
New Phenylspirodrimanes from the Sponge-Associated Fungus Stachybotrys chartarum MUT 3308

 ,
,  ,
, 
 and
and 
Abstract
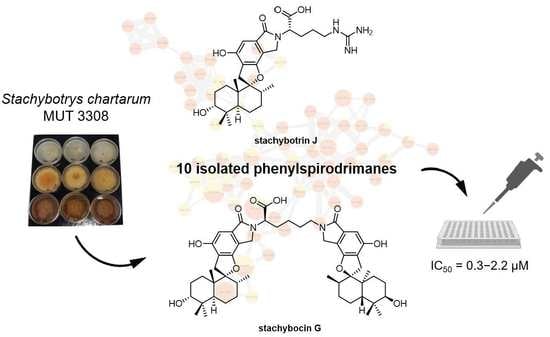
1. Introduction
2. Results and Discussion
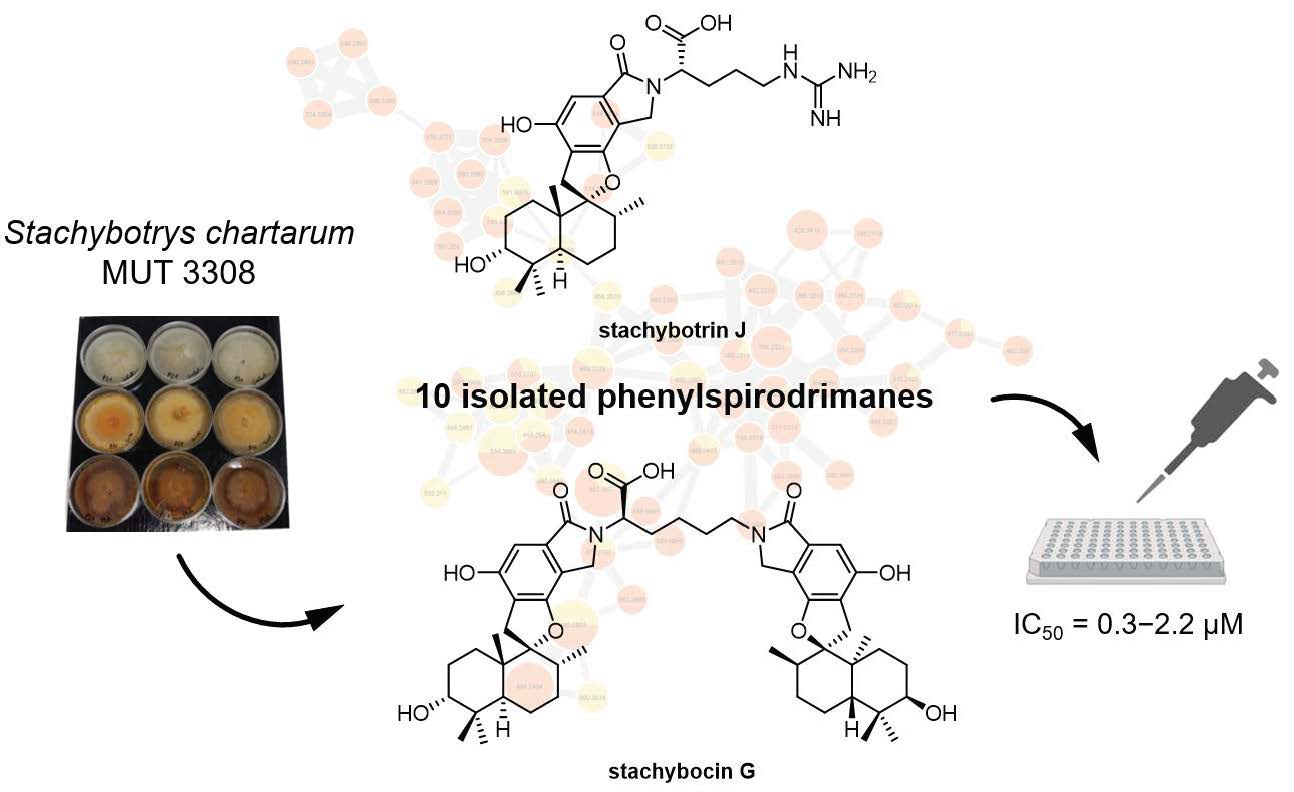
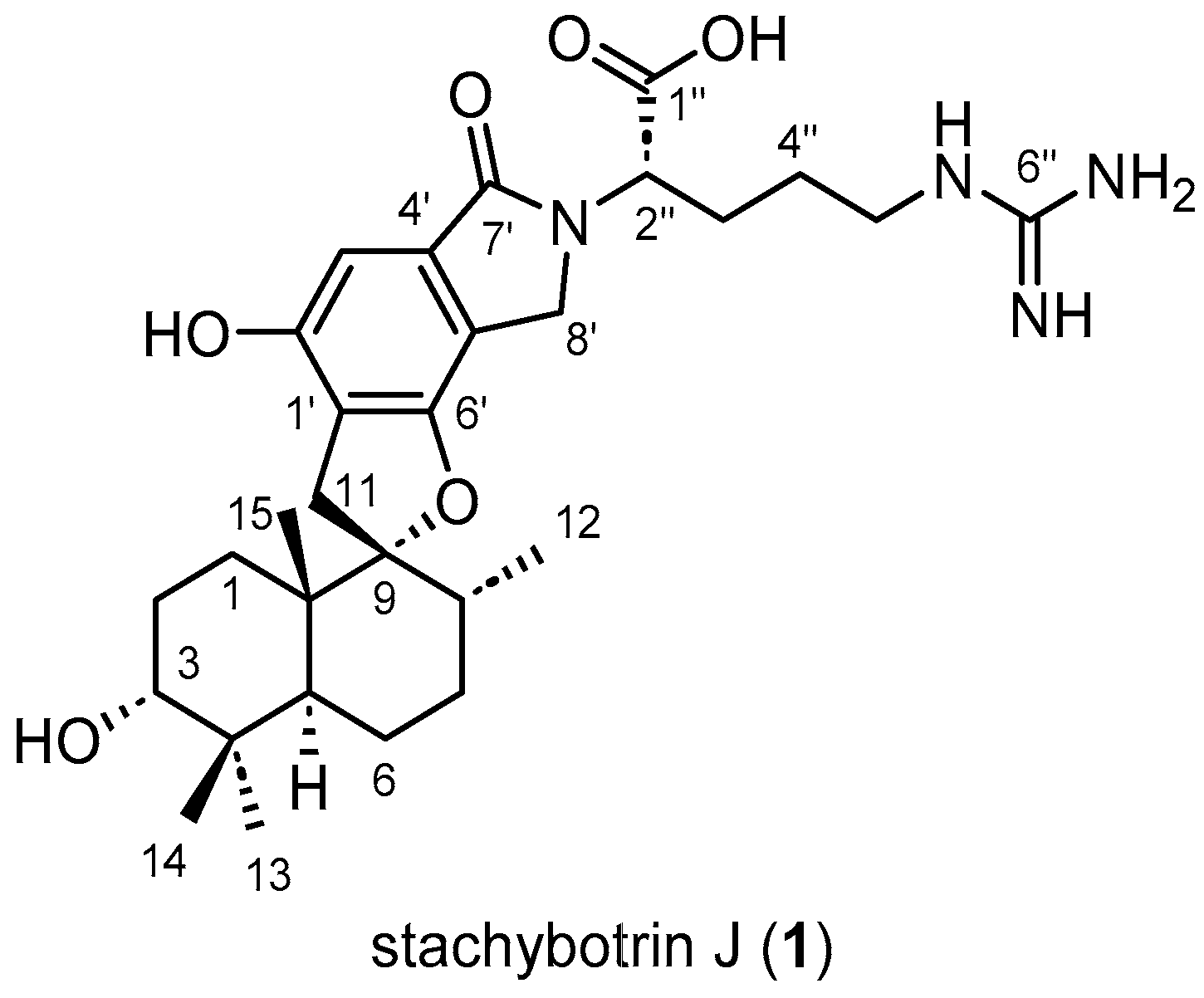
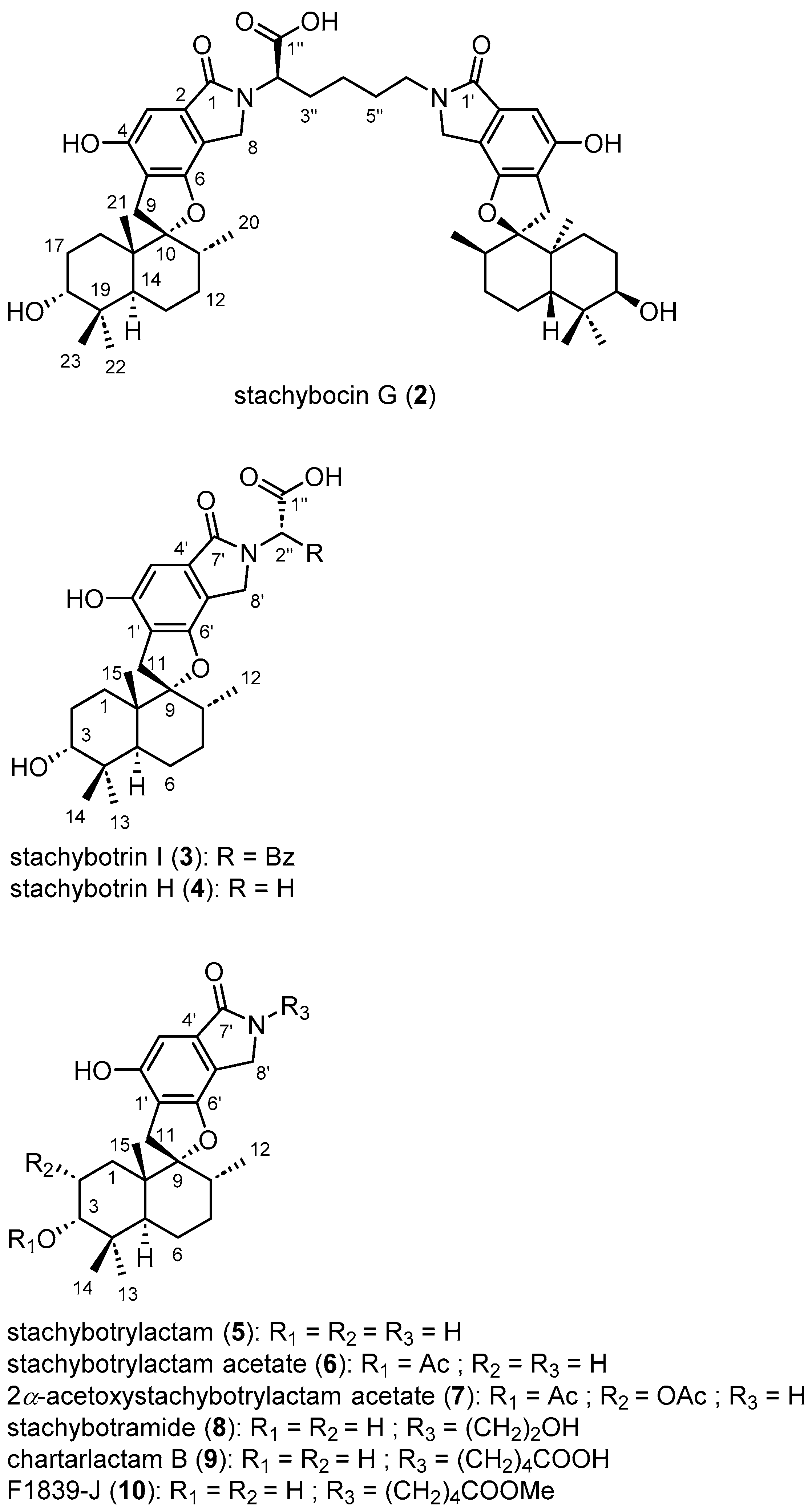
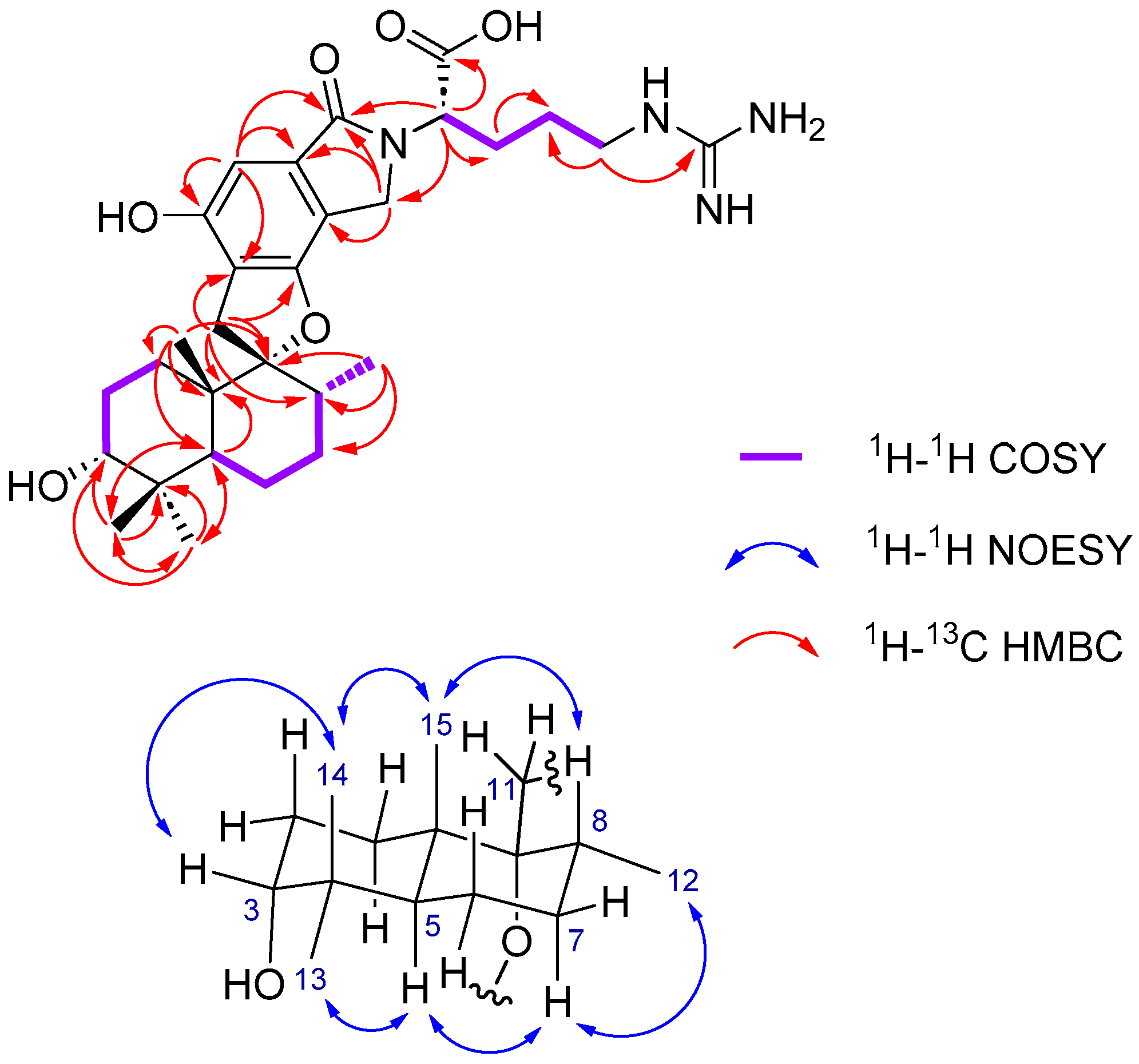
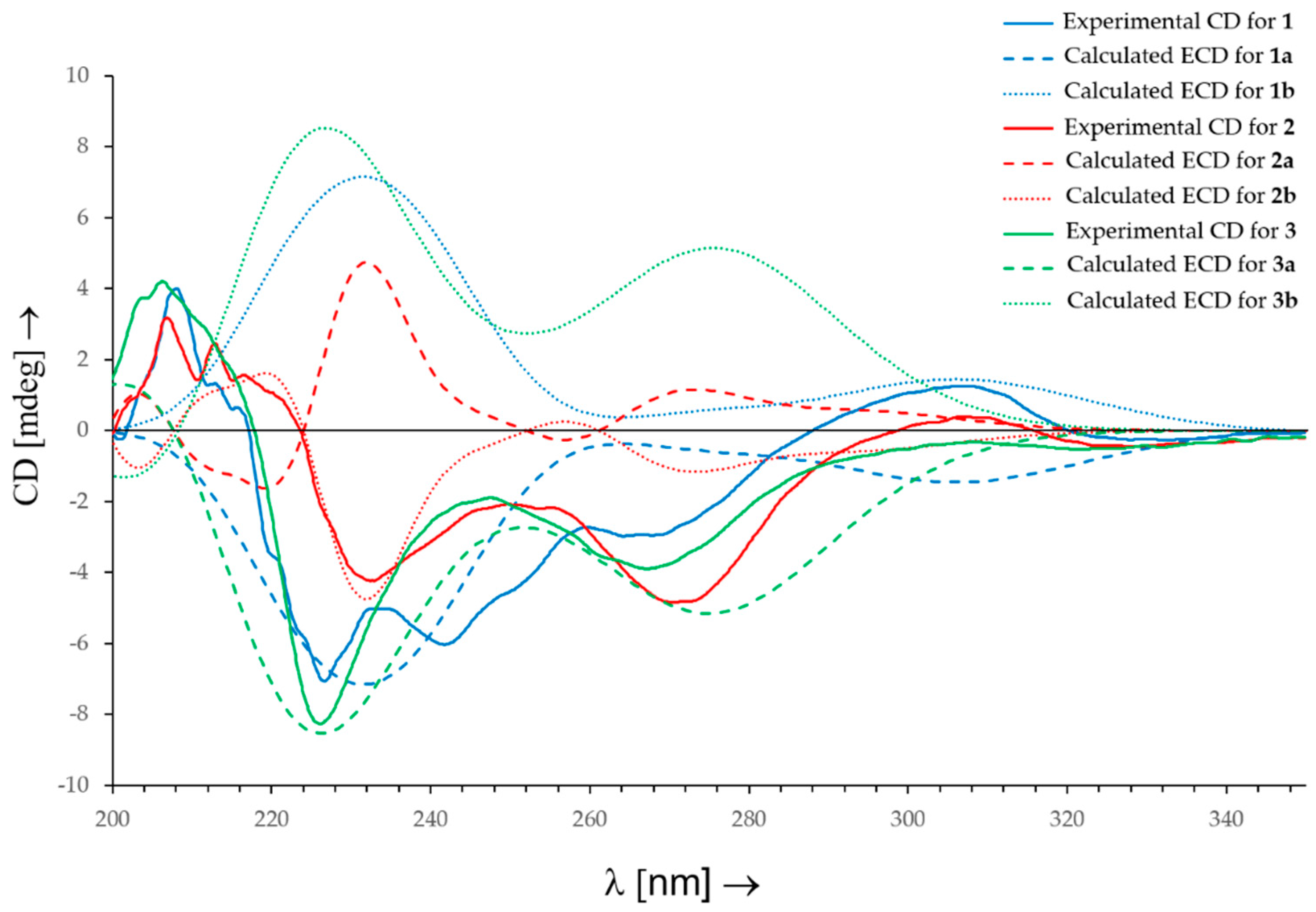
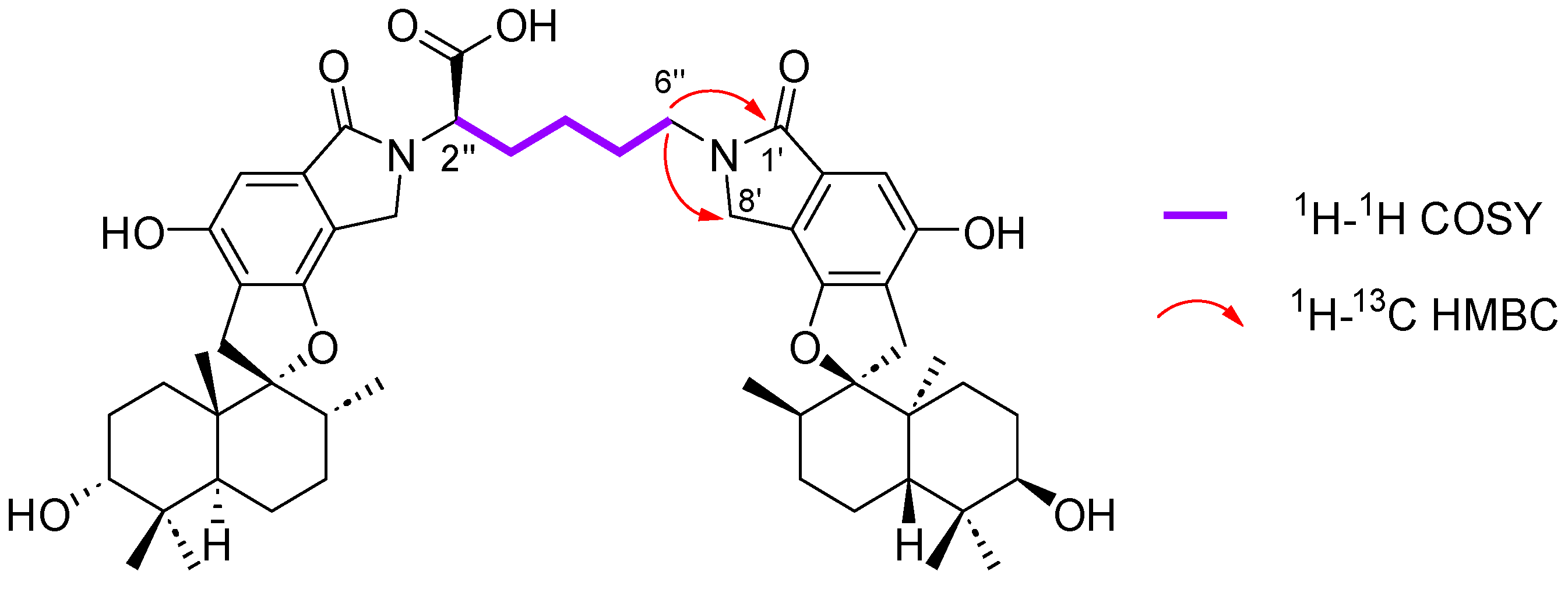
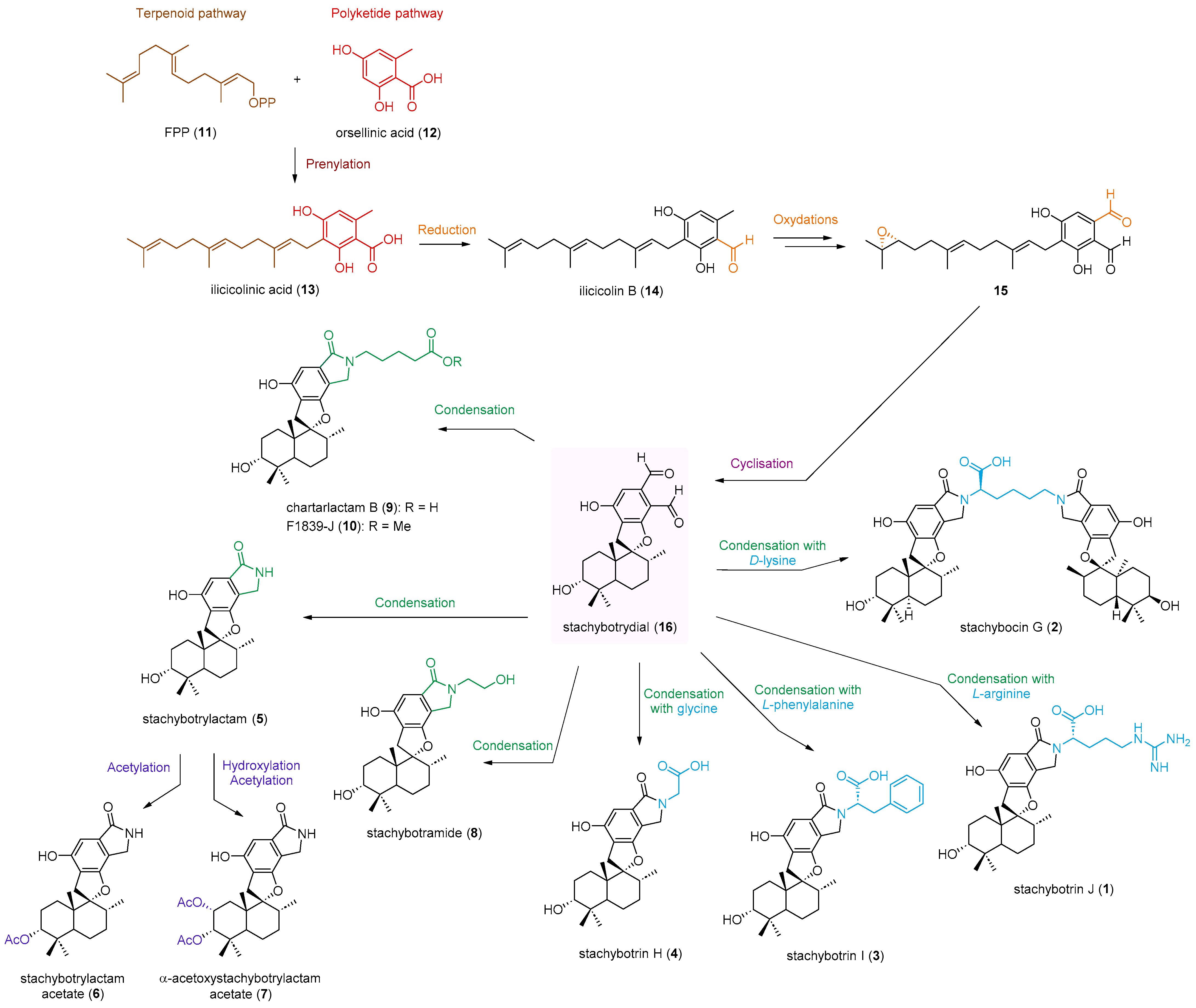
2.1. Structure Elucidation
2.2. Feature-Based Molecular Networking Analysis
2.3. Biological Assays
3. Materials and Methods
3.1. General
3.2. Fungal Material and Fermentation
3.3. Extraction and Isolation
3.4. Feature-Based Molecular Networking Analysis
3.5. Computational Analysis
3.6. Cell Culture
3.7. Cytotoxicity Measurement (XTT)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarvis, B.B. Stachybotrys chartarum: A Fungus for Our Time. Phytochemistry 2003, 64, 53–60. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Choudhry, H.; Asseri, A.H.; Elfaky, M.A.; Mohamed, S.G.A.; Mohamed, G.A. Stachybotrys chartarum—A Hidden Treasure: Secondary Metabolites, Bioactivities, and Biotechnological Relevance. J. Fungi 2022, 8, 504. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, R.; Dobashi, K.; Kojima, I.; Sakai, K.; Shibamoto, N.; Yoshioka, T.; Nishida, H.; Okamoto, R.; Akagawa, H.; Mizuno, S. Mer-NF5003B, E and F, Novel Sesquiterpenoids as Avian Myeloblastosis Virus Protease Inhibitors Produced by Stachybotrys sp. J. Antibiot. 1994, 47, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.K.T.; Wichmann, C.F.; Meinz, M.S.; Guariglia, L.; Giacobbe, R.A.; Mochales, S.; Kong, L.; Honeycutt, S.S.; Zink, D.; Bills, G.F.; et al. A Novel Inositol Mono-Phosphatase Inhibitor from Memnoniella echinata Producing Organism, Fermentation, Isolation, Physico-Chemical and In Vitro Biological Properties. J. Antibiot. 1992, 45, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, M.J.; Vega, A.; Rivera-Sagredo, A.; Jiménez-Alfaro, M.D.; Díez, E.; Hueso-Rodríguez, J.A. Novel Sesquiterpenoids as Tyrosine Kinase Inhibitors Produced by Stachybotrys chartarum. Tetrahedron 2004, 60, 2379–2385. [Google Scholar] [CrossRef]
- Sakai, K.; Watanabe, K.; Masuda, K.; Tsuji, M.; Hasumi, K.; Endo, A. Isolation, Characterization and Biological Activities of Novel Triprenyl Phenols as Pancreatic Cholesterol Esterase Inhibitors Produced by Stachybotrys sp. F-1839. J. Antibiot. 1995, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Kaise, H.; Shinohara, M.; Miyazaki, W.; Izawa, T.; Nakano, Y.; Sugawara, M.; Sugiura, K.; Sasaki, K. Structure of K-76, a Complement Inhibitor Produced by Stachybotrys complementi nov. sp. K-76. J. Chem. Soc. Chem. Commun. 1979, 16, 726–727. [Google Scholar] [CrossRef]
- Ayer, W.A.; Miao, S. Secondary Metabolites of the Aspen Fungus Stachybotrys cylindrospora. Can. J. Chem. 1993, 71, 487–493. [Google Scholar] [CrossRef]
- Lin, T.-W.; Chang, W.-W.; Chen, C.-C.; Tsai, Y.-C. Stachybotrydial, a Potent Inhibitor of Fucosyltransferase and Sialyltransferase. Biochem. Biophys. Res. Commun. 2005, 331, 953–957. [Google Scholar] [CrossRef]
- Kamalov, L.S.; Aripova, S.F.; Isaev, M.I. Low-Molecular-Mass Metabolites of Fungi I. Stachybotrin from Stachybotrys alternans. Chem. Nat. Compd. 1997, 33, 462–468. [Google Scholar] [CrossRef]
- Kamalov, L.S.; Aripova, S.F.; Isaev, M.I. Low-Molecular-Mass Metabolites of Fungi IV. The Structures of Stachybotrin A and Stachybotral. Chem. Nat. Compd. 1999, 35, 82–85. [Google Scholar] [CrossRef]
- Minagawa, K.; Kouzuki, S.; Yoshimoto, J.; Kawamura, Y.; Tani, H.; Iwata, T.; Terui, Y.; Nakai, H.; Yagi, S.; Hattori, N.; et al. Stachyflin and Acetylstachyflin, Novel Anti-Influenza A Virus Substances, Produced by Stachybotrys sp. RF-7260. I. Isolation, Structure Elucidation and Biological Activities. J. Antibiot. 2002, 55, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, K.; Kouzuki, S.; Kamigauchi, T. Stachyflin and Acetylstachyflin, Novel Anti-Influenza A Virus Substances, Produced by Stachybotrys Sp. RF-7260 II. Synthesis and Preliminary Structure-Activity Relationships of Stachyflin Derivatives. J. Antibiot. 2002, 55, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Ko, S.-K.; Kim, H.-M.; Kim, G.-H.; Son, S.; Kim, G.S.; Hwang, G.J.; Jeon, E.S.; Shin, K.-S.; Ryoo, I.-J.; et al. Stachybotrysin, an Osteoclast Differentiation Inhibitor from the Marine-Derived Fungus Stachybotrys Sp. KCB13F013. J. Nat. Prod. 2016, 79, 2703–2708. [Google Scholar] [CrossRef]
- Sawadjoon, S.; Kittakoop, P.; Isaka, M.; Kirtikara, K.; Madla, S.; Thebtaranonth, Y. Antiviral and Antiplasmodial Spirodihydrobenzofuran Terpenes from the Fungus Stachybotrys nephrospora. Planta Med. 2004, 70, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, C.; Liu, D.; Proksch, P.; Guo, P.; Lin, W. Chartarlactams A–P, Phenylspirodrimanes from the Sponge-Associated Fungus Stachybotrys chartarum with Antihyperlipidemic Activities. J. Nat. Prod. 2014, 77, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, Y.; Ogawa, K.; Michisuji, Y.; Sato, S.-I.; Takada, M.; Hayashi, M.; Yaginuma, S.; Yamamoto, S. Stachybocins, Novel Endothelin Receptor Antagonists, Produced by Stachybotrys sp. M6222. I. Taxonomy, Fermentation, Isolation and Characterization. J. Antibiot. 1995, 48, 1389–1395. [Google Scholar] [CrossRef]
- Wu, B.; Oesker, V.; Wiese, J.; Malien, S.; Schmaljohann, R.; Imhoff, J.F. Spirocyclic Drimanes from the Marine Fungus Stachybotrys sp. Strain MF347. Mar. Drugs 2014, 12, 1924–1938. [Google Scholar] [CrossRef]
- Roggo, B.E.; Hug, P.; Moss, S.; Stämpfli, A.; Kriemler, H.-P.; Peter, H.H. Novel Spirodihydrobenzofuranlactams as Antagonists of Endothelin and as Inhibitors of HIV-1 Protease Produced by Stachybotrys sp. II. Structure Determination. J. Antibiot. 1996, 49, 374–379. [Google Scholar] [CrossRef]
- Liu, J.; Jia, X.; Zhao, J.; Feng, J.; Chen, M.; Chen, R.; Xie, K.; Chen, D.; Li, Y.; Zhang, D.; et al. Bistachybotrysins L–V, Bioactive Phenylspirodrimane Dimers from the Fungus Stachybotrys chartarum. Org. Chem. Front. 2020, 7, 531–542. [Google Scholar] [CrossRef]
- Ma, X.; Li, L.; Zhu, T.; Ba, M.; Li, G.; Gu, Q.; Guo, Y.; Li, D. Phenylspirodrimanes with Anti-HIV Activity from the Sponge-Derived Fungus Stachybotrys Chartarum MXH-X73. J. Nat. Prod. 2013, 76, 2298–2306. [Google Scholar] [CrossRef]
- Zhao, J.; Feng, J.; Tan, Z.; Liu, J.; Zhao, J.; Chen, R.; Xie, K.; Zhang, D.; Li, Y.; Yu, L.; et al. Stachybotrysins A–G, Phenylspirodrimane Derivatives from the Fungus Stachybotrys chartarum. J. Nat. Prod. 2017, 80, 1819–1826. [Google Scholar] [CrossRef]
- Giugliano, F.; Crimini, E.; Tarantino, P.; Zagami, P.; Uliano, J.; Corti, C.; Trapani, D.; Curigliano, G.; Ascierto, P.A. First Line Treatment of BRAF Mutated Advanced Melanoma: Does One Size Fit All? Cancer Treat. Rev. 2021, 99, 102253. [Google Scholar] [CrossRef] [PubMed]
- Al Hamad, M. Contribution of BCR-ABL Molecular Variants and Leukemic Stem Cells in Response and Resistance to Tyrosine Kinase Inhibitors: A Review. F1000Research 2021, 10, 1288. [Google Scholar] [CrossRef] [PubMed]
- Morales, S.; Gasol, A.; Sanchez, D.R. Her2-Positive Cancers and Antibody-Based Treatment: State of the Art and Future Developments. Cancers 2021, 13, 5771. [Google Scholar] [CrossRef] [PubMed]
- Comandone, A.; Vana, F.; Comandone, T.; Tucci, M. Antiangiogenic Therapy in Clear Cell Renal Carcinoma (CCRC): Pharmacological Basis and Clinical Results. Cancers 2021, 13, 5896. [Google Scholar] [CrossRef]
- Moskovitz, J.; Moy, J.; Ferris, R.L. Immunotherapy for Head and Neck Squamous Cell Carcinoma. Curr Oncol Rep 2018, 20, 22. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Killock, D. Tebentafusp for Uveal Melanoma. Nat. Rev. Clin. Oncol. 2021, 18, 747. [Google Scholar] [CrossRef]
- Bovio, E.; Sfecci, E.; Poli, A.; Gnavi, G.; Prigione, V.; Lacour, T.; Mehiri, M.; Varese, G.C. The Culturable Mycobiota Associated with the Mediterranean Sponges Aplysina cavernicola, Crambe crambe and Phorbas tenacior. FEMS Microbiol. Lett. 2019, 366, fnaa014. [Google Scholar] [CrossRef] [PubMed]
- Steinert, K.; Berg, N.; Kalinin, D.V.; Jagels, A.; Würthwein, E.-U.; Humpf, H.-U.; Kalinina, S. Semisynthetic Approach toward Biologically Active Derivatives of Phenylspirodrimanes from S. chartarum. ACS Omega 2022, 7, 45215–45230. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Nakamura, M.; Hayashi, M.; Yaginuma, S.; Yamamoto, S.; Furihata, K.; Shin-Ya, K.; Seto, H. Stachybocins, Novel Endothelin Receptor Antagonists, Produced by Stachybotrys sp. M6222. II. Structure Determination of Stachybocins A, B and C. J. Antibiot. 1995, 48, 1396–1400. [Google Scholar] [CrossRef]
- Kaiser, K.; Benner, R. Hydrolysis-Induced Racemization of Amino Acids. Limnol. Oceanogr. Methods 2005, 3, 318–325. [Google Scholar] [CrossRef]
- Eder, C.; Kurz, M.; Toti, L. Novel Spirobenzofuranlactams and the Derivatives Thereof, Methods for the Production Thereof, and Use Thereof 2004. CA 2508229A1, 1 June 2005. [Google Scholar]
- Jarvis, B.B.; Salemme, J.; Morais, A. Stachybotrys Toxins. 1. Nat. Toxins 1995, 3, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zheng, W.; Sun, K.; Gu, X.; Zeng, X.; Zhang, H.; Zhong, T.; Shao, Z.; Zhang, Y. Two New Phenylspirodrimanes from the Deep-Sea Derived Fungus Stachybotrys sp. MCCC 3A00409. Nat. Prod. Res. 2019, 33, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Jagels, A.; Lindemann, V.; Ulrich, S.; Gottschalk, C.; Cramer, B.; Hübner, F.; Gareis, M.; Humpf, H.-U. Exploring Secondary Metabolite Profiles of Stachybotrys spp. by LC-MS/MS. Toxins 2019, 11, 133. [Google Scholar] [CrossRef]
- Geris, R.; Simpson, T.J. Meroterpenoids Produced by Fungi. Nat. Prod. Rep. 2009, 26, 1063–1094. [Google Scholar] [CrossRef]
- Yin, Y.; Fu, Q.; Wu, W.; Cai, M.; Zhou, X.; Zhang, Y. Producing Novel Fibrinolytic Isoindolinone Derivatives in Marine Fungus Stachybotrys longispora FG216 by the Rational Supply of Amino Compounds According to Its Biosynthesis Pathway. Mar. Drugs 2017, 15, 214. [Google Scholar] [CrossRef]
- Zhang, P.; Li, Y.; Jia, C.; Lang, J.; Niaz, S.-I.; Li, J.; Yuan, J.; Yu, J.; Chen, S.; Liu, L. Antiviral and Anti-Inflammatory Meroterpenoids: Stachybonoids A–F from the Crinoid-Derived Fungus Stachybotrys chartarum 952. RSC Adv. 2017, 7, 49910–49916. [Google Scholar] [CrossRef]
- Augner, D.; Gerbino, D.C.; Slavov, N.; Neudörfl, J.-M.; Schmalz, H.-G. N-Capping of Primary Amines with 2-Acyl-Benzaldehydes to Give Isoindolinones. Org. Lett. 2011, 13, 5374–5377. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Hohn, T.M. Metabolic Pathway Gene Clusters in Filamentous Fungi. Fungal Genet. Biol. 1997, 21, 17–29. [Google Scholar] [CrossRef]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-Based Molecular Networking in the GNPS Analysis Environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, J.; Shen, Y.; Tan, Z.; Zhang, M.; Chen, R.; Zhao, J.; Zhang, D.; Yu, L.; Dai, J. Stachybotrysams A–E, Prenylated Isoindolinone Derivatives with Anti-HIV Activity from the Fungus Stachybotrys chartarum. Phytochem. Lett. 2017, 20, 289–294. [Google Scholar] [CrossRef]
- Chunyu, W.-X.; Ding, Z.-G.; Li, M.-G.; Zhao, J.-Y.; Gu, S.-J.; Gao, Y.; Wang, F.; Ding, J.-H.; Wen, M.-L. Stachartins A–E, Phenylspirodrimanes from the Tin Mine Tailings-Associated Fungus Stachybotrys chartarum. Helv. Chim. Acta 2016, 99, 583–587. [Google Scholar] [CrossRef]
- Feng, J.-M.; Li, M.; Zhao, J.-L.; Jia, X.-N.; Liu, J.-M.; Zhang, M.; Chen, R.-D.; Xie, K.-B.; Chen, D.-W.; Yu, H.-B.; et al. Three New Phenylspirodrimane Derivatives with Inhibitory Effect towards Potassium Channel Kv1.3 from the Fungus Stachybotrys chartarum. J. Asian Nat. Prod. Res. 2019, 21, 887–894. [Google Scholar]
- Yang, B.; Long, J.; Pang, X.; Lin, X.; Liao, S.; Wang, J.; Zhou, X.; Li, Y.; Liu, Y. Structurally Diverse Polyketides and Phenylspirodrimanes from the Soft Coral-Associated Fungus Stachybotrys chartarum SCSIO41201. J. Antibiot. 2021, 74, 190–198. [Google Scholar] [CrossRef]
- Dufies, M.; Grytsai, O.; Ronco, C.; Camara, O.; Ambrosetti, D.; Hagege, A.; Parola, J.; Mateo, L.; Ayrault, M.; Giuliano, S.; et al. New CXCR1/CXCR2 Inhibitors Represent an Effective Treatment for Kidney or Head and Neck Cancers Sensitive or Refractory to Reference Treatments. Theranostics 2019, 9, 5332–5346. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Gioanni, J.; Fischel, J.-L.; Lambert, J.-C.; Demard, F.; Mazeau, C.; Zanghellini, E.; Ettore, F.; Formento, P.; Chauvel, P.; Lalanne, C.-M.; et al. Two New Human Tumor Cell Lines Derived from Squamous Cell Carcinomas of the Tongue: Establishment, Characterization and Response to Cytotoxic Treatment. Eur. J. Cancer Clin. Oncol. 1988, 24, 1445–1455. [Google Scholar] [CrossRef]
- Hagege, A.; Saada-Bouzid, E.; Ambrosetti, D.; Rastoin, O.; Boyer, J.; He, X.; Rousset, J.; Montemagno, C.; Doyen, J.; Pedeutour, F.; et al. Targeting of C-MET and AXL by Cabozantinib Is a Potential Therapeutic Strategy for Patients with Head and Neck Cell Carcinoma. Cell Rep. Med. 2022, 3, 100659. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Giuliano, S.; Ambrosetti, D.; Claren, A.; Ndiaye, P.D.; Mastri, M.; Moghrabi, W.; Cooley, L.S.; Ettaiche, M.; Chamorey, E.; et al. Sunitinib Stimulates Expression of VEGFC by Tumor Cells and Promotes Lymphangiogenesis in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017, 77, 1212–1226. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
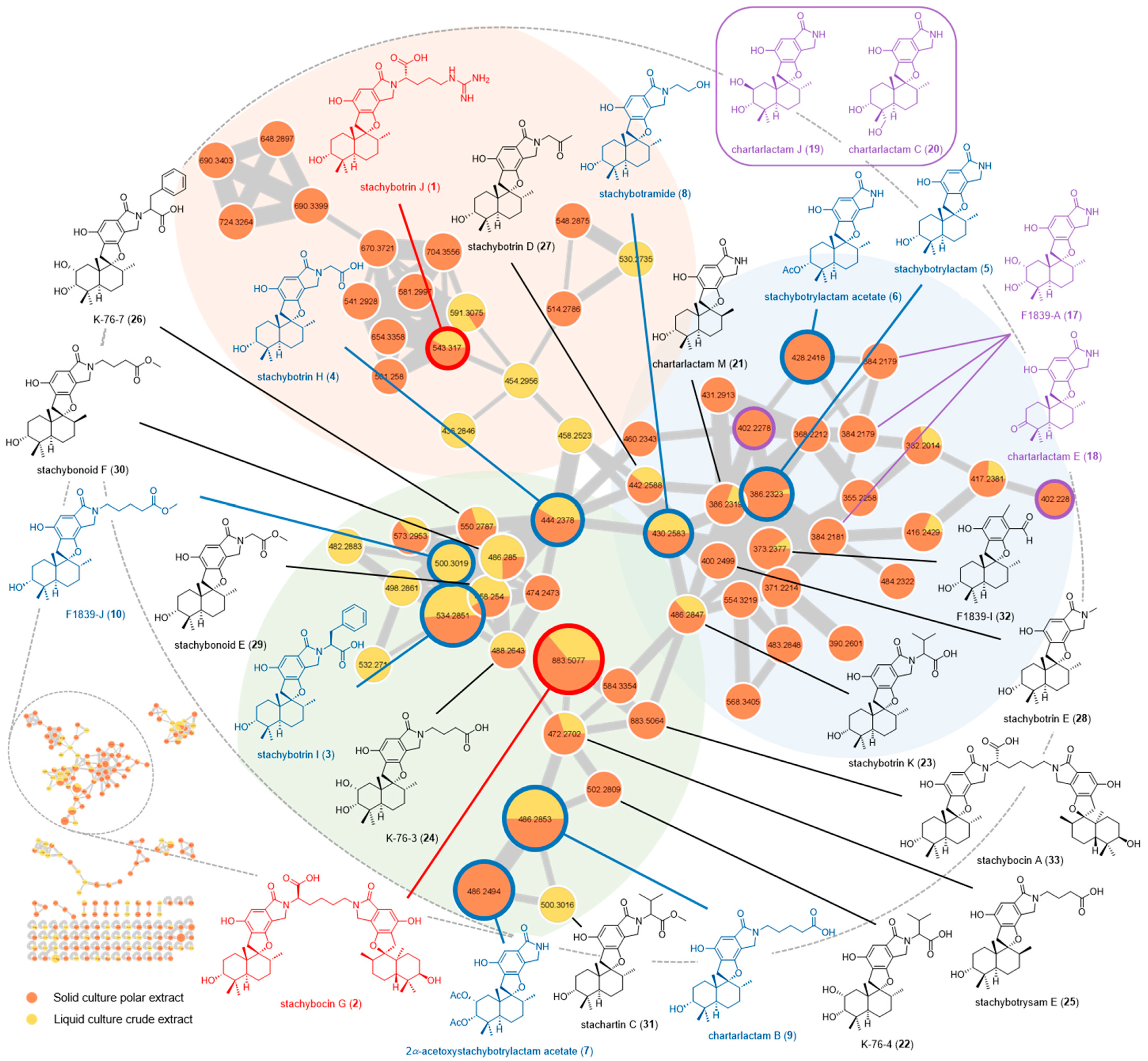{kind=link}
| N° | δC (ppm)/Mult. | δH (ppm)/Mult./J(Hz) | 1H-1H COSY | 1H-13C HMBC |
|---|---|---|---|---|
| 1α | 25.3, CH2 | 1.09, m | 1β, 2, 15 | - |
| 1β | 1.90, m | 1α, 2, 15 | ||
| 2 | 22.1, CH2 | 1.56, m | 1α, 1β, 3 | - |
| 3 | 76.3, CH | 3.33, s | - | - |
| 4 | 38.5, C | - | - | - |
| 5 | 41.3, CH | 2.15, m | 6α, 6β | 13, 14 |
| 6α | 26.1, CH | 1.90, m | 5, 7 | - |
| 6β | 1.56, m | |||
| 7 | 32.3, CH2 | 1.56, m | 6α, 6β | - |
| 8 | 38.6, CH | 1.90, m | 7, 12 | - |
| 9 | 99.7, C | - | - | - |
| 10 | 43.5, C | - | - | - |
| 11α | 33.0, CH | 3.24, d, 16.9 | 11β | 8, 9, 10, 1′, 6′ |
| 11β | 2.86, d, 16.9 | 11α | ||
| 12 | 16.0, CH3 | 0.74, d, 6.5 | 8 | 7, 8, 9 |
| 13 | 29.0, CH3 | 0.98, s | - | 3, 4, 5, 14 |
| 14 | 23.0, CH3 | 0.89, s | - | 3, 4, 5, 13 |
| 15 | 16.6, CH3 | 1.06, s | 1α, 1β | 1, 5, 9, 10 |
| 1′ | 118.7, C | - | - | - |
| 2′ | 155.1, C | - | - | - |
| 3′ | 102.1, CH | 6.69, s | - | 1′, 2′, 4′, 7′ |
| 4′ | 114.9, C | - | - | - |
| 5′ | 135.1, C | - | - | - |
| 6′ | 157.6, C | - | - | - |
| 7′ | 171.7, C | - | - | - |
| 8′a | 45.9, CH | 4.74, d, 17.2 | 8′b | 4′, 5′, 7′ |
| 8′b | 4.29, d, 17.2 | 8′a | ||
| 1″ | 170.3, C | - | - | - |
| 2″ | 57.6, CH | 4.76, m | 3″a, 3″b | 7′, 8′, 1″, 3″ |
| 3″a | 28.8, CH2 | 2.15, m | 2″, 4″ | 4″ |
| 3″b | 1.90, m | |||
| 4″ | 27.2, CH2 | 1.56, m | 3″a, 3″b, 5″ | - |
| 5″ | 41.9, CH2 | 3.24, m | 4″ | 4″, 6″ |
| 6″ | 158.6, C | - | - | - |
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| Molecule | MP41 | 786 | 786R | CAL33 | CAL33RR |
| 1 | >100 | 100 | >100 | 20 ± 4 | 100 ± 7 |
| 2 | >100 | 85 ± 7 | >100 | 30 ± 4 | >100 |
| 3 | >100 | >100 | >100 | 40 ± 4 | 97 ± 5 |
| 4 | >100 | 20 ± 4 | 80 ± 6 | 40 ± 4 | 100 ± 7 |
| 5 | <1 | 0.7 ± 0.1 | 0.8 ± 0.2 | 0.3 ± 0.1 | 0.6 ± 0.2 |
| 6 | 0.6 ± 0.1 | 1.5 ± 0.2 | 2.2 ± 0.3 | 0.6 ± 0.1 | 1.0 ± 0.2 |
| 7 | 0.4 ± 0.1 | 0.8 ± 0.2 | 2.2 ± 0.4 | 0.4 ± 0.1 | 0.7 ± 0.2 |
| 8 | 50 ± 5 | >100 | 74 ± 5 | 5 ± 5 | 97 ± 5 |
| 9 | >100 | 60 ± 6 | >100 | 30 ± 3 | 69 ± 6 |
| 10 | >100 | 30 ± 6 | 75 ± 5 | 35 ± 4 | 97 ± 6 |
| cisplatin | - | - | - | 1.5 ± 0.3 | >10 ± 1 |
| sunitinib | - | 2.5 ± 0.5 | >10 ± 1 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dayras, M.; Sfecci, E.; Bovio, E.; Rastoin, O.; Dufies, M.; Fontaine-Vive, F.; Taffin-de-Givenchy, E.; Lacour, T.; Pages, G.; Varese, G.C.; et al. New Phenylspirodrimanes from the Sponge-Associated Fungus Stachybotrys chartarum MUT 3308. Mar. Drugs 2023, 21, 135. https://doi.org/10.3390/md21030135
Dayras M, Sfecci E, Bovio E, Rastoin O, Dufies M, Fontaine-Vive F, Taffin-de-Givenchy E, Lacour T, Pages G, Varese GC, et al. New Phenylspirodrimanes from the Sponge-Associated Fungus Stachybotrys chartarum MUT 3308. Marine Drugs. 2023; 21(3):135. https://doi.org/10.3390/md21030135
Chicago/Turabian StyleDayras, Marie, Estelle Sfecci, Elena Bovio, Olivia Rastoin, Maeva Dufies, Fabien Fontaine-Vive, Elisabeth Taffin-de-Givenchy, Thierry Lacour, Gilles Pages, Giovanna Cristina Varese, and et al. 2023. "New Phenylspirodrimanes from the Sponge-Associated Fungus Stachybotrys chartarum MUT 3308" Marine Drugs 21, no. 3: 135. https://doi.org/10.3390/md21030135
APA StyleDayras, M., Sfecci, E., Bovio, E., Rastoin, O., Dufies, M., Fontaine-Vive, F., Taffin-de-Givenchy, E., Lacour, T., Pages, G., Varese, G. C., & Mehiri, M. (2023). New Phenylspirodrimanes from the Sponge-Associated Fungus Stachybotrys chartarum MUT 3308. Marine Drugs, 21(3), 135. https://doi.org/10.3390/md21030135