
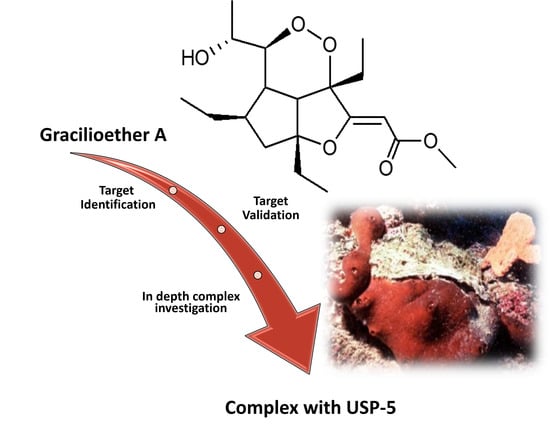
Chemoproteomics Reveals USP5 (Ubiquitin Carboxyl-Terminal Hydrolase 5) as Promising Target of the Marine Polyketide Gracilioether A

 ,
, 
 ,
,  ,
,  ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
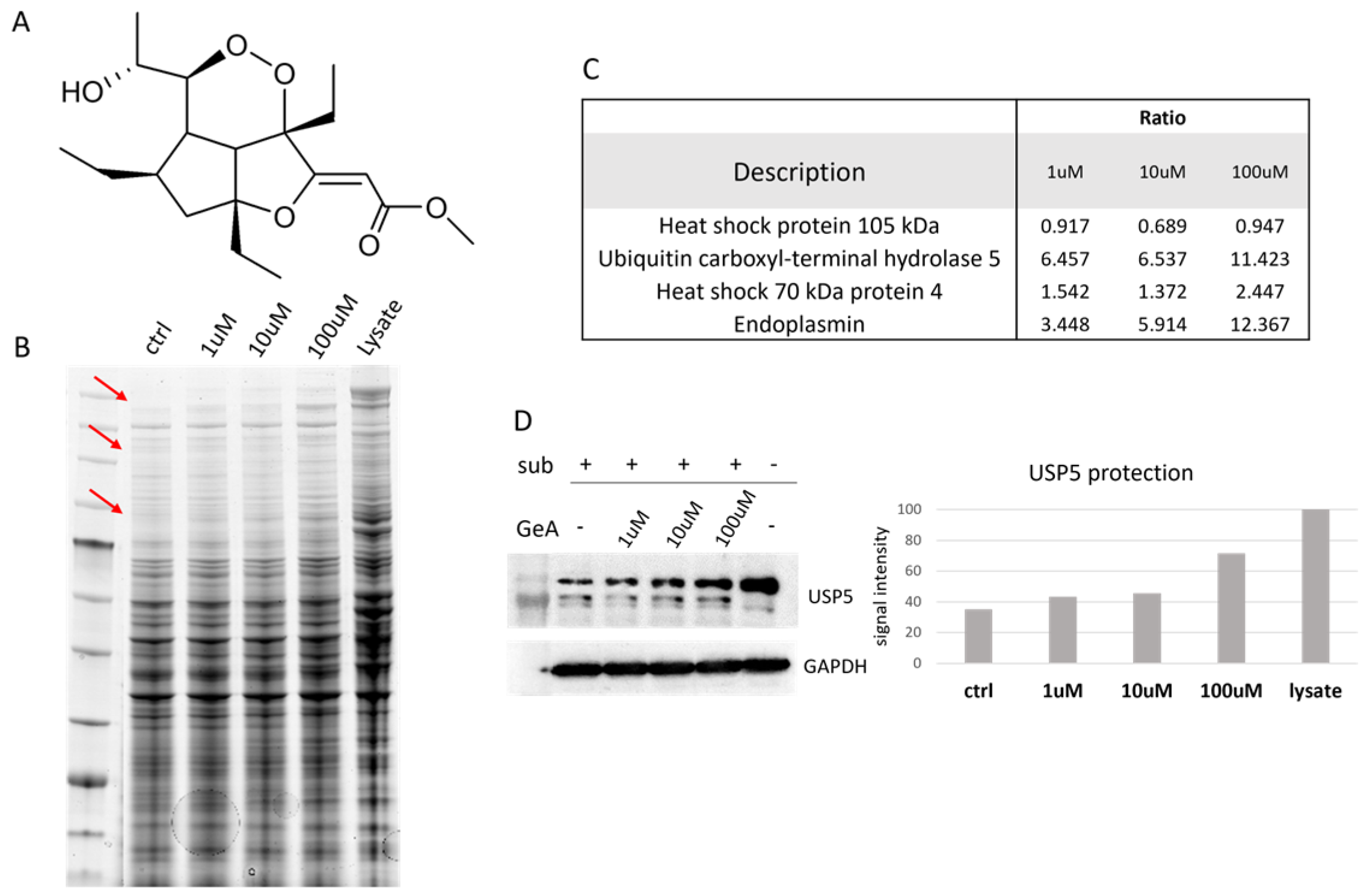
2.1. USP5 Identification as Main GeA Protein Partner
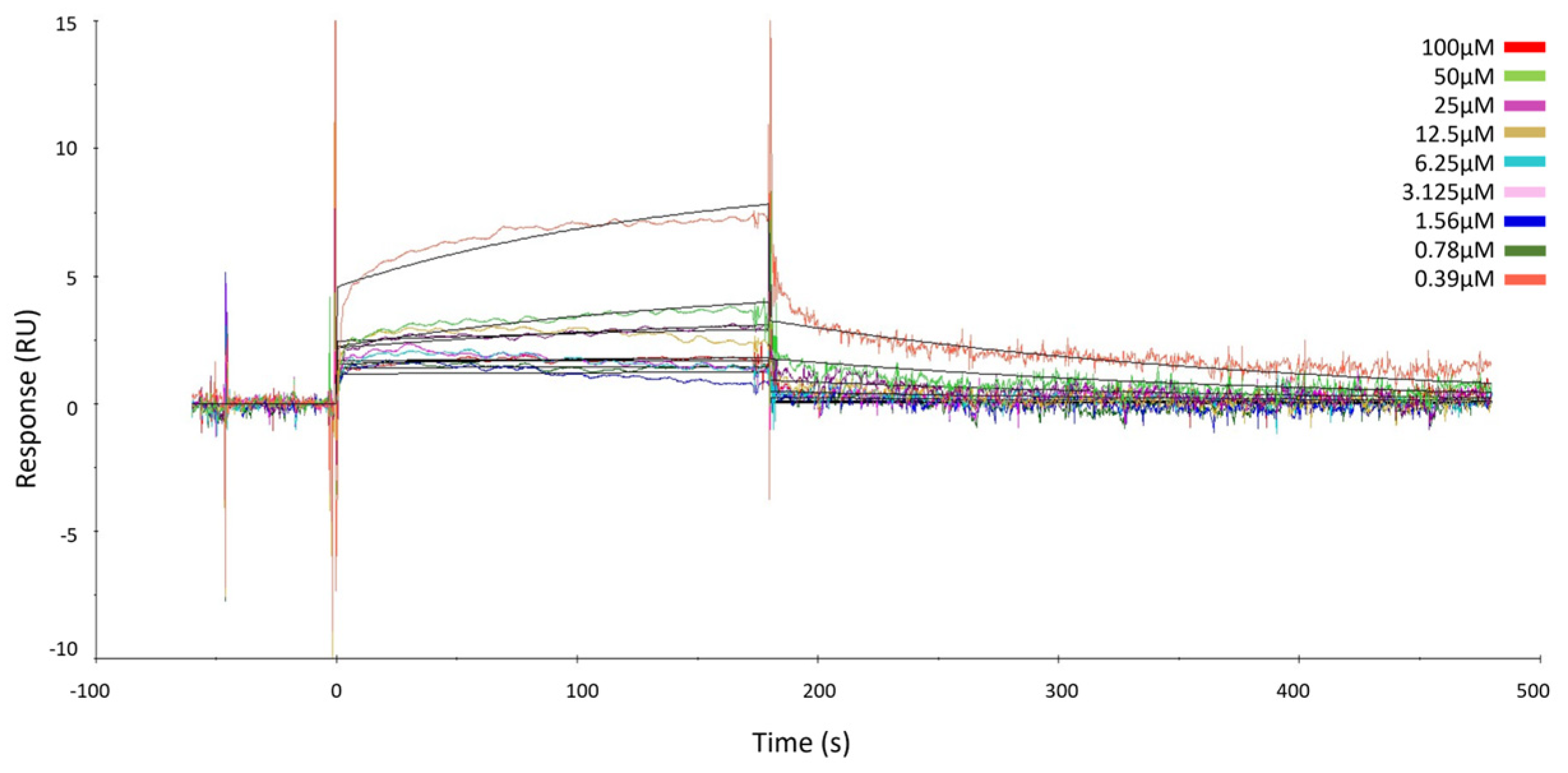
2.2. Surface Plasmon Resonance of GeA and USP5
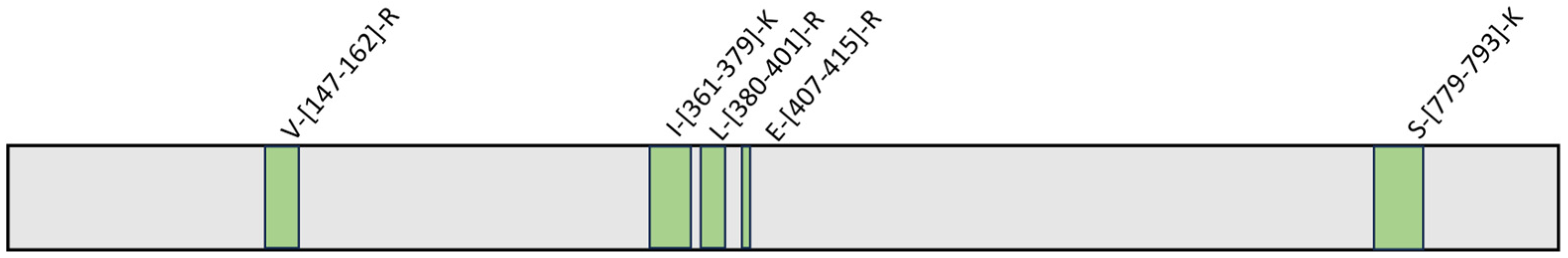
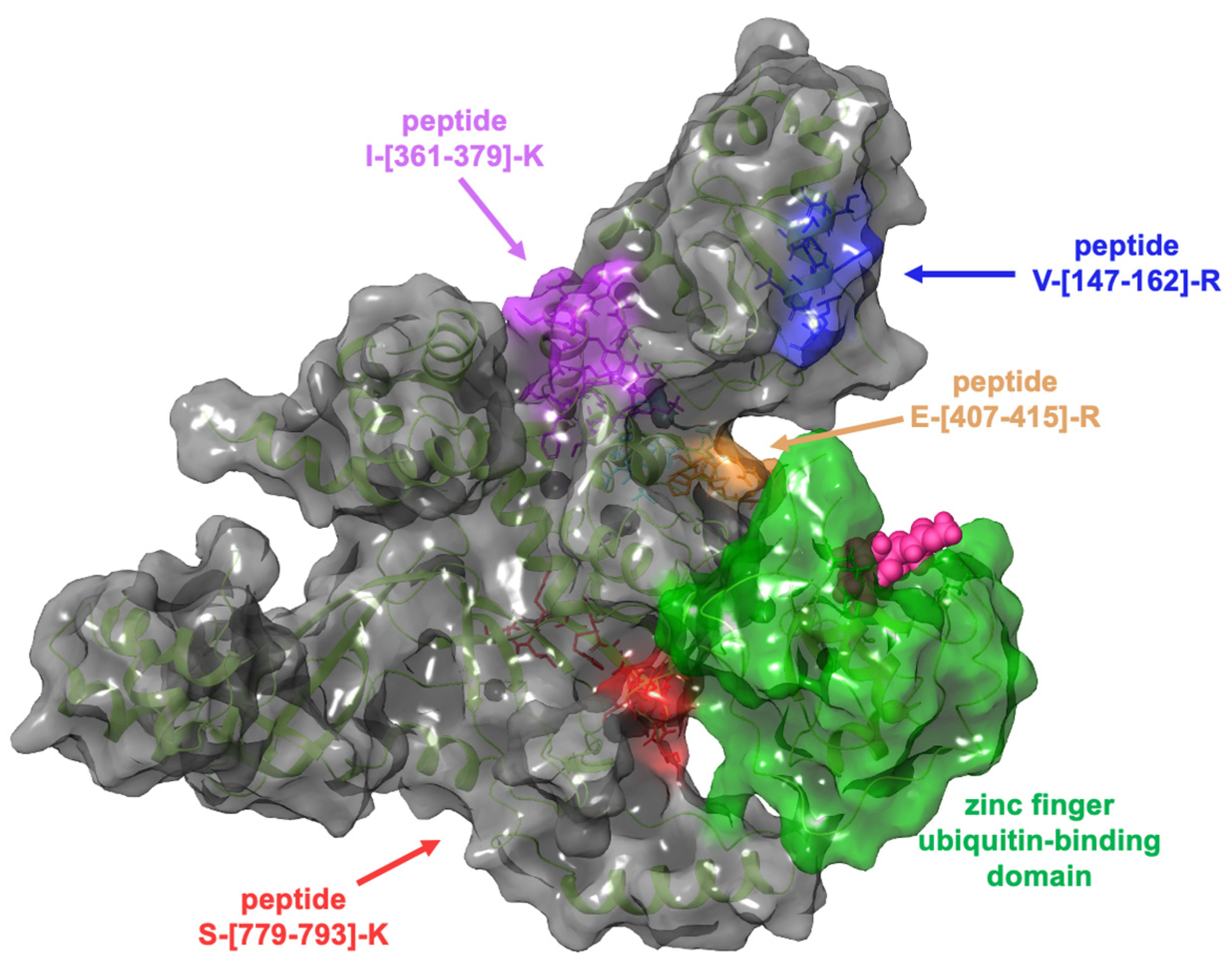
2.3. Evaluation of the Interaction Features between GeA and USP5 by t-LiP-MRM-MS
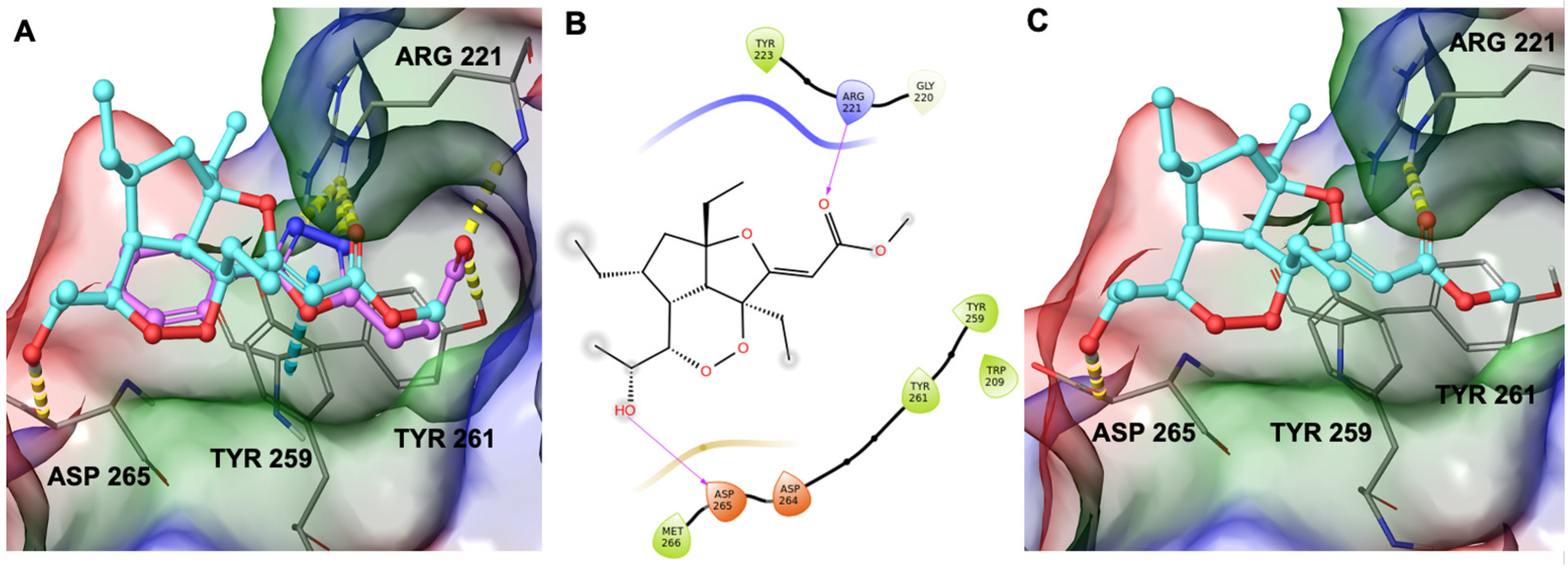
2.4. Computational Study on the Interactions between GeA and USP5
3. Discussion
4. Materials and Methods
4.1. Gracilioether A Extraction
4.2. Cell Culture
4.3. DARTS Experiment
4.4. Western Blotting
4.5. Surface Plasmon Resonance
4.6. t-LiP-MRM-MS: Methods Fine-Tuning
4.7. Interactions Study of the Complex GeA/USP5: t-LiP-MRM-MS
4.8. Interactions of the Complex GeA/USP5: Computational Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, Q.G.; Zhao, K.; Peuronen, A.; Rissanen, K.; Enders, D.; Tang, Y.F. Enantioselective Total Syntheses of (+)-Hippolachnin A, (+)-Gracilioether A, (−)-Gracilioether E, and (−)-Gracilioether F. J. Am. Chem. Soc. 2018, 140, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.G.; Yang, H.Z.; Tang, Y.F. Recent advances in the synthesis of plakortin-type polyketides. Org. Biomol. Chem. 2020, 18, 9371–9384. [Google Scholar] [CrossRef]
- Ueoka, R.; Nakao, Y.; Kawatsu, S.; Yaegashi, J.; Matsumoto, Y.; Matsunaga, S.; Furihata, K.; van Soest, R.W.M.; Fusetani, N. Gracilioethers A-C, Antimalarial Metabolites from the Marine Sponge Agelas gracilis. J. Org. Chem. 2009, 74, 4203–4207. [Google Scholar] [CrossRef]
- Festa, C.; D’Amore, C.; Renga, B.; Lauro, G.; Marino, S.D.; D’Auria, M.V.; Bifulco, G.; Zampella, A.; Fiorucci, S. Oxygenated polyketides from Plakinastrella mamillaris as a new chemotype of PXR agonists. Mar. Drugs 2013, 11, 2314–2327. [Google Scholar] [CrossRef]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.N.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of Direct Protein Targets of Small Molecules. ACS Chem. Biol. 2011, 6, 34–46. [Google Scholar] [CrossRef]
- Fontana, A.; de Laureto, P.P.; Spolaore, B.; Frare, E.; Picotti, P.; Zambonin, M. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 2004, 51, 299–321. [Google Scholar] [CrossRef]
- Feng, Y.H.; De Franceschi, G.; Kahraman, A.; Soste, M.; Melnik, A.; Boersema, P.J.; de Laureto, P.P.; Nikolaev, Y.; Oliveira, A.P.; Picotti, P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014, 32, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef]
- Sun, T.S.; Liu, Z.N.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 146. [Google Scholar] [CrossRef]
- Snyder, N.A.; Silva, G.M. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response. J. Biol. Chem. 2021, 297, 101077. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D.; Tashayev, V.L.; Oconnor, L.B.; Larsen, C.N.; Kasperek, E.; Pickart, C.M. Metabolism of the Polyubiquitin Degradation Signal: Structure, Mechanism, and Role of Isopeptidase-T. Biochemistry 1995, 34, 14535–14546. [Google Scholar] [CrossRef]
- Ning, F.; Xin, H.; Liu, J.; Lv, C.; Xu, X.; Wang, M.; Wang, Y.; Zhang, W.; Zhang, X. Structure and function of USP5: Insight into physiological and pathophysiological roles. Pharmacol. Res. 2020, 157, 104557. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, W.; Huang, H.; Jiang, J.; Ding, Y.; Li, X.; Ma, J.; Hou, M.; Pu, X.; Qian, G.; et al. Kawasaki disease: Ubiquitin-specific protease 5 promotes endothelial inflammation via TNFα-mediated signaling. Pediatr. Res. 2023, 93, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Gadotti, V.M.; Zamponi, G.W. Disrupting USP5/Cav3.2 interactions protects female mice from mechanical hypersensitivity during peripheral inflammation. Mol. Brain 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol. Biol. 2015, 1263, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Kusebauch, U.; Campbell, D.S.; Deutsch, E.W.; Chu, C.S.; Spicer, D.A.; Brusniak, M.Y.; Slagel, J.; Sun, Z.; Stevens, J.; Grimes, B.; et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell 2016, 166, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Allali-Hassani, A.; Asinas, A.; Nair, U.B.; Fang, X.Y.; Zuo, X.B.; Wane, Y.X.; Wilkinson, K.D.; et al. Two ZnF-UBP Domains in Isopeptidase T (USP5). Biochemistry 2012, 51, 1188–1198. [Google Scholar] [CrossRef]
- Mann, M.K.; Franzoni, I.; de Freitas, R.F.; Tempel, W.; Houliston, S.; Smith, L.; Vedadi, M.; Arrowsmith, C.H.; Harding, R.J.; Schapira, M. Discovery of Small Molecule Antagonists of the USP5 Zinc Finger Ubiquitin-Binding Domain. J. Med. Chem. 2019, 62, 10144–10155. [Google Scholar] [CrossRef]
- Halgren, T.A.M.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Ruggiero, D.; Terracciano, S.; Lauro, G.; Pecoraro, M.; Franceschelli, S.; Bifulco, G.; Bruno, I. Structural Refinement of 2,4-Thiazolidinedione Derivatives as New Anticancer Agents Able to Modulate the BAG3 Protein. Molecules 2022, 27, 665. [Google Scholar] [CrossRef]
- Meng, J.; Ai, X.; Lei, Y.; Zhong, W.; Qian, B.; Qiao, K.; Wang, X.; Zhou, B.; Wang, H.; Huai, L. USP5 promotes epithelial-mesenchymal transition by stabilizing SLUG in hepatocellular carcinoma. Theranostics 2019, 9, 573. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger. Release 2021-1: Protein Preparation Wizard; Epik, Schrödinger, LLC; Impact, Schrödinger, LLC; Prime, Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger. Release 2021-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger. Release 2021-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger. Release 2021 Glide; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capuano, A.; D’Urso, G.; Aliberti, M.; Ruggiero, D.; Terracciano, S.; Festa, C.; Tosco, A.; Chini, M.G.; Lauro, G.; Bifulco, G.; et al. Chemoproteomics Reveals USP5 (Ubiquitin Carboxyl-Terminal Hydrolase 5) as Promising Target of the Marine Polyketide Gracilioether A. Mar. Drugs 2024, 22, 41. https://doi.org/10.3390/md22010041
Capuano A, D’Urso G, Aliberti M, Ruggiero D, Terracciano S, Festa C, Tosco A, Chini MG, Lauro G, Bifulco G, et al. Chemoproteomics Reveals USP5 (Ubiquitin Carboxyl-Terminal Hydrolase 5) as Promising Target of the Marine Polyketide Gracilioether A. Marine Drugs. 2024; 22(1):41. https://doi.org/10.3390/md22010041
Chicago/Turabian StyleCapuano, Alessandra, Gilda D’Urso, Michela Aliberti, Dafne Ruggiero, Stefania Terracciano, Carmen Festa, Alessandra Tosco, Maria Giovanna Chini, Gianluigi Lauro, Giuseppe Bifulco, and et al. 2024. "Chemoproteomics Reveals USP5 (Ubiquitin Carboxyl-Terminal Hydrolase 5) as Promising Target of the Marine Polyketide Gracilioether A" Marine Drugs 22, no. 1: 41. https://doi.org/10.3390/md22010041
APA StyleCapuano, A., D’Urso, G., Aliberti, M., Ruggiero, D., Terracciano, S., Festa, C., Tosco, A., Chini, M. G., Lauro, G., Bifulco, G., & Casapullo, A. (2024). Chemoproteomics Reveals USP5 (Ubiquitin Carboxyl-Terminal Hydrolase 5) as Promising Target of the Marine Polyketide Gracilioether A. Marine Drugs, 22(1), 41. https://doi.org/10.3390/md22010041