
Proteomic and Transcriptomic Analyses to Decipher the Chitinolytic Response of Jeongeupia spp.



Abstract
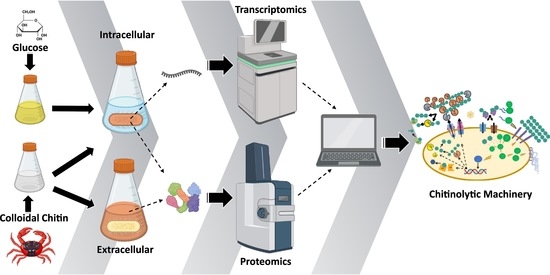
1. Introduction
2. Results and Discussion
2.1. Extracellular Proteomics
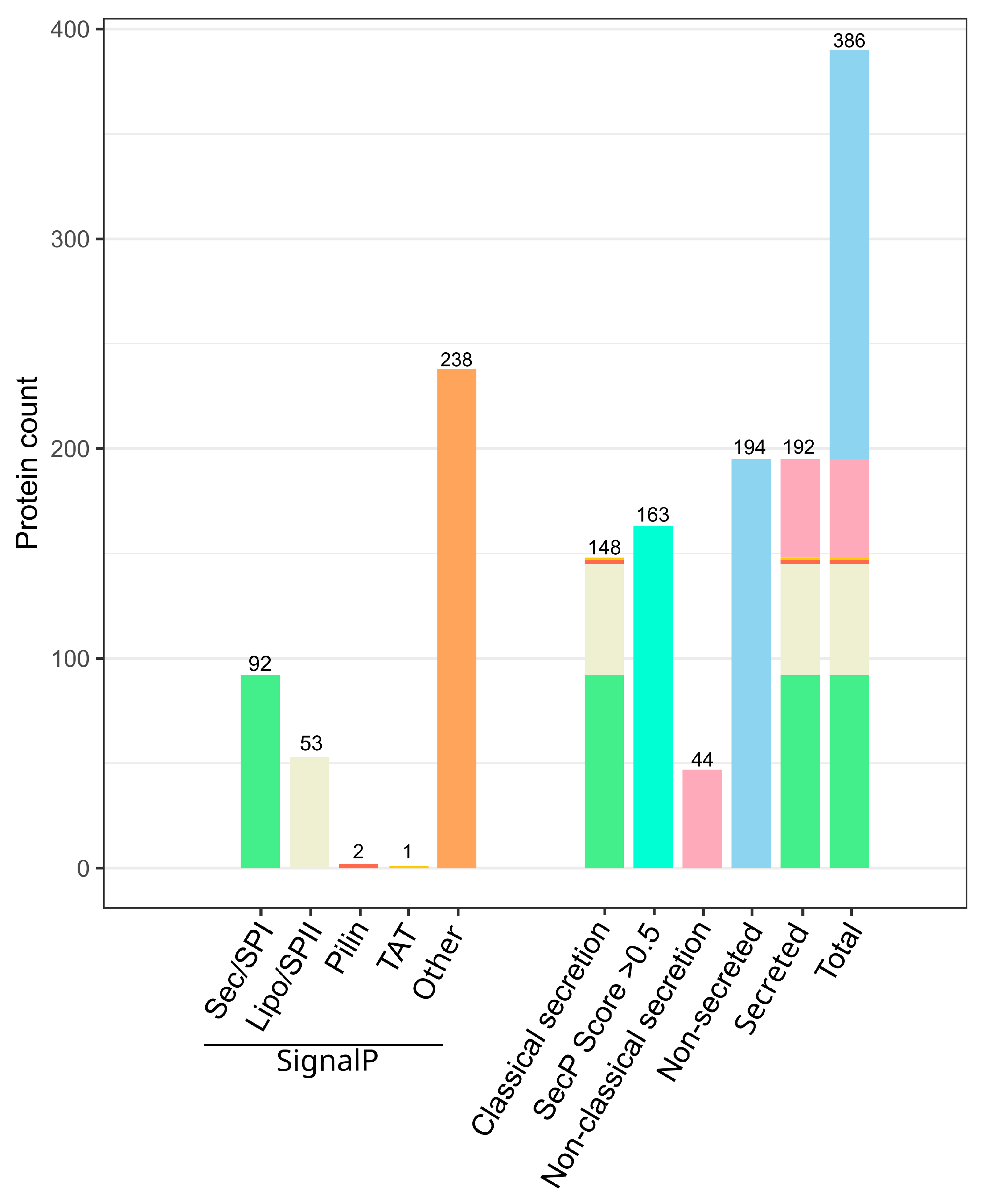
2.1.1. Predicted Chitinolytic Machinery of J. wiesaeckerbachi and Subcellular Localization
2.1.2. The Vast Majority of the Chitinolytic System Was Detected Extracellularly
2.1.3. The Lytic Polysaccharide Monooxygenase of the Auxiliary Activity Enzyme Family 10 Seems to Play a Minor Role in α-Chitin Hydrolysis of Jeongeupia spp.
2.1.4. Promising Candidate Proteins for Recombinant Expression Studies
2.1.5. In Silico Analyses May Aid in Reduction of Cell-Lysis Derived False-Positives
2.1.6. Highly Abundant Chitinases Exhibit Two Carbohydrate-Binding Modules
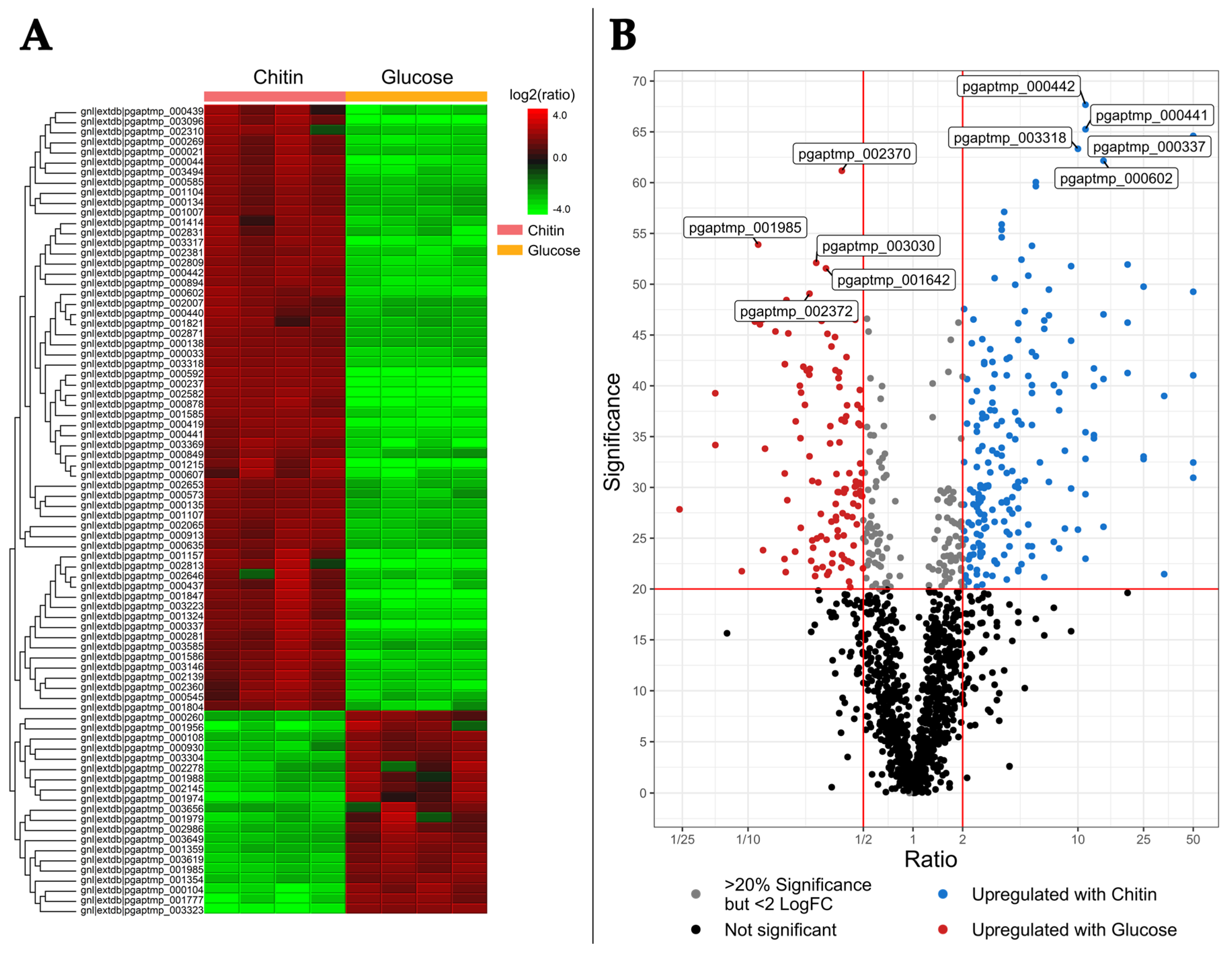
2.2. Differential Intracellular Protein Expression Using Chitin and Glucose Media
2.2.1. The Intracellular Chitin Response Specializes in Glucosamine Utilization and Cell Maintenance over Hydrolysis
2.2.2. Comparison of Intra- and Extracellular Chitin-Induced Proteomics
2.2.3. Challenges and Benefits of Biocomputational Approaches
2.3. Differential Transcriptomics
2.3.1. Distinct Transcription Patterns Highlight the Increased Burden of the Metabolic Chitin Response
2.3.2. Low Protein-mRNA Correlation between the Intracellular Datasets
2.3.3. Chitin Metabolism Transcript Upregulation Is Time Dependent
3. Materials and Methods
3.1. Chemicals and Consumables
3.2. Colloidal Chitin and Media Preparation
3.3. Bacterial Strains
3.4. Proteomics
3.4.1. Culture Conditions
3.4.2. Whole Cell Protein Extraction
3.4.3. Tryptic In-Gel Digestion and LC-MS/MS Analysis
3.4.4. Bioinformatic Analysis
3.5. Differential Transcriptomics
3.5.1. Culture Conditions
3.5.2. RNA Extraction and Quality Control
3.5.3. Next Generation Sequencing and Bioinformatic Analysis
4. Conclusions
4.1. Chitin-Metabolism Causes Profound Genetic Changes
4.2. Potential Role of Redox Enzymes in Chitin Hydrolysis
4.3. Chitin-Rich Environments Prompt a Multitude of Methyl-Accepting Chemotaxis and Motility Proteins
4.4. Schematic Summary of the Cumulative Systems Biology Approach
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Protein Accession Identifier | log2Fold Change | Adjusted p-Value | Annotation (PGAP and dbCAN 3.0) |
|---|---|---|---|---|
| 7 | pgaptmp_002582 | 4.04 | 4.84 × 10−46 | inclusion body family protein |
| 26 | pgaptmp_000680 | 3.45 | 2.02 × 10−34 | peptidoglycan-binding protein (GH19) |
| 34 | pgaptmp_002996 | 3.48 | 4 × 10−30 | FAD binding oxidoreductase (AA7) |
| 52 | pgaptmp_000371 | 2.77 | 1.15 × 10−26 | glycosyl hydrolase family 18 protein (GH18) |
| 66 | pgaptmp_003576 | 2.71 | 4.4 × 10−24 | methyl-accepting chemotaxis protein |
| 69 | pgaptmp_001506 | 2.77 | 2.06 × 10−23 | PAS domain-containing methyl-accepting chemotaxis protein |
| 102 | pgaptmp_000306 | 2.54 | 4.18 × 10−20 | carbohydate-binding domain-containing protein (GH20) |
| 105 | pgaptmp_001215 | 2.40 | 5.45 × 10−20 | SDR family oxidoreductase |
| 127 | pgaptmp_003096 | 3.04 | 1.03 × 10−18 | DUF1631 domain-containing protein |
| 169 | pgaptmp_002942 | 2.09 | 4.56 × 10−17 | cellulase family glycosylhydrolase |
| 228 | pgaptmp_003580 | 2.47 | 4.69 × 10−15 | hydrolase |
| 239 | pgaptmp_001847 | 2.20 | 1.4 × 10−14 | substrate-binding domain-containing protein |
| 294 | pgaptmp_000836 | 1.80 | 8.38 × 10−13 | glycosyl hydrolase family 18 protein (GH18) |
| 309 | pgaptmp_001841 | 1.90 | 2.1 × 10−12 | glycosyl hydrolase family 18 protein (GH18) |
| 314 | pgaptmp_000237 | 2.21 | 2.66 × 10−12 | hypothetical protein |
| 348 | pgaptmp_002137 | 2.25 | 1.08 × 10−11 | glycosyl hydrolase family 18 protein (GH18) |
| 370 | pgaptmp_000470 | 1.69 | 2.48 × 10−11 | N-acetylglucosamine-6-phosphate deacetylase |
| 394 | pgaptmp_000419 | 2.06 | 8.64 × 10−11 | glutamine amidotransferase |
| 438 | pgaptmp_000602 | 1.60 | 2.97 × 10−10 | methyl-accepting chemotaxis protein |
| 448 | pgaptmp_000372 | 2.92 | 4.33 × 10−10 | glycosyl hydrolase family 18 protein (GH18) |
| 453 | pgaptmp_000302 | 1.66 | 4.9 × 10−10 | chitinase (GH19) |
| 507 | pgaptmp_000337 | 1.64 | 3.04 × 10−9 | nitrate reductase subunit beta |
| 521 | pgaptmp_001552 | 1.43 | 4.87 × 10−9 | FAD-dependent monooxygenase |
| 589 | pgaptmp_002133 | 1.37 | 3.47 × 10−8 | alpha/beta hydrolase-fold protein |
| 678 | pgaptmp_001746 | 1.32 | 2.83 × 10−7 | glycosyl hydrolase family 18 protein (GH18) |
| 691 | pgaptmp_000878 | 1.84 | 3.37 × 10−7 | hypothetical protein |
| 869 | pgaptmp_003318 | 1.82 | 5.51 × 10−6 | PrkA family serine protein kinase |
| 981 | pgaptmp_002310 | 1.04 | 2.73 × 10−5 | basic amino acid ABC transporter substrate-binding protein |
| 1057 | pgaptmp_000437 | 1.15 | 6.33 × 10−5 | polysaccharide deacetylase family protein |
| 1334 | pgaptmp_001731 | −0.93 | 8.50 × 10−4 | carbohydate-binding domain-containing protein (GH20) |
| 1353 | pgaptmp_001323 | −0.94 | 9.96 × 10−4 | beta-N-acetylhexosaminidase (GH3) |
| 1512 | pgaptmp_001504 | −0.79 | 2.89 × 10−3 | glycosyl hydrolase family 18 protein (GH18) |
| 1900 | pgaptmp_000440 | −0.60 | 2.47 × 10−2 | sugar ABC transporter permease |
| 3390 | pgaptmp_000441 | −0.03 | 9.32 × 10−1 | sugar ABC transporter substrate-binding protein |
| Gene ID | HMMER | dbCAN_sub | DIAMOND | SignalP | Dataset |
|---|---|---|---|---|---|
| pgaptmp_000203 | N | N | CBM12+CBM5 | Y(1–21) | EC |
| pgaptmp_000437 | CE4(62–182) | CE4_e257 | CE4 | Y(1–21) | IC/EC |
| pgaptmp_000464 | N | N | CBM50+GH25 | N | IC |
| pgaptmp_001157 | N | AA2_e1 | AA0 | N | IC/EC |
| pgaptmp_001255 | N | CBM12_e3 | N | Y(1–28) | IC/EC |
| pgaptmp_001722 | GH23(474–603) | GH23_e952 | GH23 | Y(1–19) | EC |
| pgaptmp_001840 | N | N | CBM12 | Y(1–21) | T/EC |
| pgaptmp_001854 | N | CBM5_e49 | CBM5 | Y(1–24) | T/EC |
| pgaptmp_002133 | CE1(152–377) | N | CBM5 | Y(1–27) | T |
| pgaptmp_002212 | CE2(195–406) | CBM5_e51+CE2_e8 | CBM5+CE2 | Y(1–21) | T |
| pgaptmp_002996 | AA7(31–458) | AA7_e0 | N | N | T/EC |
| pgaptmp_003133 | N | CBM50_e508 | CBM50 | Y(1–26) | IC/EC |
| pgaptmp_003266 | GH23(302–444) | GH23_e322 | GH23 | N | IC |
| pgaptmp_003309 | GH23(334–469) | GH23_e69 | GH23 | Y(1–32) | T |
| pgaptmp_003521 | N | N | CBM50 | Y(1–24) | IC |
| pgaptmp_003567 | N | CBM50_e665 | N | N | EC |
References
- Gooday, G.W. The Ecology of Chitin Degradation BT-Advances in Microbial Ecology; Marshall, K.C., Ed.; Springer: Boston, MA, USA, 1990; pp. 387–430. ISBN 978-1-4684-7612-5. [Google Scholar]
- Younes, I.; Hajji, S.; Frachet, V.; Rinaudo, M.; Jellouli, K.; Nasri, M. Chitin Extraction from Shrimp Shell Using Enzymatic Treatment. Antitumor, Antioxidant and Antimicrobial Activities of Chitosan. Int. J. Biol. Macromol. 2014, 69, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and Chitosan: Properties and Applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- FAO. The State of World Fisheries and Aquaculture 2022; Food & Agriculture Organization: Rome, Italy, 2022; ISBN 9789251363645. [Google Scholar]
- Yan, N.; Chen, X. Don’t Waste Seafood Waste: Turning Cast-off Shells into Nitrogen-Rich Chemicals Would Benefit Economies and the Environment. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef]
- Xie, X.H.; Fu, X.; Yan, X.Y.; Peng, W.F.; Kang, L.X. A Broad-Specificity Chitinase from Penicillium Oxalicum K10 Exhibits Antifungal Activity and Biodegradation Properties of Chitin. Mar. Drugs 2021, 19, 356. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.; Muralisankar, T.; Jayakumar, R.; Rajeevgandhi, C. A Study on Structural Comparisons of α-Chitin Extracted from Marine Crustacean Shell Waste. Carbohydr. Polym. Technol. Appl. 2021, 2, 100037. [Google Scholar] [CrossRef]
- Aklog, Y.F.; Egusa, M.; Kaminaka, H.; Izawa, H.; Morimoto, M.; Saimoto, H.; Ifuku, S. Protein/CaCO3/Chitin Nanofiber Complex Prepared from Crab Shells by Simple Mechanical Treatment and Its Effect on Plant Growth. Int. J. Mol. Sci. 2016, 17, 1600. [Google Scholar] [CrossRef] [PubMed]
- Boßelmann, F.; Romano, P.; Fabritius, H.; Raabe, D.; Epple, M. The Composition of the Exoskeleton of Two Crustacea: The American Lobster Homarus Americanus and the Edible Crab Cancer Pagurus. Thermochim. Acta 2007, 463, 65–68. [Google Scholar] [CrossRef]
- Rødde, R.H.; Einbu, A.; Vårum, K.M. A Seasonal Study of the Chemical Composition and Chitin Quality of Shrimp Shells Obtained from Northern Shrimp (Pandalus borealis). Carbohydr. Polym. 2008, 71, 388–393. [Google Scholar] [CrossRef]
- Tharanathan, R.N.; Kittur, F.S. Chitin—The Undisputed Biomolecule of Great Potential. Crit. Rev. Food Sci. Nutr. 2003, 43, 61–87. [Google Scholar] [CrossRef]
- Kerton, F.M.; Liu, Y.; Omari, K.W.; Hawboldt, K. Green Chemistry and the Ocean-Based Biorefinery. Green Chem. 2013, 15, 860–871. [Google Scholar] [CrossRef]
- Pacheco, N.; Garnica-Gonzalez, M.; Gimeno, M.; Bárzana, E.; Trombotto, S.; David, L.; Shirai, K. Structural Characterization of Chitin and Chitosan Obtained by Biological and Chemical Methods. Biomacromolecules 2011, 12, 3285–3290. [Google Scholar] [CrossRef]
- Arbia, W.; Arbia, L.; Adour, L.; Amrane, A. Chitin Extraction from Crustacean Shells Using Biological Methods—A Review. Food Technol. Biotechnol. 2013, 51, 12–25. [Google Scholar]
- Keyhani, N.O.; Roseman, S. Physiological Aspects of Chitin Catabolism in Marine Bacteria. Biochim. Biophys. Acta-Gen. Subj. 1999, 1473, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Roseman, S. The Chitinolytic Cascade in Vibrios Is Regulated by Chitin Oligosaccharides and a Two-Component Chitin Catabolic Sensor/Kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 627–631. [Google Scholar] [CrossRef]
- Souza, C.P.; Almeida, B.C.; Colwell, R.R.; Rivera, I.N.G. The Importance of Chitin in the Marine Environment. Mar. Biotechnol. 2011, 13, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Roseman, S. A Conversation with Saul Roseman. Biochem. Biophys. Res. Commun. 2003, 300, 5–8. [Google Scholar] [PubMed]
- Arnold, N.D.; Garbe, D.; Brück, T.B. Isolation, Biochemical Characterization, and Sequencing of Two High-Quality Genomes of a Novel Chitinolytic Jeongeupia Species. MicrobiologyOpen 2023, 12, e1372. [Google Scholar] [CrossRef]
- Yoon, J.H.; Choi, J.H.; Kang, S.J.; Choi, N.S.; Lee, J.S.; Song, J.J. Jeongeupia Naejangsanensis Gen. Nov., Sp. Nov., a Cellulose-Degrading Bacterium Isolated from Forest Soil from Naejang Mountain in Korea. Int. J. Syst. Evol. Microbiol. 2010, 60, 615–619. [Google Scholar] [CrossRef]
- Lopes, M.A.; Gomes, D.S.; Koblitz, M.G.B.; Pirovani, C.P.; de Mattos Cascardo, J.C.; Góes-Neto, A.; Micheli, F. Use of Response Surface Methodology to Examine Chitinase Regulation in the Basidiomycete Moniliophthora Perniciosa. Mycol. Res. 2008, 112, 399–406. [Google Scholar] [CrossRef]
- Itoh, T.; Kimoto, H. Bacterial Chitinase System as a -Model of Chitin Biodegradation. In Targeting Chitin-Containing Organisms; Springer: Singapore, 2019; Volume 1142, pp. 131–151. ISBN 978-981-13-7317-6. [Google Scholar]
- Trost, M.; Wehmhöner, D.; Kärst, U.; Dieterich, G.; Wehland, J.; Jänsch, L. Comparative Proteome Analysis of Secretory Proteins from Pathogenic and Nonpathogenic Listeria Species. Proteomics 2005, 5, 1544–1557. [Google Scholar] [CrossRef]
- Hamilton, J.J.; Marlow, V.L.; Owen, R.A.; Costa, M.d.A.A.; Guo, M.; Buchanan, G.; Chandra, G.; Trost, M.; Coulthurst, S.J.; Palmer, T.; et al. A Holin and an Endopeptidase Are Essential for Chitinolytic Protein Secretion in Serratia Marcescens. J. Cell Biol. 2014, 207, 615–626. [Google Scholar] [CrossRef]
- Freudl, R. Signal Peptides for Recombinant Protein Secretion in Bacterial Expression Systems. Microb. Cell Factories 2018, 17, 52. [Google Scholar] [CrossRef]
- Davies, G.; Henrissat, B. Structures and Mechanisms of Glycosyl Hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Armenteros, J.J.A.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Gíslason, M.H.; Nielsen, H.; Armenteros, J.J.A.; Johansen, A.R. Prediction of GPI-Anchored Proteins with Pointer Neural Networks. Curr. Res. Biotechnol. 2021, 3, 6–13. [Google Scholar] [CrossRef]
- Tuveng, T.R.; Arntzen, M.Ø.; Bengtsson, O.; Gardner, J.G.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Proteomic Investigation of the Secretome of Cellvibrio Japonicus during Growth on Chitin. Proteomics 2016, 16, 1904–1914. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.; Xia, Y.; Cui, J.; Gu, Z.; Song, Y.; Chen, Y.Q.; Zhang, H.; Chen, W. How Are the Non-Classically Secreted Bacterial Proteins Released into the Extracellular Milieu? Curr. Microbiol. 2013, 67, 688–695. [Google Scholar] [CrossRef]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. DbCAN3: Automated Carbohydrate-Active Enzyme and Substrate Annotation. Nucleic Acids Res. 2023, 51, gkad328. [Google Scholar] [CrossRef]
- Forsberg, Z.; Nelson, C.E.; Dalhus, B.; Mekasha, S.; Loose, J.S.M.; Crouch, L.I.; Røhr, Å.K.; Gardner, J.G.; Eijsink, V.G.H.; Vaaje-Kolstad, G. Structural and Functional Analysis of a Lytic Polysaccharide Monooxygenase Important for Efficient Utilization of Chitin in Cellvibrio Japonicus. J. Biol. Chem. 2016, 291, 7300–7312. [Google Scholar] [CrossRef]
- Mekasha, S.; Byman, I.R.; Lynch, C.; Toupalová, H.; Anděra, L.; Næs, T.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Development of Enzyme Cocktails for Complete Saccharification of Chitin Using Mono-Component Enzymes from Serratia Marcescens. Process Biochem. 2017, 56, 132–138. [Google Scholar] [CrossRef]
- Hemsworth, G.R.; Johnston, E.M.; Davies, G.J.; Walton, P.H. Lytic Polysaccharide Monooxygenases in Biomass Conversion. Trends Biotechnol. 2015, 33, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Várnai, A.; Johansen, K.S.; Eijsink, V.G.H.; Horn, S.J. Harnessing the Potential of LPMO-Containing Cellulase Cocktails Poses New Demands on Processing Conditions. Biotechnol. Biofuels 2015, 8, 187. [Google Scholar] [CrossRef]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2O2. Nat. Chem. Biol. 2017, 13, 1123–1128. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful Software for Peptide de Novo Sequencing by Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Nakagawa, Y.S.; Kudo, M.; Loose, J.S.M.; Ishikawa, T.; Totani, K.; Eijsink, V.G.H.; Vaaje-Kolstad, G. A Small Lytic Polysaccharide Monooxygenase from Streptomyces Griseus Targeting α- And β-Chitin. FEBS J. 2015, 282, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Houston, D.R.; Riemen, A.H.K.; Eijsink, V.G.H.; van Aalten, D.M.F. Crystal Structure and Binding Properties of the Serratia Marcescens Chitin-Binding Protein CBP21. J. Biol. Chem. 2005, 280, 11313–11319. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, M.; Zhao, S.; Lin, C.; Song, J.; Yang, Q. ATP-Binding Cassette Transporter Regulates N,N′-Diacetylchitobiose Transportation and Chitinase Production in Trichoderma Asperellum T4. Int. J. Mol. Sci. 2019, 20, 2412. [Google Scholar] [CrossRef]
- Zeng, Y.; Charkowski, A.O. The Role of ATP-Binding Cassette Transporters in Bacterial Phytopathogenesis. Phytopathology® 2020, 111, 600–610. [Google Scholar] [CrossRef]
- Dassa, E.; Bouige, P. The ABC of ABCs: A Phylogenetic and Functional Classification of ABC Systems in Living Organisms. Res. Microbiol. 2001, 152, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.A.; Elie, D.; Cedric, O.; Jue, C. Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, O.; Arntzen, M.Ø.; Mathiesen, G.; Skaugen, M.; Eijsink, V.G.H. A Novel Proteomics Sample Preparation Method for Secretome Analysis of Hypocrea Jecorina Growing on Insoluble Substrates. J. Proteom. 2016, 131, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Kiemer, L.; Fausbøll, A.; Brunak, S. Non-Classical Protein Secretion in Bacteria. BMC Microbiol. 2005, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.H.; Tan, T.W.; Ranganathan, S. A Comprehensive Assessment of N-Terminal Signal Peptides Prediction Methods. BMC Bioinform. 2009, 10, S2. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Mass Spectrometry and the Search for Moonlighting Proteins. Mass Spectrom. Rev. 2005, 24, 772–782. [Google Scholar] [CrossRef]
- Brix, K.; Summa, W.; Lottspeich, F.; Herzog, V. Extracellularly Occurring Histone H1 Mediates the Binding of Thyroglobulin to the Cell Surface of Mouse Macrophages. J. Clin. Investig. 1998, 102, 283–293. [Google Scholar] [CrossRef]
- Juncker, A.S.; Willenbrock, H.; Von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of Lipoprotein Signal Peptides in Gram-Negative Bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef]
- Lorentzen, S.B.; Arntzen, M.; Hahn, T.; Tuveng, T.R.; Sørlie, M.; Zibek, S.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Genomic and Proteomic Study of Andreprevotia Ripae Isolated from an Anthill Reveals an Extensive Repertoire of Chitinolytic Enzymes. J. Proteome Res. 2021, 20, 4041–4052. [Google Scholar] [CrossRef]
- Liu, X.; Luo, Y.; Li, Z.; Wei, G. Functional Analysis of PrkA-a Putative Serine Protein Kinase from Mesorhizobium Alhagi CCNWXJ12-2-in Stress Resistance. BMC Microbiol. 2016, 16, 227. [Google Scholar] [CrossRef]
- Galinier, A.; Kravanja, M.; Engelmann, R.; Hengstenberg, W.; Kilhoffer, M.C.; Deutscher, J.; Haiech, J. New Protein Kinase and Protein Phosphatase Families Mediate Signal Transduction in Bacterial Catabolite Repression. Proc. Natl. Acad. Sci. USA 1998, 95, 1823–1828. [Google Scholar] [CrossRef]
- Kavanagh, K.L.; Jörnvall, H.; Persson, B.; Oppermann, U. Medium- and Short-Chain Dehydrogenase/Reductase Gene and Protein Families: The SDR Superfamily: Functional and Structural Diversity within a Family of Metabolic and Regulatory Enzymes. Cell. Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Bhatwa, A.; Wang, W.; Hassan, Y.I.; Abraham, N.; Li, X.-Z.; Zhou, T. Challenges Associated With the Formation of Recombinant Protein Inclusion Bodies in Escherichia Coli and Strategies to Address Them for Industrial Applications. Front. Bioeng. Biotechnol. 2021, 9, 630551. [Google Scholar] [CrossRef] [PubMed]
- Matilla, M.A.; Ortega, Á.; Krell, T. The Role of Solute Binding Proteins in Signal Transduction. Comput. Struct. Biotechnol. J. 2021, 19, 1786–1805. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- de Boer, W.; Folman, L.B.; Summerbell, R.C.; Boddy, L. Living in a Fungal World: Impact of Fungi on Soil Bacterial Niche Development. FEMS Microbiol. Rev. 2005, 29, 795–811. [Google Scholar] [CrossRef]
- Lee, J.; Hiibel, S.R.; Reardon, K.F.; Wood, T.K. Identification of Stress-related Proteins in Escherichia Coli Using the Pollutant Cis-dichloroethylene. J. Appl. Microbiol. 2010, 108, 2088–2102. [Google Scholar] [CrossRef]
- Giltner, C.L.; Nguyen, Y.; Burrows, L.L. Type IV Pilin Proteins: Versatile Molecular Modules. Microbiol. Mol. Biol. Rev. 2012, 76, 740–772. [Google Scholar] [CrossRef]
- Craig, L.; Forest, K.T.; Maier, B. Type IV Pili: Dynamics, Biophysics and Functional Consequences. Nat. Rev. Microbiol. 2019, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Güell, M.; Serrano, L. Correlation of MRNA and Protein in Complex Biological Samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Choi, P.J.; Li, G.-W.; Chen, H.; Babu, M.; Hearn, J.; Emili, A.; Xie, X.S. Quantifying, E. coli Proteome and Transcriptome with Single-Molecule Sensitivity in Single Cells. Science 2010, 329, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the Regulation of Protein Abundance from Proteomic and Transcriptomic Analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Delogu, F.; Kunath, B.J.; Evans, P.N.; Arntzen, M.; Hvidsten, T.R.; Pope, P.B. Integration of Absolute Multi-Omics Reveals Dynamic Protein-to-RNA Ratios and Metabolic Interplay within Mixed-Domain Microbiomes. Nat. Commun. 2020, 11, 4708. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on MRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. MRNAs, Proteins and the Emerging Principles of Gene Expression Control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Monge, E.C.; Tuveng, T.R.; Vaaje-Kolstad, G.; Eijsink, V.G.H.; Gardner, J.G. Systems Analysis of the Glycoside Hydrolase Family 18 Enzymes from Cellvibrio Japonicus Characterizes Essential Chitin Degradation Functions. J. Biol. Chem. 2018, 293, 3849–3859. [Google Scholar] [CrossRef]
- Dekel, E.; Alon, U. Optimality and Evolutionary Tuning of the Expression Level of a Protein. Nature 2005, 436, 588–592. [Google Scholar] [CrossRef]
- KOCH, A.L.; LEVY, H.R. Protein Turnover in Growing Cultures of Escherichia Coli. J. Biol. Chem. 1955, 217, 947–957. [Google Scholar] [CrossRef]
- Saito, A.; Ishizaka, M.; Francisco, J.; Fujii, T.; Miyashita, K. Transcriptional Co-Regulation of Five Chitinase Genes Scattered on the Streptomyces Coelicolor A3(2) Chromosome. Microbiology 2000, 146, 2937–2946. [Google Scholar] [CrossRef]
- Saito, A.; Fujii, T.; Miyashita, K. Distribution and Evolution of Chitinase Genes in Streptomyces Species: Involvement of Gene-Duplication and Domain-Deletion. Antonie Van Leeuwenhoek 2003, 84, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Eijsink, V.G.H.; Kielak, A.M.; van Veen, J.A.; de Boer, W. Genomic Comparison of Chitinolytic Enzyme Systems from Terrestrial and Aquatic Bacteria. Environ. Microbiol. 2016, 18, 38–49. [Google Scholar] [CrossRef]
- Wessely, F.; Bartl, M.; Guthke, R.; Li, P.; Schuster, S.; Kaleta, C. Optimal Regulatory Strategies for Metabolic Pathways in Escherichia Coli Depending on Protein Costs. Mol. Syst. Biol. 2011, 7, 515. [Google Scholar] [CrossRef]
- Suvorova, I.A.; Korostelev, Y.D.; Gelfand, M.S. GntR Family of Bacterial Transcription Factors and Their DNA Binding Motifs: Structure, Positioning and Co-Evolution. PLoS ONE 2015, 10, e0132618. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Murthy, N.; Bleakley, B. Simplified Method of Preparing Colloidal Chitin Used For Screening of Chitinase- Producing Microorganisms. Internet J. Microbiol. 2012, 10, e2bc3. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lee, H.-J.; Choi, S.-W.; Her, S.; Oh, D.-H. Purification and Characterization of Antifungal Chitinase from Pseudomonas Sp. YHS-A2. J. Microbiol. Biotechnol. 1997, 7, 107–113. [Google Scholar]
- Singh, P.P.; Shin, Y.C.; Park, C.S.; Chung, Y.R. Biological Control of Fusarium Wilt of Cucumber by Chitinolytic Bacteria. Phytopathology 1998, 89, 92–99. [Google Scholar] [CrossRef]
- Engelhart-Straub, S.; Cavelius, P.; Hölzl, F.; Haack, M.; Awad, D.; Brueck, T.; Mehlmer, N. Effects of Light on Growth and Metabolism of Rhodococcus Erythropolis. Microorganisms 2022, 10, 1680. [Google Scholar] [CrossRef]
- Fuchs, T.; Melcher, F.; Rerop, Z.S.; Lorenzen, J.; Shaigani, P.; Awad, D.; Haack, M.; Prem, S.A.; Masri, M.; Mehlmer, N.; et al. Identifying Carbohydrate-Active Enzymes of Cutaneotrichosporon Oleaginosus Using Systems Biology. Microb. Cell Factories 2021, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.-L.; Münch, P.C.; Mreches, R.; McHardy, A.C. Rapid and Accurate Identification of Ribosomal RNA Sequences via Deep Learning. Nucleic Acids Res. 2022, 50, e60. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Sentieon Sentieon Provides Complete Solutions for Secondary Dna/Rna Analysis for a Variety of Sequencing Platforms, Including Short and Long Reads. Available online: https://www.sentieon.com/ (accessed on 24 July 2023).
- Sun, X.; Li, Y.; Tian, Z.; Qian, Y.; Zhang, H.; Wang, L. A Novel Thermostable Chitinolytic Machinery of Streptomyces Sp. F-3 Consisting of Chitinases with Different Action Modes. Biotechnol. Biofuels 2019, 12, 136. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Bøhle, L.A.; Gåseidnes, S.; Dalhus, B.; Bjørås, M.; Mathiesen, G.; Eijsink, V.G.H. Characterization of the Chitinolytic Machinery of Enterococcus Faecalis V583 and High-Resolution Structure of Its Oxidative CBM33 Enzyme. J. Mol. Biol. 2012, 416, 239–254. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Horn, S.J.; van Aalten, D.M.F.; Synstad, B.; Eijsink, V.G.H. The Non-Catalytic Chitin-Binding Protein CBP21 from Serratia Marcescens Is Essential for Chitin Degradation. J. Biol. Chem. 2005, 280, 28492–28497. [Google Scholar] [CrossRef]
- Paspaliari, D.K.; Loose, J.S.M.; Larsen, M.H.; Vaaje-Kolstad, G. Listeria Monocytogenes Has a Functional Chitinolytic System and an Active Lytic Polysaccharide Monooxygenase. FEBS J. 2015, 282, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Leisner, J.J.; Larsen, M.H.; Jørgensen, R.L.; Brøndsted, L.; Thomsen, L.E.; Ingmer, H. Chitin Hydrolysis by Listeria Spp., Including L. monocytogenes. Appl. Environ. Microbiol. 2008, 74, 3823–3830. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Gantner, B.N.; Ye, R.D.; Cianciotto, N.P.; Freitag, N.E. The Listeria Monocytogenes ChiA Chitinase Enhances Virulence through Suppression of Host Innate Immunity. mBio 2013, 4, e00617-12. [Google Scholar] [CrossRef]
- Ohnuma, T.; Onaga, S.; Murata, K.; Taira, T.; Katoh, E. LysM Domains from Pteris Ryukyuensis Chitinase-A: A Stability Study and Characterization of the Chitin-Binding Site. J. Biol. Chem. 2008, 283, 5178–5187. [Google Scholar] [CrossRef]
- Arimori, T.; Kawamoto, N.; Shinya, S.; Okazaki, N.; Nakazawa, M.; Miyatake, K.; Fukamizo, T.; Ueda, M.; Tamada, T. Crystal Structures of the Catalytic Domain of a Novel Glycohydrolase Family 23 Chitinase from Ralstonia Sp. A-471 Reveals a Unique Arrangement of the Catalytic Residues for Inverting Chitin Hydrolysis. J. Biol. Chem. 2013, 288, 18696–18706. [Google Scholar] [CrossRef] [PubMed]
- Klancher, C.A.; Yamamoto, S.; Dalia, T.N.; Dalia, A.B. ChiS Is a Noncanonical DNA-Binding Hybrid Sensor Kinase That Directly Regulates the Chitin Utilization Program in Vibrio Cholerae. Proc. Natl. Acad. Sci. USA 2020, 117, 20180–20189. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.E.; Gevers, D.; Vahora, N.M.; Polz, M.F. Conservation of the Chitin Utilization Pathway in the Vibrionaceae. Appl. Environ. Microbiol. 2008, 74, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Salah Ud-Din, A.I.M.; Roujeinikova, A. Methyl-Accepting Chemotaxis Proteins: A Core Sensing Element in Prokaryotes and Archaea. Cell. Mol. Life Sci. 2017, 74, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.I.; Biemann, H.-P.; Privé, G.G.; Pandit, J.; Koshland Jr, D.E.; Kim, S.-H. High-Resolution Structures of the Ligand Binding Domain of the Wild-Type Bacterial Aspartate Receptor. J. Mol. Biol. 1996, 262, 186–201. [Google Scholar] [CrossRef]
- Ashby, M.K. Survey of the Number of Two-Component Response Regulator Genes in the Complete and Annotated Genome Sequences of Prokaryotes. FEMS Microbiol. Lett. 2004, 231, 277–281. [Google Scholar] [CrossRef]
- Yu, E.W.; Koshland, J. Propagating Conformational Changes over Long (and Short) Distances in Proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9517–9520. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.P.; Zhulin, I.B. Evolutionary Genomics Reveals Conserved Structural Determinants of Signaling and Adaptation in Microbial Chemoreceptors. Proc. Natl. Acad. Sci. USA 2007, 104, 2885–2890. [Google Scholar] [CrossRef] [PubMed]





| Rank | Gene ID | Significance Score −10logP (Average) | Annotation (PGAP and dbCAN 3.0) | Complementary Annotation (KO) |
|---|---|---|---|---|
| 1 | pgaptmp_000837 | 631.37 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 2 | pgaptmp_001746 | 602.58 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 3 | pgaptmp_002871 | 595.18 | branched-chain amino acid ABC transporter substrate-binding protein | livK; branched-chain amino acid transport system substrate-binding protein |
| 4 | pgaptmp_001064 | 582.92 | TonB-dependent receptor | xylulose-5-phosphate/fructose-6-phosphate phosphoketolase |
| 5 | pgaptmp_000389 | 582.37 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 6 | pgaptmp_000021 | 560.63 | MBL fold metallo-hydrolase | sdsA1; linear primary-alkylsulfatase [EC:3.1.6.21] |
| 7 | pgaptmp_002723 | 550.72 | TonB-dependent siderophore receptor | TC.FEV.OM; iron complex outermembrane recepter protein |
| 8 | pgaptmp_000441 | 548.21 | sugar ABC transporter substrate-binding protein | chiE; putative chitobiose transport system substrate-binding protein |
| 9 | pgaptmp_001471 | 525.48 | porin | NA |
| 10 | pgaptmp_000366 | 523.85 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 49 | pgaptmp_002996 | 394,47 | FAD-binding oxidoreductase (AA7) | NA |
| 56 | pgaptmp_001732 | 385.83 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 57 | pgaptmp_000635 | 384.40 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 58 | pgaptmp_000444 | 383.76 | ATPase | gspK; glucosamine kinase [EC:2.7.1.8] |
| 91 | pgaptmp_003083 | 326.35 | glycosyl hydrolase family 18 protein | E3.2.1.14; chitinase [EC:3.2.1.14] |
| 107 | pgaptmp_000269 | 308.55 | carbohydrate-binding domain-containing protein (GH20) | HEXA_B; hexosaminidase [EC:3.2.1.52] |
| 134 | pgaptmp_000148 | 284.47 | lytic polysaccharide monooxygenase | cpbD; chitin-binding protein |
| 198 | Pgaptmp_000371 | 238.27 | glycosyl hydrolase family 18 protein | NA |
| Gene ID | Significance | Log2 Fold Change | Annotation (PGAP and dbCAN3.0) | Complementary Annotation (KO) |
|---|---|---|---|---|
| pgaptmp_000442 | 67.67 | 11.11 | sugar ABC transporter substrate-binding protein | chiE; putative chitobiose transport system substrate-binding protein |
| pgaptmp_000441 | 65.26 | 11.11 | sugar ABC transporter substrate-binding protein | chiE; putative chitobiose transport system substrate-binding protein |
| pgaptmp_000337 | 64.6 | 50 | nitrate reductase subunit beta | Identical |
| pgaptmp_003318 | 63.33 | 10 | PrkA family serine protein kinase | identical |
| pgaptmp_000602 | 62.16 | 14.29 | methyl-accepting chemotaxis protein | identical |
| pgaptmp_001215 | 49.26 | 50 | SDR family oxidoreductase | NA |
| pgaptmp_002582 | 41.03 | 50 | inclusion body family protein | aidA; nematocidal protein AidA |
| pgaptmp_001847 | 32.45 | 50 | substrate-binding domain-containing protein | rbsB; ribose transport system substrate-binding protein |
| ggaptmp_000878 | 30.95 | 50 | hypothetical protein | NA |
| pgaptmp_000237 | 39 | 33.33 | hypothetical protein | NA |
| pgaptmp_000269 | 47.03 | 14.29 | carbohydate-binding domain-containing protein (GH20) | HEXA_B; hexosaminidase [EC:3.2.1.52] |
| pgaptmp_000439 | 29.33 | 11.11 | carbohydrate ABC transporter permease | chiG; putative chitobiose transport system permease protein |
| pgaptmp_000437 | 25.84 | 10 | polysaccharide deacetylase family protein | pgdA; peptidoglycan-N-acetylglucosamine deacetylase [EC:3.5.1.104] |
| pgaptmp_000281 | 41.14 | 8.33 | polysaccharide deacetylase family protein | NA |
| pgaptmp_001323 | 29.95 | 8.33 | beta-N-acetylhexosaminidase (GH3) | nagZ; beta-N-acetylhexosaminidase [EC:3.2.1.52] |
| pgaptmp_000635 | 60.05 | 5.56 | glycoside hydrolase family 18 protein | chitinase [EC:3.2.1.14] |
| pgaptmp_002871 | 59.65 | 5.56 | branched-chain amino acid ABC transporter substrate-binding protein | identical |
| pgaptmp_000440 | 39.29 | 5.26 | Sugar ABC transporter permease | chiF; putative chitobiose transport system permease protein |
| pgaptmp_003368 | 4.62 | 1.30 | N-acetylglucosamine-specific PTS transporter subunit IIBC | nagE; N-acetylglucosamine PTS system EIICBA or EIICB component [EC:2.7.1.193] |
| Carbon Source | Rank | Adjusted p-Value | log2 Fold Change | Gene ID | Annotation (PGAP and dbCAN3.0) |
|---|---|---|---|---|---|
| Chitin | 1 | 1.44 × 10−72 | 6.34 | pgaptmp_000221 | NirD/YgiW/YdeI family stress tolerance protein |
| 2 | 6.13 × 10−69 | 6.83 | pgaptmp_001590 | Flp family type IVb pilin | |
| 3 | 8.63 × 10−61 | 6.58 | pgaptmp_001589 | Flp family type IVb pilin | |
| 4 | 5.63 × 10−48 | 4.22 | pgaptmp_001089 | FtsX-like permease family protein | |
| 5 | 3.56 × 10−47 | 3.93 | pgaptmp_000589 | hypothetical protein | |
| Chitin | 26 | 2.02 × 10−34 | 3.45 | pgaptmp_000680 | peptidoglycan-binding protein (GH19) |
| 52 | 1.15 × 10−26 | 2.77 | pgaptmp_000371 | glycosyl hydrolase family 18 protein | |
| 102 | 4.18 × 10−20 | 2.54 | pgaptmp_000306 | carbohydate-binding domain-containing protein (GH20) | |
| 294 | 8.38 × 10−13 | 1.80 | pgaptmp_000836 | glycosyl hydrolase family 18 protein | |
| 309 | 2.1 × 10−12 | 1.90 | pgaptmp_001841 | glycosyl hydrolase family 18 protein | |
| 348 | 1.08 × 10−11 | 2.25 | pgaptmp_002137 | glycosyl hydrolase family 18 protein | |
| 448 | 4.33 × 10−10 | 2.92 | pgaptmp_000372 | glycosyl hydrolase family 18 protein | |
| 453 | 4.9 × 10−10 | 1.66 | pgaptmp_000302 | chitinase (GH19) | |
| 678 | 2.83 × 10−7 | 1.32 | pgaptmp_001746 | glycosyl hydrolase family 18 protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, N.D.; Garbe, D.; Brück, T.B. Proteomic and Transcriptomic Analyses to Decipher the Chitinolytic Response of Jeongeupia spp. Mar. Drugs 2023, 21, 448. https://doi.org/10.3390/md21080448
Arnold ND, Garbe D, Brück TB. Proteomic and Transcriptomic Analyses to Decipher the Chitinolytic Response of Jeongeupia spp. Marine Drugs. 2023; 21(8):448. https://doi.org/10.3390/md21080448
Chicago/Turabian StyleArnold, Nathanael D., Daniel Garbe, and Thomas B. Brück. 2023. "Proteomic and Transcriptomic Analyses to Decipher the Chitinolytic Response of Jeongeupia spp." Marine Drugs 21, no. 8: 448. https://doi.org/10.3390/md21080448
APA StyleArnold, N. D., Garbe, D., & Brück, T. B. (2023). Proteomic and Transcriptomic Analyses to Decipher the Chitinolytic Response of Jeongeupia spp. Marine Drugs, 21(8), 448. https://doi.org/10.3390/md21080448