
Marine-Derived Anticancer Agents Targeting Apoptotic Pathways: Exploring the Depths for Novel Cancer Therapies

 , , and
, , and 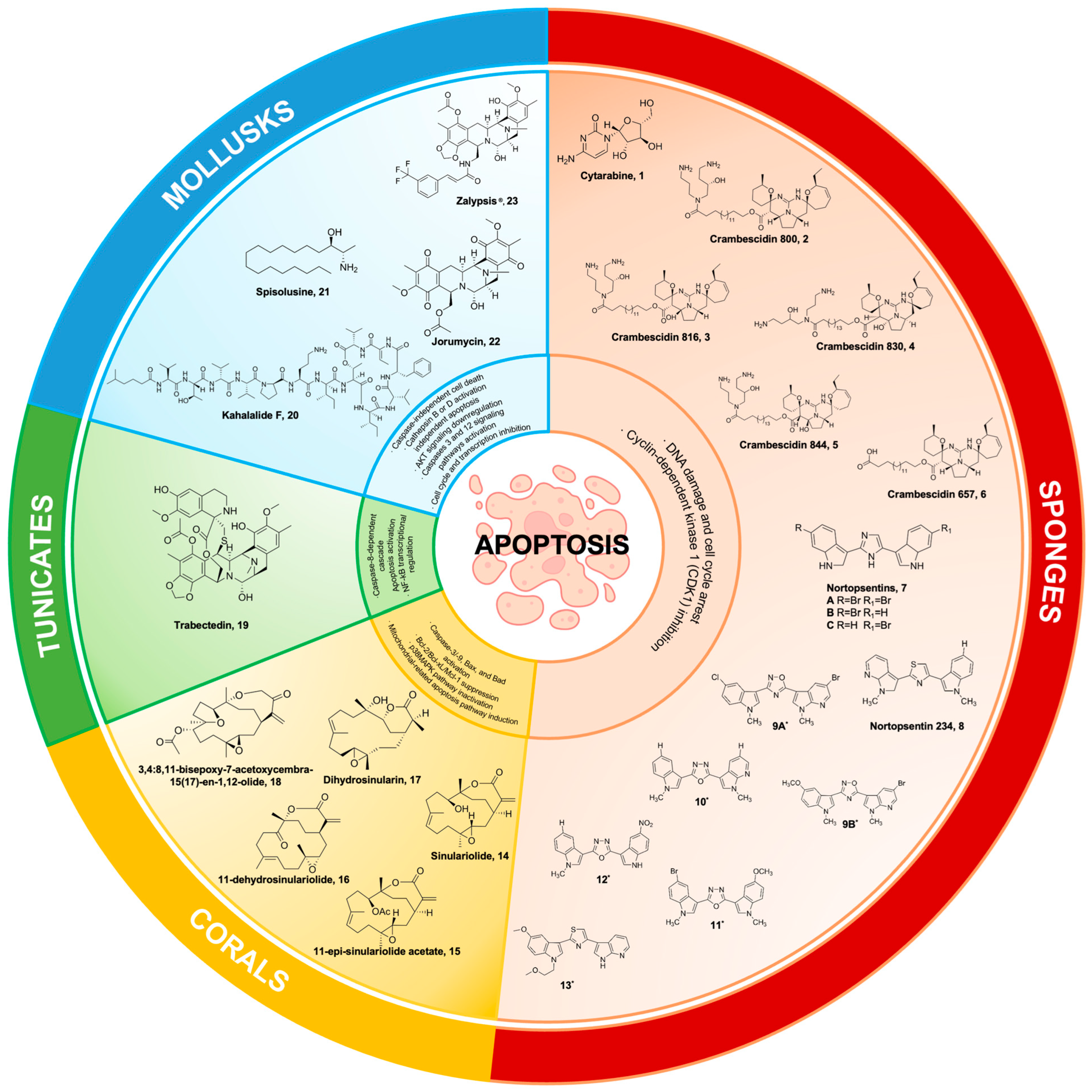{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Marine Sponges
2.1. Cytarabine (Ara-C)
2.2. Crambescidin and Its Derivatives
2.3. Nortopsentins
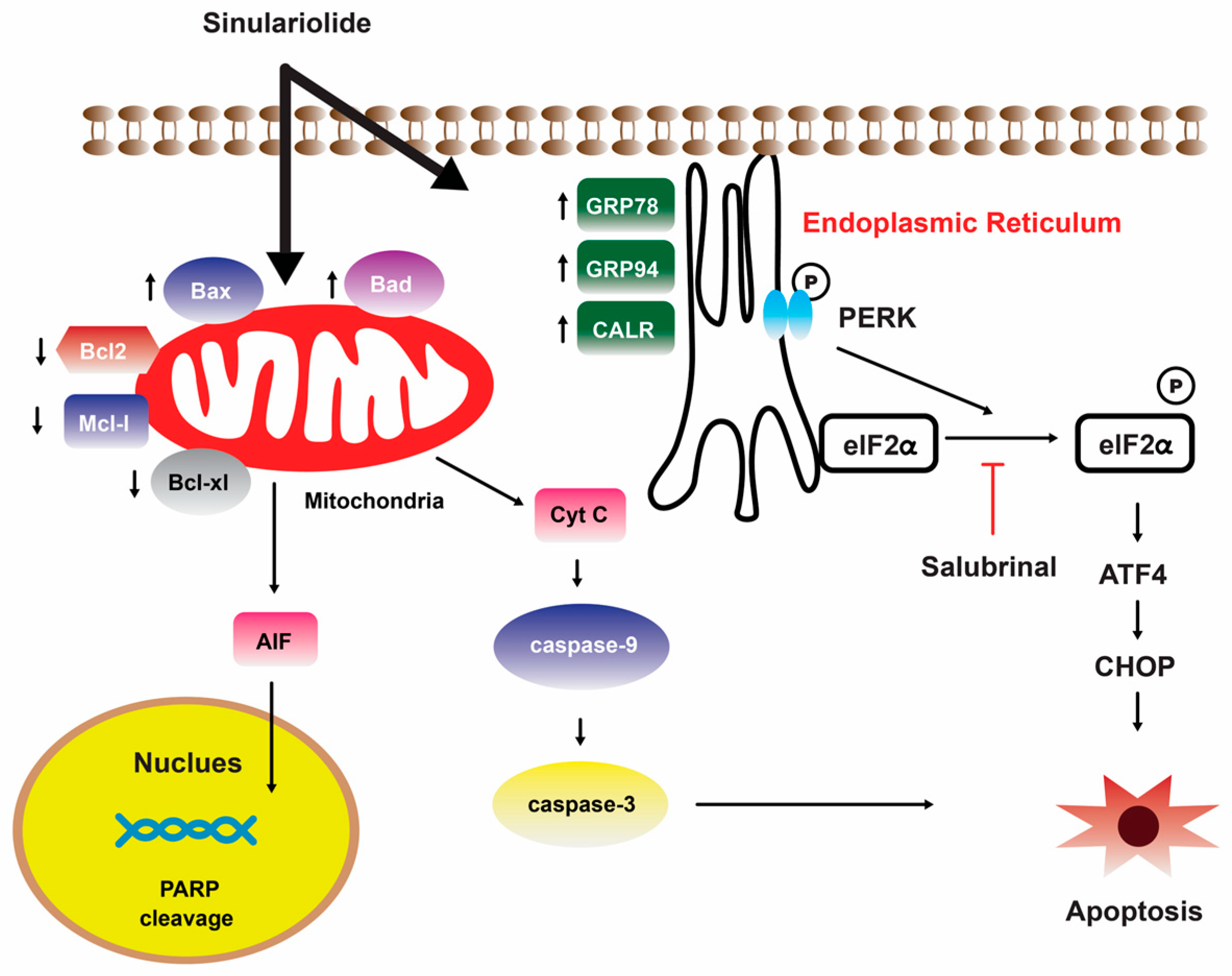
3. Marine Corals
Sinulariolide
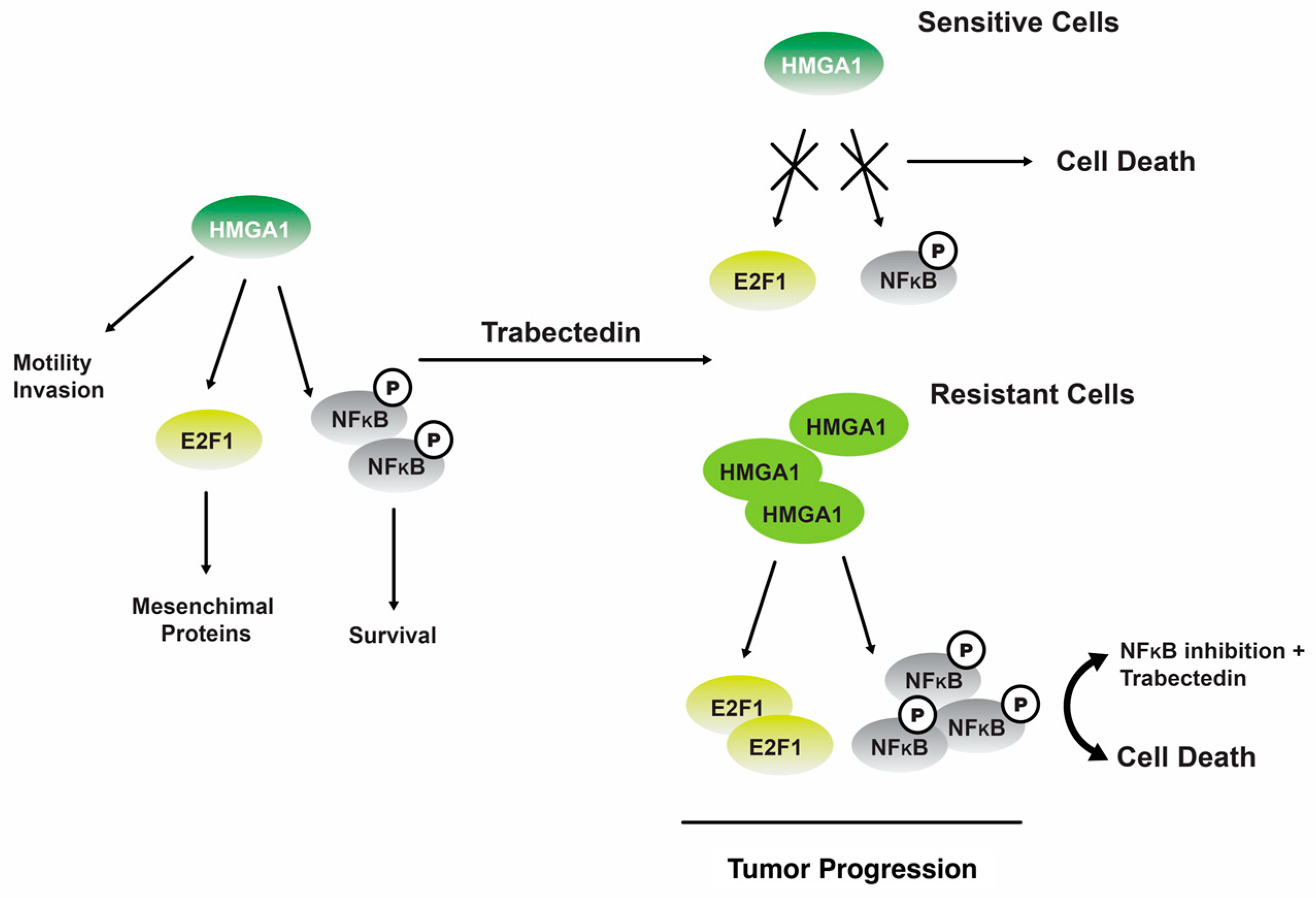
4. Tunicates
Trabectedin
5. Marine Mollusks
5.1. Kahalalide F
5.2. Spisulosine
5.3. Jorumycin
6. Ascidians
6.1. Meridine
6.2. Didemnins
7. Marine Algae
7.1. Fucoxanthin
7.2. Fucoidans
7.3. Laminarin
7.4. Tuberatolide B
7.5. Sargaquinoic Acid
7.6. Docosahexaenoic Acid
8. Marine Cyanobacteria
Apratoxins
9. Marine Fungi
9.1. Penicitrinine A
9.2. Aspergiolide
9.3. Ophiobolin O
9.4. Physcion
10. Marine Actinobacteria
Salinosporamide A
11. Future Perspective and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Voss, A.K.; Strasser, A. The essentials of developmental apoptosis. F1000Research 2020, 9, F1000 Faculty Rev-148. [Google Scholar] [CrossRef]
- Norbury, C.J.; Hickson, I.D. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef]
- Nainu, F.; Shiratsuchi, A.; Nakanishi, Y. Induction of apoptosis and subsequent phagocytosis of virus-infected cells as an antiviral mechanism. Front. Immunol. 2017, 8, 1220. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Levine, A.J. The p53 functional circuit. J. Cell Sci. 2001, 114, 4139–4140. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, S.; Vasudevan, K.M.; Rangnekar, V.M. Regulation of apoptosis in prostate cancer. Cancer Metastasis Rev. 2001, 20, 225–243. [Google Scholar] [CrossRef]
- Ganesher, A.; Chaturvedi, P.; Sahai, R.; Meena, S.; Mitra, K.; Datta, D.; Panda, G. New spisulosine derivative promotes robust autophagic response to cancer cells. Eur. J. Med. Chem. 2020, 188, 112011. [Google Scholar] [CrossRef]
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Khalifa, S.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef]
- Dayanidhi, D.L.; Thomas, B.C.; Osterberg, J.S.; Vuong, M.; Vargas, G.; Kwartler, S.K.; Schmaltz, E.; Dunphy-Daly, M.M.; Schultz, T.F.; Rittschof, D.; et al. Exploring the diversity of the marine environment for new anti-cancer compounds. Front. Mar. Sci. 2021, 7, 614766. [Google Scholar] [CrossRef]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef]
- Kluza, J.; Gallego, M.A.; Loyens, A.; Beauvillain, J.C.; Sousa-Faro, J.M.; Cuevas, C.; Marchetti, P.; Bailly, C. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin D. Cancer Res. 2006, 66, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Petek, B.J.; Loggers, E.T.; Pollack, S.M.; Jones, R.L. Trabectedin in soft tissue sarcomas. Mar. Drugs 2015, 13, 974–983. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother. Pharmacol. 2016, 77, 663–671. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Alves, C.; Silva, J.; Pinteus, S.; Gaspar, H.; Alpoim, M.C.; Botana, L.M.; Pedrosa, R. From marine origin to therapeutics: The antitumor potential of marine algae-derived compounds. Front. Pharmacol. 2018, 9, 777. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, W.; Feeney, R. The isolation of a new thymine pentoside from sponges. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R. Contributions to the study of marine products. XXXII. the nucleosides of sponges. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Cohen, S.S. Introduction to the biochemistry of D-arabinosyl nucleosides. Prog. Nucleic Acid Res. Mol. Biol. 1966, 5, 1–88. [Google Scholar] [CrossRef]
- Wu, L.; Ye, K.; Jiang, S.; Zhou, G. Marine power on cancer: Drugs, lead compounds, and mechanisms. Mar. Drugs 2021, 19, 488. [Google Scholar] [CrossRef]
- Tattersall, M.H.; Ganeshaguru, K.; Hoffbrand, A.V. Deoxyribonucleoside triphosphate pools in human bone marrow and leukaemic cells. Antibiot. Chemother. 1980, 28, 94–101. [Google Scholar] [CrossRef]
- Kimball, A.P.; Wilson, M.J. Inhibition of DNA polymerase by beta-D-arabinosylcytosine and reversal of inhibition by deoxycytidine-5’-triphosphate. Proc. Soc. Exp. Biol. Med. 1968, 127, 429–432. [Google Scholar] [CrossRef]
- Horber, D.H.; von Ballmoos, P.; Schott, H.; Schwendener, R.A. Cell cycle-dependent cytotoxicity and induction of apoptosis by liposomal N4-hexadecyl-1-beta-D-arabinofuranosylcytosine. Br. J. Cancer 1995, 72, 1067–1073. [Google Scholar] [CrossRef]
- Matsuda, Y.; Tobari, I. Radiosensitivity and effects of repair inhibitors for X-ray-induced chromosomal damage in mouse zygotes in S and G2 phases. Int. J. Radiat. Biol. 1995, 68, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, S.; Gandhi, V.; Plunkett, W.; Zwelling, L.A. The effect of 9-beta-D-arabinofuranosyl-2-fluoroadenine and 1-beta-D-arabinofuranosylcytosine on the cell cycle phase distribution, topoisomerase II level, mitoxantrone cytotoxicity, and DNA strand break production in K562 human leukemia cells. Cancer Chemother. Pharmacol. 1996, 38, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Thomas, X. Chemotherapy of acute leukemia in adults. Expert Opin. Pharmacother. 2009, 10, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Absalon, M.J.; Smith, F.O. Treatment strategies for pediatric acute myeloid leukemia. Expert Opin. Pharmacother. 2009, 10, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Kadan-Lottick, N.S.; Brouwers, P.; Breiger, D.; Kaleita, T.; Dziura, J.; Northrup, V.; Chen, L.; Nicoletti, M.; Bostrom, B.; Stork, L.; et al. Comparison of neurocognitive functioning in children previously randomly assigned to intrathecal methotrexate compared with triple intrathecal therapy for the treatment of childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2009, 27, 5986–5992. [Google Scholar] [CrossRef] [PubMed]
- Spina, M.; Chimienti, E.; Martellotta, F.; Vaccher, E.; Berretta, M.; Zanet, E.; Lleshi, A.; Canzonieri, V.; Bulian, P.; Tirelli, U. Phase 2 study of intrathecal, long-acting liposomal cytarabine in the prophylaxis of lymphomatous meningitis in human immunodeficiency virus-related non-Hodgkin lymphoma. Cancer 2010, 116, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Omura, G.A.; Vogler, W.R.; Lefante, J.; Silberman, H.; Knospe, W.; Gordon, D.; Jarrell, R. Treatment of acute myelogenous leukemia: Influence of three induction regimens and maintenance with chemotherapy or BCG immunotherapy. Cancer 1982, 49, 1530–1536. [Google Scholar] [CrossRef]
- Goldstone, A.H.; Burnett, A.K.; Wheatley, K.; Smith, A.G.; Hutchinson, R.M.; Clark, R.E.; Medical Research Council Adult Leukemia Working Party. Attempts to improve treatment outcomes in acute myeloid leukemia (AML) in older patients: The results of the United Kingdom Medical Research Council AML11 trial. Blood 2001, 98, 1302–1311. [Google Scholar] [CrossRef]
- Dufour, P.; Mors, R.; Berthaud, P.; Lamy, T.; Bergerat, J.P.; Herbrecht, R.; Maloisel, F.; Audhuy, B.; Lioure, B.; Giron, C.; et al. Idarubicin and high dose cytarabine: A new salvage treatment for refractory or relapsing non-Hodgkin’s lymphoma. Leuk. Lymphoma 1996, 22, 329–334. [Google Scholar] [CrossRef]
- Fenaux, P.; Chomienne, C.; Degos, L. Treatment of acute promyelocytic leukaemia. Best Pract. Res. Clin. Haematol. 2001, 14, 153–174. [Google Scholar] [CrossRef]
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk. Res. 2009, 33, 129–139. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.M.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Estrov, Z.; Uehara, T.; Owa, T.; et al. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer 2018, 124, 2758–2765. [Google Scholar] [CrossRef]
- Yokoi, A.; Kuromitsu, J.; Kawai, T.; Nagasu, T.; Sugi, N.H.; Yoshimatsu, K.; Yoshino, H.; Owa, T. Profiling novel sulfonamide antitumor agents with cell-based phenotypic screens and array-based gene expression analysis. Mol. Cancer Ther. 2002, 1, 275–286. [Google Scholar]
- Dean-Colomb, W.; Esteva, F.J. Emerging agents in the treatment of anthracycline- and taxane-refractory metastatic breast cancer. Semin. Oncol. 2008, 35, S31–S38. [Google Scholar] [CrossRef]
- Scatena, C.D.; Kumer, J.L.; Arbitrario, J.P.; Howlett, A.R.; Hawtin, R.E.; Fox, J.A.; Silverman, J.A. Voreloxin, a first-in-class anticancer quinolone derivative, acts synergistically with cytarabine in vitro and induces bone marrow aplasia in vivo. Cancer Chemother. Pharmacol. 2010, 66, 881–888. [Google Scholar] [CrossRef]
- Kufe, D.W.; Major, P.P.; Egan, E.M.; Beardsley, G.P. Correlation of cytotoxicity with incorporation of ara-C into DNA. J. Biol. Chem. 1980, 255, 8900–8997. [Google Scholar] [CrossRef]
- Ravandi, F.; Ritchie, E.K.; Sayar, H.; Lancet, J.E.; Craig, M.D.; Vey, N.; Strickland, S.A.; Schiller, G.J.; Jabbour, E.; Erba, H.P.; et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): A randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol. 2015, 16, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Roboz, G.J.; Cripe, L.D.; Michelson, G.C.; Fox, J.A.; Leavitt, R.D.; Chen, T.; Hawtin, R.; Craig, A.R.; Ravandi, F.; et al. A phase 1b/2 study of vosaroxin in combination with cytarabine in patients with relapsed or refractory acute myeloid leukemia. Haematologica 2015, 100, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Sedov, V.; Stuart, R.K. Vosaroxin in relapsed/refractory acute myeloid leukemia: Efficacy and safety in the context of the current treatment landscape. Ther. Adv. Hematol. 2017, 8, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Ravandi, F. The safety and efficacy of vosaroxin in patients with first relapsed or refractory acute myeloid leukemia—A critical review. Expert Rev. Hematol. 2016, 9, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Paubelle, E.; Zylbersztejn, F.; Thomas, X. The preclinical discovery of vosaroxin for the treatment of acute myeloid leukemia. Expert Opin. Drug Discov. 2017, 12, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Ritchie, E.K.; Sayar, H.; Lancet, J.E.; Craig, M.D.; Vey, N.; Strickland, S.A.; Schiller, G.J.; Jabbour, E.; Pigneux, A.; et al. Phase 3 results for vosaroxin/cytarabine in the subset of patients ≥60 years old with refractory/early relapsed acute myeloid leukemia. Haematologica 2018, 103, e514–e518. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Hu, X.; Jiang, W.; Liu, R.; Zhang, D.; Zhang, J.; Li, Z.; Luan, Y. Rational Design of a New Self-Codelivery System from Redox-Sensitive Camptothecin-Cytarabine Conjugate Assembly for Effectively Synergistic Anticancer Therapy. Adv. Healthc. Mater. 2017, 6, 1700829. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, S.Y.; Liu, Y.Q.; Wu, X.B.; Zhu, G.X.; Zhang, Y.; Wei, W.; Liu, H.X.; Chen, A.L. Synthesis, biological activities, and quantitative structure-activity relationship (QSAR) study of novel camptothecin analogues. Molecules 2015, 20, 8634–8653. [Google Scholar] [CrossRef]
- Hansch, C.; Verma, R.P. 20-(S)-camptothecin analogues as DNA topoisomerase I inhibitors: A QSAR study. ChemMedChem 2007, 2, 1807–1813. [Google Scholar] [CrossRef]
- Lu, A.J.; Zhang, Z.S.; Zheng, M.Y.; Zou, H.J.; Luo, X.M.; Jiang, H.L. 3D-QSAR study of 20 (S)-camptothecin analogs. Acta Pharmacol. Sin. 2007, 28, 307–314. [Google Scholar] [CrossRef]
- Blower, P.E.; Chung, J.H.; Verducci, J.S.; Lin, S.; Park, J.K.; Dai, Z.; Liu, C.G.; Schmittgen, T.D.; Reinhold, W.C.; Croce, C.M.; et al. MicroRNAs modulate the chemosensitivity of tumor cells. Mol. Cancer Ther. 2008, 7, 1–9. [Google Scholar] [CrossRef]
- Blower, P.E.; Verducci, J.S.; Lin, S.; Zhou, J.; Chung, J.H.; Dai, Z.; Liu, C.G.; Reinhold, W.; Lorenzi, P.L.; Kaldjian, E.P.; et al. MicroRNA expression profiles for the NCI-60 cancer cell panel. Mol. Cancer Ther. 2007, 6, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.G.; Yang, J.M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, X.; Gu, J.; Hu, H.; Dong, D.; Yao, J.; Lin, C.; Fei, J. Anti-miR-21 oligonucleotide enhances chemosensitivity of leukemic HL60 cells to arabinosylcytosine by inducing apoptosis. Hematology 2010, 15, 215–221. [Google Scholar] [CrossRef]
- Chen, K.; Chen, Y.; Chen, Z.; Shi, Y.; He, Z.; Ding, B.; Wang, C.; Yu, L. miR-134 increases the antitumor effects of cytarabine by targeting Mnks in acute myeloid leukemia cells. Oncol. Targets Ther. 2018, 11, 3141–3147. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, W.; Jiang, H.Y.; Hickey, R.J.; Malkas, L.H. Ara-C affects formation of cancer cell DNA synthesome replication intermediates. Cancer Chemother. Pharmacol. 2000, 45, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Füller, M.; Klein, M.; Schmidt, E.; Rohde, C.; Göllner, S.; Schulze, I.; Qianli, J.; Berdel, W.E.; Edemir, B.; Müller-Tidow, C.; et al. 5-azacytidine enhances efficacy of multiple chemotherapy drugs in AML and lung cancer with modulation of CpG methylation. Int. J. Oncol. 2015, 46, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ye, X.; Ji, D.; Zhang, H.; Sun, F.; Shang, C.; Zhang, Y.; Wu, E.; Wang, F.; Wu, F.; et al. Inhibition of lung tumor growth by targeting EGFR/VEGFR-Akt/NF-κB pathways with novel theanine derivatives. Oncotarget 2014, 5, 8528–8543. [Google Scholar] [CrossRef]
- Phadke, M.; Krynetskaia, N.; Krynetskiy, E. Cytotoxicity of chemotherapeutic agents in glyceraldehyde-3-phosphate dehydrogenase-depleted human lung carcinoma A549 cells with the accelerated senescence phenotype. Anticancer Drugs 2013, 24, 366–374. [Google Scholar] [CrossRef]
- Rechkoblit, O.; Choudhury, J.R.; Buku, A.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural basis for polymerase η-promoted resistance to the anticancer nucleoside analog cytarabine. Sci. Rep. 2018, 8, 12702. [Google Scholar] [CrossRef]
- Qi, W.; Yan, X.; Xu, X.; Song, B.; Sun, L.; Zhao, D.; Sun, L. The effects of cytarabine combined with ginsenoside compound K synergistically induce DNA damage in acute myeloid leukemia cells. Biomed. Pharmacother. 2020, 132, 110812. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vázquez, G.O.; Diaz-Quiñones, A.O.; Chorna, N.; Salgado-Villanueva, I.K.; Tang, J.; Ortiz, W.I.S.; Maldonado, H.M. Synergistic interactions of cytarabine-adavosertib in leukemic cell lines proliferation and metabolomic endpoints. Biomed. Pharmacother. 2023, 166, 115352. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Xiu, M.; Li, S.; Shi, Y.; Wang, X.; Lin, X.; Cai, H.; Liu, Y.; He, J. Exposure to cytarabine causes side effects on adult development and physiology and induces intestinal damage via apoptosis in Drosophila. Biomed. Pharmacother. 2023, 159, 114265. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic guanidine alkaloids from the marine sponge Crambe crambe and Ca++ channel blocker activity of crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef]
- Jares-Erijman, E.; Sakai, R.; Rinehar, K. Crambescidins: New antiviral and cytotoxic compounds from the sponge Crambe crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Aron, Z.D.; Pietraszkiewicz, H.; Overman, L.E.; Valeriote, F.; Cuevas, C. Synthesis and anticancer activity of side chain analogs of the crambescidin alkaloids. Bioorg. Med. Chem. Lett. 2004, 14, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Aron, Z.D.; Overman, L.E. Total synthesis and properties of the crambescidin core zwitterionic acid and crambescidin 359. J. Am. Chem. Soc. 2005, 127, 3380–3390. [Google Scholar] [CrossRef] [PubMed]
- Overman, L.E.; Rhee, Y.H. Total synthesis of (−)-crambidine and definition of the relative configuration of its unique tetracyclic guanidinium core. J. Am. Chem. Soc. 2005, 127, 15652–15658. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K. Total synthesis of marine cyclic guanidine compounds and development of novel guanidine type asymmetric organocatalysts. Yakugaku Zasshi 2003, 123, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Hashimoto, Y. Synthesis of marine guanidine alkaloids and their application as chemical/biological tools. Chem. Rec. 2003, 3, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Georgieva, A.; Koshino, H.; Nakata, T.; Kita, T.; Hashimoto, Y. Total synthesis of crambescidin 359. Org. Lett. 2002, 4, 177–180. [Google Scholar] [CrossRef]
- Braekman, J.C.; Daloze, D.; Tavares, R.; Hajdu, E.; Van Soest, R.W.M. Novel polycyclic guanidine alkaloids from two marine sponges of the genus Monanchora. J. Nat. Prod. 2000, 63, 193–196. [Google Scholar] [CrossRef]
- Chang, L.; Whittaker, N.F.; Bewley, C.A. Crambescidin 826 and dehydrocrambine A: New polycyclic guanidine alkaloids from the marine sponge Monanchora sp. that inhibit HIV-1 fusion. J. Nat. Prod. 2003, 66, 1490–1494. [Google Scholar] [CrossRef]
- Olszewski, A.; Sato, K.; Aron, Z.D.; Cohen, F.; Harris, A.; McDougall, B.R.; Robinson, W.E.; Overman, L.E.; Weiss, G.A. Guanidine alkaloid analogs as inhibitors of HIV-1 Nef interactions with p53, actin, and p56lck. Proc. Natl. Acad. Sci. USA 2004, 101, 14079–14084. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid differentiation in K562 chronic myelogenous cells induced by crambescidin 800, a pentacyclic guanidine alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Kasmiati, K.; Yoshioka, Y.; Okamoto, T.; Ojika, M. New crambescidin-type alkaloids from the Indonesian marine sponge Clathria bulbotoxa. Mar. Drugs 2018, 16, 84. [Google Scholar] [CrossRef]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Crambescidin 800, isolated from the marine sponge Monanchora viridis, induces cell cycle arrest and apoptosis in triple-negative breast cancer cells. Mar. Drugs 2018, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Martín, V.; Vale, C.; Bondu, S.; Thomas, O.P.; Vieytes, M.R.; Botana, L.M. Differential effects of crambescins and crambescidin 816 in voltage-gated sodium, potassium and calcium channels in neurons. Chem. Res. Toxicol. 2013, 26, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.G.; Juncal, A.B.; Silva, S.B.L.; Thomas, O.P.; Martín Vázquez, V.; Alfonso, A.; Vieytes, M.R.; Vale, C.; Botana, L.M. The marine guanidine alkaloid crambescidin 816 induces calcium influx and cytotoxicity in primary cultures of cortical neurons through glutamate receptors. ACS Chem. Neurosci. 2017, 8, 1609–1617. [Google Scholar] [CrossRef]
- Shubina, L.K.; Makarieva, T.N.; von Amsberg, G.; Denisenko, V.A.; Popov, R.S.; Dyshlovoy, S.A. Monanchoxymycalin C with anticancer properties, new analogue of crambescidin 800 from the marine sponge Monanchora pulchra. Nat. Prod. Res. 2019, 33, 1415–1422. [Google Scholar] [CrossRef]
- Rubiolo, J.A.; Ternon, E.; López-Alonso, H.; Thomas, O.P.; Vega, F.V.; Vieytes, M.R.; Botana, L.M. Crambescidin-816 acts as a fungicidal with more potency than crambescidin-800 and -830, inducing cell cycle arrest, increased cell size and apoptosis in Saccharomyces cerevisiae. Mar. Drugs 2013, 11, 4419–4434. [Google Scholar] [CrossRef]
- Roel, M.; Rubiolo, J.A.; Guerra-Varela, J.; Silva, S.B.; Thomas, O.P.; Cabezas-Sainz, P.; Sánchez, L.; López, R.; Botana, L.M. Marine guanidine alkaloids crambescidins inhibit tumor growth and activate intrinsic apoptotic signaling inducing tumor regression in a colorectal carcinoma zebrafish xenograft model. Oncotarget 2016, 7, 83071–83087. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Pecoraro, C.; Carbone, D.; Aiello, D.; Carbone, A. Synthesis and cytotoxic activity of 3-[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-c]pyridine hydrobromides, analogues of the marine alkaloid nortopsentin. Arkivoc 2022, ii, 30–42. [Google Scholar] [CrossRef]
- Pecoraro, C.; Terrana, F.; Panzeca, G.; Parrino, B.; Cascioferro, S.; Diana, P.; Giovannetti, E.; Carbone, D. Nortopsentins as Leads from Marine Organisms for Anticancer and Anti-Inflammatory Agent Development. Molecules 2023, 28, 6450. [Google Scholar] [CrossRef]
- Carbone, D.; Vestuto, V.; Ferraro, M.R.; Ciaglia, T.; Pecoraro, C.; Sommella, E.; Cascioferro, S.; Salviati, E.; Novi, S.; Tecce, M.F.; et al. Metabolomics-assisted discovery of a new anticancer GLS-1 inhibitor chemotype from a nortopsentin-inspired library: From phenotype screening to target identification. Eur. J. Med. Chem. 2022, 234, 114233. [Google Scholar] [CrossRef] [PubMed]
- Parrino, B.; Attanzio, A.; Spanò, V.; Cascioferro, S.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Diana, P.; Cirrincione, G.; Carbone, A. Synthesis, antitumor activity and CDK1 inhibition of new thiazole nortopsentin analogues. Eur. J. Med. Chem. 2017, 138, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Vita, G.D.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Livrea, M.A.; Diana, P.; et al. Synthesis and antiproliferative activity of thiazolyl-bis-pyrrolo[2,3-b]pyridines and indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, nortopsentin analogues. Mar. Drugs 2015, 13, 460–492. [Google Scholar] [CrossRef] [PubMed]
- Sofi, S.; Mehraj, U.; Qayoom, H.; Aisha, S.; Almilaibary, A.; Alkhanani, M.; Mir, M.A. Targeting cyclin-dependent kinase 1 (CDK1) in cancer: Molecular docking and dynamic simulations of potential CDK1 inhibitors. Med. Oncol. 2022, 39, 133. [Google Scholar] [CrossRef]
- Di Franco, S.; Parrino, B.; Gaggianesi, M.; Pantina, V.D.; Bianca, P.; Nicotra, A.; Mangiapane, L.R.; Lo Iacono, M.; Ganduscio, G.; Veschi, V.; et al. CHK1 inhibitor sensitizes resistant colorectal cancer stem cells to nortopsentin. iScience 2021, 24, 102664. [Google Scholar] [CrossRef]
- Cascioferro, S.; Attanzio, A.; Di Sarno, V.; Musella, S.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. New 1,2,4-oxadiazole nortopsentin derivatives with cytotoxic activity. Mar. Drugs 2019, 17, 35. [Google Scholar] [CrossRef]
- Carbone, D.; Pecoraro, C.; Panzeca, G.; Xu, G.; Roeten, M.S.F.; Cascioferro, S.; Giovannetti, E.; Diana, P.; Parrino, B. 1,3,4-Oxadiazole and 1,3,4-thiadiazole nortopsentin derivatives against pancreatic ductal adenocarcinoma: Synthesis, cytotoxic activity, and inhibition of CDK1. Mar. Drugs 2023, 21, 412. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) inhibition as a novel therapeutic strategy against pancreatic ductal adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, R.; Tej, M.B.; Jadav, S.S.; Sujitha, P.; Kumar, C.G.; Raju, R.R. Synthesis, anticancer evaluation and molecular docking studies of 2,5-bis(indolyl)-1,3,4-oxadiazoles, Nortopsentin analogues. J. Mol. Struct. 2020, 1208, 127875. [Google Scholar] [CrossRef]
- Spanò, V.; Attanzio, A.; Cascioferro, S.; Carbone, A.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. Synthesis and antitumor activity of new thiazole nortopsentin analogs. Mar. Drugs 2016, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Tursch, B.; Braekman, J.; Daloze, D.; Herin, M.; Karlsson, R.; Losman, D. Chemical studies of marine invertebrates—XI: Sinulariolide, a new cembranolide diterpene from the soft coral Sinularia flexibilis (coelenterata, octocorallia, alcyonacea). Tetrahedron Lett. 1975, 31, 129–133. [Google Scholar] [CrossRef]
- Weinheimer, A.; Matson, J.; Bilayet, H.; van der Helm, D. Marine anticancer agents: Sinularin and dihydrosinularin, new cembranolides from the soft coral, Sinularia flexibilis. Tetrahedron Lett. 1977, 18, 2923–2926. [Google Scholar] [CrossRef]
- Hsieh, P.W.; Chang, F.R.; McPhail, A.T.; Lee, K.H.; Wu, Y.C. New cembranolide analogues from the formosan soft coral Sinularia flexibilis and their cytotoxicity. Nat. Prod. Res. 2003, 17, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Neoh, C.A.; Wang, R.Y.; Din, Z.H.; Su, J.H.; Chen, Y.K.; Tsai, F.J.; Weng, S.H.; Wu, Y.J. Induction of apoptosis by sinulariolide from soft coral through mitochondrial-related and p38MAPK pathways on human bladder carcinoma cells. Mar. Drugs 2012, 10, 2893–2911. [Google Scholar] [CrossRef]
- Cheng, T.C.; Din, Z.H.; Su, J.H.; Wu, Y.J.; Liu, C.I. Sinulariolide suppresses cell migration and invasion by inhibiting matrix metalloproteinase-2/-9 and urokinase through the PI3K/AKT/mTOR signaling pathway in human bladder cancer cells. Mar. Drugs 2017, 15, 238. [Google Scholar] [CrossRef]
- Li, H.H.; Su, J.H.; Chiu, C.C.; Lin, J.J.; Yang, Z.Y.; Hwang, W.I.; Chen, Y.K.; Lo, Y.H.; Wu, Y.J. Proteomic investigation of the sinulariolide-treated melanoma cells A375: Effects on the cell apoptosis through mitochondrial-related pathway and activation of caspase cascade. Mar. Drugs 2013, 11, 2625–2642. [Google Scholar] [CrossRef]
- Chen, Y.J.; Su, J.H.; Tsao, C.Y.; Hung, C.T.; Chao, H.H.; Lin, J.J.; Liao, M.H.; Yang, Z.Y.; Huang, H.H.; Tsai, F.J.; et al. Sinulariolide induced hepatocellular carcinoma apoptosis through activation of mitochondrial-related apoptotic and PERK/eIF2α/ATF4/CHOP pathway. Molecules 2013, 18, 10146–10161. [Google Scholar] [CrossRef]
- Wu, Y.J.; Neoh, C.A.; Tsao, C.Y.; Su, J.H.; Li, H.H. Sinulariolide suppresses human hepatocellular carcinoma cell migration and invasion by inhibiting matrix metalloproteinase-2/-9 through MAPKs and PI3K/Akt signaling pathways. Int. J. Mol. Sci. 2015, 16, 16469–16482. [Google Scholar] [CrossRef]
- Rinehart, K.; Holt, T.; Fregeau, N.; Stroh, J.; Keifer, P.; Sun, F.; Li, L.; Martin, D. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Simoens, C.; Korst, A.E.; De Pooter, C.M.; Lambrechts, H.A.; Pattyn, G.G.; Faircloth, G.T.; Lardon, F.; Vermorken, J.B. In vitro interaction between ecteinascidin 743 (ET-743) and radiation, in relation to its cell cycle effects. Br. J. Cancer 2003, 89, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Serra, J.; Maffiotte, E.; Martín, J.; Bex, T.; Navarro-Palou, M.; Ros, T.; Plazas, J.M.; Vögler, O.; Gutiérrez, A.; Amat, J.C.; et al. Yondelis® (ET-743, Trabectedin) sensitizes cancer cell lines to CD95-mediated cell death: New molecular insight into the mechanism of action. Eur. J. Pharmacol. 2011, 658, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Vermeir, M.; Hemeryck, A.; Cuyckens, F.; Francesch, A.; Bockx, M.; Van Houdt, J.; Steemans, K.; Mannens, G.; Avilés, P.; De Coster, R. In vitro studies on the metabolism of trabectedin (YONDELIS) in monkey and man, including human CYP reaction phenotyping. Biochem. Pharmacol. 2009, 77, 1642–1654. [Google Scholar] [CrossRef]
- Grosso, F.; Sanfilippo, R.; Virdis, E.; Piovesan, C.; Collini, P.; Dileo, P.; Morosi, C.; Tercero, J.C.; Jimeno, J.; D’Incalci, M.; et al. Trabectedin in myxoid liposarcomas (MLS): A long-term analysis of a single-institution series. Ann. Oncol. 2009, 20, 1439–1444. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef]
- Chu, Q.; Mita, A.; Forouzesh, B.; Tolcher, A.W.; Schwartz, G.; Nieto, A.; Soto-Matos, A.; Alfaro, V.; Lebedinsky, C.; Rowinsky, E.K. Phase I and pharmacokinetic study of sequential paclitaxel and trabectedin every 2 weeks in patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 2656–2665. [Google Scholar] [CrossRef]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Grosso, F.; Scambia, G.; Raspagliesi, F.; Colombo, N.; Grignani, G.; Casali, P.; Sanfilippo, R.; Buonadonna, A.; Santoro, A.; et al. A phase II randomised (calibrated design) study on the activity of the single-agent trabectedin in metastatic or locally relapsed uterine leiomyosarcoma. Br. J. Cancer 2018, 119, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Guerrieri, M.E. Pharmacological treatment for uterine leiomyosarcomas. Expert Opin. Pharmacother. 2015, 16, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Martin-Liberal, J.; Pérez, E.; García Del Muro, X. Investigational therapies in phase II clinical trials for the treatment of soft tissue sarcoma. Expert Opin. Investig. Drugs 2019, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Camorani, S.; Cerchia, L.; Fedele, M.; Erba, E.; D’Incalci, M.; Crescenzi, E. Trabectedin modulates the senescence-associated secretory phenotype and promotes cell death in senescent tumor cells by targeting NF-κB. Oncotarget 2018, 9, 19929–19944. [Google Scholar] [CrossRef] [PubMed]
- Loria, R.; Laquintana, V.; Bon, G.; Trisciuoglio, D.; Frapolli, R.; Covello, R.; Amoreo, C.A.; Ferraresi, V.; Zoccali, C.; Novello, M.; et al. HMGA1/E2F1 axis and NFkB pathways regulate LPS progression and trabectedin resistance. Oncogene 2018, 37, 5926–5938. [Google Scholar] [CrossRef]
- Dang, V.T.; Benkendorff, K.; Green, T.; Speck, P. Marine snails and slugs: A great place to look for antiviral drugs. J. Virol. 2015, 89, 8114–8118. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Estévez-Calvar, N.; Dios, S.; Figueras, A.; Novoa, B. New insights into the apoptotic process in mollusks: Characterization of caspase genes in Mytilus galloprovincialis. PLoS ONE 2011, 6, e17003. [Google Scholar] [CrossRef] [PubMed]
- Kanagasabapathy, S.; Samuthirapandian, R.; Kumaresan, M. Preliminary studies for a new antibiotic from the marine mollusk Melo melo (Lightfoot, 1786). Asian Pac. J. Trop. Med. 2011, 4, 310–314. [Google Scholar] [CrossRef]
- Kiran, N.; Siddiqui, G.; Khan, A.N.; Ibrar, K.; Tushar, P. Extraction and screening of bioactive compounds with antimicrobial properties from selected species of mollusk and crustacean. J. Clin. Cell 2014, 5, 1. [Google Scholar] [CrossRef]
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J.; Dunbar, D.C. Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef]
- Davis, J.; Fricke, W.F.; Hamann, M.T.; Esquenazi, E.; Dorrestein, P.C.; Hill, R.T. Characterization of the bacterial community of the chemically defended Hawaiian sacoglossan Elysia rufescens. Appl. Environ. Microbiol. 2013, 79, 7073–7081. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; Horenblas, S.; Meinhardt, W.; Stokvis, E.; de Reijke, T.M.; Jimeno, J.M.; Lopez-Lazaro, L.; Lopez Martin, J.A.; Beijnen, J.H.; Schellens, J.H. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin. Cancer Res. 2005, 11, 1854–1862. [Google Scholar] [CrossRef]
- García-Rocha, M.; Bonay, P.; Avila, J. The antitumoral compound Kahalalide F acts on cell lysosomes. Cancer Lett. 1996, 99, 43–50. [Google Scholar] [CrossRef]
- Wyer, S.; Townsend, D.M.; Ye, Z.; Kourtidis, A.; Choo, Y.M.; Branco de Barros, A.L.; Donia, M.S.; Hamann, M.T. Recent advances and limitations in the application of kahalalides for the control of cancer. Biomed. Pharmacother. 2022, 148, 112676. [Google Scholar] [CrossRef]
- Zan, J.; Li, Z.; Tianero, M.D.; Davis, J.; Hill, R.T.; Donia, M.S. A microbial factory for defensive kahalalides in a tripartite marine symbiosis. Science 2019, 364, eaaw6732. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Morrissey, R.L.; Faircloth, G.T.; Levine, B.S. Preclinical toxicity studies of kahalalide F, a new anticancer agent: Single and multiple dosing regimens in the rat. Cancer Chemother. Pharmacol. 2002, 50, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Sewell, J.M.; Mayer, I.; Langdon, S.P.; Smyth, J.F.; Jodrell, D.I.; Guichard, S.M. The mechanism of action of Kahalalide F: Variable cell permeability in human hepatoma cell lines. Eur. J. Cancer 2005, 41, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, M.L.; Kruyt, F.A.; Jimeno, J.M.; Rodriguez, J.A.; Giaccone, G. Kahalalide F (KF) induces caspase-independent cytotoxicity that correlates with HER2/neu and/or HER3 expression levels and is accompanied by down-regulation of Akt signaling. Am. Assoc. Cancer Res. 2004, 64 (Suppl. S7), 1229. [Google Scholar]
- Pardo, B.; Paz-Ares, L.; Tabernero, J.; Ciruelos, E.; García, M.; Salazar, R.; López, A.; Blanco, M.; Nieto, A.; Jimeno, J.; et al. Phase I clinical and pharmacokinetic study of kahalalide F administered weekly as a 1-hour infusion to patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 1116–1123. [Google Scholar] [CrossRef]
- Salazar, R.; Cortés-Funes, H.; Casado, E.; Pardo, B.; López-Martín, A.; Cuadra, C.; Tabernero, J.; Coronado, C.; García, M.; Soto Matos-Pita, A.; et al. Phase I study of weekly kahalalide F as prolonged infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 72, 75–83. [Google Scholar] [CrossRef]
- Serova, M.; de Gramont, A.; Bieche, I.; Riveiro, M.E.; Galmarini, C.M.; Aracil, M.; Jimeno, J.; Faivre, S.; Raymond, E. Predictive factors of sensitivity to elisidepsin, a novel Kahalalide F-derived marine compound. Mar. Drugs 2013, 11, 944–959. [Google Scholar] [CrossRef]
- Sánchez, A.M.; Malagarie-Cazenave, S.; Olea, N.; Vara, D.; Cuevas, C.; Díaz-Laviada, I. Spisulosine (ES-285) induces prostate tumor PC-3 and LNCaP cell death by de novo synthesis of ceramide and PKCzeta activation. Eur. J. Pharmacol. 2008, 584, 237–245. [Google Scholar] [CrossRef]
- Dinda, S.K.; Das, S.K.; Panda, G. Asymmetric total syntheses of spisulosine, its diastereo- and regio-isomers. Tetrahedron 2010, 66, 9304–9309. [Google Scholar] [CrossRef]
- Cuadros, R.; Montejo de Garcini, E.; Wandosell, F.; Faircloth, G.; Fernández-Sousa, J.M.; Avila, J. The marine compound spisulosine, an inhibitor of cell proliferation, promotes the disassembly of actin stress fibers. Cancer Lett. 2000, 152, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Grünwald, V.; Schöffski, P.; Singer, H.; Salazar, R.; Iglesias, J.L.; Casado, E.; Cullell-Young, M.; Baselga, J.; Tabernero, J. A phase I dose-escalating study of ES-285, a marine sphingolipid-derived compound, with repeat dose administration in patients with advanced solid tumors. Investig. New Drugs 2012, 30, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, M.; Cuevas, C.; Alonso, J.L.; Otero, G.; Faircloth, G.; Fernandez-Sousa, J.M.; Avila, J.; Wandosell, F. The marine sphingolipid-derived compound ES 285 triggers an atypical cell death pathway. Apoptosis 2007, 12, 395–409. [Google Scholar] [CrossRef]
- Silveira-Dorta, G.; Sousa, I.J.; Fernandes, M.X.; Martín, V.S.; Padrón, J.M. Synthesis and identification of unprecedented selective inhibitors of CK1ε. Eur. J. Med. Chem. 2015, 96, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, P.; Shaw, A.K. An efficient total synthesis of the anticancer agent (+)-spisulosine (ES-285) from Garner’s aldehyde. Tetrahedron Lett. 2010, 51, 4140–4142. [Google Scholar] [CrossRef]
- Fabišíková, M.; Martinková, M.; Hirková, S.; Gonda, J.; Pilátová, M.B.; Gönciová, G. Total synthesis and the anticancer activity of (+)-spisulosine. Carbohydr. Res. 2016, 435, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Dumez, H.; Ruijter, R.; Miguel-Lillo, B.; Soto-Matos, A.; Alfaro, V.; Giaccone, G. Spisulosine (ES-285) given as a weekly three-hour intravenous infusion: Results of a phase I dose-escalating study in patients with advanced solid malignancies. Cancer Chemother. Pharmacol. 2011, 68, 1397–1403. [Google Scholar] [CrossRef]
- Abad, J.L.; Nieves, I.; Rayo, P.; Casas, J.; Fabrias, G.; Delgado, A. Straightforward access to spisulosine and 4,5-dehydrospisulosine stereoisomers: Probes for profiling ceramide synthase activities in intact cells. J. Org. Chem. 2013, 78, 5858–5866. [Google Scholar] [CrossRef]
- Rao, M.V.; Reddy, K.k.S.; Rao, B.V. An efficient synthesis of N-Boc-(2S,3S)-3-hydroxy-2-phenyl piperidine and N-Boc-safingol. Tetrahedron Lett. 2012, 53, 5993–5995. [Google Scholar] [CrossRef]
- Jain, V.K.; Ramapanicker, R. Diastereoselective synthesis of D-threo-sphinganine, L-erythro-sphinganine and (−)-spisulosine through asymmetric α-hydroxylation of a higher homologue of Garner’s aldehyde. Tetrahedron 2017, 73, 1568–1575. [Google Scholar] [CrossRef]
- Pilatova, M.B.; Nosalova, N.; Ockajakova, G.; Kello, M.; Kotorova, K.; Takac, P.; Petik, P.; Bohus, P.; Stankova, K.; Martinkova, M.; et al. Homospisulosine induced apoptosis in cervical carcinoma cells is associated with phosphorylation of Bcl-2 and up-regulation of p27/Kip1. J. Appl. Biomed. 2023, 21, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Cavaliere, P.; Wahidullah, S.; Naik, C.G.; Cimino, G. A new antitumor isoquinoline alkaloid from the marine nudibranch Jorunna funebris. Tetrahedron 2000, 56, 7305–7308. [Google Scholar] [CrossRef]
- Lane, J.W.; Chen, Y.; Williams, R.M. Asymmetric Total Syntheses of (−)-Jorumycin, (−)-Renieramycin G, 3-epi-Jorumycin, and 3-epi-Renieramycin G. J. Am. Chem. Soc. 2005, 127, 12684–12690. [Google Scholar] [CrossRef]
- Liu, W.; Liao, X.; Dong, W.; Yan, Z.; Wang, N.; Liu, Z. Total synthesis and cytotoxicity of (−)-jorumycin and its analogues. Tetrahedron 2012, 68, 2759–2764. [Google Scholar] [CrossRef]
- Lane, J.W.; Estevez, A.; Mortara, K.; Callan, O.; Spencer, J.R.; Williams, R.M. Antitumor activity of tetrahydroisoquinoline analogues 3-epi-jorumycin and 3-epi-renieramycin G. Bioorganic Med. Chem. Lett. 2006, 16, 3180–3183. [Google Scholar] [CrossRef]
- Ocio, E.M.; Maiso, P.; Chen, X.; Garayoa, M.; Alvarez-Fernández, S.; San-Segundo, L.; Vilanova, D.; López-Corral, L.; Montero, J.C.; Hernández-Iglesias, T.; et al. Zalypsis: A novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood 2009, 113, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.F.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; Cuevas, C.; et al. Molecular pharmacology and antitumor activity of Zalypsis in several human cancer cell lines. Biochem. Pharmacol. 2009, 78, 162–170. [Google Scholar] [CrossRef]
- Petek, B.J.; Jones, R.L. PM00104 (Zalypsis®): A marine derived alkylating agent. Molecules 2014, 19, 12328–12335. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Antony, S.; Pommier, Y. Zalypsis (PM00104) is a potent inducer of γ-H2AX foci and reveals the importance of the Cring of trabectedin for transcription-coupled repair inhibition. Mol. Cancer Ther. 2009, 8, 2007–2014. [Google Scholar] [CrossRef]
- Castellano, D.E.; Bellmunt, J.; Maroto, J.P.; Font-Pous, A.; Morales-Barrera, R.; Ghanem, I.; Suarez, C.; Martín Lorente, C.; Etxaniz, O.; Capdevila, L.; et al. Phase II clinical trial of PM00104 (Zalypsis®) in urothelial carcinoma patients progressing after first-line platinum-based regimen. Cancer Chemother. Pharmacol. 2014, 73, 857–867. [Google Scholar] [CrossRef]
- Massard, C.; Margetts, J.; Amellal, N.; Drew, Y.; Bahleda, R.; Stephens, P.; Stevens, P.; Armand, J.P.; Calvert, H.; Soria, J.C.; et al. Phase I study of PM00104 (Zalypsis®) administered as a 1-hour weekly infusion resting every fourth week in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 623–630. [Google Scholar] [CrossRef]
- Jones, R.L.; Ferrari, S.; Blay, J.Y.; Navid, F.; Lardelli, P.; Alfaro, V.; Siguero, M.; Soman, N.; Chawla, S.P. A Phase II multicenter, open-label, clinical and pharmokinetic trial of PM00104 in patients with advanced Ewing Family of Tumors. Investig. New Drugs 2014, 32, 171–177. [Google Scholar] [CrossRef]
- Martin, L.P.; Krasner, C.; Rutledge, T.; Ibañes, M.L.; Fernández-García, E.M.; Kahatt, C.; Gómez, M.S.; McMeekin, S. Phase II study of weekly PM00104 (ZALYPSIS®) in patients with pretreated advanced/metastatic endometrial or cervical cancer. Med. Oncol. 2013, 30, 627. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Health, US National Library of Medicine. Available online: https://classic.clinicaltrials.gov/ct2/show/results/NCT01222767?term=PM00104&draw=1&rank=1 (accessed on 17 January 2024).
- Palanisamy, S.K.; Rajendran, N.M.; Marino, A. Natural Products Diversity of Marine Ascidians (Tunicates; Ascidiacea) and Successful Drugs in Clinical Development. Nat. Prod. Bioprospect. 2017, 7, 1–111. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Donia, M.S.; McIntosh, J.A.; Fricke, W.F.; Ravel, J. Origin and variation of tunicate secondary metabolites. J. Nat. Prod. 2012, 75, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Darro, F.; Bontemps-Subielos, N.; Decaestecker, C.; Bastide, J.; Frydman, A.; Kiss, R. Synthesis and characterization of the antitumor activities of analogues of meridine, a marine pyridoacridine alkaloid. J. Med. Chem. 2001, 44, 3275–3282. [Google Scholar] [CrossRef] [PubMed]
- Bontemps, N.; Delfourne, E.; Bastide, J.; Francisco, C.; Brachers, F. Total synthesis of the marine pentacyclic alkaloid meridine. Tetrahedron Lett. 1997, 53, 1743–1750. [Google Scholar] [CrossRef]
- Barnes, E.C.; Said, N.A.B.M.; Williams, E.D.; Hooper, J.N.A.; Davis, R. Ecionines A and B, two new cytotoxic pyridoacridine alkaloids from the Australian marine sponge, Ecionemia geodides. Tetrahedron 2010, 66, 283–287. [Google Scholar] [CrossRef]
- Guittat, L.; De Cian, A.; Rosu, F.; Gabelica, V.; De Pauw, E.; Delfourne, E.; Mergny, J.L. Ascididemin and meridine stabilise G-quadruplexes and inhibit telomerase in vitro. Biochim. Biophys. Acta 2005, 1724, 375–384. [Google Scholar] [CrossRef]
- Rinehart, K.L. Antitumor compounds from tunicates. Med. Res. Rev. 2000, 20, 1–27. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Tulla-Puche, J.; Albericio, F. “Head-to-side-chain” cyclodepsipeptides of marine origin. Mar. Drugs 2013, 11, 1693–1717. [Google Scholar] [CrossRef] [PubMed]
- Schwartsmann, G.; Da Rocha, A.B.; Mattei, J.; Lopes, R. Marine-derived anticancer agents in clinical trials. Expert Opin. Investig. Drugs 2003, 12, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.H.; Wang, Y.J.; Sheng, J.; Wang, F.; Zheng, Y.; Lin, X.K.; Sun, M. Antitumor peptides from marine organisms. Mar. Drugs 2011, 9, 1840–1859. [Google Scholar] [CrossRef]
- Potts, M.B.; McMillan, E.A.; Rosales, T.I.; Kim, H.S.; Ou, Y.H.; Toombs, J.E.; Brekken, R.A.; Minden, M.D.; MacMillan, J.B.; White, M.A. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat. Chem. Biol. 2015, 11, 401–408. [Google Scholar] [CrossRef]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef]
- Mateos, M.V.; Cibeira, M.T.; Richardson, P.G.; Prosper, F.; Oriol, A.; de la Rubia, J.; Lahuerta, J.J.; García-Sanz, R.; Extremera, S.; Szyldergemajn, S.; et al. Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. 2010, 16, 3260–3269. [Google Scholar] [CrossRef]
- Faivre, S.; Chièze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.P.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.A.; Belanger, K.; Seymour, L.; Matthews, S.; Roach, J.; Dionne, J.; Soulieres, D.; Stewart, D.; Goel, R.; Charpentier, D.; et al. Phase I study of Aplidine in a dailyx5 one-hour infusion every 3 weeks in patients with solid tumors refractory to standard therapy. A National Cancer Institute of Canada Clinical Trials Group study: NCIC CTG IND 115. Ann. Oncol. 2006, 17, 1371–1378. [Google Scholar] [CrossRef]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Micol, V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Huang, S.Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Martínez, S.; Extremera, S.; et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann. Hematol. 2019, 98, 2139–2150. [Google Scholar] [CrossRef]
- Leisch, M.; Egle, A.; Greil, R. Plitidepsin: A potential new treatment for relapsed/refractory multiple myeloma. Future Oncol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Mateos, M.V.; Prosper, F.; Martin Sánchez, J.; Ocio, E.M.; Oriol, A.; Motlló, C.; Michot, J.M.; Jarque, I.; Iglesias, R.; Solé, M.; et al. Phase I study of plitidepsin in combination with bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Cancer Med. 2023, 12, 3999–4009. [Google Scholar] [CrossRef]
- van Poppel, G.; van den Berg, H. Vitamins and cancer. Cancer Lett. 1997, 114, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yuan, J.P.; Wu, C.F.; Wang, J.H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tang, Y.; Zhang, Y.; Zhang, S.; Qu, J.; Wang, X.; Kong, R.; Han, C.; Liu, Z. Fucoxanthin: A promising medicinal and nutritional ingredient. Evid.-Based Complement. Altern. Med. 2015, 2015, 723515. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Ko, S.C.; Kang, S.M.; Kang, H.S.; Kim, J.P.; Kim, S.H.; Lee, K.W.; Cho, M.G.; Jeon, Y.J. Cytoprotective effect of fucoxanthin isolated from brown algae Sargassum siliquastrum against H2O2-induced cell damage. Eur. Food Res. Technol. 2008, 228, 145–151. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Mendonca, P.; Elhag, R.; Soliman, K.F.A. Anticancer effects of fucoxanthin through cell cycle arrest, apoptosis induction, angiogenesis inhibition, and autophagy modulation. Int. J. Mol. Sci. 2022, 23, 16091. [Google Scholar] [CrossRef] [PubMed]
- Méresse, S.; Fodil, M.; Fleury, F.; Chénais, B. Fucoxanthin, a marine-derived carotenoid from brown seaweeds and microalgae: A promising bioactive compound for cancer therapy. Int. J. Mol. Sci. 2020, 21, 9273. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Hosokawa, M.; Miyashita, K. Fucoxanthin: A marine carotenoid exerting anti-cancer effects by affecting multiple mechanisms. Mar. Drugs 2013, 11, 5130–5147. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, J.; Zhang, Y.; Wang, Z.; Yang, Y.; Bai, M.; Dai, Y. Fucoxanthin activates apoptosis via inhibition of pi3k/akt/mtor pathway and suppresses invasion and migration by restriction of p38-MMP-2/9 pathway in human glioblastoma cells. Neurochem. Res. 2016, 41, 2728–2751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Liu, T.Y.; Zhang, L.T.; Hua, Z.H.; Jin, X.A.; Xu, F.; Ji, J.C.; Xu, B.H.; Ding, H.M. Effects and mechanisms of fucoxanthin from Hizikia fusiforme on inhibiting tongue squamous cell carcinoma proliferation via AKT/mTOR-mediated glycolysis. J. Food Biochem. 2023, 2023, 7944733. [Google Scholar] [CrossRef]
- Ye, G.; Wang, L.; Yang, K.; Wang, C. Fucoxanthin may inhibit cervical cancer cell proliferation via downregulation of HIST1H3D. J. Int. Med. Res. 2020, 48, 300060520964011. [Google Scholar] [CrossRef]
- Zorofchian Moghadamtousi, S.; Karimian, H.; Khanabdali, R.; Razavi, M.; Firoozinia, M.; Zandi, K.; Abdul Kadir, H. Anticancer and antitumor potential of fucoidan and fucoxanthin, two main metabolites isolated from brown algae. Sci. World J. 2014, 2014, 768323. [Google Scholar] [CrossRef]
- Bracht, K.; Nicholls, A.M.; Liu, Y.; Bodmer, W.F. 5-Fluorouracil response in a large panel of colorectal cancer cell lines is associated with mismatch repair deficiency. Br. J. Cancer 2010, 103, 340–346. [Google Scholar] [CrossRef]
- Lopes-Costa, E.; Abreu, M.; Gargiulo, D.; Rocha, E.; Ramos, A.A. Anticancer effects of seaweed compounds fucoxanthin and phloroglucinol, alone and in combination with 5-fluorouracil in colon cells. J. Toxicol. Environ. Health A 2017, 80, 776–787. [Google Scholar] [CrossRef]
- Almeida, T.P.; Ferreira, J.; Vettorazzi, A.; Azqueta, A.; Rocha, E.; Ramos, A.A. Cytotoxic activity of fucoxanthin, alone and in combination with the cancer drugs imatinib and doxorubicin, in CML cell lines. Environ. Toxicol. Pharmacol. 2018, 59, 24–33. [Google Scholar] [CrossRef]
- Malhão, F.; Macedo, A.C.; Costa, C.; Rocha, E.; Ramos, A.A. Fucoxanthin holds potential to become a drug adjuvant in breast cancer treatment: Evidence from 2D and 3D cell cultures. Molecules 2021, 26, 4288. [Google Scholar] [CrossRef]
- Lu, J.; Wu, X.J.; Hassouna, A.; Wang, K.S.; Li, Y.; Feng, T.; Zhao, Y.; Jin, M.; Zhang, B.; Ying, T.; et al. Gemcitabine-fucoxanthin combination in human pancreatic cancer cells. Biomed. Rep. 2023, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Nova, P.; Gomes, A.M.; Costa-Pinto, A.R. It comes from the sea: Macroalgae-derived bioactive compounds with anti-cancer potential. Crit. Rev. Biotechnol. 2023, 1–15. [Google Scholar] [CrossRef]
- Tavares, J.O.; Cotas, J.; Valado, A.; Pereira, L. Algae food products as a healthcare solution. Mar. Drugs 2023, 21, 578. [Google Scholar] [CrossRef]
- Liyanage, N.M.; Nagahawatta, D.P.; Jayawardena, T.U.; Sanjeewa, K.K.A.; Jayawrdhana, H.H.A.C.K.; Kim, J.-I.; Jeon, Y.-J. Sulfated polysaccharides from seaweeds: A promising strategy for combatting viral diseases—A review. Mar. Drugs 2023, 21, 461. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, Q.; Farooqi, A.A.; Wang, J.; Yue, Y.; Geng, L.; Wu, N. Opportunities and challenges of fucoidan for tumors therapy. Carbohydr Polym. 2024, 324, 121555. [Google Scholar] [CrossRef]
- Ouyang, Y.; Qiu, Y.; Liu, Y.; Zhu, R.; Chen, Y.; El-Seedi, H.R.; Chen, X.; Zhao, C. Cancer-fighting potentials of algal polysaccharides as nutraceuticals. Food Res. Int. 2021, 147, 110522. [Google Scholar] [CrossRef]
- Jin, J.-O.; Yadav, D.; Madhwani, K.; Puranik, N.; Chavda, V.; Song, M. Seaweeds in the oncology arena: Anti-cancer potential of fucoidan as a drug—A review. Molecules 2022, 27, 6032. [Google Scholar] [CrossRef]
- Turrini, E.; Maffei, F.; Fimognari, C. Ten years of research on fucoidan and cancer: Focus on its antiangiogenic and antimetastatic effects. Mar. Drugs 2023, 21, 307. [Google Scholar] [CrossRef] [PubMed]
- Marudhupandi, T.; Ajith-Kumar, T.T.; Lakshmanasenthil, S.; Suja, G.; Vinothkumar, T. In vitro anticancer activity of fucoidan from Turbinaria conoides against A549 cell lines. Int. J. Biol. Macromol. 2015, 72, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Lee, J.-Y.; Yang, C.; Song, G.; Lim, W. Fucoidan derived from Fucus vesiculosus inhibits the development of human ovarian cancer via the disturbance of calcium homeostasis, endoplasmic reticulum stress, and angiogenesis. Mar. Drugs 2020, 18, 45. [Google Scholar] [CrossRef]
- Chen, H.; Cong, Q.; Du, Z.; Liao, W.; Zhang, L.; Yao, Y.; Ding, K. Sulfated fucoidan FP08S2 Inhibits lung cancer cell growth in vivo by disrupting angiogenesis via targeting VEGFR2/VEGF and blocking VEGFR2/Erk/VEGF signaling. Cancer Lett. 2016, 382, 44–52. [Google Scholar] [CrossRef]
- Cong, Q.; Chen, H.; Liao, W.; Xiao, F.; Wang, P.; Qin, Y.; Dong, Q.; Ding, K. Structural characterization and effect on anti-angiogenic activity of a fucoidan from Sargassum fusiforme. Carbohydr. Polym. 2016, 136, 899–907. [Google Scholar] [CrossRef]
- Shen, H.-Y.; Li, L.-Z.; Xue, K.-C.; Hu, D.-D.; Gao, Y.-J. Antitumor activity of fucoidan in anaplastic thyroid cancer via apoptosis and anti-angiogenesis. Mol. Med. Rep. 2017, 15, 2620–2624. [Google Scholar] [CrossRef]
- Oliveira, C.; Granja, S.; Neves, N.M.; Reis, R.L.; Baltazar, F.; Silva, T.H.; Martins, A. Fucoidan from fucus vesiculosus inhibits new blood vessel formation and breast tumor growth in vivo. Carbohydr. Polym. 2019, 223, 115034. [Google Scholar] [CrossRef]
- Yun, C.W.; Yun, S.; Lee, J.H.; Han, Y.-S.; Yoon, Y.M.; An, D.; Lee, S.H. Silencing prion protein in HT29 human colorectal cancer cells enhances anticancer response to fucoidan. Anticancer Res. 2016, 36, 4449–4458. [Google Scholar] [CrossRef]
- Rui, X.; Pan, H.-F.; Shao, S.-L.; Xu, X.-M. Anti-tumor and anti-angiogenic effects of fucoidan on prostate cancer: Possible JAK-STAT3 pathway. BMC Complement. Altern. Med. 2017, 17, 378. [Google Scholar] [CrossRef]
- Hsu, W.-J.; Lin, M.-H.; Kuo, T.-C.; Chou, C.-M.; Mi, F.-L.; Cheng, C.-H.; Lin, C.-W. Fucoidan from Laminaria japonica exerts antitumor effects on angiogenesis and micrometastasis in triple-negative breast cancer cells. Int. J. Biol. Macromol. 2020, 149, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.-J.; Li, L.-X.; Zhang, J.-W.; Yang, Z.-S.; Shi, D.-M.; Yang, Y.-K.; Wu, W.-Z. Antimetastatic effect of fucoidan-sargassum against liver cancer cell invadopodia formation via targeting integrin AVβ3 and mediating AVβ3/Src/E2F1 signaling. J. Cancer 2019, 10, 4777–4792. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-C.; Hsu, W.-L.; Hwang, P.-A.; Chou, T.-C. Low molecular weight fucoidan inhibits tumor angiogenesis through downregulation of HIF-1/VEGF signaling under hypoxia. Mar. Drugs 2015, 13, 4436–4451. [Google Scholar] [CrossRef] [PubMed]
- Delma, C.R.; Somasundaram, S.T.; Srinivasan, G.P.; Khursheed, M.; Bashyam, M.D.; Aravindan, N. Fucoidan from Turbinaria conoides: A multifaceted “deliverable” to combat pancreatic cancer progression. Int. J. Biol. Macromol. 2015, 74, 447–457. [Google Scholar] [CrossRef]
- Sanniyasi, E.; Gopal, R.K.; Damodharan, R.; Arumugam, A.; Sampath Kumar, M.; Senthilkumar, N.; Anbalagan, M. In vitro anticancer potential of laminarin and fucoidan from brown seaweeds. Sci. Rep. 2023, 13, 14452. [Google Scholar] [CrossRef]
- Saliba, J.; Manseur, C.; Groult, H.; Akil, H.; Tannoury, M.; Troutaud, D.; Maugard, T.; Feuillard, J.; Arnaudin, I.; Jayat-Vignoles, C. Anti-proliferative and pro-apoptotic vlmw fucoidan formulas decrease PD-L1 surface expression in EBV Latency III and DLBCL tumoral B-cells by decreasing actin network. Mar. Drugs 2023, 21, 132. [Google Scholar] [CrossRef]
- Chen, L.M.; Yang, P.P.; Al Haq, A.T.; Hwang, P.A.; Lai, Y.C.; Weng, Y.S.; Chen, M.A.; Hsu, H.L. Oligo-Fucoidan supplementation enhances the effect of Olaparib on preventing metastasis and recurrence of triple-negative breast cancer in mice. J. Biomed. Sci. 2022, 29, 70. [Google Scholar] [CrossRef]
- Cheung, N.K.; Modak, S.; Vickers, A.; Knuckles, B. Orally administered beta-glucans enhance anti-tumor effects of monoclonal antibodies. Cancer Immunol. Immunother. 2002, 51, 557–564. [Google Scholar] [CrossRef]
- Kim, E.J.; Lee, Y.J.; Shim, H.K.; YoonPark, J.H. A study on the mechanism by which the aqueous extract of Inonotus obliquus induces apoptosis and inhibits proliferation in HT-29 human colon cancer cells. J. Korean Soc. Food Sci. Nutr. 2006, 35, 516–523. [Google Scholar] [CrossRef]
- Ji, C.F.; Ji, Y.B. Laminarin-induced apoptosis in human colon cancer LoVo cells. Oncol. Lett. 2014, 7, 1728–1732. [Google Scholar] [CrossRef]
- Ji, Y.B.; Ji, C.F.; Zhang, H. Laminarin induces apoptosis of human colon cancer LOVO cells through a mitochondrial pathway. Molecules 2012, 17, 9947–9960. [Google Scholar] [CrossRef]
- Park, H.K.; Kim, I.H.; Kim, J.; Nam, T.J. Induction of apoptosis by laminarin, regulating the insulin-like growth factor-IR signaling pathways in HT-29 human colon cells. Int. J. Mol. Med. 2012, 30, 734–738. [Google Scholar] [CrossRef]
- Ji, C.F.; Ji, Y.B.; Meng, D.Y. Sulfated modification and anti-tumor activity of laminarin. Exp. Ther. Med. 2013, 6, 1259–1264. [Google Scholar] [CrossRef]
- Miao, H.Q.; Elkin, M.; Aingorn, E.; Ishai-Michaeli, R.; Stein, C.A.; Vlodavsky, I. Inhibition of heparanase activity and tumor metastasis by laminarin sulfate and synthetic phosphorothioate oligodeoxynucleotides. Int. J. Cancer 1999, 83, 424–431. [Google Scholar] [CrossRef]
- Malyarenko, O.S.; Usoltseva, R.V.; Zvyagintseva, T.N.; Ermakova, S.P. Laminaran from brown alga Dictyota dichotoma and its sulfated derivative as radioprotectors and radiosensitizers in melanoma therapy. Carbohydr. Polym. 2019, 206, 539–547. [Google Scholar] [CrossRef]
- Desamero, M.J.; Kakuta, S.; Chambers, J.K.; Uchida, K.; Hachimura, S.; Takamoto, M.; Nakayama, J.; Nakayama, H.; Kyuwa, S. Orally administered brown seaweed-derived β-glucan effectively restrained development of gastric dysplasia in A4gnt KO mice that spontaneously develop gastric adenocarcinoma. Int. Immunopharmacol. 2018, 60, 211–220. [Google Scholar] [CrossRef]
- Karasawa, F.; Shiota, A.; Goso, Y.; Kobayashi, M.; Sato, Y.; Masumoto, J.; Fujiwara, M.; Yokosawa, S.; Muraki, T.; Miyagawa, S.; et al. Essential role of gastric gland mucin in preventing gastric cancer in mice. J. Clin. Investig. 2012, 122, 923–934. [Google Scholar] [CrossRef]
- Shang, H.S.; Shih, Y.L.; Chen, C.P.; Lee, M.H.; Lu, H.F.; Chou, P.Y.; Liao, N.C.; Chen, Y.L.; Hsueh, S.C.; Chung, J.G. Laminarin promotes immune responses and normalizes glutamic oxaloacetic transaminase and glutamic pyruvic transaminase levels in leukemic mice. In Vivo 2018, 32, 783–790. [Google Scholar] [CrossRef]
- Dai, H.; Hiromasa, Y.; Takahashi, D.; VanderVelde, D.; Fabrick, J.A.; Kanost, M.R.; Krishnamoorthi, R. An initial event in the insect innate immune response: Structural and biological studies of interactions between β-1,3-glucan and the N-terminal domain of β-1,3-glucan recognition protein. Biochemistry 2013, 52, 161–170. [Google Scholar] [CrossRef]
- Yamashita, T.; Ohshima, H.; Asanuma, T.; Inukai, N.; Miyoshi, I.; Kasai, N.; Kon, Y.; Watanabe, T.; Sato, F.; Kuwabara, M. The effects of alpha-phenyl-tert-butyl nitrone (PBN) on copper-induced rat fulminant hepatitis with jaundice. Free Radic. Biol. Med. 1996, 21, 755–761. [Google Scholar] [CrossRef]
- Han, H.; Wang, L.; Liu, Y.; Shi, X.; Zhang, X.; Li, M.; Wang, T. Combination of curcuma zedoary and kelp inhibits growth and metastasis of liver cancer in vivo and in vitro via reducing endogenous H. Food Funct. 2018, 10, 224–234. [Google Scholar] [CrossRef]
- Song, K.; Xu, L.; Zhang, W.; Cai, Y.; Jang, B.; Oh, J.; Jin, J.O. Laminarin promotes anti-cancer immunity by the maturation of dendritic cells. Oncotarget 2017, 8, 38554–38567. [Google Scholar] [CrossRef]
- Fang, B. Genetic interactions of STAT3 and anticancer drug development. Cancers 2014, 6, 494–525. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef]
- Choi, H.; Hwang, H.; Chin, J.; Kim, E.; Lee, J.; Nam, S.J.; Lee, B.C.; Rho, B.J.; Kang, H. Tuberatolides, potent FXR antagonists from the Korean marine tunicate Botryllus tuberatus. J. Nat. Prod. 2011, 74, 90–94. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, J.; Lee, K.M.; Choi, Y.J.; Ye, B.R.; Kim, M.S.; Ko, S.G.; Lee, S.H.; Kang, D.H.; Heo, S.J. Tuberatolide B suppresses cancer progression by promoting ROS-mediated inhibition of STAT3 signaling. Mar. Drugs 2017, 15, 55. [Google Scholar] [CrossRef]
- Tu, W.Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.D.; Guan, H.; Zhang, S.M.; Tang, Y.; Rang, W.Q.; Zhou, P.K. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef]
- Kim, E.A.; Kim, S.-Y.; Kim, J.; Oh, J.Y.; Kim, H.S.; Yoon, W.J.; Kang, D.H.; Heo, S.J. Tuberatolide B isolated from Sargassum macrocarpum inhibited LPS-stimulated inflammatory response via MAPKs and NF-κB signaling pathway in RAW264.7 cells and zebrafish model. J. Funct. Foods 2018, 52, 109–115. [Google Scholar] [CrossRef]
- Kang, G.J.; Han, S.C.; Yoon, W.J.; Koh, Y.S.; Hyun, J.W.; Kang, H.K.; Youl Cho, J.; Yoo, E.S. Sargaquinoic acid isolated from Sargassum siliquastrum inhibits lipopolysaccharide-induced nitric oxide production in macrophages via modulation of nuclear factor-κB and c-Jun N-terminal kinase pathways. Immunopharmacol. Immunotoxicol. 2013, 35, 80–87. [Google Scholar] [CrossRef]
- Gwon, W.G.; Lee, B.; Joung, E.J.; Choi, M.W.; Yoon, N.; Shin, T.; Oh, C.W.; Kim, H.R. Sargaquinoic acid inhibits tnf-α-induced nf-κb signaling, thereby contributing to decreased monocyte adhesion to human umbilical vein endothelial cells (HUVECs). J. Agric. Food Chem. 2015, 63, 9053–9061. [Google Scholar] [CrossRef]
- Kim, J.A.; Karadeniz, F.; Ahn, B.N.; Kwon, M.S.; Mun, O.J.; Bae, M.J.; Seo, Y.; Kim, M.; Lee, S.H.; Kim, Y.Y.; et al. Bioactive quinone derivatives from the marine brown alga Sargassum thunbergii induce anti-adipogenic and pro-osteoblastogenic activities. J. Sci. Food Agric. 2016, 96, 783–790. [Google Scholar] [CrossRef]
- Tsang, C.K.; Kamei, Y. Sargaquinoic acid supports the survival of neuronal PC12D cells in a nerve growth factor-independent manner. Eur. J. Pharmacol. 2004, 488, 11–18. [Google Scholar] [CrossRef]
- Hur, S.; Lee, H.; Kim, Y.; Lee, B.H.; Shin, J.; Kim, T.Y. Sargaquinoic acid and sargachromenol, extracts of Sargassum sagamianum, induce apoptosis in HaCaT cells and mice skin: Its potentiation of UVB-induced apoptosis. Eur. J. Pharmacol. 2008, 582, 1–11. [Google Scholar] [CrossRef]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef]
- Azam, M.S.; Joung, E.; Choi, J.; Kim, H. Ethanolic extract from Sargassum serratifolium attenuates hyperpigmentation through CREB/ERK signaling pathways in α-MSH-stimulated B16F10 melanoma cells. J. Appl. Phycol. 2017, 29, 2089–2096. [Google Scholar] [CrossRef]
- Azam, M.S.; Kwon, M.; Choi, J.; Kim, H.R. Sargaquinoic acid ameliorates hyperpigmentation through cAMP and ERK-mediated downregulation of MITF in α-MSH-stimulated B16F10 cells. Biomed. Pharmacother. 2018, 104, 582–589. [Google Scholar] [CrossRef]
- de la Mare, J.A.; Lawson, J.C.; Chiwakata, M.T.; Beukes, D.R.; Edkins, A.L.; Blatch, G.L. Quinones and halogenated monoterpenes of algal origin show anti-proliferative effects against breast cancer cells in vitro. Investig. New Drugs 2012, 30, 2187–2200. [Google Scholar] [CrossRef]
- Yoon, J.H.; Youn, K.; Jun, M. Protective effect of sargahydroquinoic acid against Aβ25-35-evoked damage via PI3K/Akt mediated Nrf2 antioxidant defense system. Biomed. Pharmacother. 2021, 144, 112271. [Google Scholar] [CrossRef]
- Fu, Y.; Xie, D.; Zhu, Y.; Zhang, X.; Yue, H.; Zhu, K.; Pi, Z.; Dai, Y. Anti-colorectal cancer effects of seaweed-derived bioactive compounds. Front. Med. 2022, 9, 988507. [Google Scholar] [CrossRef]
- Xia, J.; Yang, L.; Huang, C.; Deng, S.; Yang, Z.; Zhang, Y.; Zhang, C.; Song, C. Omega-3 polyunsaturated fatty acid eicosapentaenoic acid or docosahexaenoic acid improved ageing-associated cognitive decline by regulating glial polarization. Mar. Drugs 2023, 21, 398. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Bilotto, S.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; Nabavi, S.M. Omega-3 polyunsaturated fatty acids and cancer: Lessons learned from clinical trials. Cancer Metastasis Rev. 2015, 34, 359–380. [Google Scholar] [CrossRef]
- Montecillo-Aguado, M.; Tirado-Rodriguez, B.; Huerta-Yepez, S. The involvement of polyunsaturated fatty acids in apoptosis mechanisms and their implications in cancer. Int. J. Mol. Sci. 2023, 24, 11691. [Google Scholar] [CrossRef]
- Ulmann, L.; Blanckaert, V.; Mimouni, V.; Andersson, M.X.; Schoefs, B.; Chenais, B. Microalgal fatty acids and their implication in health and disease. Mini Rev. Med. Chem. 2017, 17, 1112–1123. [Google Scholar] [CrossRef]
- Terme, N.; Chénais, B.; Fournière, M.; Bourgougnon, N.; Bedoux, G. Algal derived functional lipids and their role in promoting health. In Recent Advances in Micro and Macroalgal Processing; Rajauria, G., Yuan, Y.V., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Tsai, C.H.; Shen, Y.C.; Chen, H.W.; Liu, K.L.; Chang, J.W.; Chen, P.Y.; Lin, C.Y.; Yao, H.T.; Li, C.C. Docosahexaenoic acid increases the expression of oxidative stress-induced growth inhibitor 1 through the PI3K/Akt/Nrf2 signaling pathway in breast cancer cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2017, 108 Pt A, 276–288. [Google Scholar] [CrossRef]
- Crovella, S.; Ouhtit, A.; Rahman, S.M.; Rahman, M.M. Docosahexaenoic acid, a key compound for enhancing sensitization to drug in doxorubicin-resistant MCF-7 cell line. Nutrients 2023, 15, 1658. [Google Scholar] [CrossRef]
- Ortega, L.; Lobos-González, L.; Reyna-Jeldes, M.; Cerda, D.; De la Fuente-Ortega, E.; Castro, P.; Bernal, G.; Coddou, C. The Ω-3 fatty acid docosahexaenoic acid selectively induces apoptosis in tumor-derived cells and suppress tumor growth in gastric cancer. Eur. J. Pharmacol. 2021, 896, 173910. [Google Scholar] [CrossRef]
- Tan, L.T.; Phyo, M.Y. Marine Cyanobacteria: A source of lead compounds and their clinically-relevant molecular targets. Molecules 2020, 25, 2197. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Ma, D.; Zou, B.; Cai, G.; Hu, X.; Liu, J.O. Total synthesis of the cyclodepsipeptide apratoxin A and its analogues and assessment of their biological activities. Chemistry 2006, 12, 7615–7626. [Google Scholar] [CrossRef]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Izpisúa Belmonte, J.C.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef]
- Liu, Y.; Law, B.K.; Luesch, H. Apratoxin a reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharmacol. 2009, 76, 91–104. [Google Scholar] [CrossRef]
- Mackinnon, A.L.; Paavilainen, V.O.; Sharma, A.; Hegde, R.S.; Taunton, J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. Elife 2014, 3, e01483. [Google Scholar] [CrossRef]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin kills cells by direct blockade of the sec61 protein translocation channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar] [CrossRef]
- Kazemi, S.; Kawaguchi, S.; Badr, C.E.; Mattos, D.R.; Ruiz-Saenz, A.; Serrill, J.D.; Moasser, M.M.; Dolan, B.P.; Paavilainen, V.O.; Oishi, S.; et al. Targeting of HER/ErbB family proteins using broad spectrum Sec61 inhibitors coibamide A and apratoxin A. Biochem. Pharmacol. 2021, 183, 114317. [Google Scholar] [CrossRef]
- Huang, K.C.; Chen, Z.; Jiang, Y.; Akare, S.; Kolber-Simonds, D.; Condon, K.; Agoulnik, S.; Tendyke, K.; Shen, Y.; Wu, K.M.; et al. Apratoxin A Shows Novel Pancreas-Targeting Activity through the Binding of Sec 61. Mol. Cancer Ther. 2016, 15, 1208–1216. [Google Scholar] [CrossRef]
- Cai, W.; Chen, Q.Y.; Dang, L.H.; Luesch, H. Correction to “Apratoxin S10, a dual inhibitor of angiogenesis and cancer cell growth to treat highly vascularized tumors”. ACS Med. Chem. Lett. 2017, 8, 1342. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Liu, Y.; Cai, W.; Luesch, H. Improved total synthesis and biological evaluation of potent apratoxin S4 based anticancer agents with differential stability and further enhanced activity. J. Med. Chem. 2014, 57, 3011–3029. [Google Scholar] [CrossRef]
- Cai, W.; Ratnayake, R.; Gerber, M.H.; Chen, Q.Y.; Yu, Y.; Derendorf, H.; Trevino, J.G.; Luesch, H. Development of apratoxin S10 (Apra S10) as an anti-pancreatic cancer agent and its preliminary evaluation in an orthotopic patient-derived xenograft (PDX) model. Investig. New Drugs 2018, 37, 364–374. [Google Scholar] [CrossRef]
- Onda, Y.; Masuda, Y.; Yoshida, M.; Doi, T. Conformation-based design and synthesis of Apratoxin A mimetics modified at the α,β-unsaturated thiazoline moiety. J. Med. Chem. 2017, 60, 6751–6765. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Zhou, T.; Zhao, Y.Y.; Chen, L.; Gong, M.W.; Xia, Q.W.; Ying, M.G.; Zheng, Q.H.; Zhang, Q.Q. Antitumor Effects and Related Mechanisms of Penicitrinine A, a novel alkaloid with a unique spiro skeleton from the marine fungus Penicillium citrinum. Mar. Drugs 2015, 13, 4733–4753. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, X.; Li, D.; Zhu, T.; Mo, X.; Li, J. Anticancer efficacy and absorption, distribution, metabolism, and toxicity studies of aspergiolide A in early drug development. Drug Des. Dev. Ther. 2014, 8, 1965–1977. [Google Scholar] [CrossRef][Green Version]
- Du, L.; Zhu, T.; Liu, H.; Fang, Y.; Zhu, W.; Gu, Q. Cytotoxic polyketides from a marine-derived fungus Aspergillus glaucus. J. Nat. Prod. 2008, 71, 1837–1842. [Google Scholar] [CrossRef]
- Yuan, F.; Qiao, L.; Chen, Y.; Qi, X.; Liu, Y.; Li, D.; Gu, Q.; Li, J.; Liu, M. AS1041, a novel synthesized derivative of marine natural compound aspergiolide a, arrests cell cycle, induces apoptosis, and inhibits ERK activation in K562 cells. Mar. Drugs 2017, 15, 346. [Google Scholar] [CrossRef]
- Lu, X.; Yuan, F.; Qiao, L.; Liu, Y.; Gu, Q.; Qi, X.; Li, J.; Li, D.; Liu, M. AS1041, a novel derivative of marine natural compound Aspergiolide A, induces senescence of leukemia cells via oxidative stress-induced DNA damage and BCR-ABL degradation. Biomed. Pharmacother. 2024, 171, 116099. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 57, 268–272. [Google Scholar] [CrossRef]
- Yang, T.; Lu, Z.; Meng, L.; Wei, S.; Hong, K.; Zhu, W.; Huang, C. The novel agent ophiobolin O induces apoptosis and cell cycle arrest of MCF-7 cells through activation of MAPK signaling pathways. Bioorg. Med. Chem. Lett. 2012, 22, 579–585. [Google Scholar] [CrossRef]
- Wang, S.; He, M.; Li, L.; Liang, Z.; Zou, Z.; Tao, A. Cell-in-cell death is not restricted by caspase-3 deficiency in MCF-7 Cells. J. Breast Cancer 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Lv, C.; Qin, W.; Zhu, T.; Wei, T.; Hong, K.; Zhu, W.; Chen, R.C.H. Ophiobolin O isolated from Aspergillus ustus induces G1 arrest of MCF-7 cells through interaction with AKT/GSK3β/Cyclin D1 signaling. Mar. Drugs 2015, 13, 431–443. [Google Scholar] [CrossRef]
- Sun, W.; Lv, C.; Zhu, T.; Yang, X.; Wei, S.; Sun, J.; Hong, K.; Zhu, W.; Huang, C. Ophiobolin-O reverses adriamycin resistance via cell cycle arrest and apoptosis sensitization in adriamycin-resistant human breast carcinoma (MCF-7/ADR) cells. Mar. Drugs 2013, 11, 4570–4584. [Google Scholar] [CrossRef]
- Kai, W.; Yating, S.; Lin, M.; Kaiyong, Y.; Baojin, H.; Wu, Y.; Fangzhou, Y.; Yan, C. Natural product toosendanin reverses the resistance of human breast cancer cells to adriamycin as a novel PI3K inhibitor. Biochem. Pharmacol. 2018, 152, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.G.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can some marine-derived fungal metabolites become actual anticancer agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef]
- Wijesekara, I.; Zhang, C.; Van Ta, Q.; Vo, T.S.; Li, Y.X.; Kim, S.K. Physcion from marine-derived fungus Microsporum sp. induces apoptosis in human cervical carcinoma HeLa cells. Microbiol. Res. 2014, 169, 255–261. [Google Scholar] [CrossRef]
- Xiong, Y.; Ren, L.; Wang, Z.; Hu, Z.; Zhou, Y. Anti-proliferative Effect of physcion on human gastric cell line via inducing ROS-dependent apoptosis. Cell Biochem. Biophys. 2015, 73, 537–543. [Google Scholar] [CrossRef]
- Pang, M.J.; Yang, Z.; Zhang, X.L.; Liu, Z.F.; Fan, J.; Zhang, H.Y. Physcion, a naturally occurring anthraquinone derivative, induces apoptosis and autophagy in human nasopharyngeal carcinoma. Acta Pharmacol. Sin. 2016, 37, 1623–1640. [Google Scholar] [CrossRef]
- Chen, X.; Gao, H.; Han, Y.; Ye, J.; Xie, J.; Wang, C. Physcion induces mitochondria-driven apoptosis in colorectal cancer cells via downregulating EMMPRIN. Eur. J. Pharmacol. 2015, 764, 124–133. [Google Scholar] [CrossRef]
- Adnan, M.; Rasul, A.; Hussain, G.; Shah, M.A.; Sarfraz, I.; Nageen, B.; Riaz, A.; Khalid, R.; Asrar, M.; Selamoglu, Z.; et al. Physcion and physcion 8-O-β-D-glucopyranoside: Natural anthraquinones with potential anticancer activities. Curr. Drug Targets 2021, 22, 488–504. [Google Scholar] [CrossRef]
- Gao, F.; Liu, W.; Guo, Q.; Bai, Y.; Yang, H.; Chen, H. Physcion blocks cell cycle and induces apoptosis in human B cell precursor acute lymphoblastic leukemia cells by downregulating HOXA5. Biomed. Pharmacother. 2017, 94, 850–857. [Google Scholar] [CrossRef]
- Ding, Y.S.; Kim, W.S.; Park, S.; Kim, S.K. Apoptotic effect of physcion isolated from marine fungus Microsporum sp. in PC3 human prostate cancer cells. Fish Aquatic Sci. 2018, 21, 22. [Google Scholar] [CrossRef]
- Pan, X.P.; Jiya, B.R.; Wang, F.; Lan, Z. Physcion increases the sensitivity of hepatocellular carcinoma to sorafenib through miRNA-370/PIM1 axis-regulated glycolysis. World J. Gastrointest. Oncol. 2023, 15, 1400–1411. [Google Scholar] [CrossRef]
- Khuda, F.; Zahir, I.; Khalil, A.A.K.; Ali, S.; Ullah, N.; Albariqi, A.H.; Ahn, M.J.; Shafique, M.; Mehtap Büyüker, S.; Almawash, S. Preparation, characterization, and evaluation of physcion nanoparticles for enhanced oral bioavailability: An attempt to improve its antioxidant and anticancer potential. ACS Omega 2023, 8, 33955–33965. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R.; Palladino, M.A.; Lam, K.S.; Lloyd, G.K.; Potts, B.C. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg. Med. Chem. 2009, 17, 2175–2180. [Google Scholar] [CrossRef]
- Jensen, P.R.; Moore, B.S.; Fenical, W. The marine actinomycete genus Salinispora: A model organism for secondary metabolite discovery. Nat. Prod. Rep. 2015, 32, 738–751. [Google Scholar] [CrossRef]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–409. [Google Scholar] [CrossRef]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A. Marizomib activity as a single agent in malignant gliomas: Ability to cross the blood-brain barrier. Neuro Oncol. 2016, 18, 840–848. [Google Scholar] [CrossRef]
- Miller, C.P.; Ban, K.; Dujka, M.E.; McConkey, D.J.; Munsell, M.; Palladino, M.; Chandra, J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 2007, 110, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Badros, A.; Singh, Z.; Dhakal, B.; Kwok, Y.; MacLaren, A.; Richardson, P.; Trikha, M.; Hari, P. Marizomib for central nervous system-multiple myeloma. Br. J. Haematol. 2017, 177, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Nguyen, H.; Vellalath, S.; Romo, D. (−)-Homosalinosporamide A and its mode of proteasome inhibition: An x-ray crystallographic study. Mar. Drugs 2018, 16, 240. [Google Scholar] [CrossRef]
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase I clinical trial of marizomib (NPI-0052) in patients with advanced malignancies including multiple myeloma: Study NPI-0052-102 final results. Clin. Cancer Res. 2016, 22, 4559–4566. [Google Scholar] [CrossRef] [PubMed]
- Laporte, A.N.; Barrott, J.J.; Yao, R.J.; Poulin, N.M.; Brodin, B.A.; Jones, K.B.; Underhill, T.M.; Nielsen, T.O. HDAC and proteasome inhibitors synergize to activate pro-apoptotic factors in synovial sarcoma. PLoS ONE 2017, 12, e0169407. [Google Scholar] [CrossRef]
- Spratlin, J.L.; Pitts, T.M.; Kulikowski, G.N.; Morelli, M.P.; Tentler, J.J.; Serkova, N.J.; Eckhardt, S.G. Synergistic activity of histone deacetylase and proteasome inhibition against pancreatic and hepatocellular cancer cell lines. Anticancer Res. 2011, 31, 1093–1103. [Google Scholar]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investig. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Spencer, A.; Harrison, S.; Zonder, J.; Badros, A.; Laubach, J.; Bergin, K.; Khot, A.; Zimmerman, T.; Chauhan, D.; Levin, N.; et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): Final study results. Br. J. Haematol. 2018, 180, 41–51. [Google Scholar] [CrossRef]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Bota, D.A.; Mason, W.; Kesari, S.; Magge, R.; Winograd, B.; Elias, I.; Reich, S.D.; Levin, N.; Trikha, M.; Desjardins, A. Marizomib alone or in combination with bevacizumab in patients with recurrent glioblastoma: Phase I/II clinical trial data. Neurooncol. Adv. 2021, 3, vdab142. [Google Scholar] [CrossRef]
- Bota, D.A.; Alexandru, D.; Keir, S.T.; Bigner, D.; Vredenburgh, J.; Friedman, H.S. Proteasome inhibition with bortezomib induces cell death in GBM stem-like cells and temozolomide-resistant glioma cell lines, but stimulates GBM stem-like cells’ VEGF production and angiogenesis. J. Neurosurg. 2013, 119, 1415–1423. [Google Scholar] [CrossRef]
- Boccellato, C.; Kolbe, E.; Peters, N.; Juric, V.; Fullstone, G.; Verreault, M.; Idbaih, A.; Lamfers, M.L.M.; Murphy, B.M.; Rehm, M. Marizomib sensitizes primary glioma cells to apoptosis induced by a latest-generation TRAIL receptor agonist. Cell Death Dis. 2021, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Lin, B.; Wang, Q.; Nie, X.; Shi, Y. Combined treatment of marizomib and cisplatin modulates cervical cancer growth and invasion and enhances antitumor potential. Front. Oncol. 2022, 12, 974573. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Reslink, M.C.; Gatesman, T.A.; Halbert, M.E.; Miller, T.A.; Golbourn, B.J.; Casillo, S.M.; Mullett, S.J.; Wendell, S.G.; Obodo, U.; et al. Targeting mitochondrial energetics reverses panobinostat- and marizomib-induced resistance in pediatric and adult high-grade gliomas. Mol. Oncol. 2023, 17, 1821–1843. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Gorlia, T.; Reijneveld, J.C.; De Vos, F.Y.F.L.; Idbaih, A.; Frenel, J.-S.; Le Rhun, E.; Sepulveda Sánchez, J.M.; Perry, J.R.; Masucci, L.; et al. EORTC 1709/CCTG CE.8: A phase III trial of marizomib in combination with temozolomide-based radiochemotherapy versus temozolomide-based radiochemotherapy alone in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2021, 39, 2004. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalisay, D.S.; Tenebro, C.P.; Sabido, E.M.; Suarez, A.F.L.; Paderog, M.J.V.; Reyes-Salarda, R.; Saludes, J.P. Marine-Derived Anticancer Agents Targeting Apoptotic Pathways: Exploring the Depths for Novel Cancer Therapies. Mar. Drugs 2024, 22, 114. https://doi.org/10.3390/md22030114
Dalisay DS, Tenebro CP, Sabido EM, Suarez AFL, Paderog MJV, Reyes-Salarda R, Saludes JP. Marine-Derived Anticancer Agents Targeting Apoptotic Pathways: Exploring the Depths for Novel Cancer Therapies. Marine Drugs. 2024; 22(3):114. https://doi.org/10.3390/md22030114
Chicago/Turabian StyleDalisay, Doralyn S., Chuckcris P. Tenebro, Edna M. Sabido, Angelica Faith L. Suarez, Melissa June V. Paderog, Rikka Reyes-Salarda, and Jonel P. Saludes. 2024. "Marine-Derived Anticancer Agents Targeting Apoptotic Pathways: Exploring the Depths for Novel Cancer Therapies" Marine Drugs 22, no. 3: 114. https://doi.org/10.3390/md22030114
APA StyleDalisay, D. S., Tenebro, C. P., Sabido, E. M., Suarez, A. F. L., Paderog, M. J. V., Reyes-Salarda, R., & Saludes, J. P. (2024). Marine-Derived Anticancer Agents Targeting Apoptotic Pathways: Exploring the Depths for Novel Cancer Therapies. Marine Drugs, 22(3), 114. https://doi.org/10.3390/md22030114