruption of cell membranes [
125]. Moreover, it was also demonstrated that capsaicin derivatives inhibit algal growth by increasing the membrane permeability, without causing cell death, proving their potential as environmental friendly AF agents [
125]. Remarkably, the AF mechanism of capsaicin on algal species was related to cytosolic Ca
2+. Particularly, the increase in intracellular Ca
2+ caused the unicellular green alga
Chlamydomonas reinhardtii to lose flagella (deflagellation), loss of exercise ability, and other cell function disorders, and the same inhibition effect was confirmed among five species in
Chlorophyta sp. Moreover, it is speculated that the AF mechanism of capsaicin on
Ulva sp. may be due to the rapid increase in intracellular Ca
2+ as the result of the activation of the transient receptor potential vanilloid 1 (TRPV1) channel by capsaicin, making the spores hard to settle, or other functional disorders [
135].
To improve AF performance of phloroglucinol (
613,
Figure 31) and pyrogallol (
614,
Figure 31), eight polyphenol derivatives (
615–
622,
Figure 31) were synthesized with the purpose to improve the AF activity, being that the antibacterial activity of the derivatives was much higher than the original polyphenols [
134]. The polyphenol derivatives containing phenolic hydroxyl and amide groups demonstrated similar antibacterial activities due to their similar structures. Compounds
617 and
621, containing chlorine atoms, showed the most effective antibacterial activity, indicating that the addition of chlorine atoms could improve the antibacterial activity [
134].
The effects of polyphenol derivatives on the growth of
C. vulgaris and
N. closterium were evaluated by an algal assay. Compounds
617,
619,
620,
621, and
622 revealed superior inhibitory effects with inhibition rates higher than 93% for
C. vulgaris at 10 days. For
N. closterium, their inhibition rates were higher than 57% and the anti-algal activity followed the ascending order
622,
619,
620,
617, and
621. Compounds
615,
616, and
618 did not show promising anti-algal activity. From a structural point of view, compounds with chloroacetamide groups, such as
617 and
621, revealed better anti-algal activity for both species than compounds with acrylamide, acetamide, and benzamide groups [
134]. The inhibition rates of compounds
614,
619,
620,
621, and
622 were higher than
613,
615,
616,
617, and
618, respectively. Suggesting the importance of chlorine atoms in algal inhibition activity, EC
50 values of compounds
619,
620, and
622 were significantly higher than
617 and
621 [
134].
These polyphenol derivatives (
615–
622) were added to coatings containing acrylic resin, Cu
2O, rosin resin, and other auxiliaries and applicated in PVC panels. The test panels were submerged on seawater for six months. After three months, the control panel was covered with macroorganisms, test panels with polyphenol derivatives showed limited biofilm formation and panels with compounds
616,
617, and
620 showed a few settled barnacles. After six months, only biofilm formation and a few settled barnacles were observed in test plates. It is important to note that panels with compounds
618 and
621 were clear of macrofouling, instead of control panels, which were covered with macroalgae and barnacles [
134]. Overall, some polyphenol derivatives revealed potential for the development of environmentally friendly AF applications and show high antibacterial, anti-algal, and AF activity. The addition of chlorine and amide groups revealed to be favorable to increase the activity of these derivatives [
134]. Concerning the mechanism of action, polyphenol derivatives with more hydroxyl groups that show tannic properties can cause protein coagulation, resulting in protein denaturation, protein precipitation, and inactivation of the enzyme system [
134]. A series of six gallic acid derivatives (
623–
629,
Figure 31) containing different amine groups was also synthesized to increase potency and decrease water solubility while maintaining low toxicity of a non-toxic water soluble nature-inspired AF compound, gallic acid persulfate (compound
623), against
M. galloprovincialis larvae (EC
50 = 17.65 µM; LC
50/EC
50 = 26.61) [
36]. The lead optimization strategy comprised the introduction of amine/amide groups, triazole ring, and halogen substituents, described as molecular features correlated with AF activity. In the presence of compounds
624 and
629, larvae settlement decreased to <35%, and these compounds revealed promising AF activity against
M. galloprovincialis larvae related to the lead compound, with EC
50 values of 2.74 µM and 16.28 µM, respectively [
36]. Remarkably, removing the sulfate groups and maintaining the hydroxy groups, as well as the introduction of a carbon chain with a primary amine, led to the most potent derivative, compound
624, with a seven-fold higher potency than gallic acid persulfate, and did not cause mortality to the target species even at concentration 73-fold higher than the EC
50 [
36].
Scheme 15 summarizes the SAR for polyphenol derivatives.
Regarding the mechanism of action, the activity of enzymes AChE and tyrosinase was not significantly impacted after exposure to the most promising compounds (
624, and
629) [
36]. In the ecotoxicological evaluation, it was found that compound
624 is non-toxic to
A. salina (<10% mortality at concentrations of 25 and 50 µM), similarly to gallic acid persulfate. However, in contrast to the lead compound
623, compound
624 was classified as non-toxic for the marine diatom
P. tricornutum. Moreover, amide derivative
624 reveals a reduced value of Log octanol-water partition coefficient, which suggests its low potential for bioacummulation in marine organisms [
36].
Due to its AF performance, compound
624 (
Figure 31) was incorporated as an additive in a PU-based marine paint composed of a base resin and a curing agent to evaluate its viability as an AF agent in commercial marine coatings. Compound
624 demonstrated good compatibility with the PU-based coating, revealing a better behavior than its precursor gallic acid persulfate (
623,
Figure 31). The results show that compound
624 was effective in decreasing the fixation of mussel larvae, presenting a significant difference compared with free PU-based coating after 40 h of exposure. Concerning the leaching of compound
624, a value lower than 10% of this compound was detected in artificial seawater after 45 days [
36].
Moreover, to increase the service life of these coatings, further coating optimization was performed through chemical immobilization using the trimethylolpropane triaziridine propionate (TZA) crosslinker (CL) [
136]. In the antibiofilm assay in
P. tunicata, the biofilm biovolume was significantly lower for the new PU-based marine coating containing 2% (
w/
w)
624 and the CL (
624/PU/CL) surface when compared to coating without CL. Remarkably,
624/PU/CL provided the best long-term performance. Moreover, in
624/PU/CL coating, the number of cells only started to increase from day 28 and the number of biofilm cells was lower compared with
624 PU-based coatings, which suggests the compatibility of the compound
624 in this polymer matrix and the service life of the generated matrix were improved due to TZA [
136].
2.22. Pyridinium Salts
3-Alkylpyridinium (3-AP) compounds occur mainly in marine sponges. More than 70 structurally distinct compounds were isolated from marine sponges belonging to the order Haplosclerida. Most isolated 3-AP compounds are monomeric structures differing in length, saturation, branching, and termination of the alkyl chains. These molecules exhibit valuable potential as novel pharmaceuticals with a large spectrum of applications such as oligonucleotide and/or gene therapy, treatment of several diseases due to various biological activities, and they can be used as active compounds in AF paints [
139].
From 2003 to 2014, 19 synthetic pyridinium salts with AF activity were reported in the literature [
140,
141,
142]. Polymeric 3-alkylpyridinium salts (poly-APS,
634,
Figure 33) are a natural mixture of compounds isolated from the Mediterranean sponge
Reniera sarai (Haliclonidae) that displayed potent AF activity, both in terms of settlement inhibition of
A. amphitrite larvae and inhibition of natural marine biofilm formation [
143]. The AF efficacy of poly-APS (
634) was similar to that of CuSO
4, with the advantage of having a non-toxic mechanism towards the barnacle larvae [
143].
Synthetic 3-AP compounds with a defined number of subunits were prepared to understand the relationship between structural features and AF properties. Mono-, di-, and tetrameric 3-octylpyridinium analogs (
635–
642,
Figure 33) were obtained and the AF activity was tested against the settlement of the larvae of
A. amphitrite to understand the SAR of the natural poly-APS (
634) [
140].
The absence of AF activity of compound
635 and the corresponding pyridinium salt (not shown), when compared with compounds bearing an octyl chain at position 3 of the pyridine unit (
636–
640 and
642), highlighted the importance of the alkyl chain. A significant effect of the length of the alkyl chain was observed by direct comparison of
637 (EC
50 = 98.26 µM) with a compound with a shorter alkyl chain that did not display any AF activity (structure not shown). The presence of a tetrahydropyranyl group increased the settlement inhibition of compound
636 (EC
50 = 8.07 µM) when compared with compounds
637 and
638 (EC
50 = 98.26 and 58.9 µM, respectively). The presence of a positive charge in compound
639 (EC
50 = 73.40 µM), a compound also bearing a tetrahydropyranyl group, led to a decrease in the settlement inhibition when compared with compound
636 (EC
50 = 8.07 µM). The potent AF activity observed for compound
642 (EC
50 = 1.64 µM), higher than the natural compound
634, confirms the SAR found for compounds
635–
641. In contrast to
634, a therapeutic ratio (LC
50/EC
50) of 8.1 was found for compound
642, which might suggest that cyprid settlement is inhibited by a toxic mechanism [
140].
Poly-APS (
634) and synthetic analogs
635–
642 were also described to have antibacterial activity against a panel of 24 bacterial strains, including marine isolates associated with immersed surfaces [
144]. Compounds
635–
638 and
641 did not display significant antibacterial activity. In general, the increased antibacterial activity was correlated to the increasing number of pyridinium rings, which is directly related to the molecular weight and the presence of positive charges of the tested compounds. When the bromine of compound
640 was substituted by a tetrahydropyranyl group (compound
639), a loss in antimicrobial activity was observed [
144].
The AF potential of four compounds containing one or more 3-AP moieties (
643–
646,
Figure 33) was investigated in both anti-settlement activities against cyprids and toxicity against nauplii of the barnacle
A. amphitrite [
141]. Compounds
643 and
644 were isolated from an Antarctic sponge of the genus
Haliclona sp., while compounds
645 and
646 were obtained by synthesis. All compounds showed promising activity and generally low toxicity. Compound
643 (EC
50 = 0.28 µg·mL
−1) was almost as active as the natural Poly-APS
634 (EC
50 = 0.19 µg·mL
−1) [
141].
Six synthetic polymer analogs of poly-APS (
647–
652,
Figure 33) were synthesized, and their AF activity was explored against the settlement of the cyprids of
A. amphitrite [
142]. From the synthetic poly-APS
647–
652, compound
649 (EC
50 = 0.026 µM) inhibited the settlement of cyprids most effectively. No significant toxicity towards
A. amphitrite nauplii was observed, and a therapeutic ratio higher than the natural Poly-APS
634 was found for compound
649 (LC
50/EC
50 = 111 and 158, respectively), which suggests an even lower environmental risk. In contrast, compound
652, the second most potent synthetic polymer (EC
50 = 0.146 µM), exhibited toxicity towards
A. amphitrite nauplii. No relation between the AF activity and the length of the alkyl chains, the degree of polymerization, and the nature of their counter ions (chloride or bromide) of synthetic poly-APS was found [
142].
In contrast to synthetic polymer analogs of poly-APS (
647–
652), synthetic 3-alkylpyridinium compounds (
635–
646) possess much simpler structures, and given the potential application of this class of compounds as AF agents, larger amounts of these molecules are expected to be easily obtained. Regarding the mechanism of action, 3-APS show surfactant properties that enables the disruption or solubilization of the fouling organisms’ cell membrane. Moreover, 3-APS were shown to inhibit AChE activity, acting as cholinergic antagonists, blocking neurotransmisson and decreasing the ability of settlement of fouling organisms [
46].
2.24. Sesquiterpenes
Polygodial (
684,
Figure 35), isolated from marine sponges and nudribranchs, is a drimane sesquiterpene that acts as a nonionic surfactant to disrupt the cell membrane of microorganisms [
146,
147]. This compound shows powerful AF properties against the marine macrofoulers
C. savignyi (EC
50 = 3.4 ng·mL
−1),
Spirobranchus caraniferus (EC
50 = 5.2 ng·mL
−1), and
M. galloprovincialis (EC
50 = 2.9 ng·mL
−1) [
148]. Furthermore, compound
684 was shown to display low toxicity toward higher organisms [
149,
150]. A library of 11 polygodial analogs was prepared by semi-synthesis, intending to investigate the SAR of the drimane scaffold (
685–
695,
Figure 35) [
147]. All compounds share the bicyclic drimane, with the differences lying mainly within the degree of substitution and oxidation [
147]. This compound class was only weakly active against microfoulers. In contrast, compound
684 and its synthetic analogs (
685–
695) showed powerful AF activities against macrofouling species: the ascidian
C. savignyi (EC
50 = 0.004–1.0 µg·mL
−1) and the barnacle
A. improvisus (EC
50 = 0.1–1.5 µg·mL
−1). Nevertheless, none of the synthetic analogs were more potent than the natural compound
684 [
147]. The fact that the diterpenoid reference compound sclareol (
Figure 35) was also active, while 1,5-decalindol (
Figure 35) did not display significant inhibition at the concentrations tested, suggests that the drimane scaffold (
Figure 35) may be essential to generate the AF activity [
147]. The observed bioactivity profile of the drimane-type compounds suggested that the mode of action is probably dictated by differing mechanisms depending on target species, rather than a general biocidal effect. General surfactant properties appear partly responsible for observed activity against macrofoulers, but there was evidence that specific molecular targets also exist; elucidating these targets should be a priority for future research in this area [
147].
Regarding the mechanism of actions, it deserves to be mentioned that it is speculated that polygodial can interfere with heat shock protein 90 (HSP-90), which can lead to the inhibition of larval metamorphosis. In concrete, polygodial showed inhibit larval metamorphosis in ascidian
C. savignyi larvae with IC
99 (99% inhibition concentration) of 0.003 µg·mL
−1 [
151].
A SAR study of the tricyclic sesquiterpene subergorgic acid (
696) was performed firstly by isolating the compound from the gorgonian coral
S. suberosa and then by the synthesis of diverse derivatives (
697–
722,
Figure 36) [
152]. The good AF activity in addition to the high abundancy in
S. suberosa makes compound
696 (EC
50 = 1.25 µg·mL
−1) a good lead compound to perform SAR studies aiming to identify more potent AF compounds. Compounds
696–
722 were tested against the settlement of the barnacle
A. amphitrite. Esterification of subgorgic acid (
696) led to derivative
697 (EC
50 = 3.16 µg·mL
−1), which maintained most of the AF activity, suggesting that the acid group is not essential for the AF activity. Surprisingly, both the ketone carbonyl and the double bond groups were essential for the AF effect, as seen for the loss of AF activity of compounds
699 and
700 (EC
50 > 25 µg·mL
−1 for both compounds) [
152]. Following this, two series of subgorgic acid (
696) derivatives, one containing benzyl functional groups (
701–
710) and the other involving the methylene chain of various lengths (
711–
722), were obtained to develop a more detailed SAR [
152]. All benzyl esters exhibited good AF effects against the settlement of
A. amphitrite. The most potent compound (
701, EC
50 = 0.30 µg·mL
−1) containing a non-substituted benzyl group showed that substituents around the benzene ring decreased the AF activity as well as steric effects [
152]. The position of the substituent on the benzene ring might influence the AF effect, with substituents in the ortho or meta position being more favorable than substituents in the
para’position. Bromoalkane derivatives
711–
716 of various lengths did not exhibit better or comparable AF activity when compared with subgorgic acid (
696), and the greater the length, the lower the AF activity. For the series of aminoalkanes derivatives of subgorgic acid with various lengths (
717–
722), all the compounds were active, although less active than subgorgic acid (
696) [
152]. Interestingly, the opposite trend of compounds
711–
716 was found for compounds
717–
722 and the compound with the longest methylene chain, derivative
722 (EC
50 = 4.5 µg·mL
−1), showed the best AF activity. The differences observed were attributed to the fact that the presence of -NH
2 is better compared to -Br [
152].
Scheme 16 summarizes the SAR for subergonic acid and derivatives.
2.26. Structure-Diverse Sulfated Derivatives
Sulfation is used in nature as a metabolic strategy to avoid toxicity, and based on this consideration, a library of 13 sulfated NPs derivatives (
745–
756,
Figure 38) was developed and investigated for AF activities [
65]. Compounds
746,
750, and
623 showed a significant inhibitory effects against the settlement of
M. galloprovincialis larvae with EC
50 values of 22.59, 23.19, and 17.65 µM, respectively, without toxicity against this target organism. To find some clues about their mechanism of action, the activities of AChE and tyrosinase in the presence of compounds
746,
750, and
623 were studied, although none of them were able to inhibit these two enzymes implicated in the adhesion process of mussels [
65]. No antibacterial activity against the growth of biofilm-forming marine bacteria (
C. marina,
V. harveyi,
P. atlantica, and
H. aquamarina) was observed for compounds
745–
756. Two ecotoxicity assays were performed to gain insights about the potential toxicity of the compounds against non-target organisms, specifically,
A. salina and
Vibrio fischeri. None of the compounds showed toxicity in the non-target organisms [
65]. It is important to notice that none of the non-sulfated compounds showed AF activity, suggesting that sulfate groups are responsible for the AF activity displayed by these compounds. Some SAR could be observed for this library of sulfated compounds, namely, the nature of the scaffold (compound
750, which showed some activity compared to compound
751, a triazole-linked xanthone with no activity). For the flavonoids
745–
748, sulfate groups at positions 5, 7, 3′, and 4′ were important to the slight anti-settlement activity displayed by compound
746 (which was only active at the highest concentration tested). Additionally, the degree of acidity of the carboxylic group seems to influence the AF activity, when comparing compound
623 with an arylcarboxy group and compound
756 with an alkylcarboxy group [
65]. Compound
623 was selected for further studies concerning its immobilization in polymeric coatings [
155]. Additionally, the synthetic method to obtain compound
623 was optimized, increasing its scale-up potential, an important aspect when considering the development of a new AF agent [
155]. Compound
623 was directly incorporated and chemically immobilized in two representative PDMS and PU-based marine coatings and two commercial acrylic and room-temperature-vulcanizing polydimethylsiloxane (RTV-PDMS)-based non-marine coatings. Chemical immobilization consisted of the pre-functionalization of compound
623 with TZA [
155]. Leaching studies proved that the formulation with pre-functionalized compound
623 released less amounts of compound
623, compared with the formulation containing compound
623 directly incorporated, in both PDMS and PU-based coatings. PU, acrylic, and RTV-PDMS-based coatings formulated with pre-functionalized compound
623 maintained their AF activity against the settlement of
M. galloprovincialis larvae [
155]. These results indicate that compound
623 has great potential to be developed as an active ingredient in AF paints.
2.27. Xanthones
Xanthone is a heterocyclic molecule, with a dibenzo-
γ-pirone scaffold, known as 9
H-xanthen-9-one. The designation of rings A and B is congruent with the biosynthetic pathways for the compounds from higher plants, A-ring (carbons 1–4) being acetate-derived and the shikimic acid pathway gives B-ring (carbons 5–8); the other carbon atoms are numbered according to IUPAC [
156]. The first report of a synthetic xanthone as an AF agent was described in the previous structure-diverse sulfated derivatives section, compound
750 [
155]. Following this, a library of 19 synthetic xanthones (
757–
775,
Figure 39) was investigated for their AF potential, specifically through the anti-settlement activity toward the macrofouling species
M. galloprovincialis, the study of the potential modulation of AChE and tyrosinase, enzymes with a role in adhesive processes, and by the differential analysis of the proteome of
M. galloprovincialis plantigrade larvae after exposure [
157]. Additionally, QSAR studies were performed to understand the structural requirements to achieve AF activity. General marine ecotoxicity against a non-target organism,
A. salina, was also studied [
157].
Xanthones
760,
761,
763,
766,
771, and
773 were able to inhibit the settlement of
M. galloprovincialis larvae (EC
50 = 3.53–28.60 µM) without exerting toxicity against this target species (LC
50 > 500 µM and LC
50/EC
50 = 17.48–141.64). The QSAR model developed revealed that the complexity of the molecule, the electronic charge distribution, and inter-atomic distances influence their AF activity [
157]. Within this series, compounds
763,
766, and
773 were the most potent by inhibiting mussel larvae settlement with EC
50 values of 11.53 µM, 4.60 µM, and 3.53 µM, respectively. It deserves to be highlighted that while compound
761,
763,
766,
771, and
773 showed some toxic effects (mortality > 10%), at 50 and 25 µM to the nauplii of
A. salina, xanthones
763 and
766 demonstrated no toxicity to this non-target species, which makes these two compounds better candidates for their inclusion in AF coatings [
157].
Proteomic studies were realized to provide molecular insights on the AF properties of selected compounds. The proteome of competent
M. galloprovincialis plantigrade larvae was analyzed in response to the three most potent compounds (
763,
766, and
773) [
157]. Compound
763 showed an impact on the expression of proteins involved in cytoskeleton formation, cell-redox status, and chaperone-mediated regulation. Moreover, compound
763 may be related to the alteration in the abundance mussel collagen proteins (PreCols), specific to the byssal threads, that play an important role in elasticity, resistance to tension, and shock absorption [
157]. Remarkably, the inhibition of larvae adhesion of compound
766 may be explained by the abrupt decrease of two putative proximal thread matrix proteins (TMPs), proteins that are localized in byssal threads to provide them viscoelasticity [
157]. Compound
773 caused an alteration in the abundance of five proteins which are involved in major metabolic processes such as glycolysis/gluconeogenesis, gene translation, lysosomal protein degradation, or are constituents of the cytoskeleton [
157].
In what concerns SAR, it was later shown that while the presence of hydroxyl groups at positions 3 and 4 was not crucial for activity, the presence of 3 and 4 oxygenated groups were important for the antisettlement activity on mussel larvae [
158].
The xanthone derivative (3,4-dihydroxyxanthone)
763 (
Figure 39) was immobilized in representative coating components of marine PU-based coatings using a 4,4 diphenyl diisocyanate-monomeric (MDI) crosslinker, allowing its chemical binding with the polymeric matrix of the coating. The water solubility of compound
763 is similar to both booster biocides irgarol 1051
® (6.0–7.0 mg·L
−1) and sea nine 211
® (4.7–6.5 mg·L
−1), demonstrating to be a suitable AF agent to be incorporated in marine coatings. Laboratory tests showed that compound
763 maintained its AF activity against the settlement of
M. galloprovincialis after being chemically immobilized following this crosslinking strategy, and its release from the formulation was lower than the conventional releasing systems of PU-based marine coatings [
158].
In another study, 24 synthetic xanthones with three substitution patterns—1,3,4-trisubstituted xanthones, 1,3,4,6-tetrasubstituted xanthones, and 1,2,3,4,6-pentasubstituted xanthone—were analyzed [
159]. To evaluate the AF potential of these compounds, anti-settlement bioassays against the larvae of
M. galloproviancialis and antibacterial assays against
C. marina,
V. harveyi,
P. atlantica,
H. aquamarina, and
R. litoralis were performed. None of tested compounds were considered antibacterial agents against these marine biofilm-forming strains because they did not inhibit significantly (>30%) the growth of bacteria. Moreover, promising AF compounds were submitted to a marine ecotoxicity evaluation using
A. salina and proteomic studies were performed with the purpose to identify putative AF molecular targets. Particularly, compounds
776,
778,
780,
782 790,
793,
795, and
797 presented highly significant anti-settlement responses. In what concerns SAR of xanthone derivatives, comparing compounds
776,
781, and
785 with
780, and
784, it appears that the introduction of an additional methoxy group at position
780 does not affect the anti-settlement activity. The introduction of a chlorine atom in compound
777 nullified the pronounced anti-settlement activity of compound
776. Comparing compounds
784,
795, and
793 with compounds
783,
789, and
786, respectively, it is possible to infer that the presence of methoxy groups at positions 3 and 4 increase the anti-settlement effect, while the presence of a hydroxy group at position 4 and a methoxy group at position 3 has not so much significant impact in the
M. galloprovincialis larvae settlement inhibition [
159]. In respect to the variety of substituents at C-1, the chloromethyl group in compound
778 increased the anti-settlement activity if compared with the bromomethyl group in xanthone
779 and the hydroxymethyl group in xanthone
782. Relative to the aminated alkyl groups in xanthones
790–
798, derivatives
790,
793,
797, and
795 that present cyclic amine moieties showed better AF performance [
159]. Xanthones
776,
778,
780,
782,
790,
793,
795, and
797 were the most promising candidates as AF agents, but is important to emphasize that compounds
795 and
797, both containing pyridine moiety at C-1, were considered the most effective larval settlement inhibitors with EC
50 levels of 1.26 µg·mL
−1 and 3.03 µg·mL
−1, respectively. It is valuable to note that, compound
773 containing a pyridine group was related to the enhancement of anti-settlement activity [
159]. According to the SAR analysis, it is possible to infer the importance of a 3,4-dimethoxy substitution in the xanthone core with an additional cyclic amine moiety, preferably a pyridine to hit optimization from 3,4-dihydroxyxanthone (compound
763).
Scheme 17 summarizes the SAR for xanthone derivatives. Preliminary ecotoxicity evaluation in
A. salina showed that xanthones
778,
780,
782,
790,
793,
795, and
797 may have lower toxicity to the environment than some of the current biocides in use, as in the case of econea
® [
159]. Regarding the mechanism of action, proteomic studies indicated that xanthones
795 and
797 show similar molecular targets and impact similar molecular processes, which include diverse miosines from pedal refractor muscle. In conclusion, xanthones
795 and
797 emerged as hit AF compounds revealing higher anti-settlement effectiveness against
M. galloprovincialis larvae compared with
763 [
159].
2.28. Zosteric Acid and Other Cinnamic Acid Derivatives
Zosteric acid or
p-(sulfoxy) cinnamic acid (ZA,
799,
Figure 40) is a specialized metabolite produced by the seagrass
Zostera marina capable of preventing biofilm formation [
160]. This natural product is one of the most studied natural AF compound [
161]. To identify important structural determinants for the ZA antibiofilm activity, a library of ZA analogs with improved antibiofilm activity was developed (
800–
841,
Figure 40). The
E. coli maximum specific growth rates in the presence of several concentrations of each compound were calculated and compared to bacterial growth in the absence of compound. Compounds
800–
805,
807–
808,
812–
814,
819,
820,
822,
825,
827, and
840 had no biological activity at low concentrations, but showed antibiofilm activity at middle concentrations and induced biocidal effect at the maximum concentration. ZA and compounds
815,
816,
821, and
832–
835 had a biocidal effect at middle and higher concentrations, while at low concentrations, these compounds promoted biofilm formation [
161]. A peculiar trend was observed for compounds
831,
836, and
837: compound
830 showed antibiofilm activity at the lowest concentration, no biological activity at middle concentrations, and biocidal effect at the highest concentration; compound
836 promoted biofilm formation at 0.183, 1.83, and 183 μM, inhibited biofilm formation at 18.3 μM, and induced a biocidal effect at the highest concentration; finally, compound
837 promoted biofilm formation at all tested concentrations except for the 183 μM, where no biological activity effect was observed [
161].
According to the substitutions performed, the
p-sulfoxy ester group is not necessary for the antibiofilm activity; however, the cinnamic acid scaffold is responsible for the antibiofilm activity; the antibiofilm activity of ZA derivatives is dependent on the presence of a carboxylic acid and consequently on its hydrogen-donating ability, which is influenced by the type of groups present in the phenyl ring; the conjugated aromatic system is essential for the antibiofilm activity of ZA and its analogs, since the presence of the double bond stabilizes the carboxylate anion [
160].
Scheme 18 summarizes the SAR for zosteric acid derivatives. It was shown that 500 mg·L
−1 of the sodium salt of zosteric acid causes alterations in the whole proteomic signature of
E. coli, including stress-associated, motility-related, quorum-sensing-associated (LuxS enzyme), and metabolism/biosynthesis-related proteins [
162].
Interestingly, it was speculated that the AF effect of zosteric acid on algae
Ulva sp. could be due to the combination with the adhesive secreted by spores, or the attachment to the surface of the glass substrate. The strong hydrophilicity of the sulfonic acid group prevented or reduced the exclusion of the water between the adhesive and the substratum, making it difficult to form the adhesive-substratum interface [
135].
A series of sinapic acid (
842) derivatives was synthesized (
843–
849,
Figure 41) by click chemistry [
36]. For SAR purposes, similar derivatives of syringic acid (
850) were also obtained (
851–
857,
Figure 41). However, only the syringic acid derivative
856 was active, decreasing the settlement of
M. galloprovincialis larvae with an EC
50 of 12.91 μM, a lower value than the previous obtained lead compound gallic acid persulfate (EC
50 = 18 μM,
623,
Figure 31,
Section 2.20). Regarding the mechanism of action, the compound
856 was tested for the inhibition of AChE and tyrosinase activities, but the compound did not affect these two pathways [
36].


 ,
, 



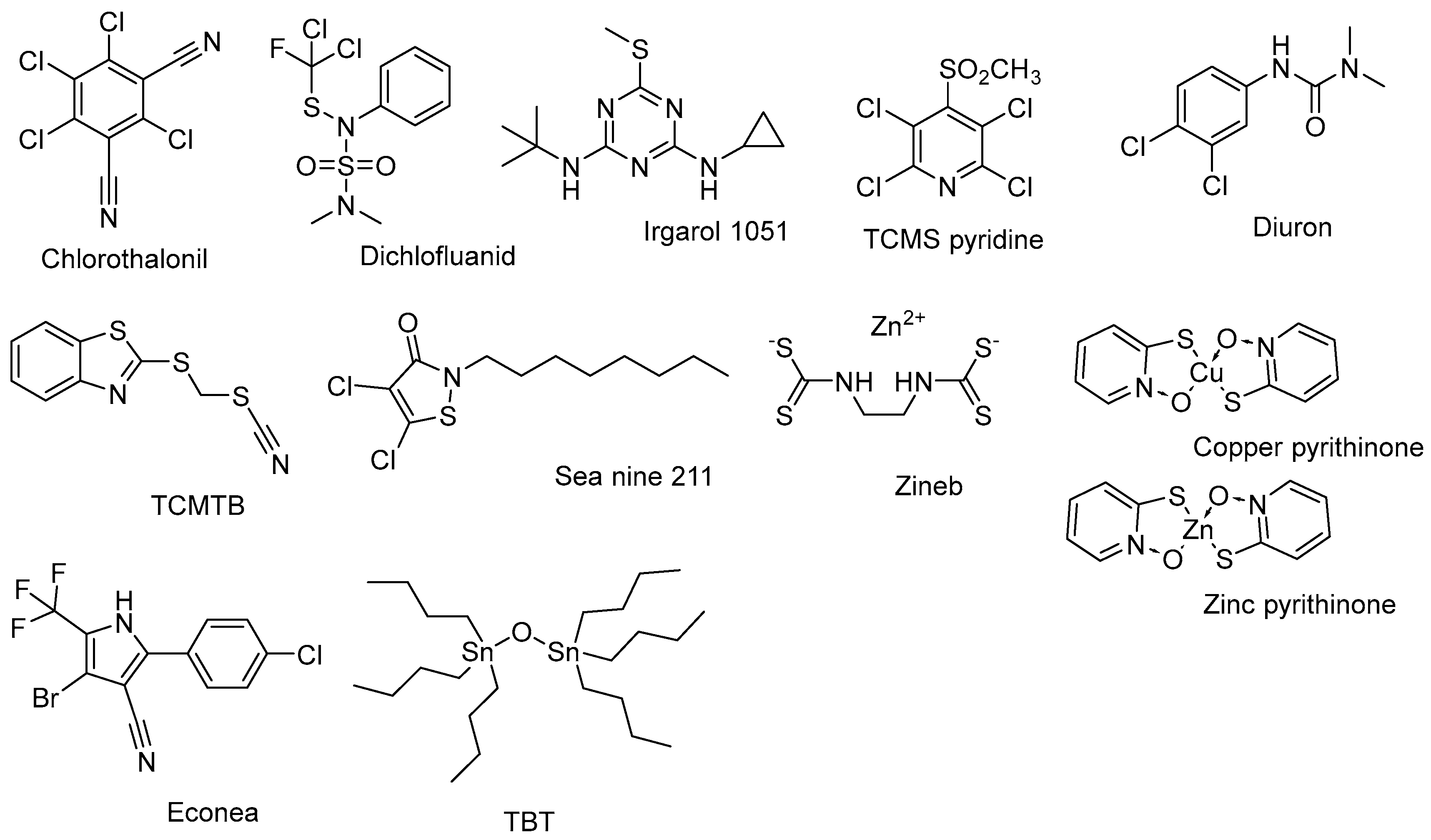
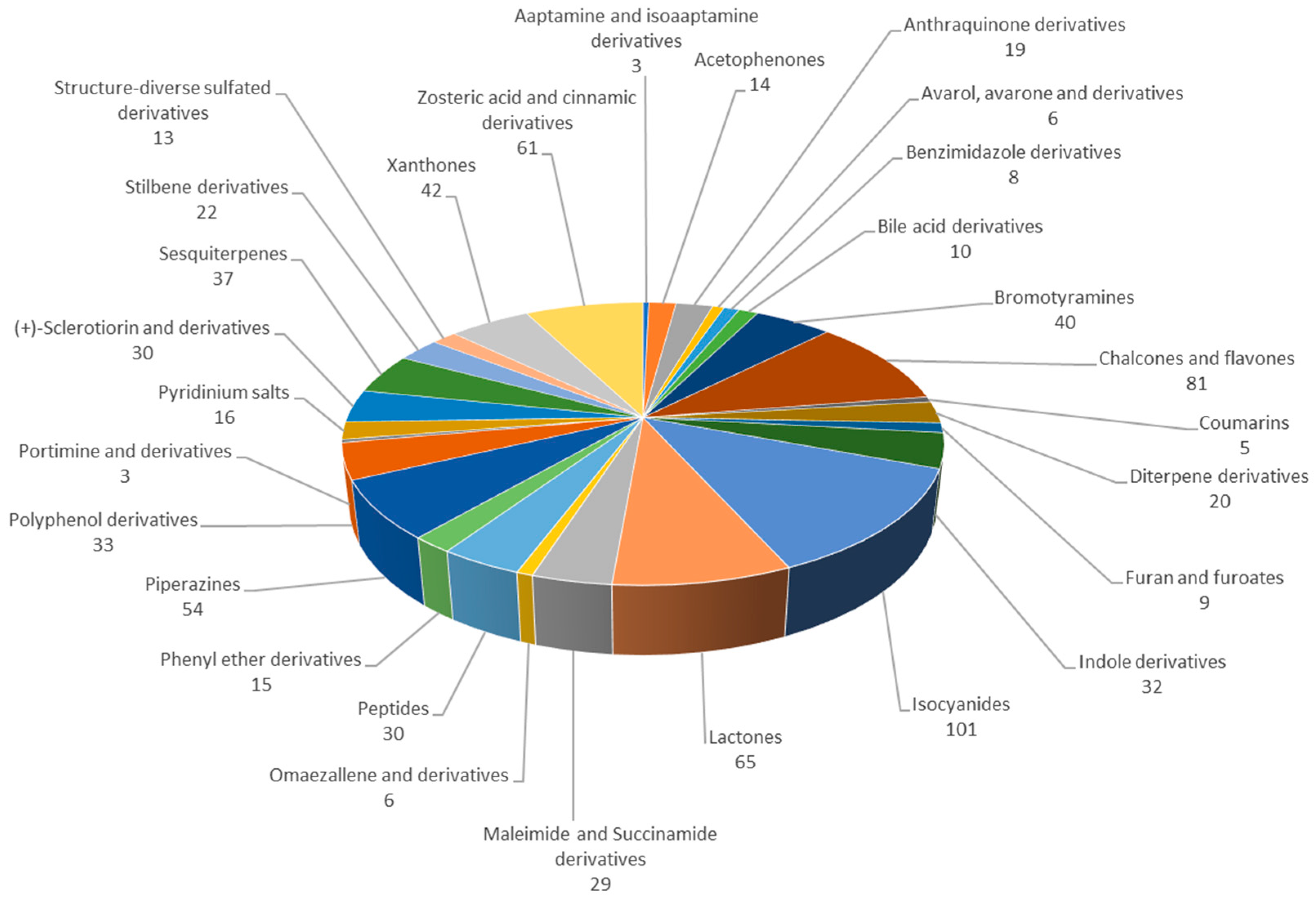


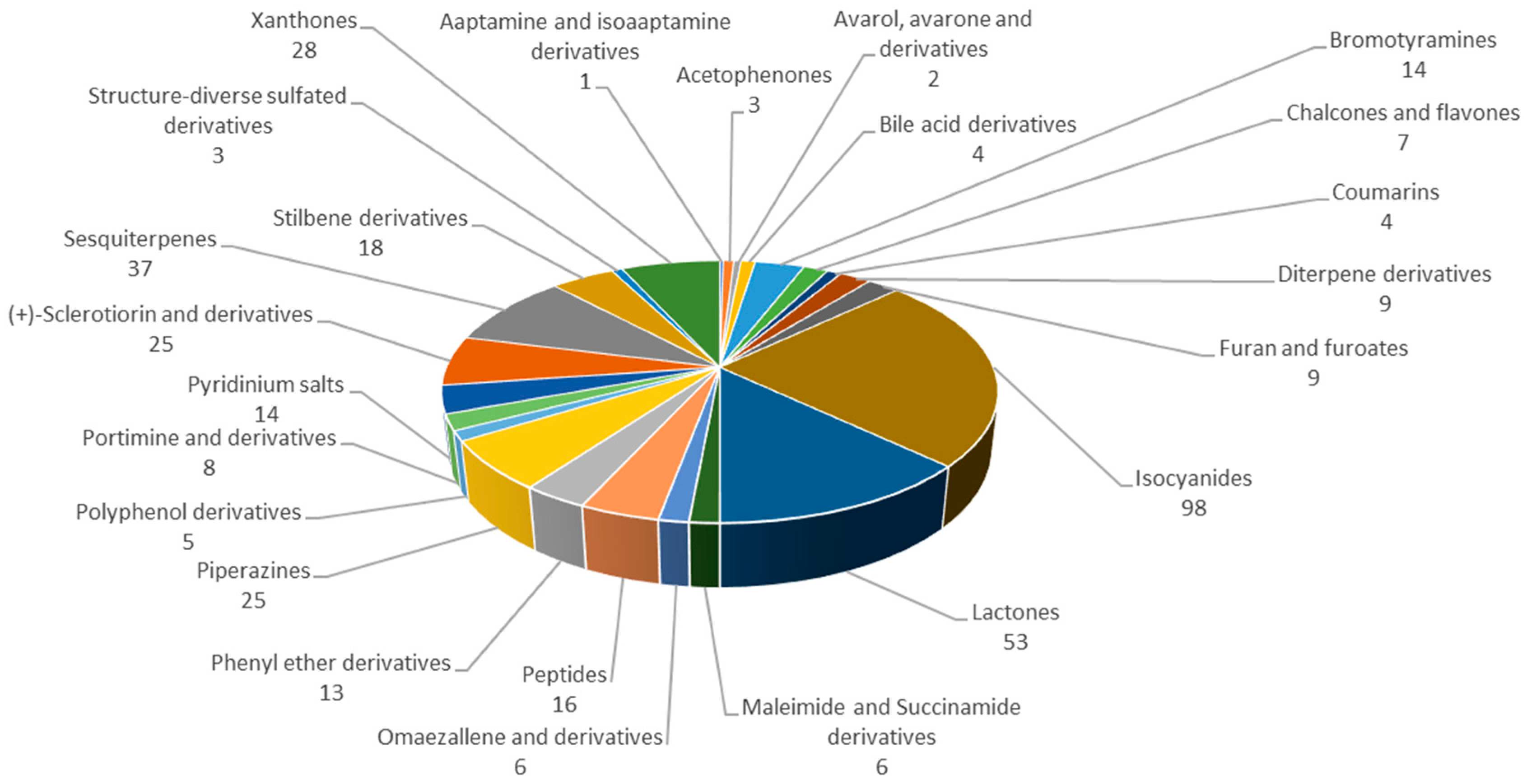

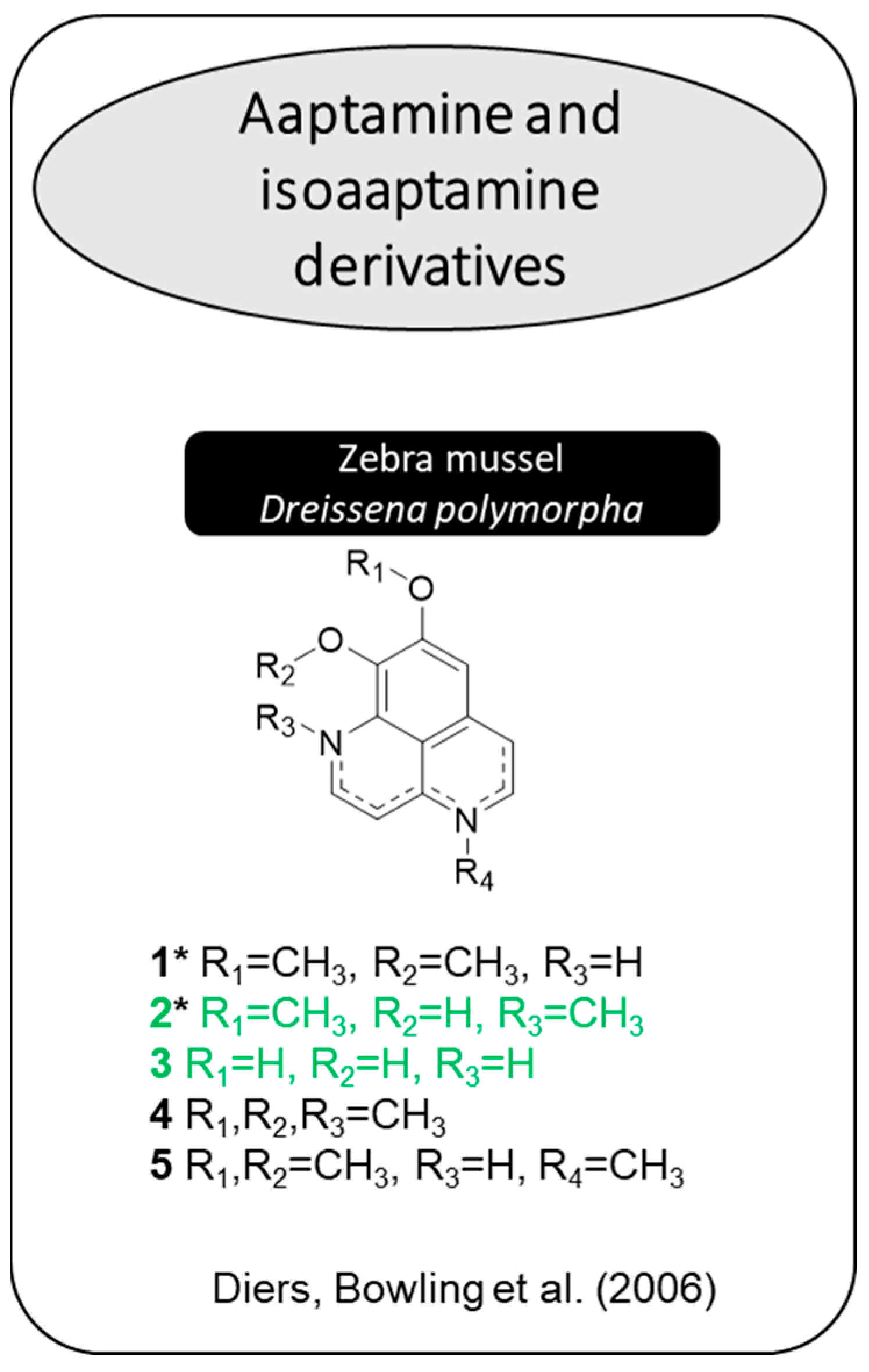



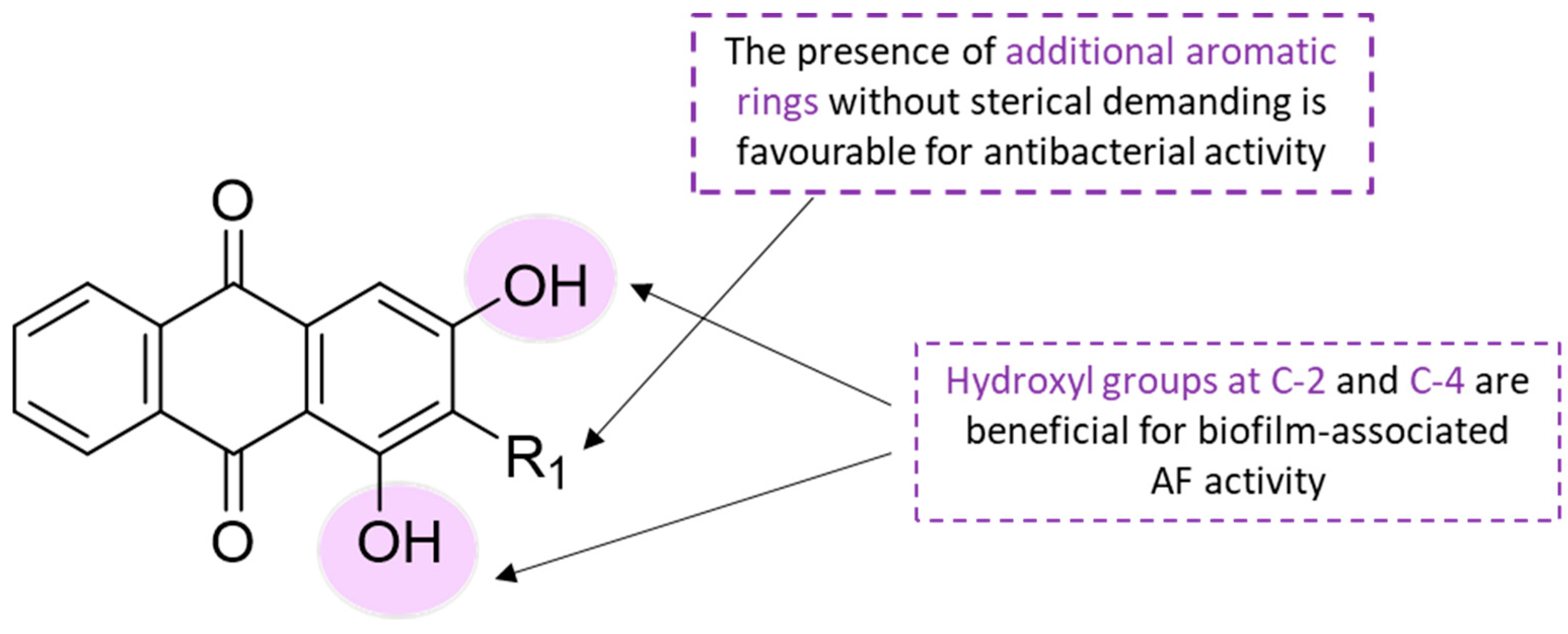
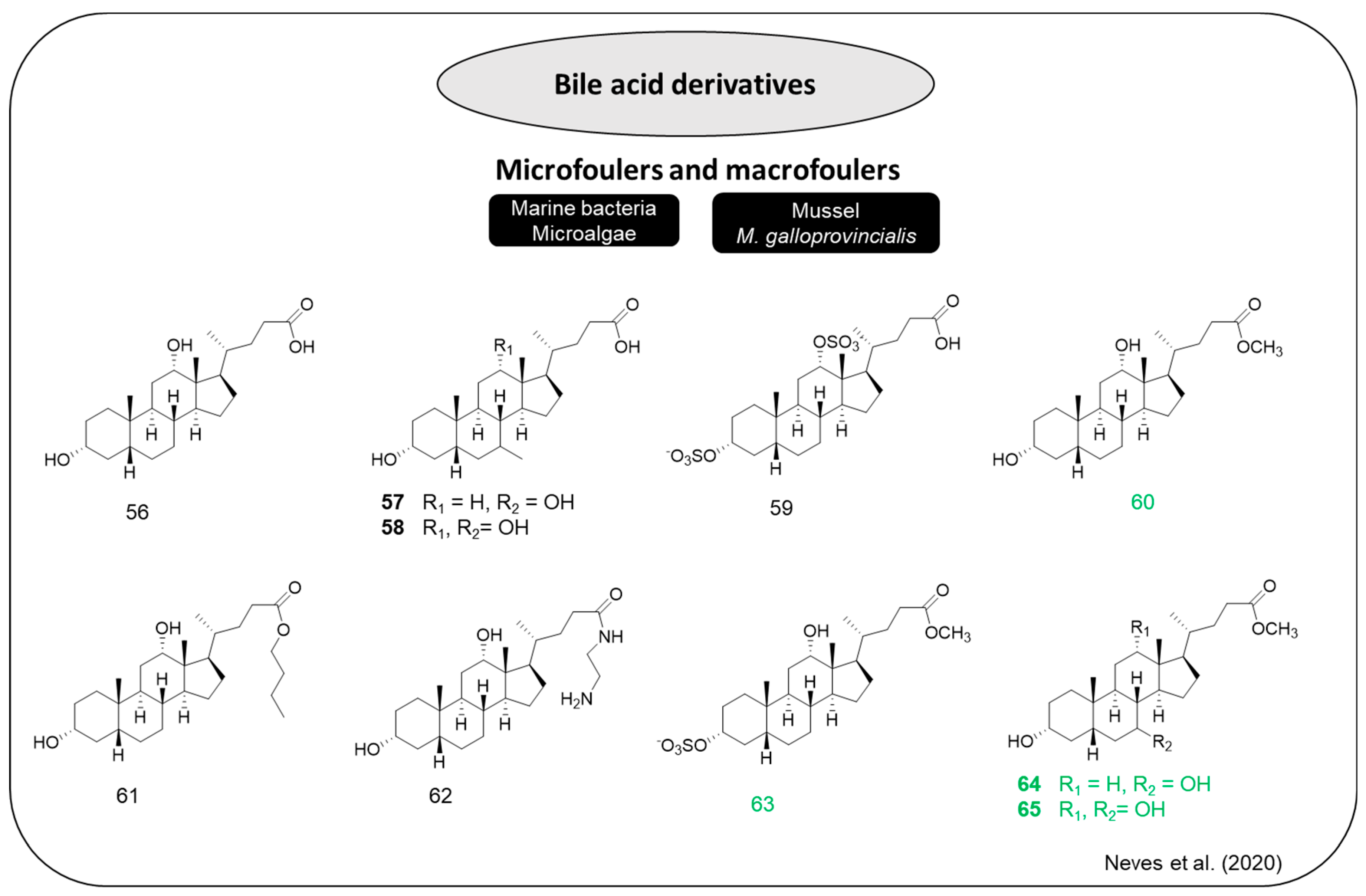

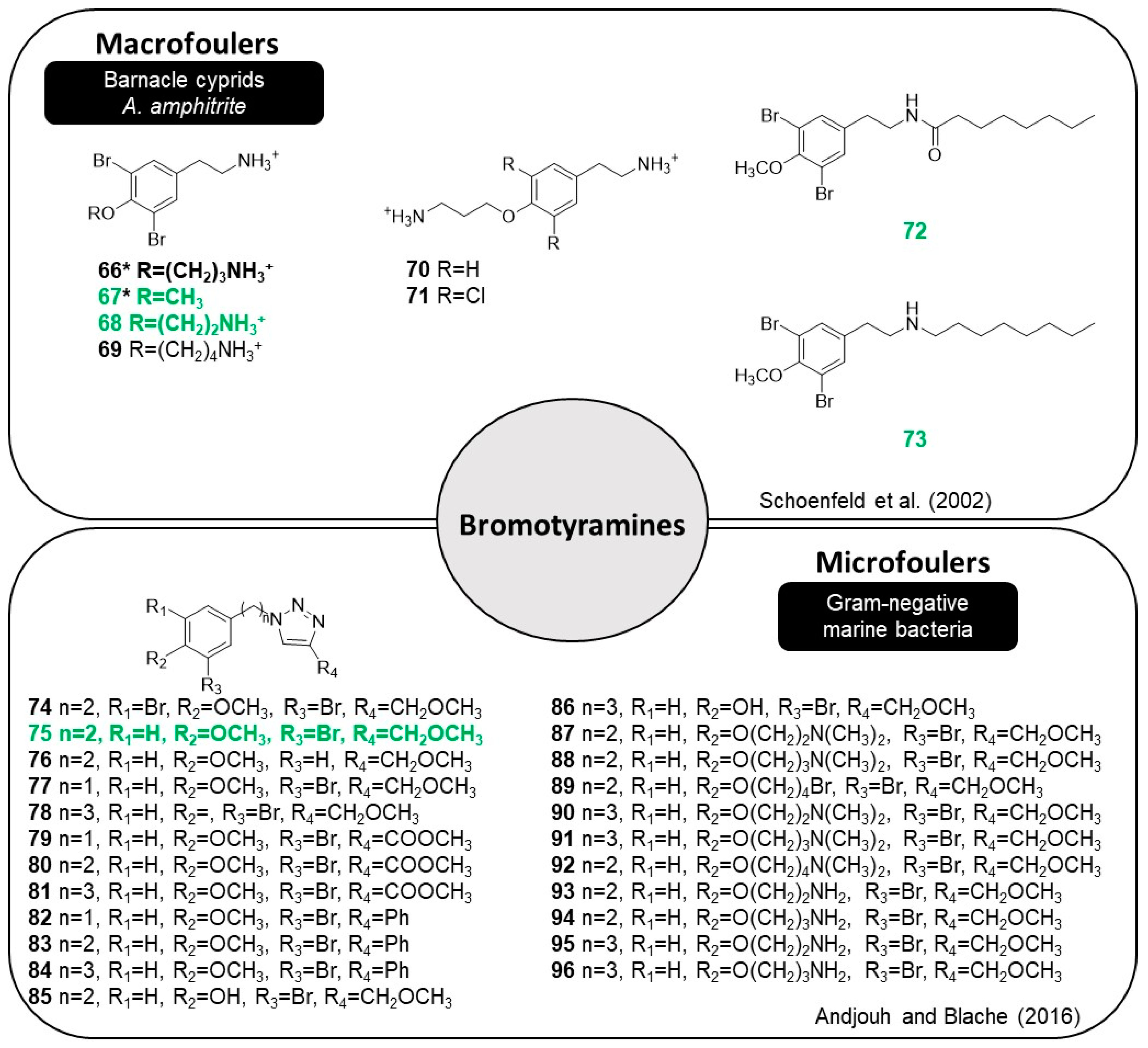
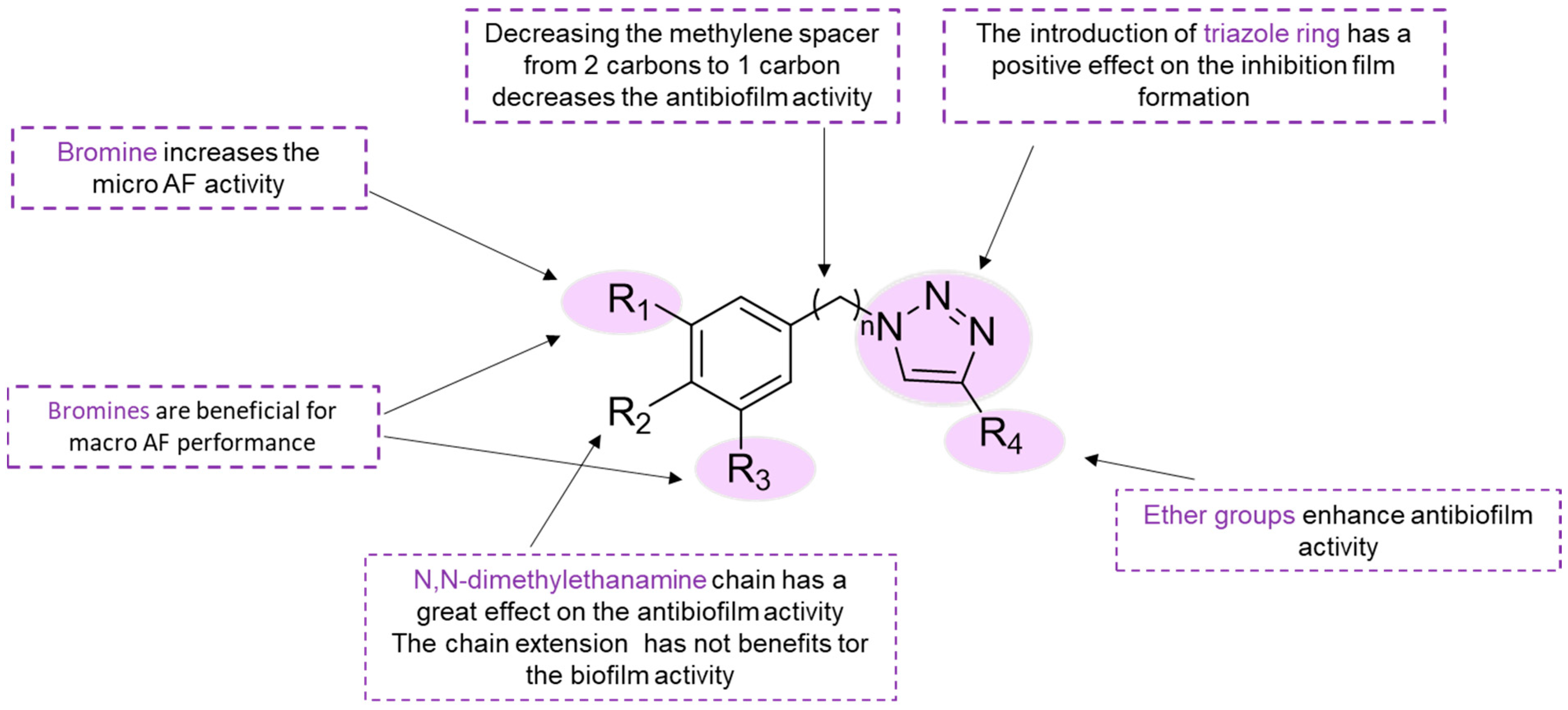

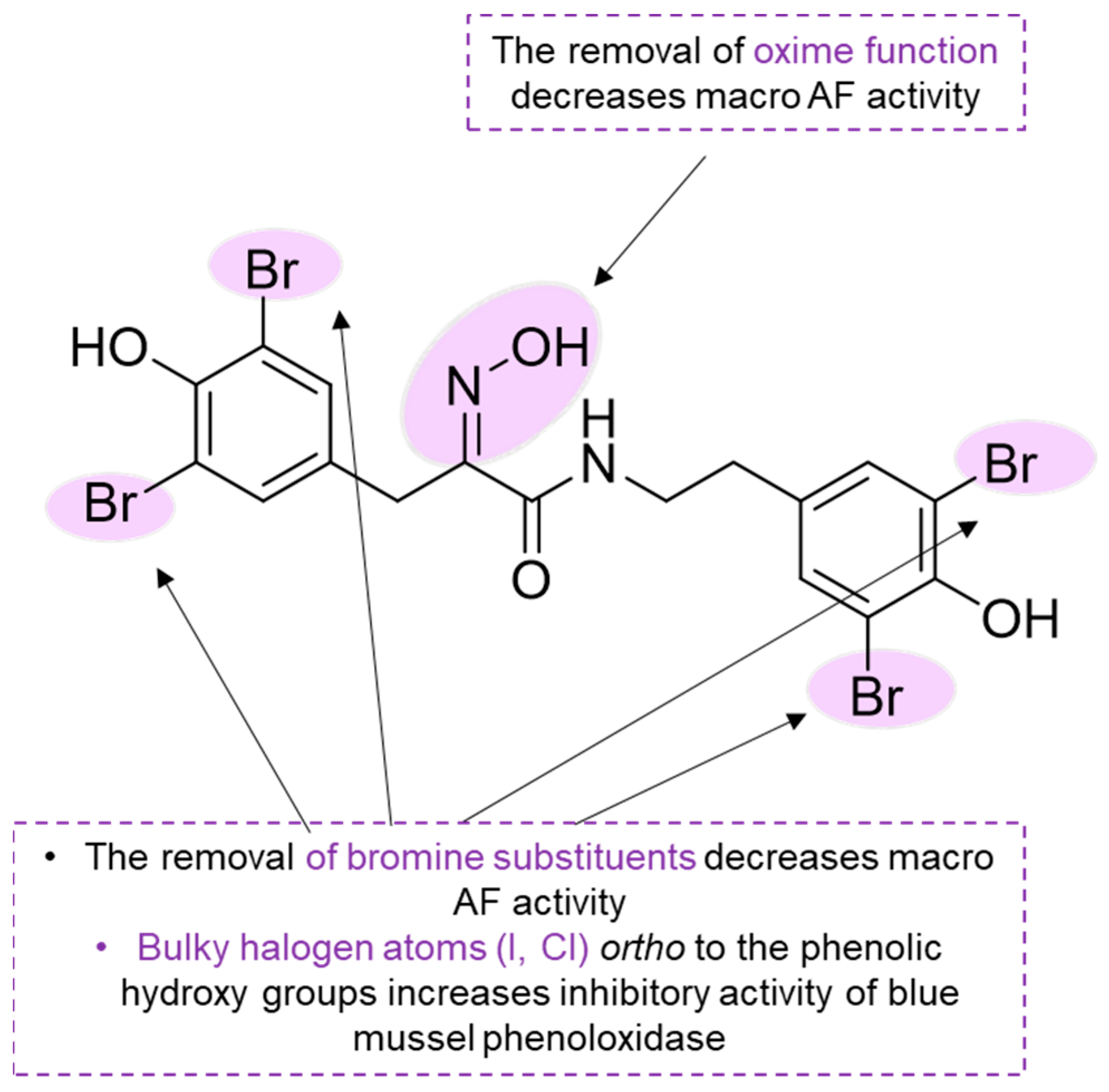


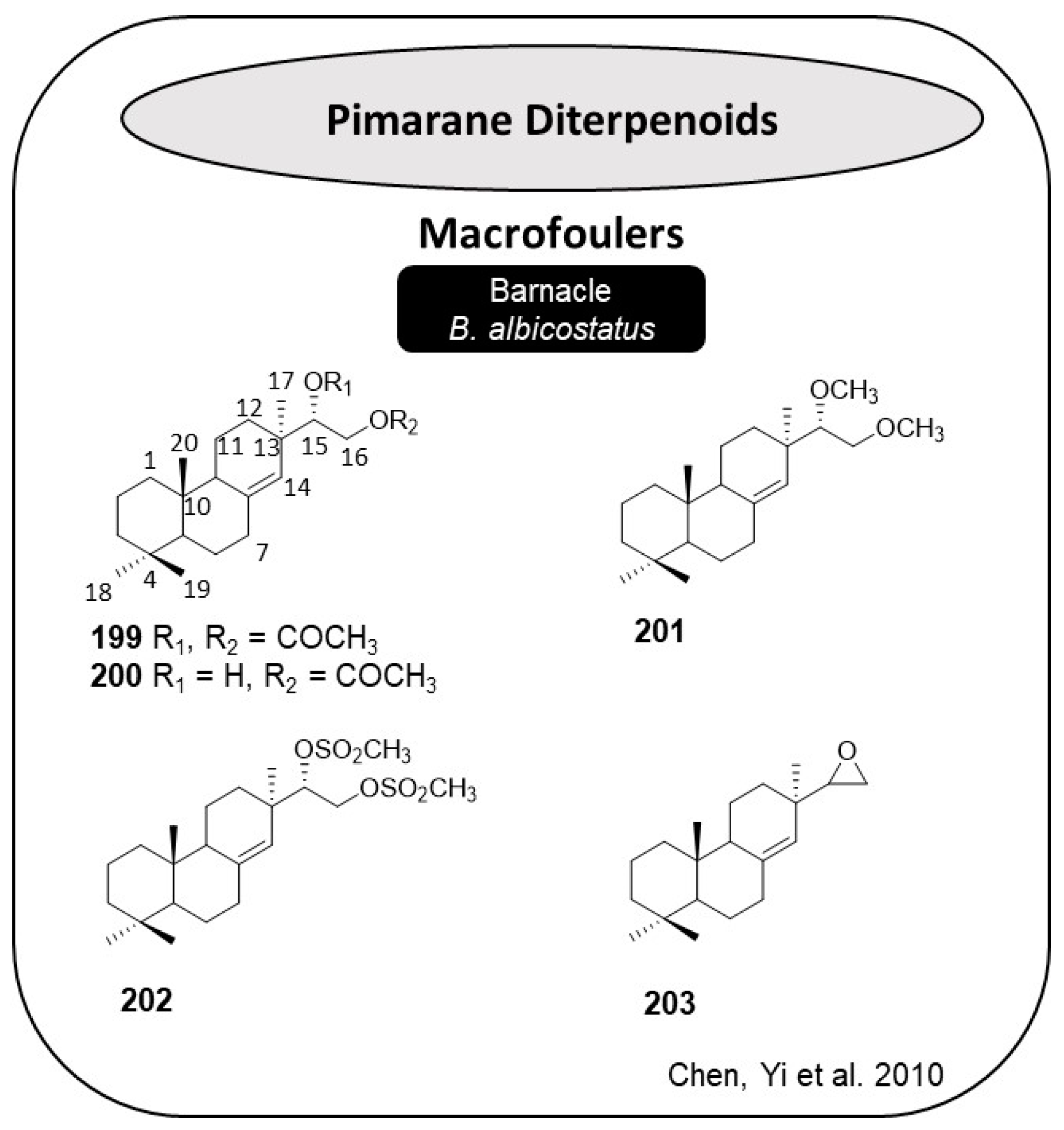
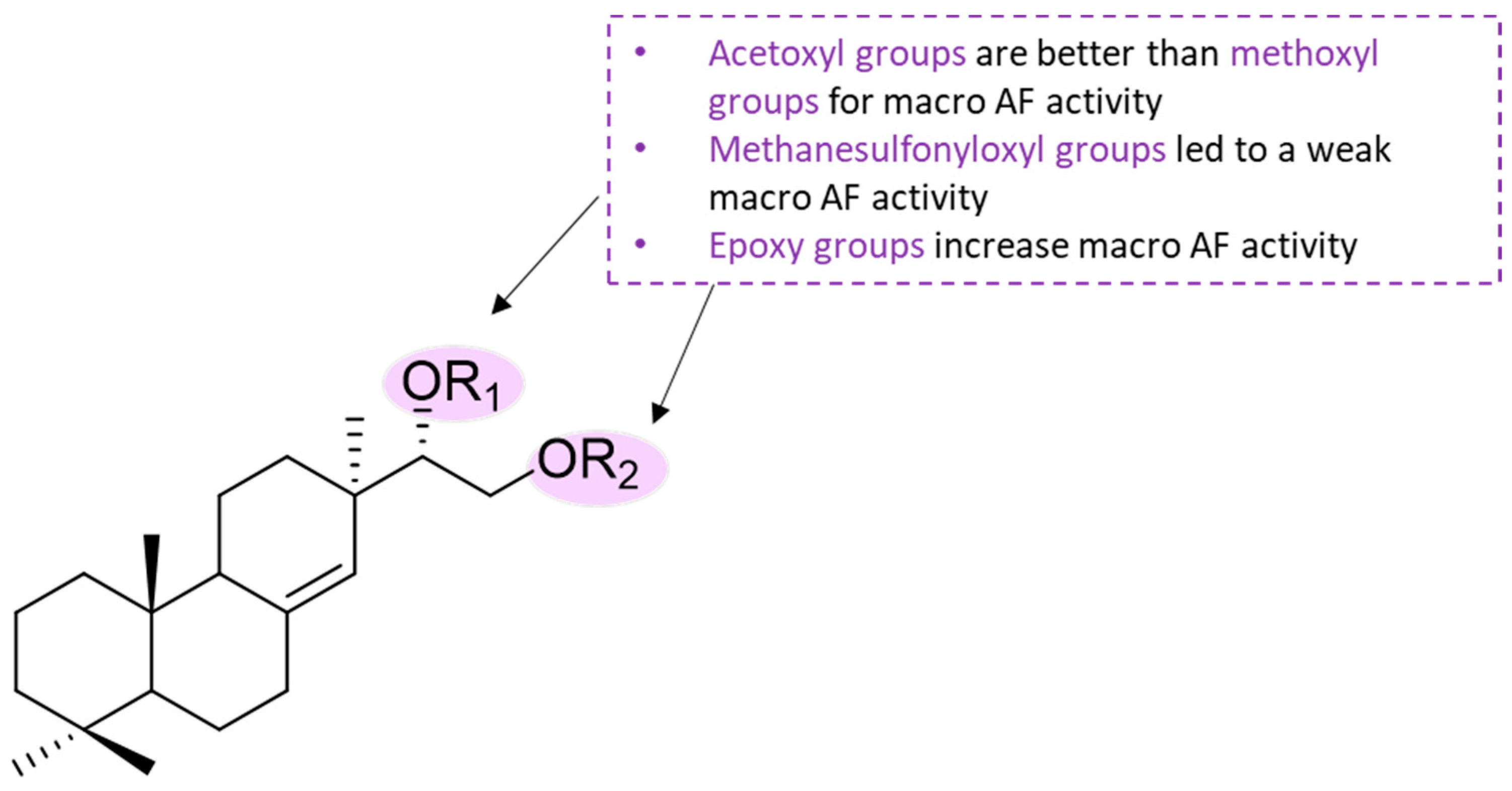
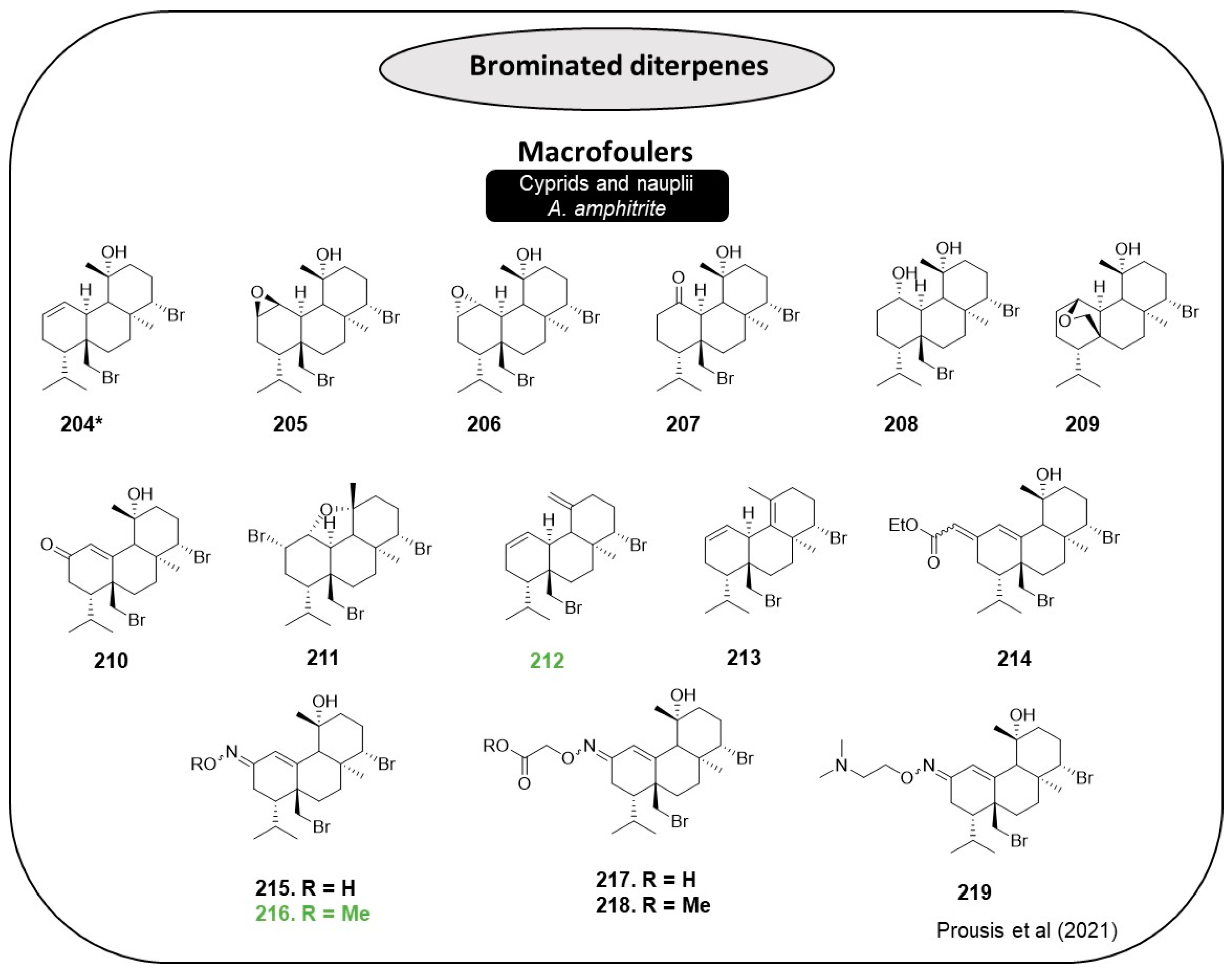
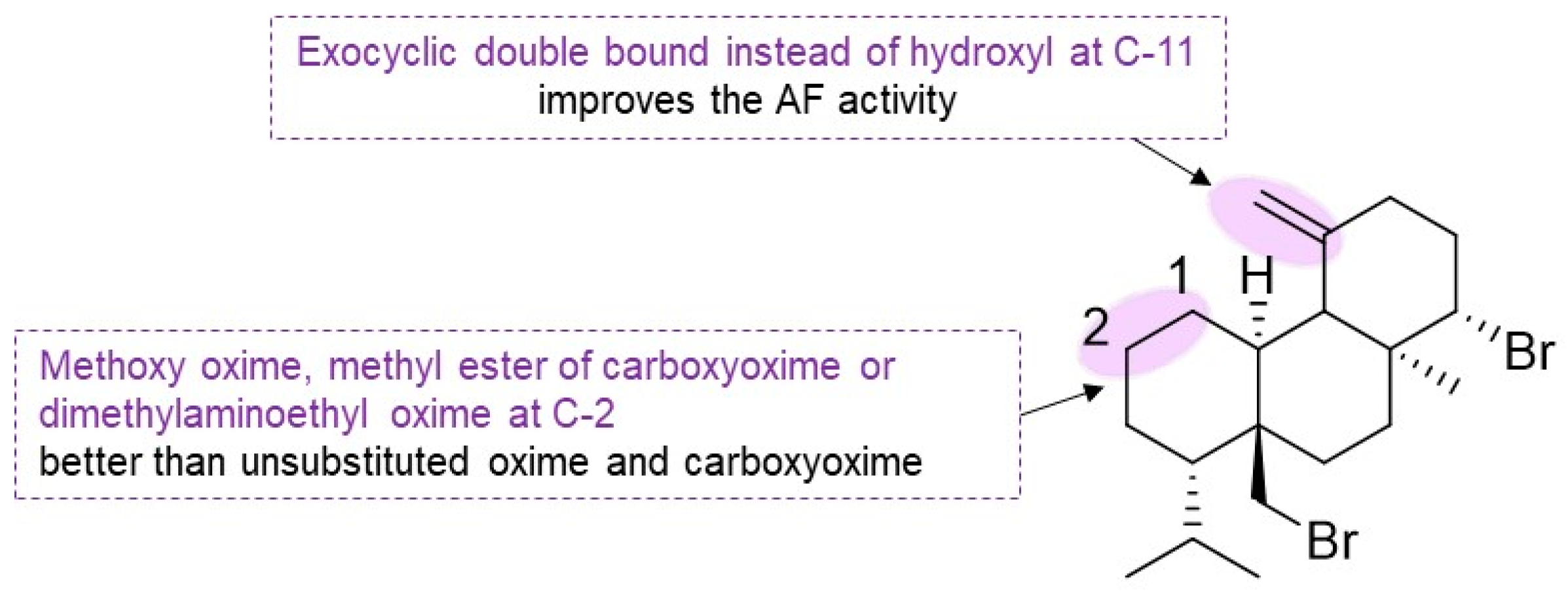

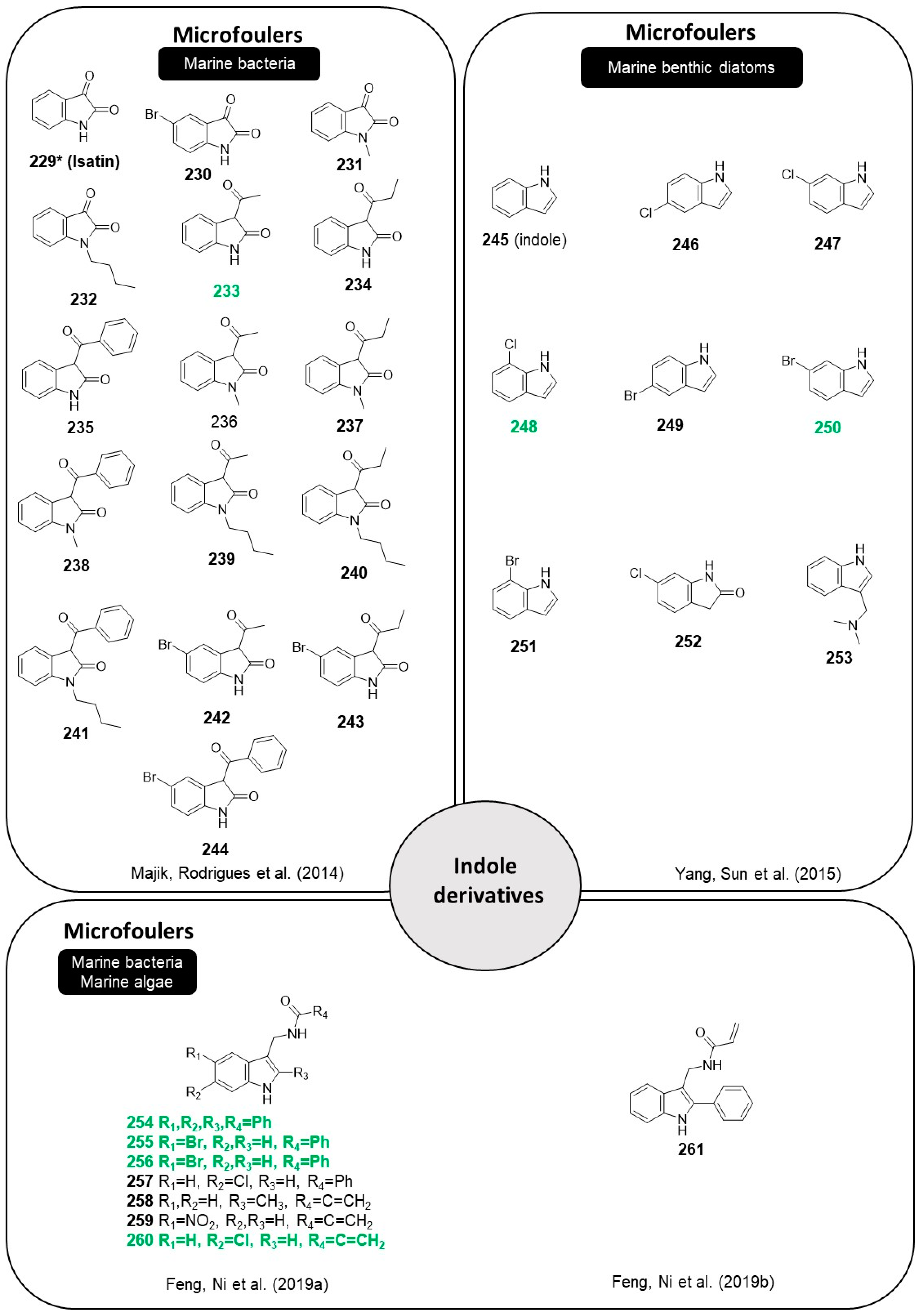
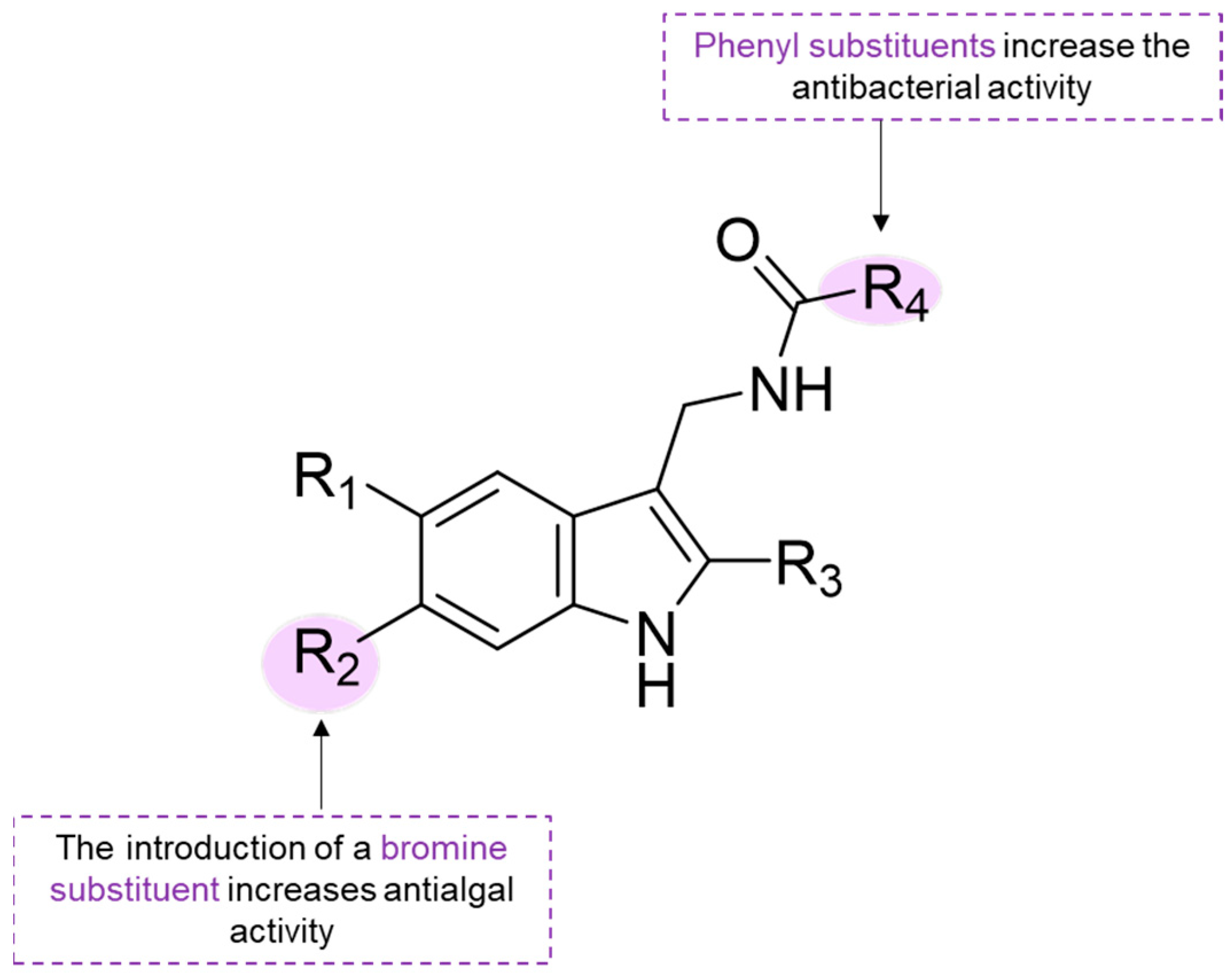



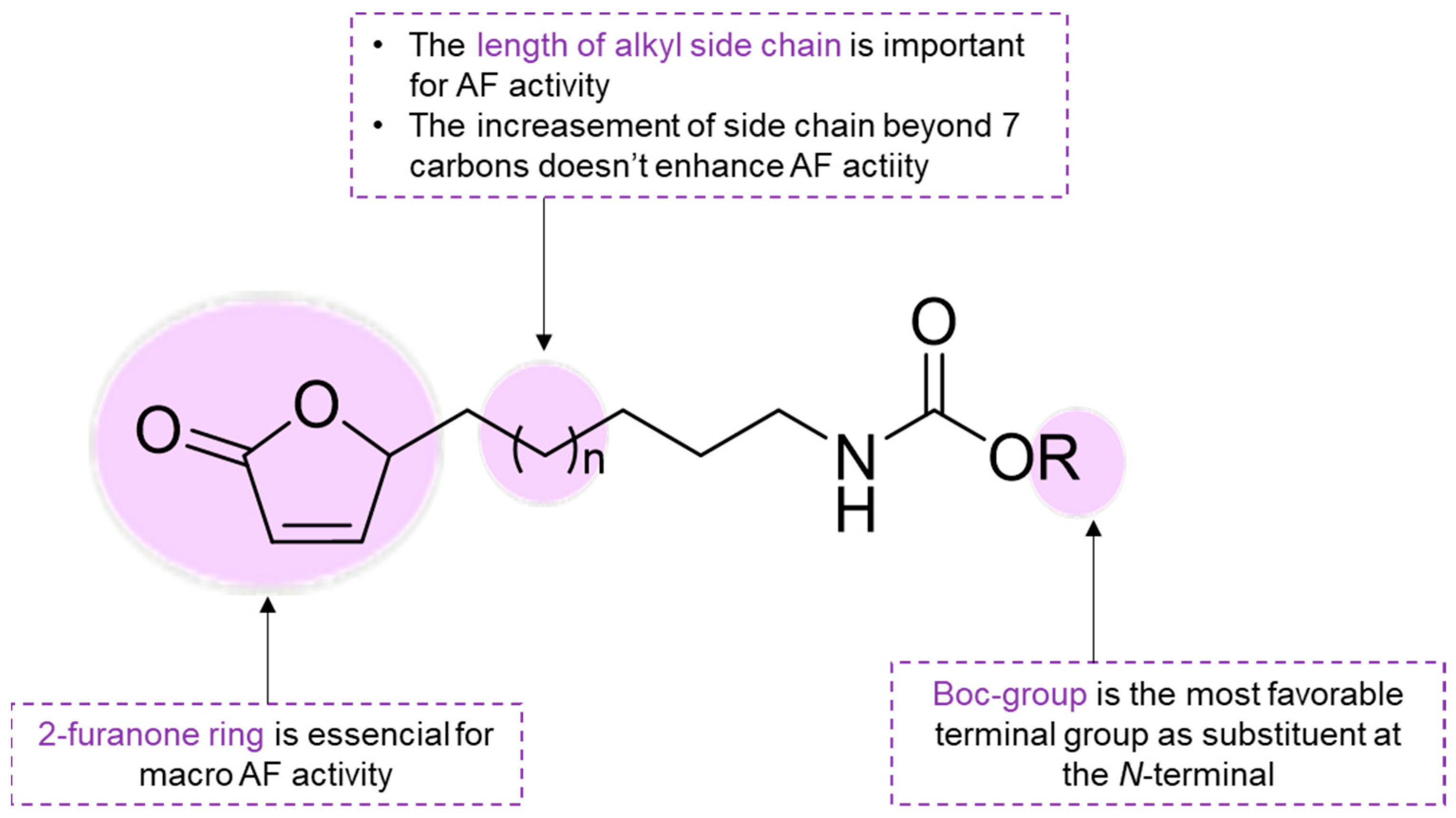


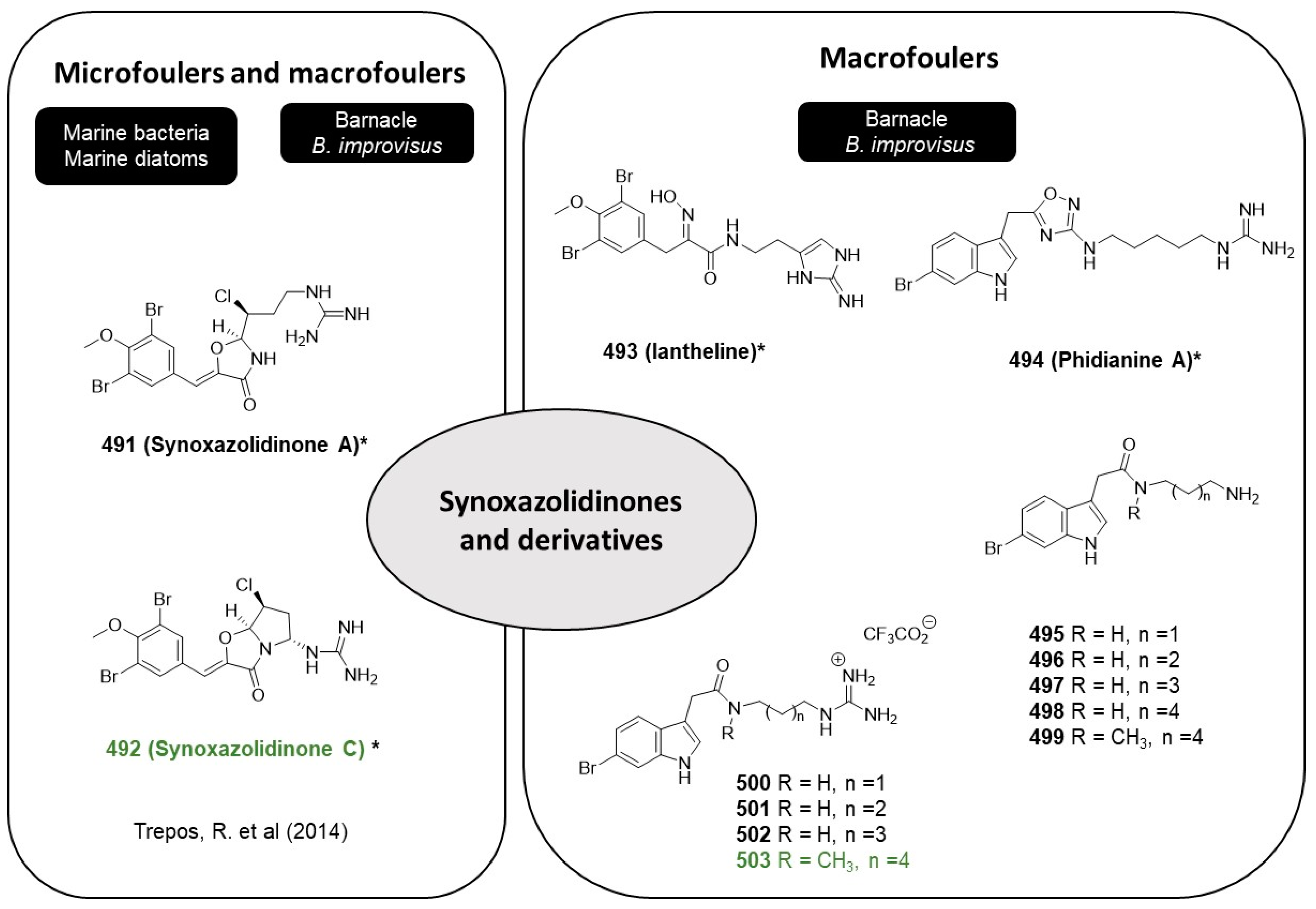





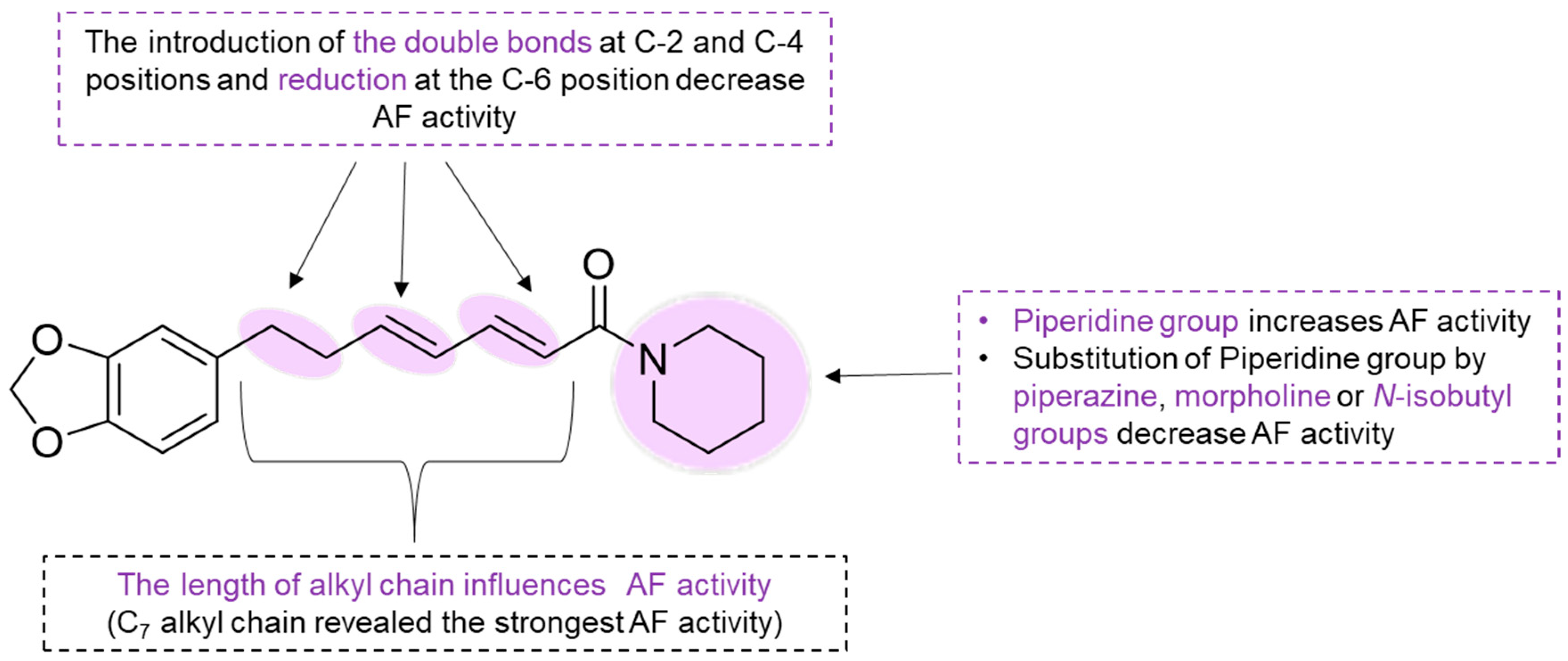
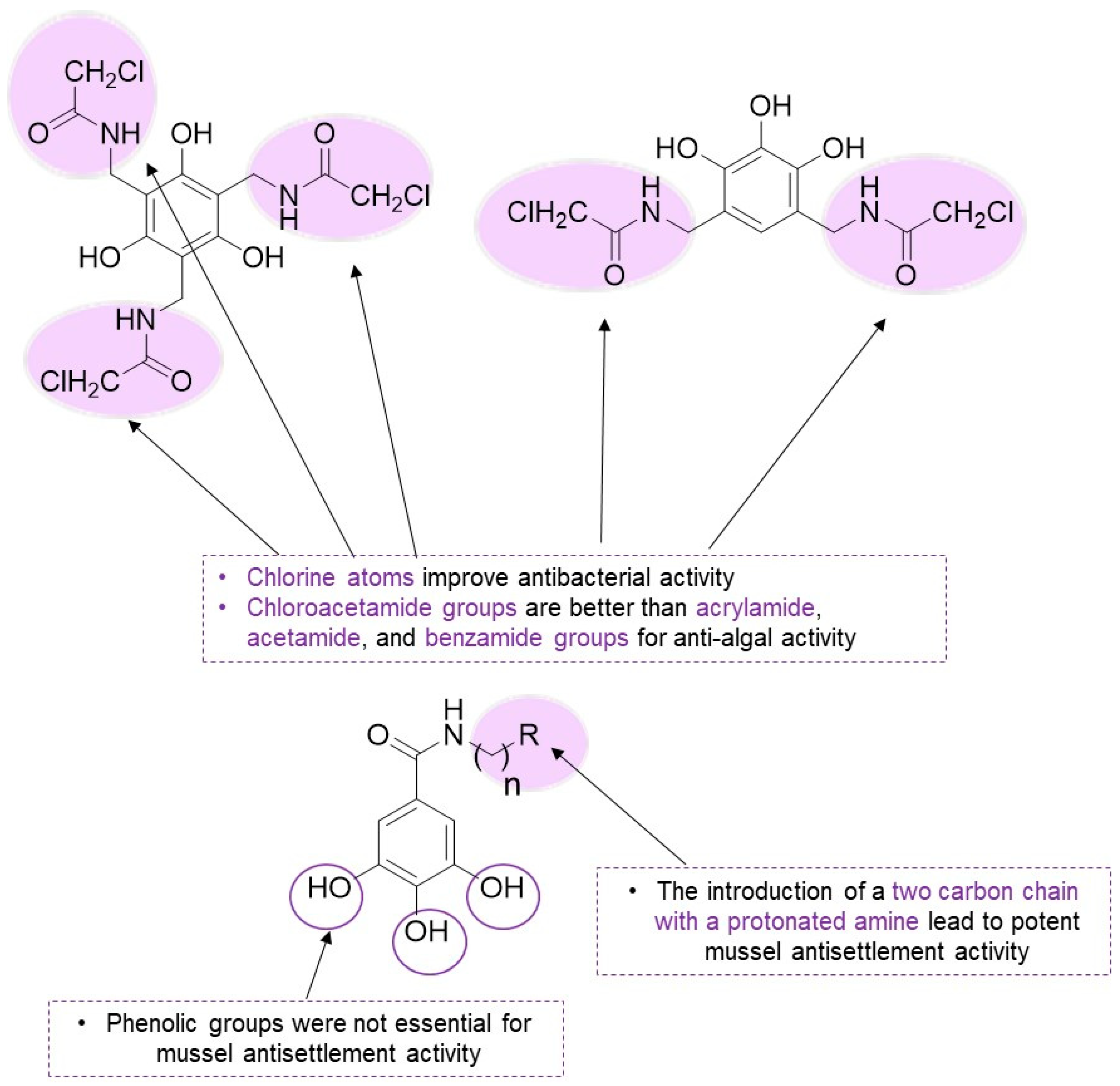



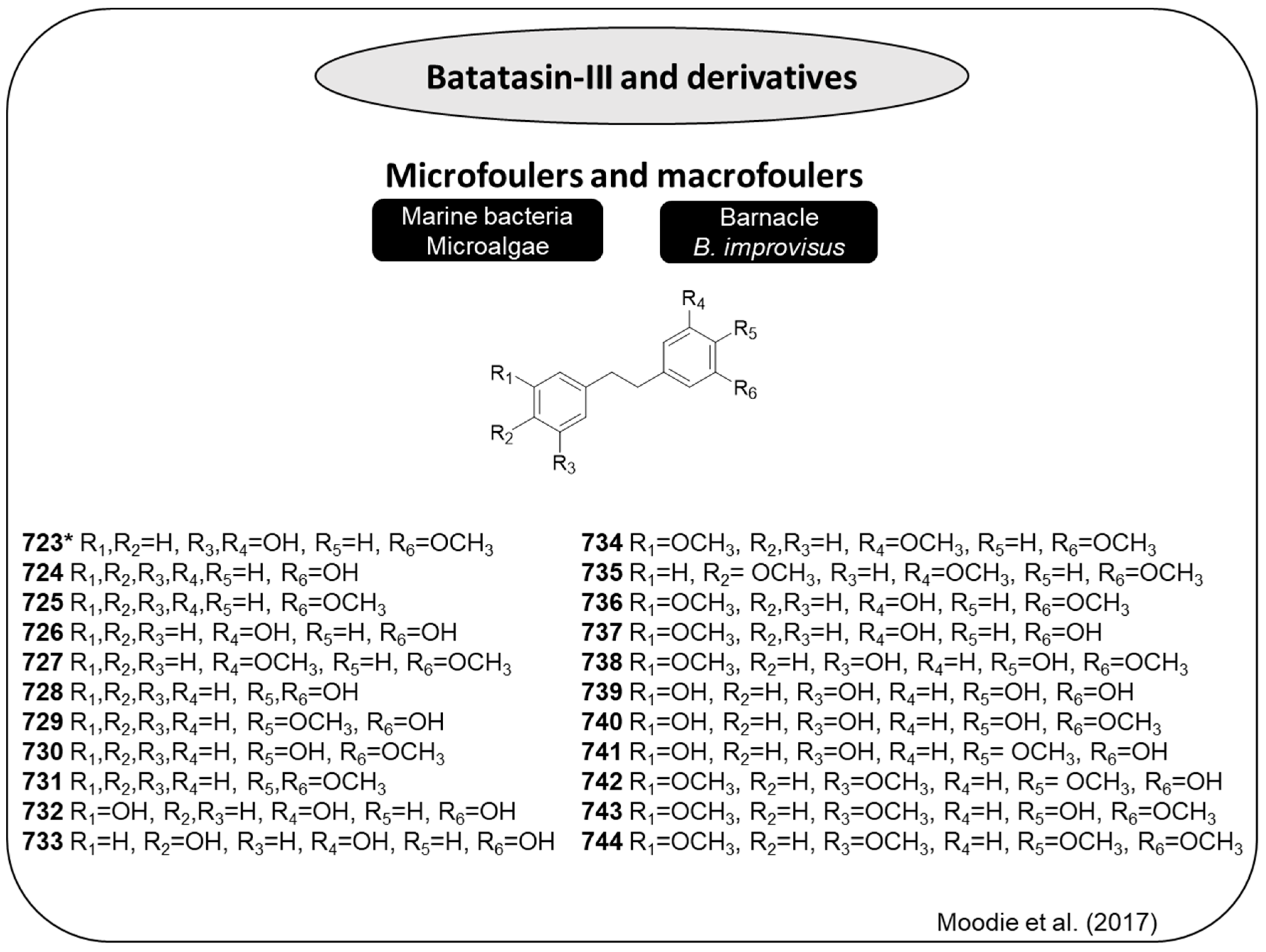
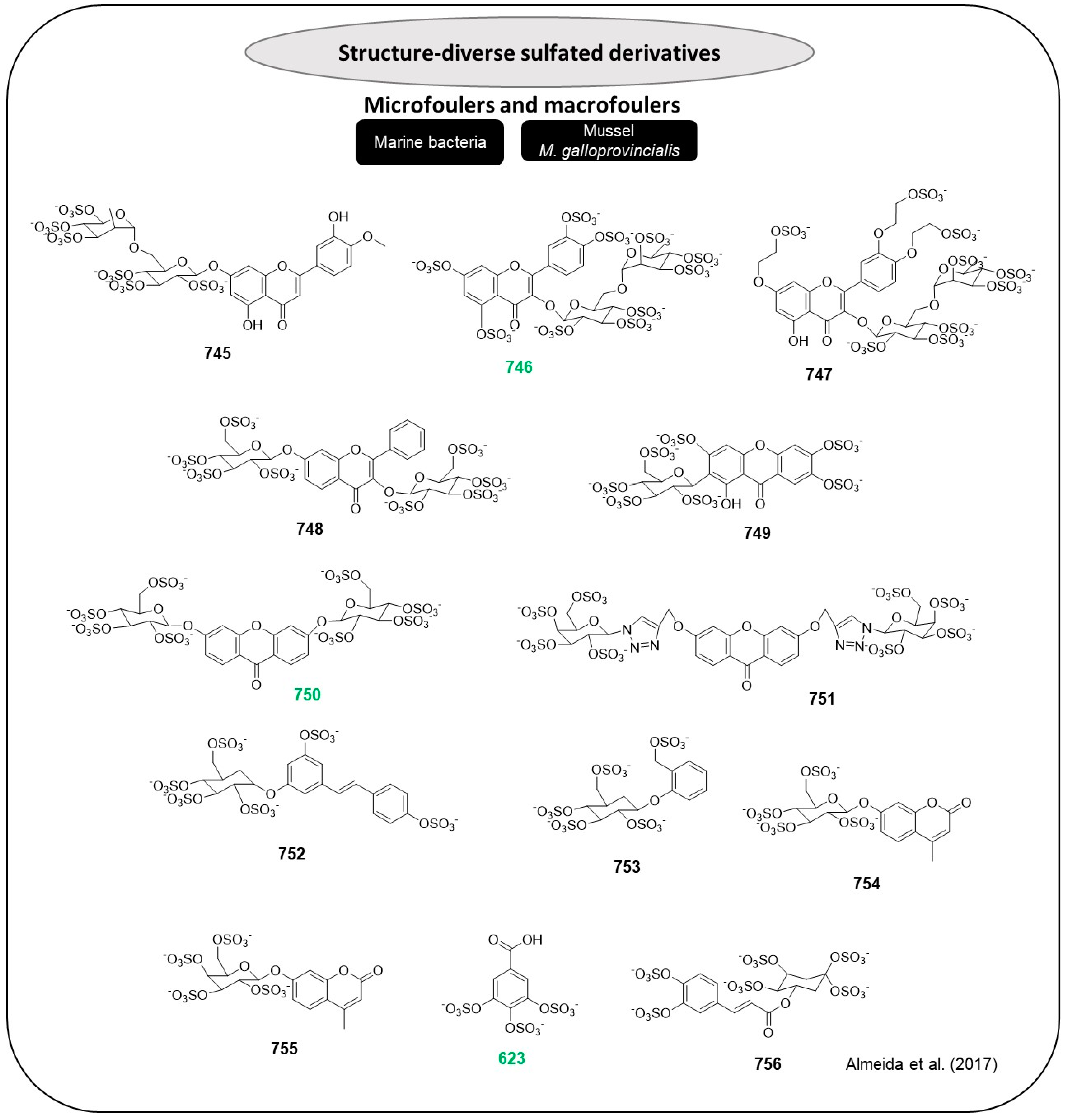
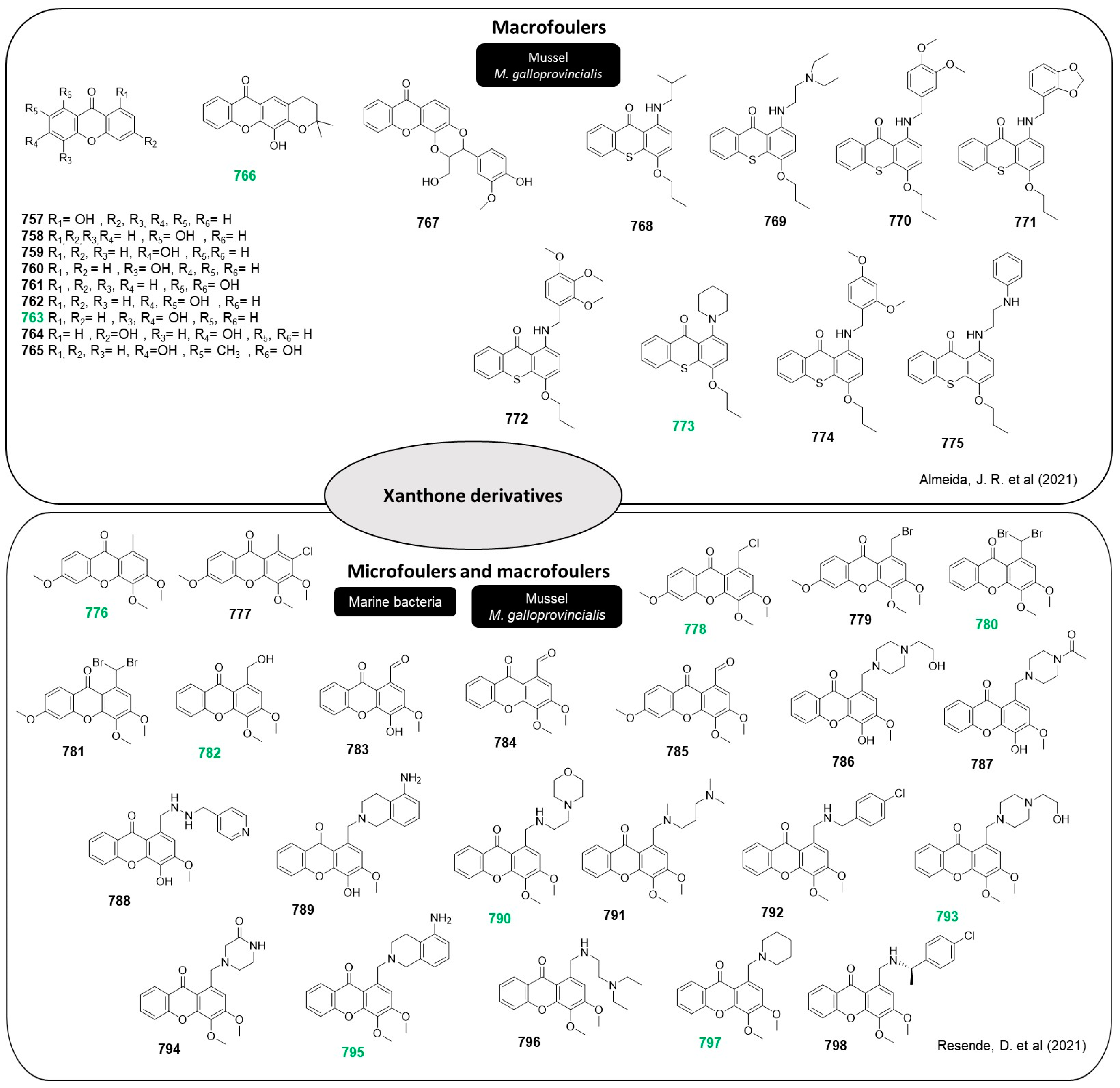





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}