

Identification of PLK1-PBD Inhibitors from the Library of Marine Natural Products: 3D QSAR Pharmacophore, ADMET, Scaffold Hopping, Molecular Docking, and Molecular Dynamics Study


Abstract
1. Introduction
2. Results
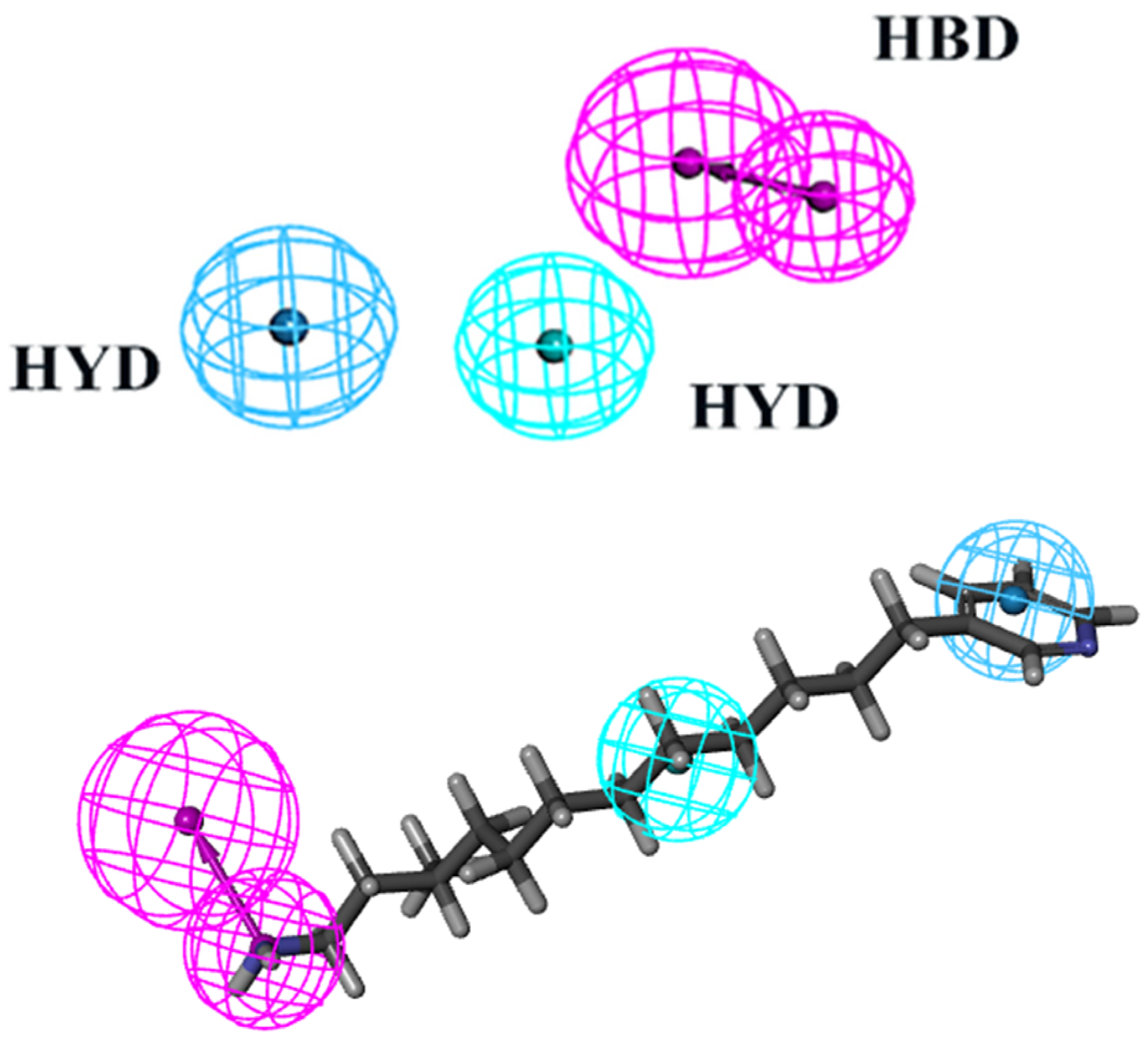
2.1. Construction, Selection, and Validation of Pharmacophore Model
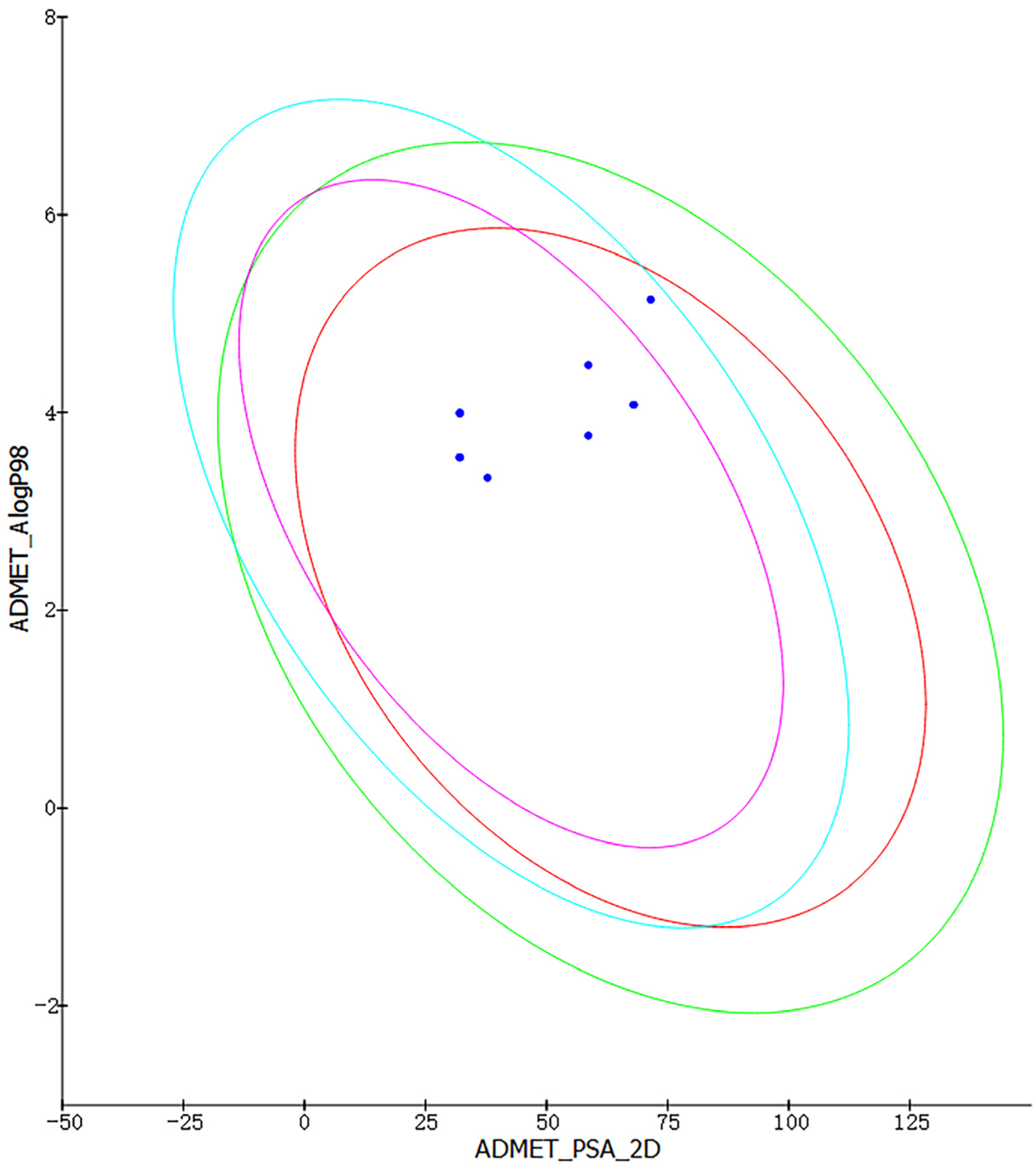
2.2. ADMET Analysis
2.3. Replace Fragment Protocol
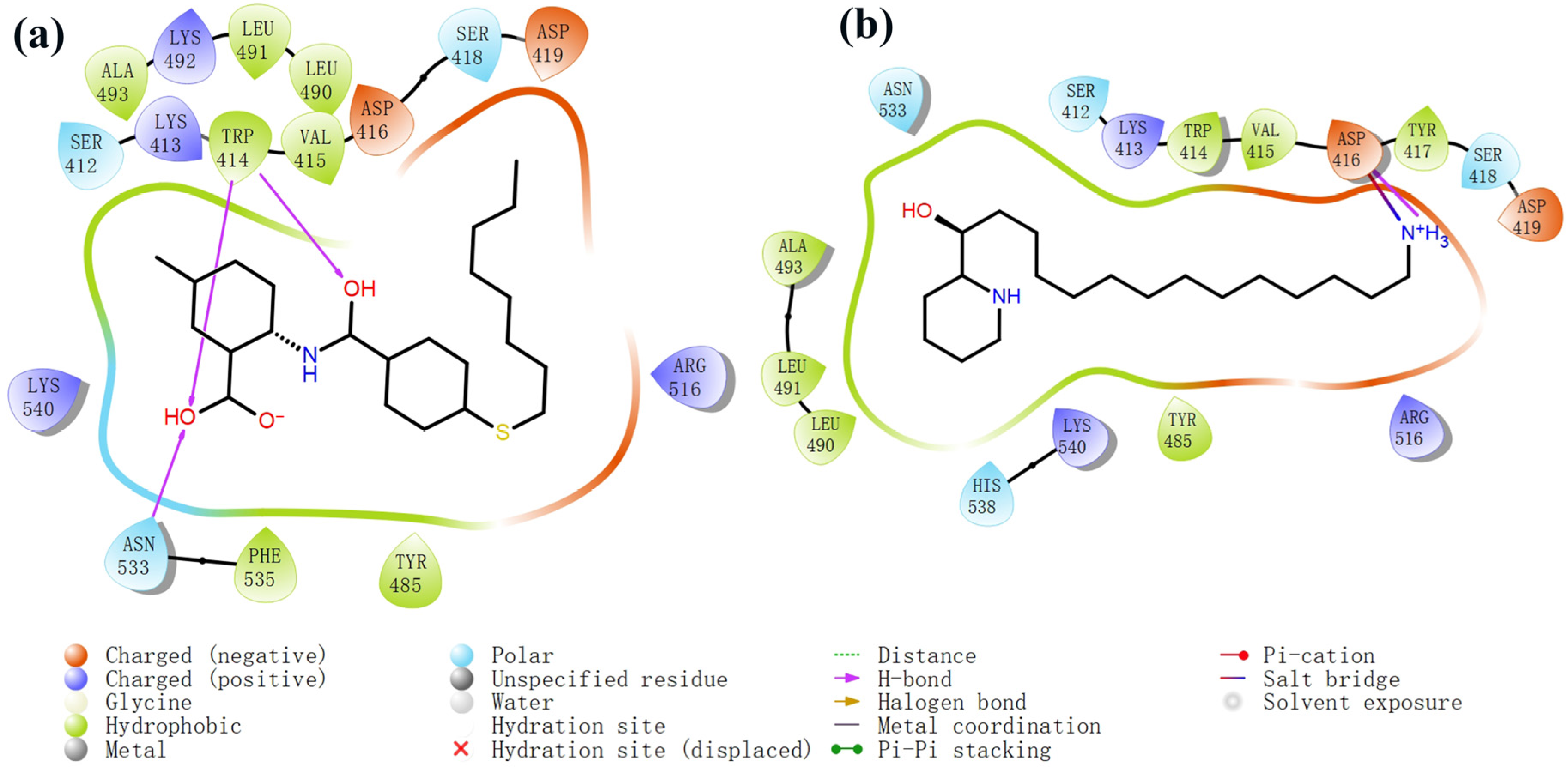
2.4. Molecular Docking
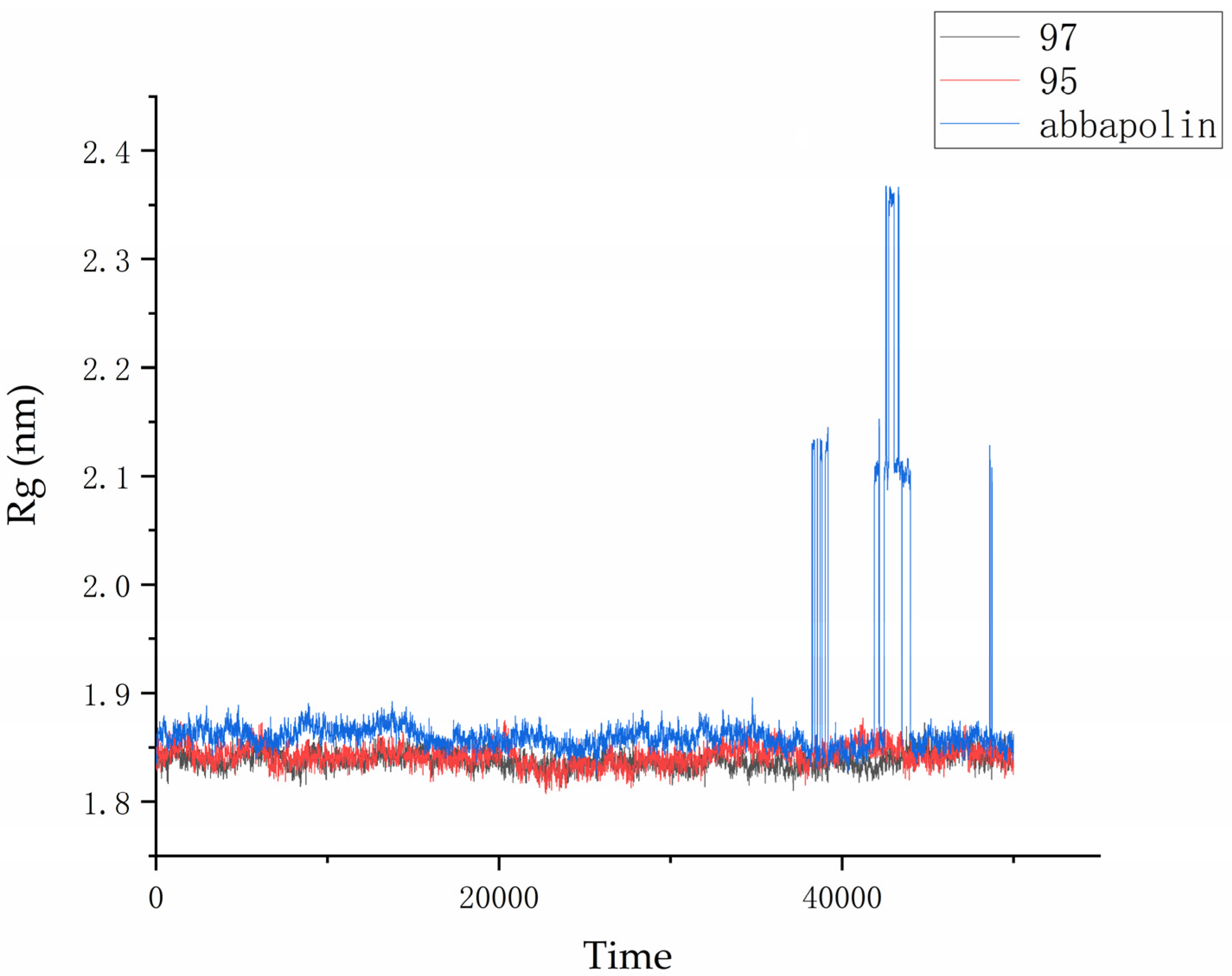
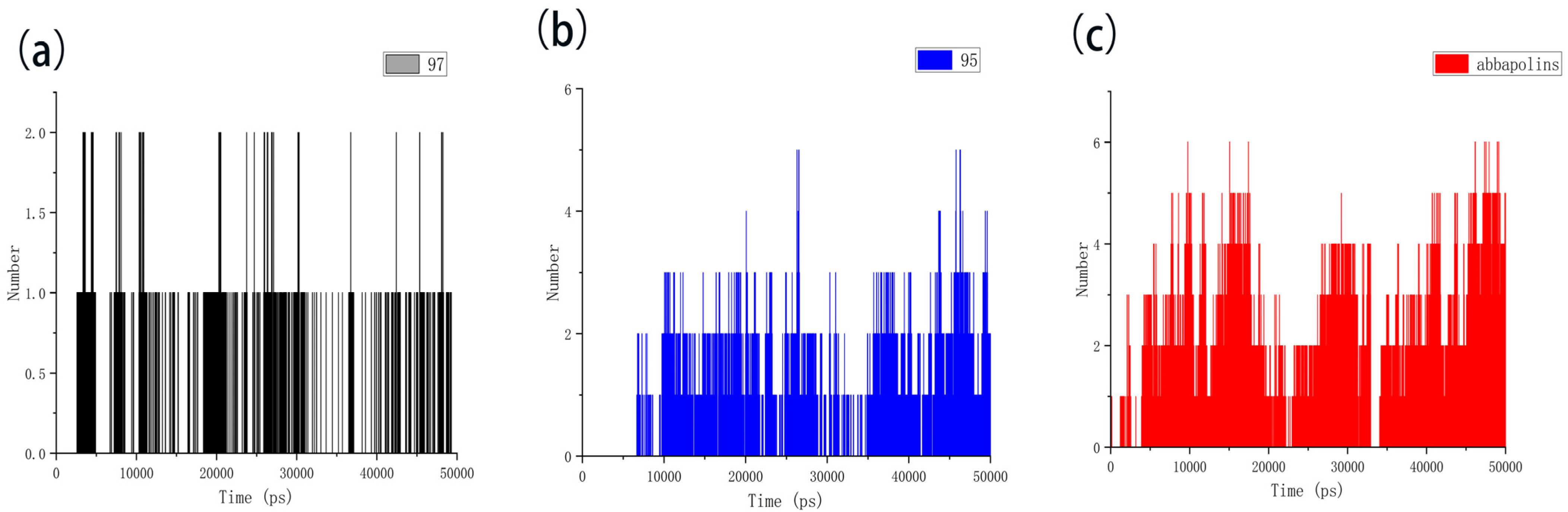
2.5. Molecular Dynamics
2.6. Calculation of Binding Free Energy
3. Discussion
4. Materials and Methods
4.1. Compound Preparations
4.2. Pharmacophore Model Generation
4.3. Validation of a Pharmacophore Model
4.4. Database Searching
4.5. ADMET
4.6. Scaffold Hopping Replace Fragment Protocol
4.7. Molecular Docking
4.8. Molecular Dynamics
4.9. MM-PBSA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.; Zhang, M.; Pan, X.; Yuan, X.; Zhou, L.; Yan, L.; Zeng, L.-H.; Xu, J.; Yang, B.; Zhang, L.; et al. Bub1 and CENP-U Redundantly Recruit Plk1 to Stabilize Kinetochore-Microtubule Attachments and Ensure Accurate Chromosome Segregation. Cell Rep. 2021, 36, 109740. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liu, Y.; Zheng, R.; Ye, Y.; Fu, Y.; Yin, L.; Gao, Y.; Fu, Y.; Qi, X.; Deng, T.; et al. PLK1 Maintains DNA Methylation and Cell Viability by Regulating Phosphorylation-Dependent UHRF1 Protein. Cell Death Discov. 2023, 9, 367. Available online: https://pubmed.ncbi.nlm.nih.gov/37788997/ (accessed on 28 December 2023). [CrossRef] [PubMed]
- Luo, P.; Yan, H.; Du, J.; Chen, X.; Shao, J.; Zhang, Y.; Xu, Z.; Jin, Y.; Lin, N.; Yang, B.; et al. PLK1 (Polo like Kinase 1)-Dependent Autophagy Facilitates Gefitinib-Induced Hepatotoxicity by Degrading COX6A1 (Cytochrome c Oxidase Subunit 6A1). Autophagy 2021, 17, 3221–3237. Available online: https://www.tandfonline.com/doi/full/10.1080/15548627.2020.1851492 (accessed on 28 December 2023). [CrossRef] [PubMed]
- Zhang, J.; Zhang, L.; Wang, J.; Ouyang, L.; Wang, Y. Polo-like Kinase 1 Inhibitors in Human Cancer Therapy: Development and Therapeutic Potential. J. Med. Chem. 2022, 65, 10133–10160. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Vaughan, C.A.; Singh, S.; Subler, M.A.; Windle, J.J.; Inoue, K.; Fry, E.A.; Pillappa, R.; Grossman, S.R.; Windle, B.; Andrew Yeudall, W.; et al. The Oncogenicity of Tumor-Derived Mutant P53 Is Enhanced by the Recruitment of PLK3. Nat. Commun. 2021, 12, 704. [Google Scholar] [CrossRef]
- Gao, Y.; Kabotyanski, E.B.; Shepherd, J.H.; Villegas, E.; Acosta, D.; Hamor, C.; Sun, T.; Montmeyor-Garcia, C.; He, X.; Dobrolecki, L.E.; et al. Tumor Suppressor PLK2 May Serve as a Biomarker in Triple-Negative Breast Cancer for Improved Response to PLK1 Therapeutics. Cancer Res. Commun. 2021, 1, 178–193. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current Trends in Computer Aided Drug Design and a Highlight of Drugs Discovered via Computational Techniques: A Review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Huang, W.; Zheng, X.; Liyan, C.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis Targeting Chimera (PROTAC) in Drug Discovery Paradigm: Recent Progress and Future Challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spanò, V.; Montalbano, A.; Cueto, M.; Díaz Marrero, A.R.; Deniz, I.; Erdoğan, A.; Lukić Bilela, L.; Moulin, C.; Taffin-de-Givenchy, E.; et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching Cancer Pharmacology with Drugs of Marine Origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef]
- Alves, C.; Silva, J.; Pinteus, S.; Gaspar, H.; Alpoim, M.C.; Botana, L.M.; Pedrosa, R. From Marine Origin to Therapeutics: The Antitumor Potential of Marine Algae-Derived Compounds. Front. Pharmacol. 2018, 9, 777. [Google Scholar] [CrossRef]
- Luo, L.; Wang, Q.; Liao, Y. The Inhibitors of CDK4/6 from a Library of Marine Compound Database: A Pharmacophore, ADMET, Molecular Docking and Molecular Dynamics Study. Mar. Drugs 2022, 20, 319. [Google Scholar] [CrossRef]
- Lyu, C.; Chen, T.; Qiang, B.; Liu, N.; Wang, H.; Zhang, L.; Liu, Z. CMNPD: A Comprehensive Marine Natural Products Database towards Facilitating Drug Discovery from the Ocean. Nucleic Acids Res. 2021, 49, D509–D515. [Google Scholar] [CrossRef]
- Chapagai, D.; Merhej, G.; McInnes, C.; Wyatt, M.D. Structural Basis for Variations in Polo-like Kinase 1 Conformation and Intracellular Stability Induced by ATP-Competitive and Novel Noncompetitive Abbapolin Inhibitors. ACS Chem. Biol. 2023, 18, 1642–1652. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The Molecular Basis for Phosphodependent Substrate Targeting and Regulation of Plks by the Polo-Box Domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and Function of Polo-like Kinases. Oncogene 2005, 24, 248–259. [Google Scholar] [CrossRef]
- Yun, S.-M.; Moulaei, T.; Lim, D.; Bang, J.K.; Park, J.-E.; Shenoy, S.R.; Liu, F.; Kang, Y.H.; Liao, C.; Soung, N.-K.; et al. Structural and Functional Analyses of Minimal Phosphopeptides Targeting the Polo-Box Domain of Polo-like Kinase 1. Nat. Struct. Mol. Biol. 2009, 16, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.M.; Wyatt, M.D.; McInnes, C. Inhibitors of the PLK1 Polo-Box Domain: Drug Design Strategies and Therapeutic Opportunities in Cancer. Expert Opin. Drug Discov. 2023, 18, 65–81. Available online: https://www.tandfonline.com/doi/full/10.1080/17460441.2023.2159942?scroll=top&needAccess=true (accessed on 29 December 2023). [CrossRef] [PubMed]
- Yekeen, A.A.; Durojaye, O.A.; Idris, M.O.; Muritala, H.F.; Arise, R.O. CHAPERONg: A Tool for Automated GROMACS-Based Molecular Dynamics Simulations and Trajectory Analyses. Comput. Struct. Biotechnol. J. 2023, 21, 4849–4858. [Google Scholar] [CrossRef]
- AlAjmi, M.F.; Rehman, M.T.; Hussain, A.; Rather, G.M. Pharmacoinformatics Approach for the Identification of Polo-like Kinase-1 Inhibitors from Natural Sources as Anti-Cancer Agents. Int. J. Biol. Macromol. 2018, 116, 173–181. [Google Scholar] [CrossRef]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like Kinases: Structural Variations Lead to Multiple Functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide Chemistry Toolbox—Transforming Natural Peptides into Peptide Therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Murugan, R.N.; Park, J.-E.; Kim, E.-H.; Shin, S.Y.; Cheong, C.; Lee, K.S.; Bang, J.K. Plk1-Targeted Small Molecule Inhibitors: Molecular Basis for Their Potency and Specificity. Mol. Cells 2011, 32, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.; Qin, T.; Liu, Y.; Lai, L. Identification of Acylthiourea Derivatives as Potent Plk1 PBD Inhibitors. Eur. J. Med. Chem. 2016, 124, 229–236. [Google Scholar] [CrossRef]
- Matulja, D.; Wittine, K.; Malatesti, N.; Laclef, S.; Turks, M.; Markovic, M.K.; Ambrožić, G.; Marković, D. Marine Natural Products with High Anticancer Activities. Curr. Med. Chem. 2020, 27, 1243–1307. [Google Scholar] [CrossRef]
- Song, C.; Yang, J.; Zhang, M.; Ding, G.; Jia, C.; Qin, J.; Guo, L. Marine Natural Products: The Important Resource of Biological Insecticide. Chem. Biodivers. 2021, 18, e2001020. [Google Scholar] [CrossRef]
- Luo, L.; Zhong, A.; Wang, Q.; Zheng, T. Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products. Mar. Drugs 2021, 20, 29. [Google Scholar] [CrossRef]
- Craig, S.N.; Baxter, M.; Chapagai, D.; Stafford, J.M.; Nurmemmedov, E.; Altomare, D.; Wyatt, M.D.; McInnes, C. Structure-Activity and Mechanistic Studies of Non-Peptidic Inhibitors of the PLK1 Polo Box Domain Identified through REPLACE. Eur. J. Med. Chem. 2022, 227, 113926. [Google Scholar] [CrossRef]
- Abdelfatah, S.; Berg, A.; Huang, Q.; Yang, L.J.; Hamdoun, S.; Klinger, A.; Greten, H.J.; Fleischer, E.; Berg, T.; Wong, V.K.W.; et al. MCC1019, a Selective Inhibitor of the Polo-Box Domain of Polo-like Kinase 1 as Novel, Potent Anticancer Candidate. Acta Pharm. Sin. B 2019, 9, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Tsuji, K.; Hymel, D.; Burke, T.R. Development of Highly Selective 1,2,3-Triazole-Containing Peptidic Polo-like Kinase 1 Polo-Box Domain-Binding Inhibitors. Molecules 2019, 24, 1488. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Hymel, D.; Burke, T.R. Enhancing Polo-like Kinase 1 Selectivity of Polo-Box Domain-Binding Peptides. Bioorg. Med. Chem. 2017, 25, 5041–5049. [Google Scholar] [CrossRef] [PubMed]
- Hymel, D.; Grant, R.A.; Tsuji, K.; Yaffe, M.B.; Burke, T.R. Histidine N(τ)-Cyclized Macrocycles as a New Genre of Polo-like Kinase 1 Polo-Box Domain-Binding Inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3202–3205. [Google Scholar] [CrossRef] [PubMed]
- Hymel, D.; Burke, T.R. Phosphatase-Stable Phosphoamino Acid Mimetics That Enhance Binding Affinities with the Polo-Box Domain of Polo-like Kinase 1. ChemMedChem 2017, 12, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Raafat, A.; Mowafy, S.; Abouseri, S.M.; Fouad, M.A.; Farag, N.A. Lead Generation of Cysteine Based Mesenchymal Epithelial Transition (c-Met) Kinase Inhibitors: Using Structure-Based Scaffold Hopping, 3D-QSAR Pharmacophore Modeling, Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation. Comput. Biol. Med. 2022, 146, 105526. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. Available online: https://pubs.acs.org/doi/10.1021/jp003020w (accessed on 25 January 2024). [CrossRef]
- Hosseini, F.S.; Amanlou, M. Anti-HCV and Anti-Malaria Agent, Potential Candidates to Repurpose for Coronavirus Infection: Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Study. Life Sci. 2020, 258, 118205. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. Available online: https://onlinelibrary.wiley.com/doi/10.1002/jcc.20291 (accessed on 25 January 2024). [CrossRef] [PubMed]
- Nava, M. Implementing Dimer Metadynamics Using Gromacs. J. Comput. Chem. 2018, 39, 2126–2132. Available online: https://onlinelibrary.wiley.com/doi/10.1002/jcc.25386 (accessed on 25 January 2024). [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. Available online: https://pubs.acs.org/doi/10.1021/acs.chemrev.9b00055?ref=PDF (accessed on 25 January 2024). [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore | Features | Total Cost (USD) | Cost Difference (USD) | Correlation | RMSD | Max.Fit |
|---|---|---|---|---|---|---|
| Phar01 | HHD | 363.483 | 45.313 | 0.964 | 1.216 | 7.222 |
| Phar02 | HHD | 370.353 | 38.443 | 0.448 | 1.274 | 4.30 |
| Phar03 | HHD | 370.457 | 38.339 | 0.364 | 1.290 | 6.375 |
| Phar04 | HAA | 372.205 | 36.591 | 0.828 | 1.290 | 4.239 |
| Phar05 | HDD | 374.201 | 34.595 | 0.383 | 1.326 | 5.840 |
| Phar06 | HAD | 374.325 | 34.471 | 0.165 | 1.323 | 5.146 |
| Phar07 | HAD | 374.473 | 34.323 | 0.137 | 1.325 | 5.234 |
| Phar08 | ADD | 375.973 | 32.823 | 0.457 | 1.340 | 6.509 |
| Phar09 | HAA | 376.062 | 32.734 | 0.229 | 1.319 | 3.930 |
| Phar10 | HAAD | 376.754 | 32.042 | 0.400 | 1.309 | 4.436 |
| Compound | BBB Level | Solubility | Absorption Level | Hepatotoxic | CYP2D6 Inhibit | Plasma Protein-Binding Prediction |
|---|---|---|---|---|---|---|
 | ||||||
| 1 | 1 | 3 | 0 | −7.00099 | −3.01411 | False |
 | ||||||
| 2 | 1 | 3 | 0 | −6.3594 | −4.63198 | False |
 | ||||||
| 3 | 1 | 3 | 0 | −7.1642 | −5.51768 | True |
 | ||||||
| 4 | 1 | 3 | 0 | −7.42021 | −1.2257 | False |
 | ||||||
| 5 | 1 | 3 | 0 | −7.25533 | −5.44008 | True |
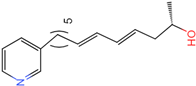 | ||||||
| 6 | 1 | 3 | 0 | −6.13045 | −3.11628 | True |
 | ||||||
| 7 | 1 | 3 | 0 | −7.34797 | −2.16912 | True |
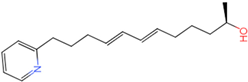 | ||||||
| 8 | 1 | 3 | 0 | −6.43977 | −1.8575 | True |
| Molecules | -CDOCKER_ENERGY |
|---|---|
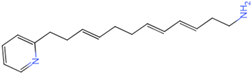
| Molecule 90 [(14S)-14-hydroxy-14-(pyridin-2-yl)tetradecyl]ammonium | 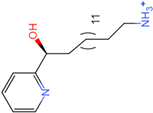 44.0031 |
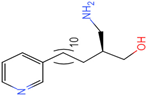
| Molecule 95 [(14S)-15-(2-furyl)-14-hydroxypentadecyl]ammonium | 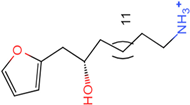 43.9085 |
| Molecule 97 [(14S)-14-hydroxy-14-phenyltetradecyl]ammonium | 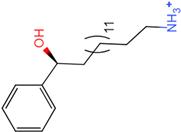 39.3765 |
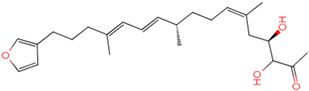
| Abbapolin | 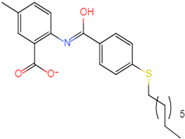 41.8117 |
| Molecule | Chemical Formula | Exact Mass | Mol Weight | m/z | Elemental Analysis | Log P | Henry’s Law | tPSA | Clog P | CMR | LOG S | pKa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 90 | C19H35N2O+ | 307.274 | 307.501 | 307.274 (100.0%), 308.277 (20.5%), 309.281 (2.0%) | C,74.21;H,11.47:N,9.11:; O,5.20 | 4.71 | 0.69 | 60.23 | 4.417 | 9.3151 | −4.652 | 10.324.13.885 |
| 95 | C19H36NO2+ | 310.274 | 310.501 | 310.274 (100.0%), 311.277 (20.5%), 312.280 (2.0%) | C,73.50;H,11.69:N,4.51: 0,10.31 | _ | −1.32 | 57.1 | 5.319 | 9.2039 | −4.45 | 10.324.14.308 |
| 97 | C20H36NO+ | 306.279 | 306.513 | 306.279 (100.0%), 307.282 (21.6%), 308.285 (2.2%) | C,78.37:H,11.84:N,4.57: 0,5.22 | 5.59 | −0.67 | 47.87 | 5.914 | 9.5262 | −5.429 | 10.324.14.257 |
| Molecules | ΔVDWAALS | ΔEEL | ΔGSOLV | ΔGGAS | ΔTOTAL |
|---|---|---|---|---|---|
| Molecule 95 | −18.41 ± 0.93 | −30.2 ± 12.86 | 24.04 ± 12.90 | −48.6 ± 12.90 | −24.57 ± 18.24 |
| Molecule 97 | −28.53 ± 0.47 | 52.38 ± 0.31 | 23.85 ± 0.57 | −49.82 ± 0.95 | −25.97 ± 1.11 |
| Abbapolin | −47.15 ± 0.94 | −175.79 ± 4.90 | 171.96 ± 3.48 | −210.94 ± 4.98 | −38.98 ± 6.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, N.; Zheng, C.; Tan, H.; Luo, L. Identification of PLK1-PBD Inhibitors from the Library of Marine Natural Products: 3D QSAR Pharmacophore, ADMET, Scaffold Hopping, Molecular Docking, and Molecular Dynamics Study. Mar. Drugs 2024, 22, 83. https://doi.org/10.3390/md22020083
Zhou N, Zheng C, Tan H, Luo L. Identification of PLK1-PBD Inhibitors from the Library of Marine Natural Products: 3D QSAR Pharmacophore, ADMET, Scaffold Hopping, Molecular Docking, and Molecular Dynamics Study. Marine Drugs. 2024; 22(2):83. https://doi.org/10.3390/md22020083
Chicago/Turabian StyleZhou, Nan, Chuangze Zheng, Huiting Tan, and Lianxiang Luo. 2024. "Identification of PLK1-PBD Inhibitors from the Library of Marine Natural Products: 3D QSAR Pharmacophore, ADMET, Scaffold Hopping, Molecular Docking, and Molecular Dynamics Study" Marine Drugs 22, no. 2: 83. https://doi.org/10.3390/md22020083
APA StyleZhou, N., Zheng, C., Tan, H., & Luo, L. (2024). Identification of PLK1-PBD Inhibitors from the Library of Marine Natural Products: 3D QSAR Pharmacophore, ADMET, Scaffold Hopping, Molecular Docking, and Molecular Dynamics Study. Marine Drugs, 22(2), 83. https://doi.org/10.3390/md22020083