
Oxidative Cyclization at ortho-Position of Phenol: Improved Total Synthesis of 3-(Phenethylamino)demethyl(oxy)aaptamine
Abstract

1. Introduction
2. Results and Discussion
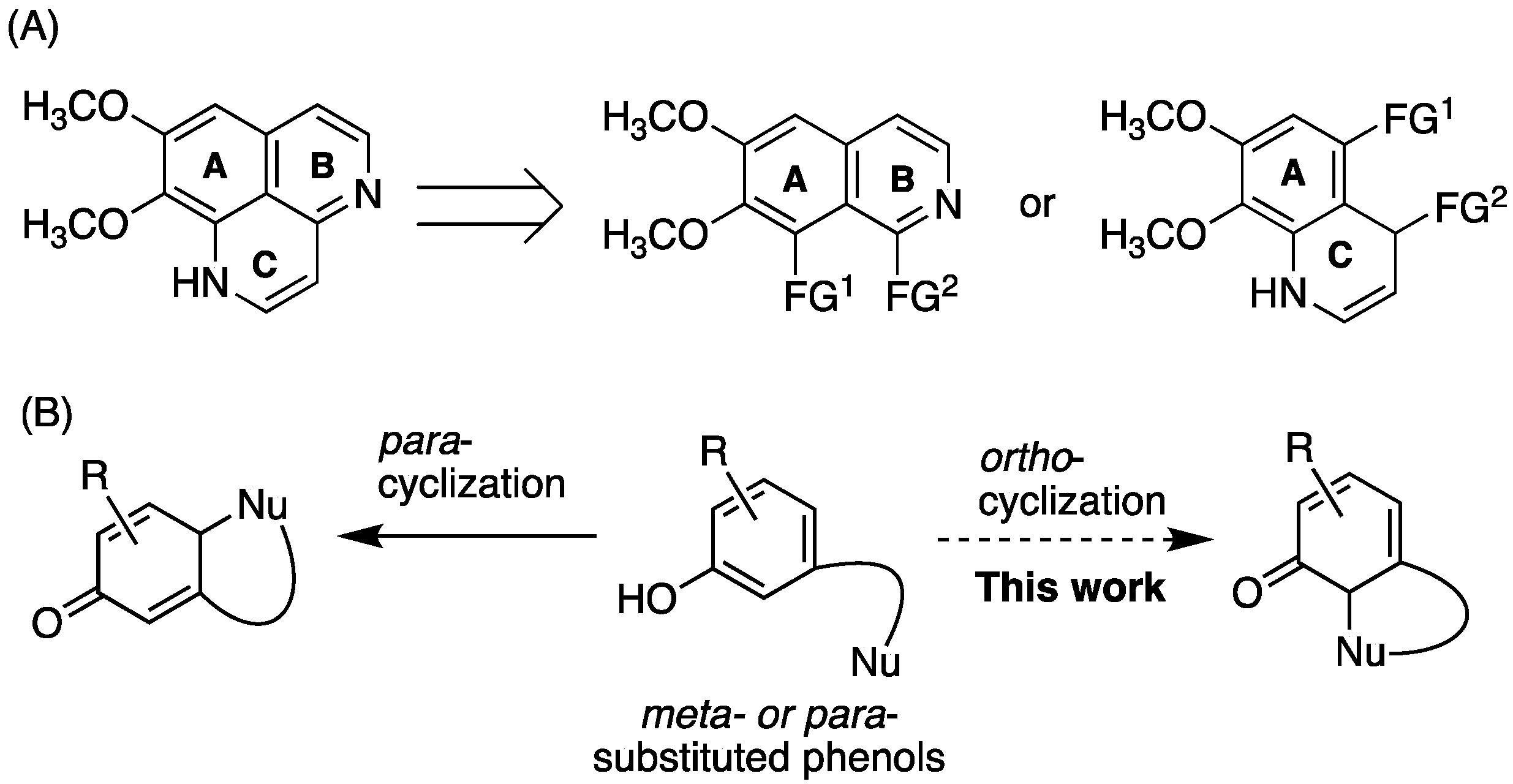
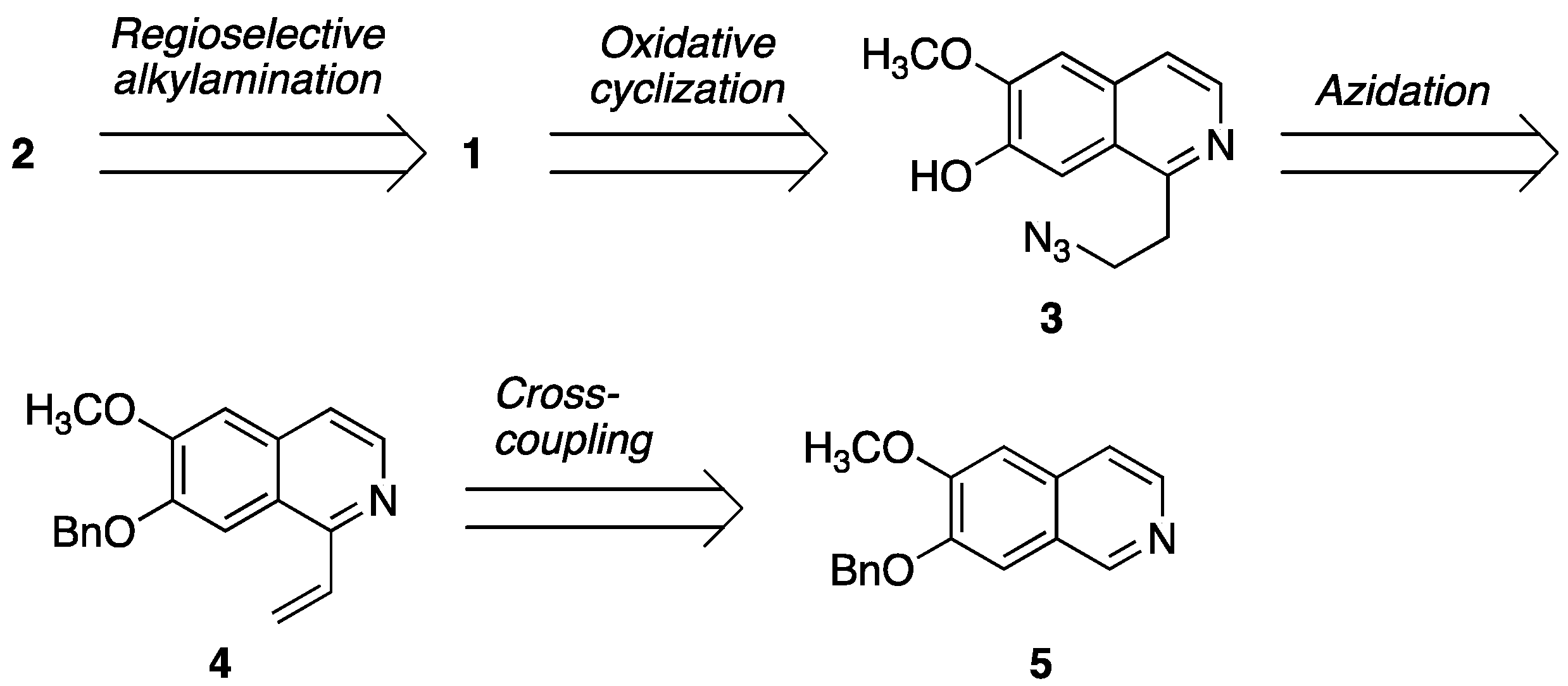
2.1. Retrosynthesis
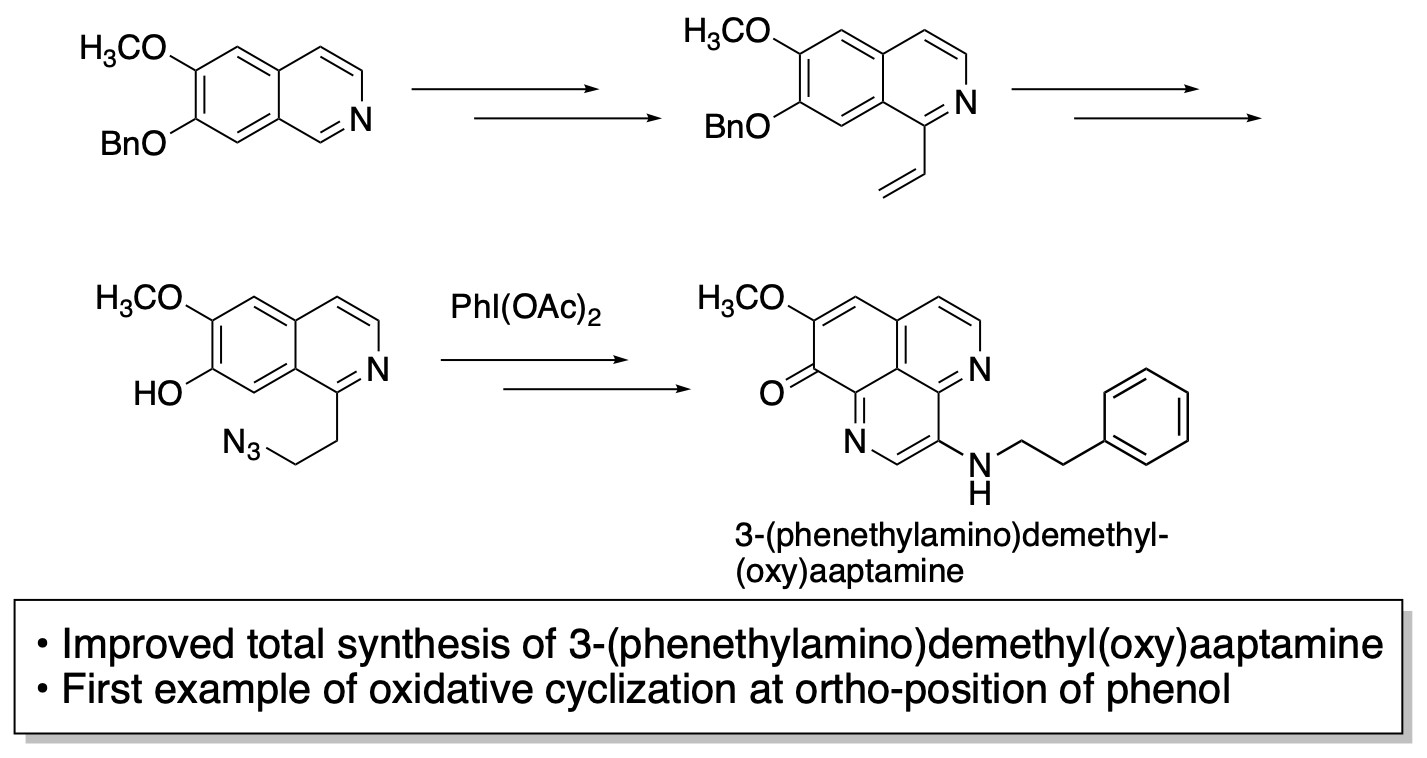
2.2. Total Synthesis of 3-(Phenethylamino)demethyl(oxy)aaptamine
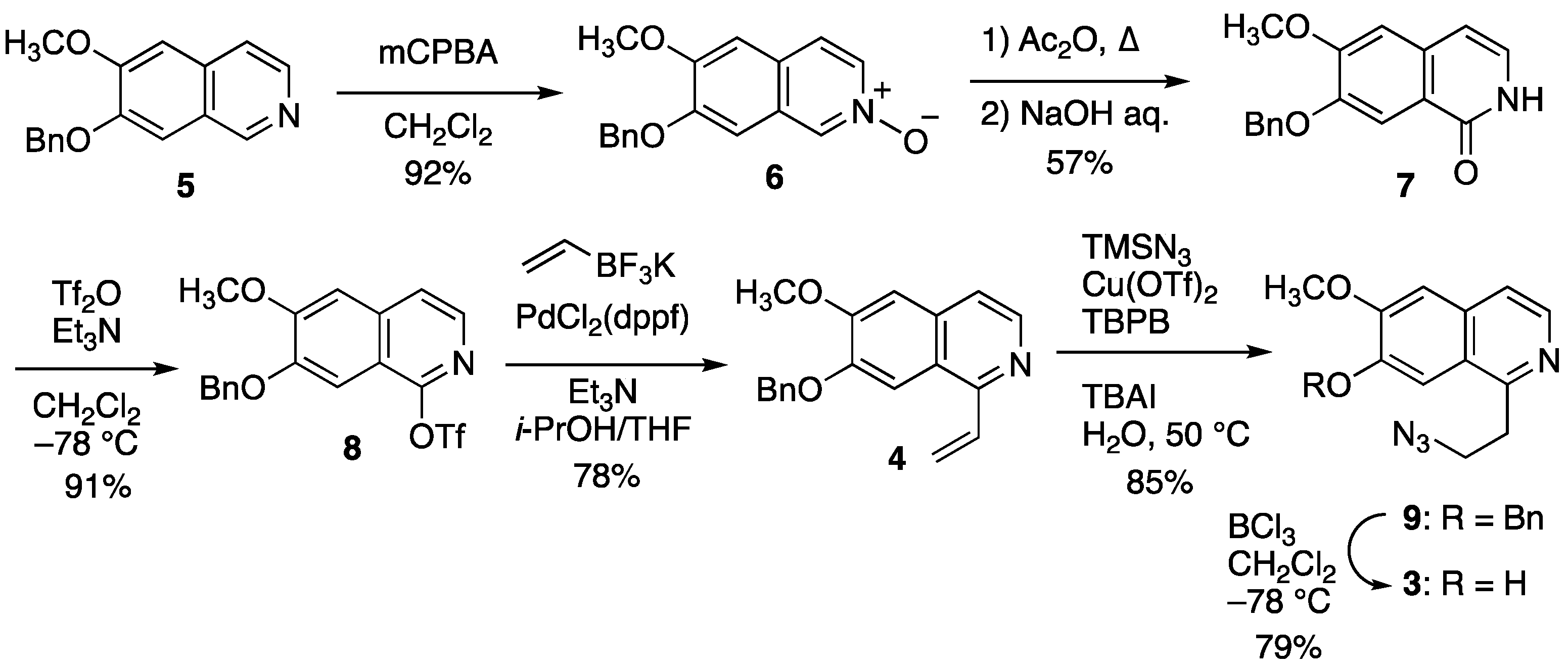
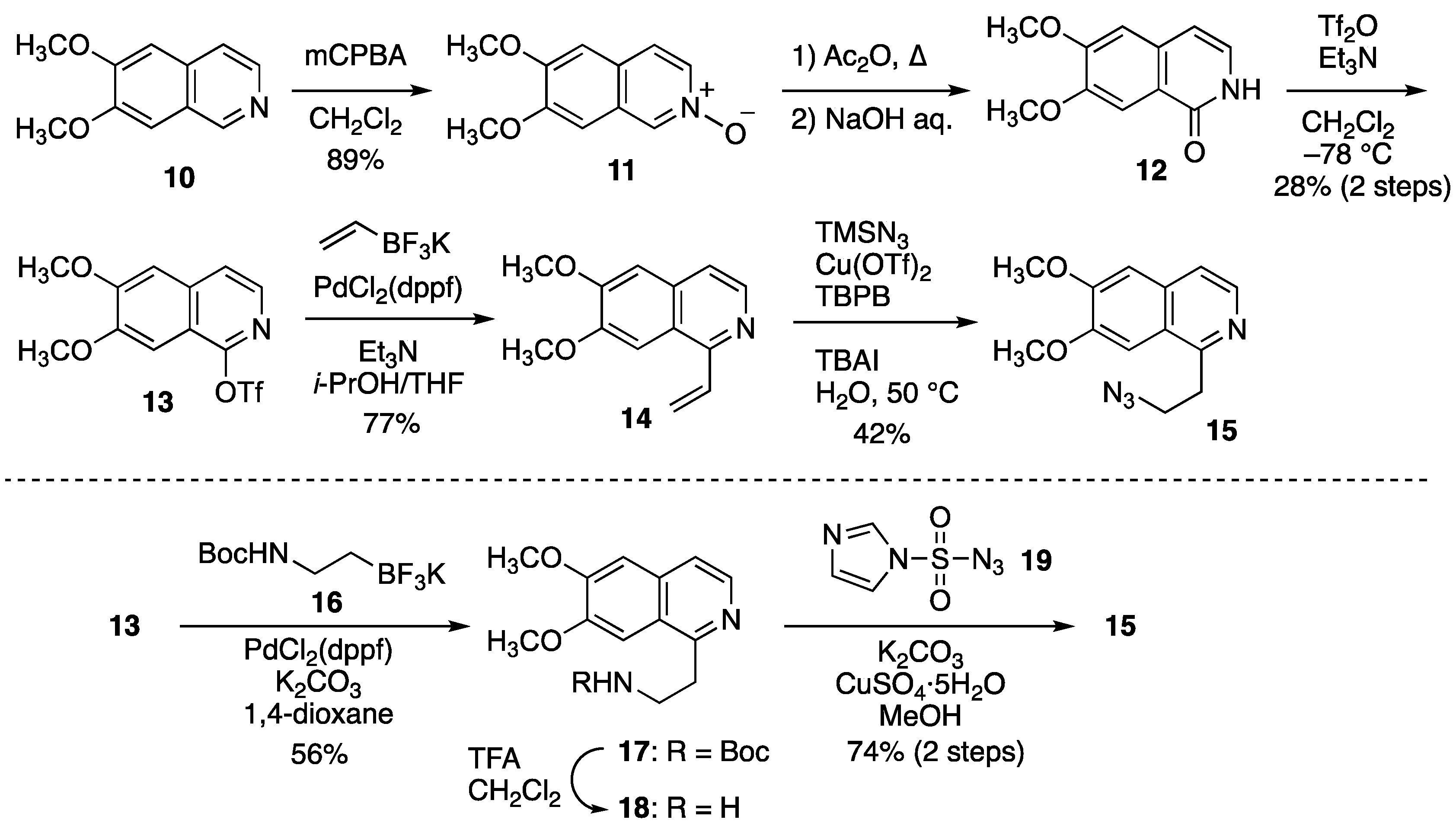
2.2.1. Synthesis of Cyclization Precursors
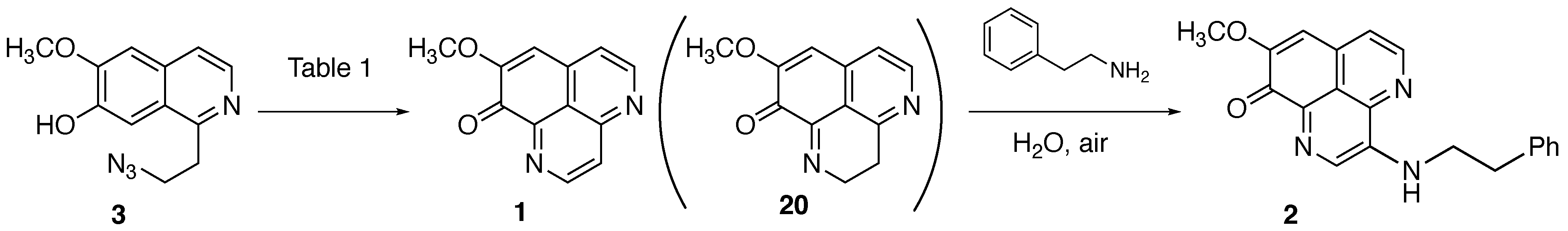
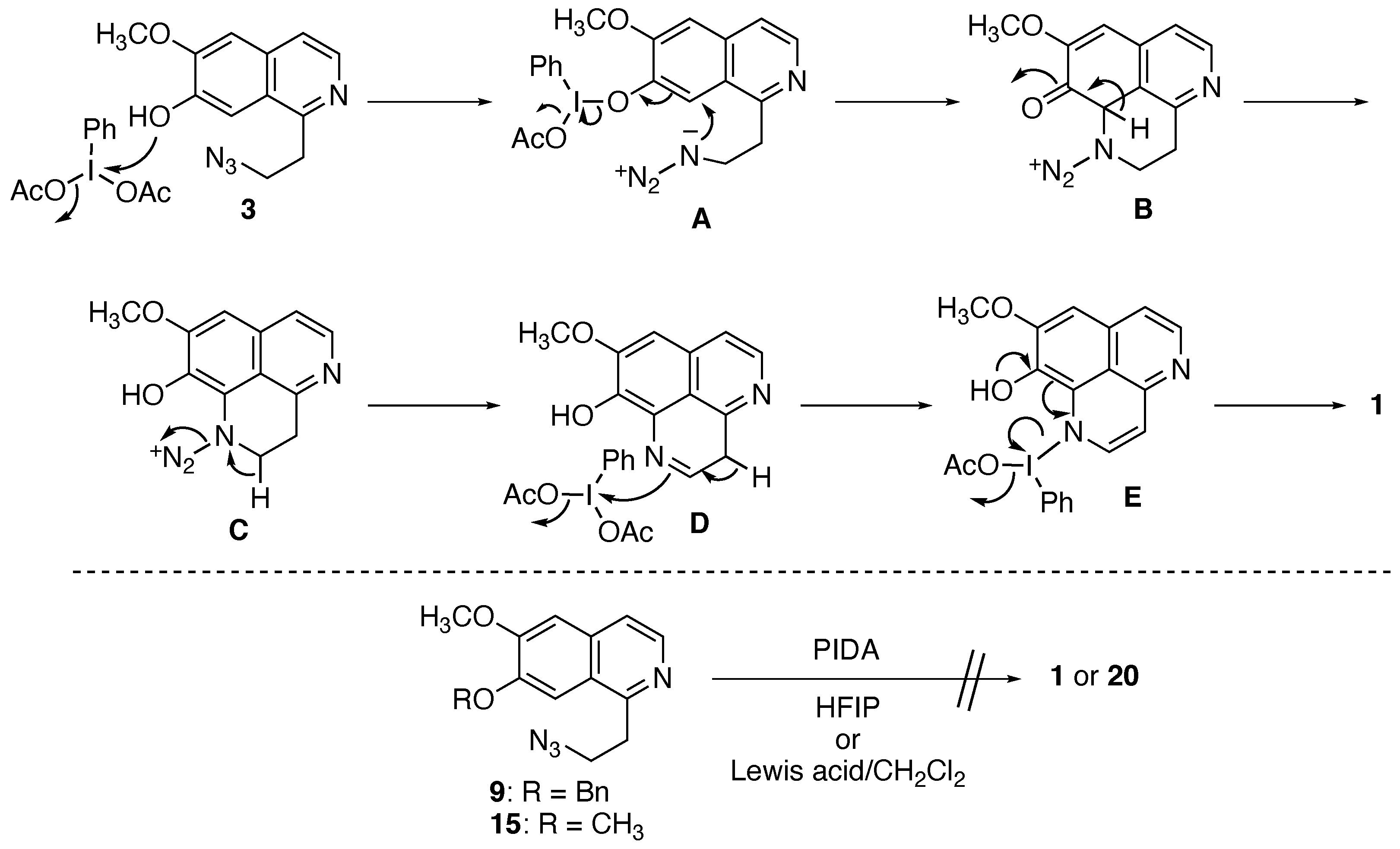
2.2.2. Oxidative Cyclization and Completion of Total Synthesis
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. 7-(Benzyloxy)-6-methoxyisoquinoline 2-oxide (6)
3.2.2. 7-(Benzyloxy)-6-methoxyisoquinoline-1(2H)-one (7)
3.2.3. 7-(Benzyloxy)-6-methoxyisoquinolin-1-yl Trifluoromethanesulfonate (8)
3.2.4. 7-(Benzyloxy)-6-methoxy-1-vinylisoquinoline (4)
3.2.5. 1-(2-Azidoethyl)-7-(benzyloxy)-6-methoxyisoquinoline (9)
3.2.6. 1-(2-Azidoethyl)-6-methoxyisoquinoline-7-ol (3)
3.2.7. 6,7-Dimethoxyisoquinoline 2-oxide (11)
3.2.8. 6,7-Dimethoxyisoquinolin-1-yl Trifluoromethanesulfonate (13)
3.2.9. 6,7-Dimethoxy-1-vinylisoquinoline (14)
3.2.10. 1-(Azidomethyl)-6,7-dimethoxyisoquinoline (15)
3.2.11. tert-Butyl (2-(6,7-Dimethoxyisoquinolin-1-yl)ethyl)carbamate (17)
3.2.12. Reaction of Compound 17 to 15
3.2.13. Demethyl(oxy)aaptamine (1)
3.2.14. 3-(Phenethylamino)demethyl(oxy)aaptamine (2)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nweze, J.A.; Mbaoji, F.N.; Huang, G.; Li, Y.; Yang, L.; Zhang, Y.; Huang, S.; Pan, L.; Yang, D. Antibiotics Development and the Potentials of Marine-Derived Compounds to Stem the Tide of Multidrug-Resistant Pathogenic Bacteria, Fungi, and Protozoa. Mar. Drugs 2020, 18, 145. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2023, 40, 275–325. [Google Scholar] [CrossRef] [PubMed]
- Larghi, E.L.; Bohn, M.L.; Kaufman, T.S. Aaptamine and related products. Their isolation, chemical syntheses, and biological activity. Tetrahedron 2009, 65, 4257–4282. [Google Scholar] [CrossRef]
- Sung, C.-S.; Cheng, H.-J.; Chen, N.-F.; Tang, S.-H.; Kuo, H.-M.; Sung, P.-J.; Chen, W.-F.; Wen, Z.-H. Antinociceptive Effects of Aaptamine, a Sponge Component, on Peripheral Neuropathy in Rats. Mar. Drugs 2023, 21, 113. [Google Scholar] [CrossRef]
- Miao, S.; He, Q.; Li, C.; Wu, Y.; Liu, M.; Chen, Y.; Qi, S.; Gong, K. Aaptamine—A dual acetyl- and butyrylcholinesterase inhibitor as potential anti-Alzheimer’s disease agent. Pharm. Biol. 2022, 60, 1502–1510. [Google Scholar] [CrossRef]
- Nakamura, H.; Kobayashi, J.; Ohizumi, Y.; Hirata, Y. Physiologically active marine natural products from Prolifera. Part 10. Aaptamines. Novel benzo[de][1,6]naphthyridines from the Okinawan marine sponge Aaptos aaptos. J. Chem. Soc. Perkin Trans. 1 1987, 173–176. [Google Scholar] [CrossRef]
- Shaari, K.; Ling, K.C.; Rashid, Z.M.; Jean, T.P.; Abas, F.; Raof, S.M.; Zainal, Z.; Lajis, N.H.; Mohamad, H.; Ali, A.M. Cytotoxic Aaptamines from Malaysian Aaptos aaptos. Mar. Drugs 2009, 7, 1–8. [Google Scholar] [CrossRef]
- Arai, M.; Han, C.; Yamano, Y.; Setiawan, A.; Kobayashi, M. Aaptamines, marine spongean alkaloids, as anti-dormant mycobacterial substances. J. Nat. Med. 2014, 68, 372–376. [Google Scholar] [CrossRef]
- Sumii, Y.; Kotoku, N.; Han, C.; Kamiya, K.; Setiawan, A.; Vilchèze, C.; Jacobs, W.R., Jr.; Arai, M. 3-(Phenethylamino)demethyl(oxy)aaptamine as an anti-dormant mycobacterial substance: Isolation, evaluation and total synthesis. Tetrahedron Lett. 2020, 61, 151924. [Google Scholar] [CrossRef]
- Han, J.; Liu, X.; Zhang, L.; Quinn, R.J.; Feng, Y. Anti-mycobacterial natural products and mechanisms of action. Nat. Prod. Rep. 2022, 39, 77–89. [Google Scholar] [CrossRef]
- Sumii, Y.; Kamiya, K.; Nakamura, T.; Tanaka, K.; Kaji, T.; Mukomura, J.; Kotoku, N.; Arai, M. Study of structure-activity relationship of an anti-dormant mycobacterial substance 3-(phenethylamino)demethyl(oxy)aaptamine to create a probe molecule for detecting its target protein. Mar. Drugs 2022, 20, 98. [Google Scholar] [CrossRef]
- Kelly, T.R.; Maguire, M.P. A synthesis of aaptamine. Tetrahedron 1985, 41, 3033–3036. [Google Scholar] [CrossRef]
- Puvvala, S.; Jadhav, V.D.; Narkhede, U.C.; Karun, M.A.; Reddy, C.V.R. First Total Synthesis of a Cytotoxic Derivative of the Natural Product Aaptamine. Synthesis 2017, 49, 2768–2774. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, F.; Sun, F.; Liu, L.; Liu, B.; Wang, S.-P.; Cheng, C.-W.; Liao, H.; Lin, H.-W. Total Synthesis of Aaptamine, Demethyloxyaaptamine, and Their 3-Alkylamino Derivatives. Org. Lett. 2019, 21, 1430–1433. [Google Scholar] [CrossRef]
- Odagi, M.; Nagasawa, K. Total Synthesis of Fused Polycyclic Alkaloids Based on Oxidative Phenolic Couplings and Aza-Michael Reactions. Synlett 2023. [Google Scholar] [CrossRef]
- Dohi, T.; Kita, Y. Hypervalent Iodine-Induced Oxidative Couplings (New Metal-Free Coupling Advances and Their Applications in Natural Product Syntheses). Top. Curr. Chem. 2016, 373, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, F.V.; Takenaga, N.; Dohi, T. Asymmetric direct/stepwise dearomatization reactions involving hypervalent iodine reagents. Chem.-Asian J. 2022, 17, e202101115. [Google Scholar] [CrossRef]
- Kita, Y.; Egi, M.; Okajima, A.; Ohtsubo, M.; Takada, T.; Tohma, H. Hypervalent iodine(III) induced intramolecular cyclization of substituted phenol ethers bearing an alkyl azido sidechain–a novel synthesis of quinone imine ketals. Chem. Commun. 1996, 13, 1491–1492. [Google Scholar] [CrossRef]
- Kita, Y.; Watanabe, H.; Egi, M.; Saiki, T.; Fukuoka, Y.; Tohma, H. Novel and efficient synthesis of pyrroloiminoquinones using a hypervalent iodine(III) reagent. J. Chem. Soc., Perkin Trans. 1 1998, 4, 635–636. [Google Scholar] [CrossRef]
- Reimann, E.; Renz, H. Protoberberine aus Reissert-Verbindungen, 2. Mitt.: Eine neue Synthese von 8-Methyldibenzo[a,g]chinolizidinen. Arch. Pharm. 1993, 326, 253–258. [Google Scholar] [CrossRef]
- Gary, A.; Molander, G.A.; Rivero, M.R. Suzuki Cross-Coupling Reactions of Potassium Alkenyltrifluoroborates. Org. Lett. 2002, 4, 107–109. [Google Scholar] [CrossRef]
- Zhou, H.; Jian, W.; Qian, B.; Ye, C.; Li, D.; Zhou, J.; Bao, H. Copper-Catalyzed Ligand-Free Diazidation of Olefins with TMSN3 in CH3CN or in H2O. Org. Lett. 2017, 19, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhao, J.; Wang, Y.; Hu, J.; Li, L.; Miao, L.; Feng, H.; Désaubry, L.; Yu, P. A General and Efficient Synthesis of 2-Pyridones, 2-Quinolinones, and 1-Isoquinolinones from Azine N-Oxides. Asian J. Org. Chem. 2016, 5, 1442–1446. [Google Scholar] [CrossRef]
- Molander, A.G.; Gerard, J.L. Scope of the Suzuki−Miyaura Aminoethylation Reaction Using Organotrifluoroborates. J. Org. Chem. 2007, 72, 8422–8426. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Stick, R.V. An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef]
- Ward, R.S.; Pelter, A.; Abd-El-Ghani, A. Preparation of tetrahydrodibenzocyclooctene lignans and spirodienones by hypervalent iodine oxidation of phenolic dibenzylbutyrolactones. Tetrahedron 1996, 52, 1303–1336. [Google Scholar] [CrossRef]








{kind=link}
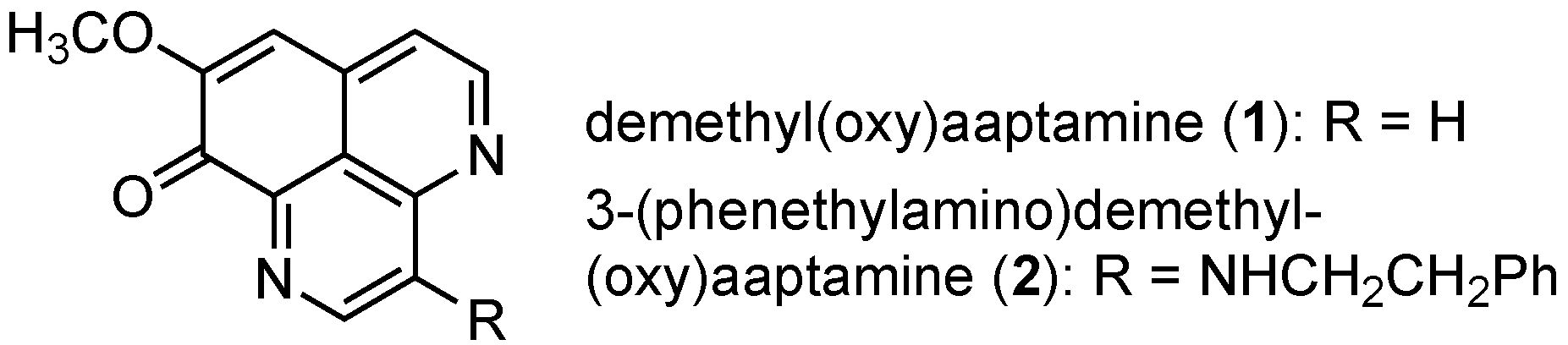{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent b | Solvent c | Time | Yield d |
|---|---|---|---|---|
| 1 | PIFA | CH3CN (0.05) | 1 h | trace |
| 2 | PIFA | TFE (0.05) | 1 h | 10% |
| 3 | PIFA | HFIP (0.05) | 1 h | 22% |
| 4 | PIDA | CH3CN (0.05) | 6 h | trace |
| 5 | PIDA | TFE (0.05) | 6 h | 40% |
| 6 | PIDA | HFIP (0.05) | 6 h | 52% |
| 7 | PIDA | HFIP (0.1) | 1 h | 31% |
| 8 | PIDA | HFIP (0.025) | 12 h | 70% |
| 9 | IBX | HFIP (0.05) | 1 h | 0% |
| 10 | IBX | DMF (0.025) | 1 h | 0% |
| 11 | CAN | CH3CN/H2O (0.1) | 1 h | 0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakatani, Y.; Kimura, R.; Kimata, T.; Kotoku, N. Oxidative Cyclization at ortho-Position of Phenol: Improved Total Synthesis of 3-(Phenethylamino)demethyl(oxy)aaptamine. Mar. Drugs 2023, 21, 311. https://doi.org/10.3390/md21050311
Nakatani Y, Kimura R, Kimata T, Kotoku N. Oxidative Cyclization at ortho-Position of Phenol: Improved Total Synthesis of 3-(Phenethylamino)demethyl(oxy)aaptamine. Marine Drugs. 2023; 21(5):311. https://doi.org/10.3390/md21050311
Chicago/Turabian StyleNakatani, Yuki, Risa Kimura, Tomoyo Kimata, and Naoyuki Kotoku. 2023. "Oxidative Cyclization at ortho-Position of Phenol: Improved Total Synthesis of 3-(Phenethylamino)demethyl(oxy)aaptamine" Marine Drugs 21, no. 5: 311. https://doi.org/10.3390/md21050311
APA StyleNakatani, Y., Kimura, R., Kimata, T., & Kotoku, N. (2023). Oxidative Cyclization at ortho-Position of Phenol: Improved Total Synthesis of 3-(Phenethylamino)demethyl(oxy)aaptamine. Marine Drugs, 21(5), 311. https://doi.org/10.3390/md21050311