
Marine Natural and Nature-Inspired Compounds Targeting Peroxisome Proliferator Activated Receptors (PPARs)


Abstract
1. Introduction
2. Domain Organization of PPARs and Their Mechanism of Action
PPAR-Ligand Binding Domain (LBD)
3. Marine Natural and Nature-Inspired Compounds as PPAR Modulators
3.1. Oxygenated/Unsaturated FFAs
3.2. Mero/Nor-Terpenoids
3.3. Polyketides and Lactones
3.4. Phtalides
3.5. Phosphonates
3.6. Miscellaneous
4. Conclusions
Funding
Conflicts of Interest
References
- Poulsen, L.L.C.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor induces fatty acid β-Oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef]
- Rial, S.A.; Karelis, A.D.; Bergeron, K.-F.; Mounier, C. Gut Microbiota and Metabolic Health: The Potential Beneficial Effects of a Medium Chain Triglyceride Diet in Obese Individuals. Nutrients 2016, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Vitale, R.M.; Iannotti, F.A.; Silvestri, C.; Flamand, N.; Druart, C.; Everard, A.; Pelicaen, R.; Maiter, D.; Thissen, J.-P.; et al. Beneficial Effects of Akkermansia muciniphila Are Not Associated with Major Changes in the Circulating Endocannabinoidome but Linked to Higher Mono-Palmitoyl-Glycerol Levels as New PPARα Agonists. Cells 2021, 10, 185. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Iannotti, F.; Vitale, R. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef]
- D’Aniello, E.; Fellous, T.; Iannotti, F.A.; Gentile, A.; Allarà, M.; Balestrieri, F.; Gray, R.; Amodeo, P.; Vitale, R.M.; Di Marzo, V. Identification and characterization of phytocannabinoids as novel dual PPARα/γ agonists by a computational and in vitro experimental approach. Biochim. Biophys. Acta-Gen. Subj. 2019, 1863, 586–597. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- Bao, W.; Kong, R.; Wang, N.; Han, W.; Lu, J. PPAR-Alpha Agonist Fenofibrate Combined with Octreotide Acetate in the Treatment of Acute Hyperlipidemia Pancreatitis. PPAR Res. 2021, 2021, 6629455. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the Promise of Insulin Sensitization in Type 2 Diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, C.; Barbier, O.; Fruchart, J.C.; Staels, B.; Glineur, C. Peroxisome proliferator-activated receptors: Regulation of transcriptional activities and roles in inflammation. J. Steroid Biochem. Mol. Biol. 2003, 85, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed]
- Strosznajder, A.K.; Wójtowicz, S.; Jeżyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. Neuromol. Med. 2021, 23, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Govindarajulu, M.; Pinky, P.D.; Bloemer, J.; Ghanei, N.; Suppiramaniam, V.; Amin, R. Signaling Mechanisms of Selective PPARγ Modulators in Alzheimer’s Disease. PPAR Res. 2018, 2018, 2010675. [Google Scholar] [CrossRef]
- Shen, L.Q. Rosiglitazone and Delayed Onset of Proliferative Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 793–799. [Google Scholar] [CrossRef]
- Hodel, C. Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicol. Lett. 2002, 128, 159–168. [Google Scholar] [CrossRef]
- Sica, D.A. Fibrate therapy and renal function. Curr. Atheroscler. Rep. 2009, 11, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Zinman, B.; Lachin, J.M.; Haffner, S.M.; Herman, W.H.; Holman, R.R.; Kravitz, B.G.; Yu, D.; Heise, M.A.; Aftring, R.P.; et al. Rosiglitazone-Associated Fractures in Type 2 Diabetes. Diabetes Care 2008, 31, 845–851. [Google Scholar] [CrossRef]
- Artis, D.R.; Lin, J.J.; Zhang, C.; Wang, W.; Mehra, U.; Perreault, M.; Erbe, D.; Krupka, H.I.; England, B.P.; Arnold, J.; et al. Scaffold-based discovery of indeglitazar, a PPAR pan-active anti-diabetic agent. Proc. Natl. Acad. Sci. USA 2009, 106, 262–267. [Google Scholar] [CrossRef]
- Chigurupati, S.; Dhanaraj, S.A.; Balakumar, P. A step ahead of PPARγ full agonists to PPARγ partial agonists: Therapeutic perspectives in the management of diabetic insulin resistance. Eur. J. Pharmacol. 2015, 755, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, M.; Wright, M.B.; Bopst, M.; Balas, B. Examining the safety of PPAR agonists—Current trends and future prospects. Expert Opin. Drug Saf. 2012, 12, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef]
- Aranda, A.; Pascual, A. Nuclear Hormone Receptors and Gene Expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef]
- Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell. Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-Regulated Gene Expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγPartial Agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef]
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARγ. Elife 2018, 7, e43320. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα Ligand-Binding Domain Structures with Endogenous Fatty Acids and Fibrates. Iscience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial Agonists Activate PPARγ Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef]
- Kojetin, D.J.; Burris, T.P. Small Molecule Modulation of Nuclear Receptor Conformational Dynamics: Implications for Function and Drug Discovery. Mol. Pharmacol. 2013, 83, 1–8. [Google Scholar] [CrossRef]
- Jang, J.Y.; Koh, M.; Bae, H.; An, D.R.; Im, H.N.; Kim, H.S.; Yoon, J.Y.; Yoon, H.-J.; Han, B.W.; Park, S.B.; et al. Structural basis for differential activities of enantiomeric PPARγ agonists: Binding of S35 to the alternate site. Biochim. Biophys. Acta-Proteins Proteom. 2017, 1865, 674–681. [Google Scholar] [CrossRef]
- Igarashi, Y.; Ikeda, M.; Miyanaga, S.; Kasai, H.; Shizuri, Y.; Matsuura, N. Two butenolides with PPARα agonistic activity from a marine-derived Streptomyces. J. Antibiot. 2014, 68, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hu, S.; Pang, X.; Wang, J.; Yin, J.; Li, F.; Wang, J.; Yang, X.; Xia, B.; Liu, Y.; et al. The marine-derived furanone reduces intracellular lipid accumulation in vitro by targeting LXRα and PPARα. J. Cell. Mol. Med. 2020, 24, 3384–3398. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Jang, D.M.; Park, S.C.; An, S.; Shin, J.; Han, B.W.; Noh, M. Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ. Biomolecules 2020, 10, 275. [Google Scholar] [CrossRef]
- Liu, S.; Su, M.; Song, S.-J.; Hong, J.; Chung, H.Y.; Jung, J.H. An Anti-Inflammatory PPAR-γ Agonist from the Jellyfish-Derived Fungus Penicillium chrysogenum J08NF-4. J. Nat. Prod. 2018, 81, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; Lauro, G.; De Marino, S.; D’Auria, M.V.; Monti, M.C.; Casapullo, A.; D’Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; et al. Plakilactones from the Marine Sponge Plakinastrella mamillaris. Discovery of a New Class of Marine Ligands of Peroxisome Proliferator-Activated Receptor γ. J. Med. Chem. 2012, 55, 8303–8317. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Yin, J.; Park, M.; Liu, J.; Li, J.L.; La Kim, E.; Hong, J.; Chung, H.Y.; Jung, J.H. Design and synthesis of marine fungal phthalide derivatives as PPAR-γ agonists. Bioorganic Med. Chem. 2012, 20, 4954–4961. [Google Scholar] [CrossRef]
- Mora, F.D.; Jones, D.K.; Desai, P.V.; Patny, A.; Avery, M.A.; Feller, D.R.; Smillie, T.; Zhou, Y.-D.; Nagle, D.G. Bioassay for the Identification of Natural Product-Based Activators of Peroxisome Proliferator-Activated Receptor-γ (PPARγ): The Marine Sponge Metabolite Psammaplin A Activates PPARγ and Induces Apoptosis in Human Breast Tumor Cells. J. Nat. Prod. 2006, 69, 547–552. [Google Scholar] [CrossRef]
- Chianese, G.; Yu, H.-B.; Yang, F.; Sirignano, C.; Luciano, P.; Han, B.-N.; Khan, S.; Lin, H.-W.; Taglialatela-Scafati, O. PPAR Modulating Polyketides from a Chinese Plakortis simplex and Clues on the Origin of Their Chemodiversity. J. Org. Chem. 2016, 81, 5135–5143. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chin, J.; Choi, H.; Baek, K.; Lee, T.-G.; Park, S.E.; Wang, W.; Hahn, D.; Yang, I.; Lee, J.; et al. Phosphoiodyns A and B, Unique Phosphorus-Containing Iodinated Polyacetylenes from a Korean Sponge Placospongia sp. Org. Lett. 2012, 15, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Otton, R.; Marin, D.P.; Bolin, A.P.; De Cássia Macedo Dos Santos, R.; Polotow, T.G.; Sampaio, S.C.; de Barros, M.P. Astaxanthin ameliorates the redox imbalance in lymphocytes of experimental diabetic rats. Chem. Interact. 2010, 186, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Moldes-Anaya, A.; Sæther, T.; Uhlig, S.; Nebb, H.I.; Larsen, T.; Eilertsen, H.C.; Paulsen, S.M. Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ. Mar. Drugs 2017, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.M.; D’Aniello, E.; Gorbi, S.; Martella, A.; Silvestri, C.; Giuliani, M.E.; Fellous, T.; Gentile, A.; Carbone, M.; Cutignano, A.; et al. Fishing for Targets of Alien Metabolites: A Novel Peroxisome Proliferator-Activated Receptor (PPAR) Agonist from a Marine Pest. Mar. Drugs 2018, 16, 431. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, E.; Iannotti, F.A.; Falkenberg, L.G.; Martella, A.; Gentile, A.; De Maio, F.; Ciavatta, M.L.; Gavagnin, M.; Waxman, J.S.; Di Marzo, V.; et al. In Silico Identification and Experimental Validation of (−)-Muqubilin A, a Marine Norterpene Peroxide, as PPARα/γ-RXRα Agonist and RARα Positive Allosteric Modulator. Mar. Drugs 2019, 17, 110. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-N.; Choi, H.Y.; Lee, W.; Park, G.M.; Shin, W.S.; Kim, Y.K. Sargaquinoic acid and sargahydroquinoic acid from Sargassum yezoense stimulate adipocyte differentiation through PPARα/γ activation in 3T3-L1 cells. FEBS Lett. 2008, 582, 3465–3472. [Google Scholar] [CrossRef]
- Ko, J.; Hwang, H.; Chin, J.; Hahn, D.; Lee, J.; Yang, I.; Shin, K.; Ham, J.; Kang, H. A stereo-controlled synthesis of 2,4-dimethyl-4-hydroxy-16-phenylhexadecanoic acid 1,4-lactone and its PPAR activities. Bioorg. Med. Chem. Lett. 2010, 20, 6017–6019. [Google Scholar] [CrossRef]
- Bouarab, K.; Adas, F.; Gaquerel, E.; Kloareg, B.; Salaün, J.-P.; Potin, P. The Innate Immunity of a Marine Red Alga Involves Oxylipins from Both the Eicosanoid and Octadecanoid Pathways. Plant Physiol. 2004, 135, 1838–1848. [Google Scholar] [CrossRef]
- Ritter, A.; Cabioch, L.; Brillet-Guéguen, L.; Corre, E.; Cosse, A.; Dartevelle, L.; Duruflé, H.; Fasshauer, C.; Goulitquer, S.; Thomas, F.; et al. Herbivore-induced chemical and molecular responses of the kelps Laminaria digitata and Lessonia spicata. PLoS ONE 2017, 12, e0173315. [Google Scholar] [CrossRef]
- Sethi, S.; Ziouzenkova, O.; Ni, H.; Wagner, D.D.; Plutzky, J.; Mayadas, T.N. Oxidized omega-3 fatty acids in fish oil inhibit leukocyte-endothelial interactions through activation of PPARα. Blood 2002, 100, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty Acids, Eicosanoids, and Hypolipidemic Agents Identified as Ligands of Peroxisome Proliferator-Activated Receptors by Coactivator-Dependent Receptor Ligand Assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Nosjean, O.; Boutin, J.A. Natural ligands of PPARγ:: Are prostaglandin J2 derivatives really playing the part? Cell. Signal. 2002, 14, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Archambault, A.-S.; Tinto, F.; Dumais, É.; Rakotoarivelo, V.; Kostrzewa, M.; Plante, P.-L.; Martin, C.; Simard, M.; Silvestri, C.; Pouliot, R.; et al. Biosynthesis of the Novel Endogenous 15-Lipoxygenase Metabolites N-13-Hydroxy-octodecadienoyl-ethanolamine and 13-Hydroxy-octodecadienoyl-glycerol by Human Neutrophils and Eosinophils. Cells 2021, 10, 2322. [Google Scholar] [CrossRef]
- Arnesen, H.; Haj-Yasein, N.N.; Tungen, J.E.; Soedling, H.; Matthews, J.; Paulsen, S.M.; Nebb, H.I.; Sylte, I.; Hansen, T.V.; Sæther, T. Molecular modelling, synthesis, and biological evaluations of a 3,5-disubstituted isoxazole fatty acid analogue as a PPARα-selective agonist. Bioorg. Med. Chem. 2019, 27, 4059–4068. [Google Scholar] [CrossRef]
- Jia, Y.; Kim, J.-Y.; Jun, H.-J.; Kim, S.-J.; Lee, J.-H.; Hoang, M.H.; Hwang, K.-Y.; Um, S.-J.; Chang, H.I.; Lee, S.-J. The natural carotenoid astaxanthin, a PPAR-α agonist and PPAR-γ antagonist, reduces hepatic lipid accumulation by rewiring the transcriptome in lipid-loaded hepatocytes. Mol. Nutr. Food Res. 2012, 56, 878–888. [Google Scholar] [CrossRef]
- Hussein, G.; Nakagawa, T.; Goto, H.; Shimada, Y.; Matsumoto, K.; Sankawa, U.; Watanabe, H. Astaxanthin ameliorates features of metabolic syndrome in SHR/NDmcr-cp. Life Sci. 2007, 80, 522–529. [Google Scholar] [CrossRef]
- Yoshida, H.; Yanai, H.; Ito, K.; Tomono, Y.; Koikeda, T.; Tsukahara, H.; Tada, N. Administration of natural astaxanthin increases serum HDL-cholesterol and adiponectin in subjects with mild hyperlipidemia. Atherosclerosis 2010, 209, 520–523. [Google Scholar] [CrossRef]
- Xiao, B.; Su, M.; La Kim, E.; Hong, J.; Chung, H.Y.; Kim, H.S.; Yin, J.; Jung, J.H. Synthesis of PPAR-γ Activators Inspired by the Marine Natural Product, Paecilocin, A. Mar. Drugs 2014, 12, 926–939. [Google Scholar] [CrossRef]
- Su, M.; Cao, J.; Huang, J.; Liu, S.; Im, D.S.; Yoo, J.-W.; Jung, J.H. The In Vitro and In Vivo Anti-Inflammatory Effects of a Phthalimide PPAR-γ Agonist. Mar. Drugs 2017, 15, 7. [Google Scholar] [CrossRef]
- Kinarivala, N.; Suh, J.H.; Botros, M.; Webb, P.; Trippier, P.C. Pharmacophore elucidation of phosphoiodyn A—Potent and selective peroxisome proliferator-activated receptor β/δ agonists with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 1889–1893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, X.; Chen, L.; Chen, J.; Hu, L.; Jiang, H.; Shen, X. Molecular Determinants of Magnolol Targeting Both RXRα and PPARγ. PLoS ONE 2011, 6, e28253. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
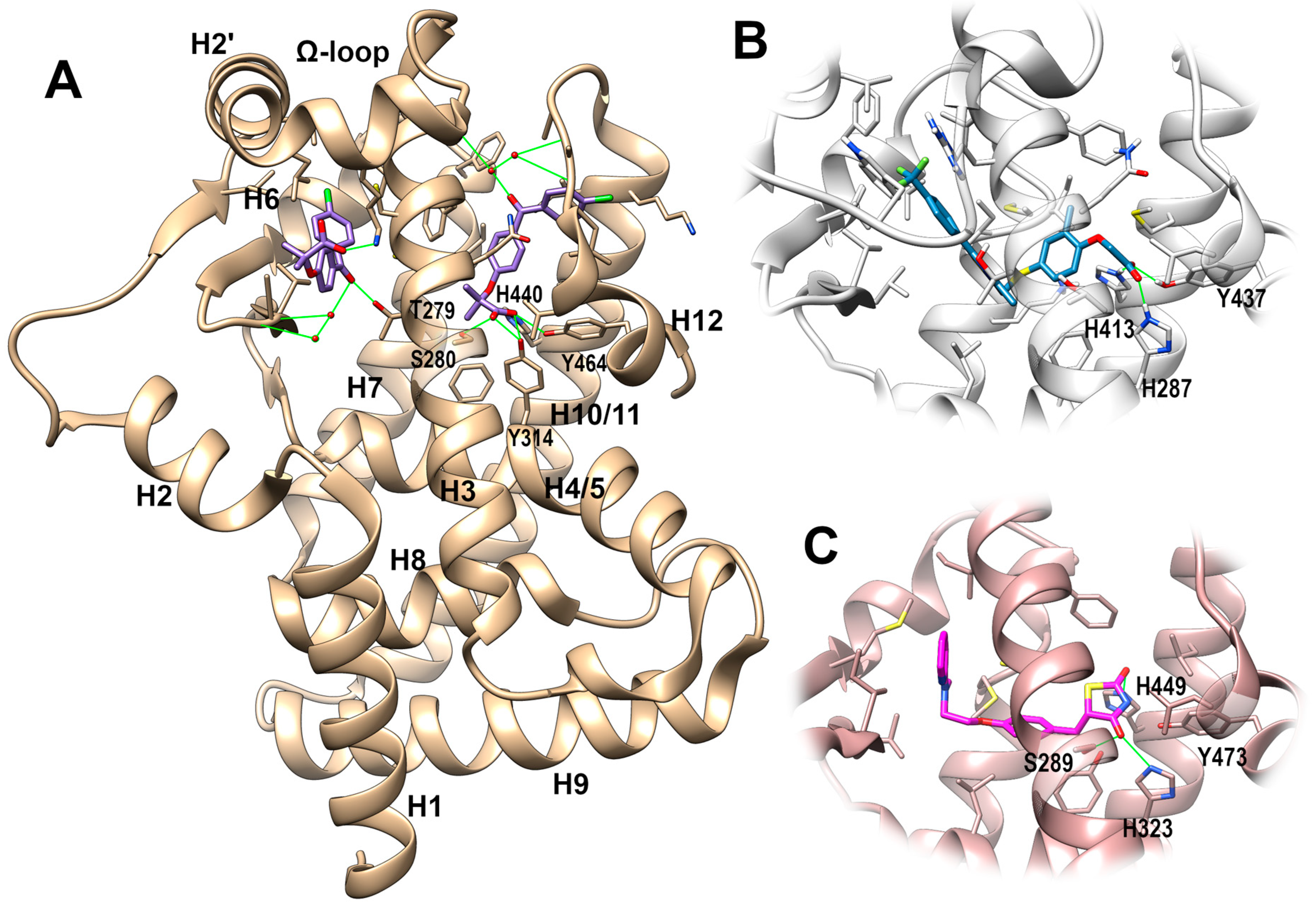{kind=link}
{kind=link}
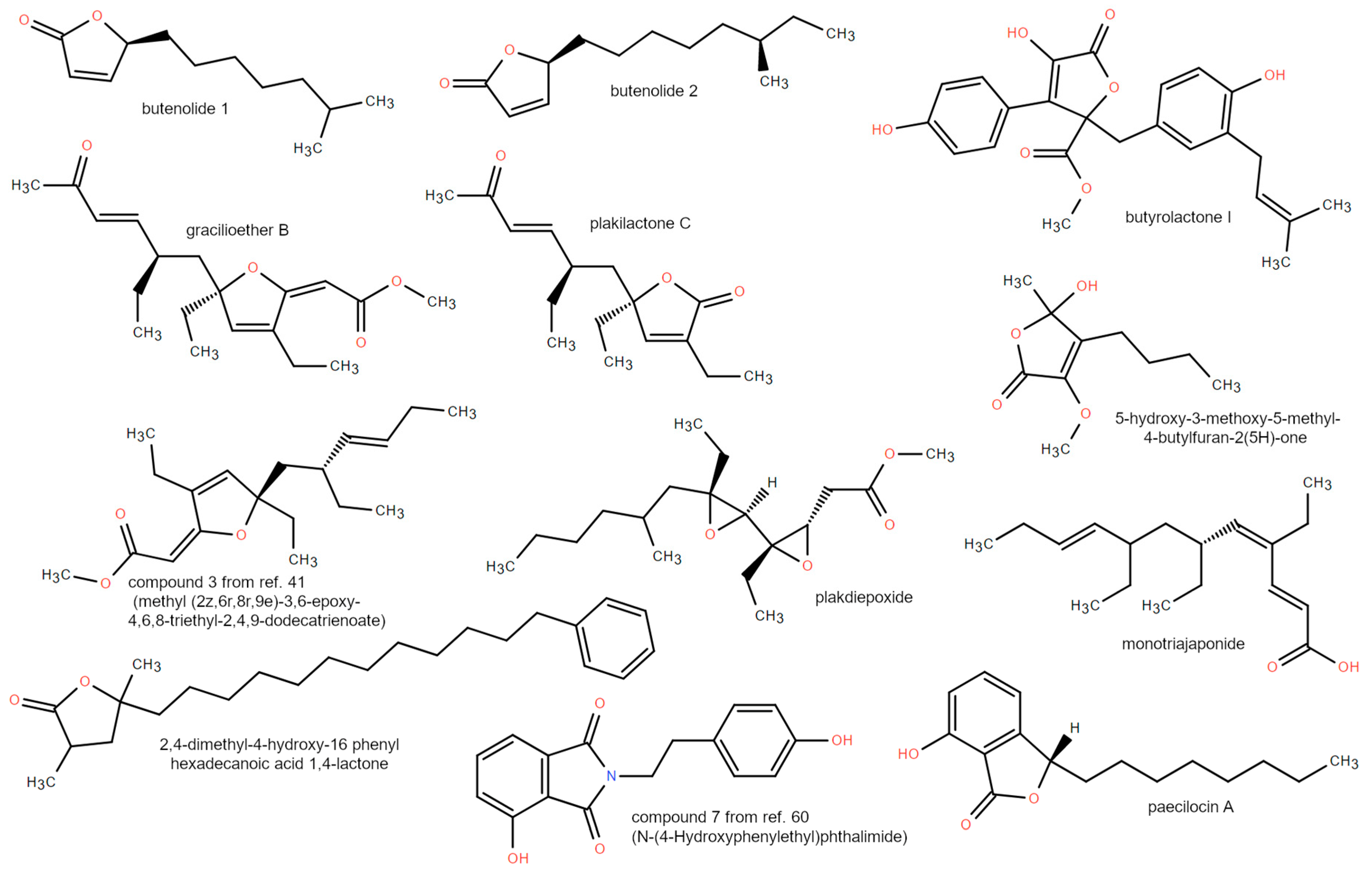{kind=link}
{kind=link}
{kind=link}
| PPAR Isotypes | Compounds | Marine Source |
|---|---|---|
| PPARα | butenolides | marine-derived Streptomyces strain [34] |
| PPARα | furanone | fungus Setosphaeria sp SCSIO41009.2 [35] |
| PPARγ | butyrolactone I | marine fungus A. terreus [36] |
| PPARγ | chrysogenester | jellyfish-derived Fungus Penicillium chrysogenum J08NF-4 [37] |
| PPARγ | gracilioether B plakilactone C | marine sponge Plakinastrella mamillaris [38] |
| PPARγ | paecilocin A | jellyfish-derived fungus Paecilomyces variotii [39] |
| PPARγ | psammaplin A | marine sponge Pseudoceratina rhax (Aplysinelliae) [40] |
| PPARγ | furanylidene acetates | marine sponge Plakortis simplex [41] |
| PPARγ | plakdiepoxide | marine sponge Plakortis simplex [41] |
| PPARδ | phosphoiodyns A and B | Korean sponge Placospongia sp. [42] |
| PPARα/γ | astaxanthin | Seafood [43] |
| PPARα/γ | C16 Oxo-Fatty Acids | diatom Chaetoceros karianus [44] |
| PPARα/γ | caulerpin | green alga Caulerpa cylindracea [45] |
| PPARα/γ | muqubilin | sponge Diacarnus cf. spinopoculum [46] |
| PPARα/γ | sargaquinoic and sargahydroquinoic acids | brown alga Sargassum yezoense [47] |
| PPARα/γ | monotriajaponide | marine sponge Plakortis simplex [41] |
| PPARα/δ | 2,4-dimethyl-4-hydroxy-16-phenylhexadecanoic acid 1,4-lactone | deep-water sponge Plakortis nigra [48] |
| PPARα/γ/δ | PUFA | red alga Chondrus crispus/ [49] brown alga Lessonia spicata [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Aniello, E.; Amodeo, P.; Vitale, R.M. Marine Natural and Nature-Inspired Compounds Targeting Peroxisome Proliferator Activated Receptors (PPARs). Mar. Drugs 2023, 21, 89. https://doi.org/10.3390/md21020089
D’Aniello E, Amodeo P, Vitale RM. Marine Natural and Nature-Inspired Compounds Targeting Peroxisome Proliferator Activated Receptors (PPARs). Marine Drugs. 2023; 21(2):89. https://doi.org/10.3390/md21020089
Chicago/Turabian StyleD’Aniello, Enrico, Pietro Amodeo, and Rosa Maria Vitale. 2023. "Marine Natural and Nature-Inspired Compounds Targeting Peroxisome Proliferator Activated Receptors (PPARs)" Marine Drugs 21, no. 2: 89. https://doi.org/10.3390/md21020089
APA StyleD’Aniello, E., Amodeo, P., & Vitale, R. M. (2023). Marine Natural and Nature-Inspired Compounds Targeting Peroxisome Proliferator Activated Receptors (PPARs). Marine Drugs, 21(2), 89. https://doi.org/10.3390/md21020089