
In Vitro Anticancer and Cancer-Preventive Activity of New Triterpene Glycosides from the Far Eastern Starfish Solaster pacificus

 ,
,  , ,
, ,  and
and 
Abstract
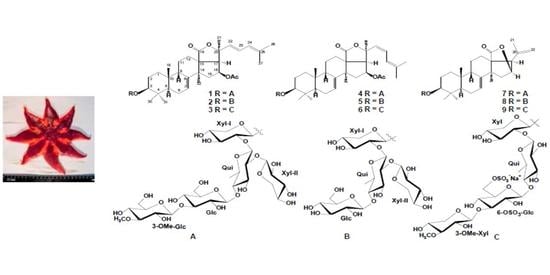
:
1. Introduction
2. Results and Discussion
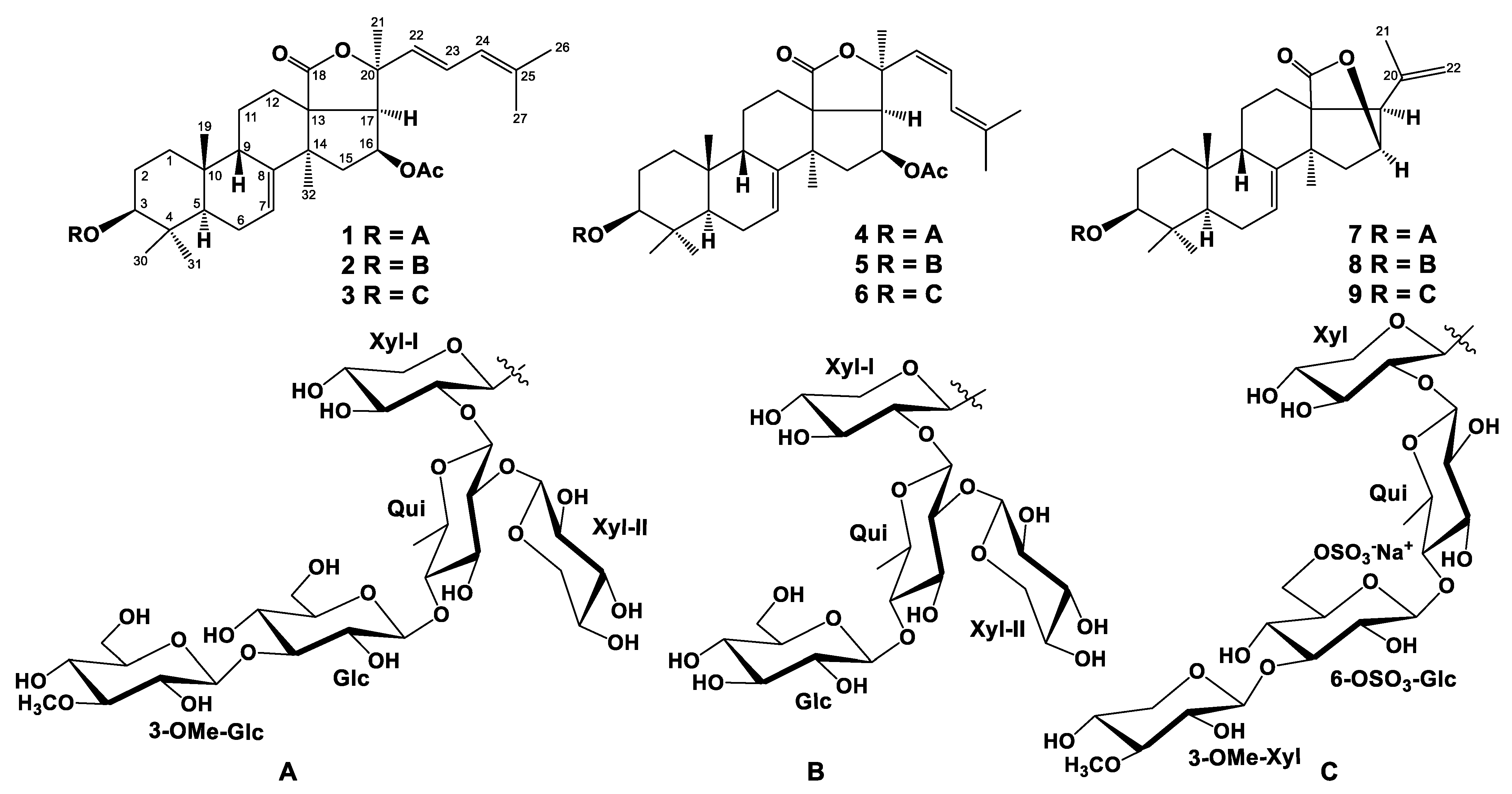
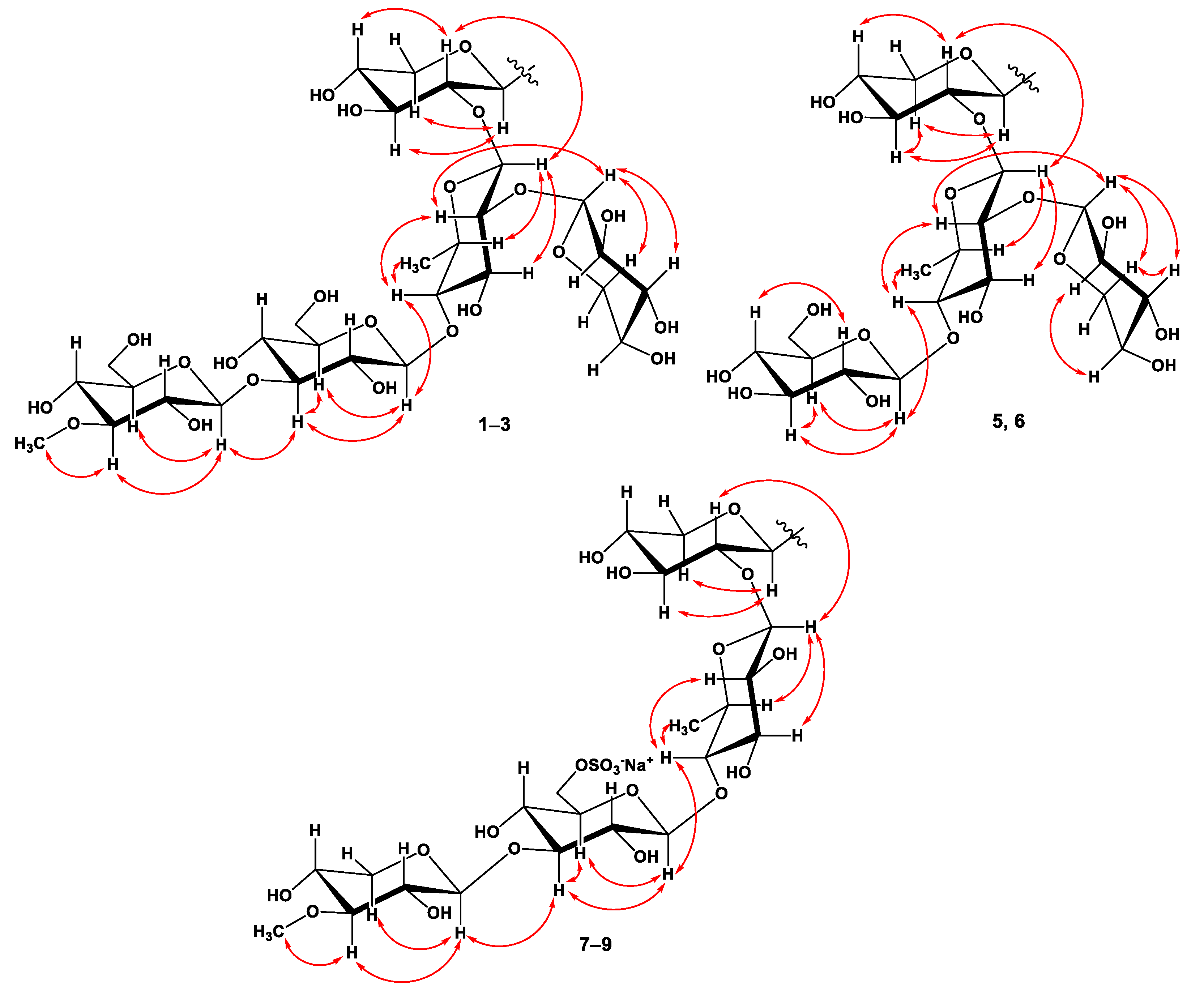
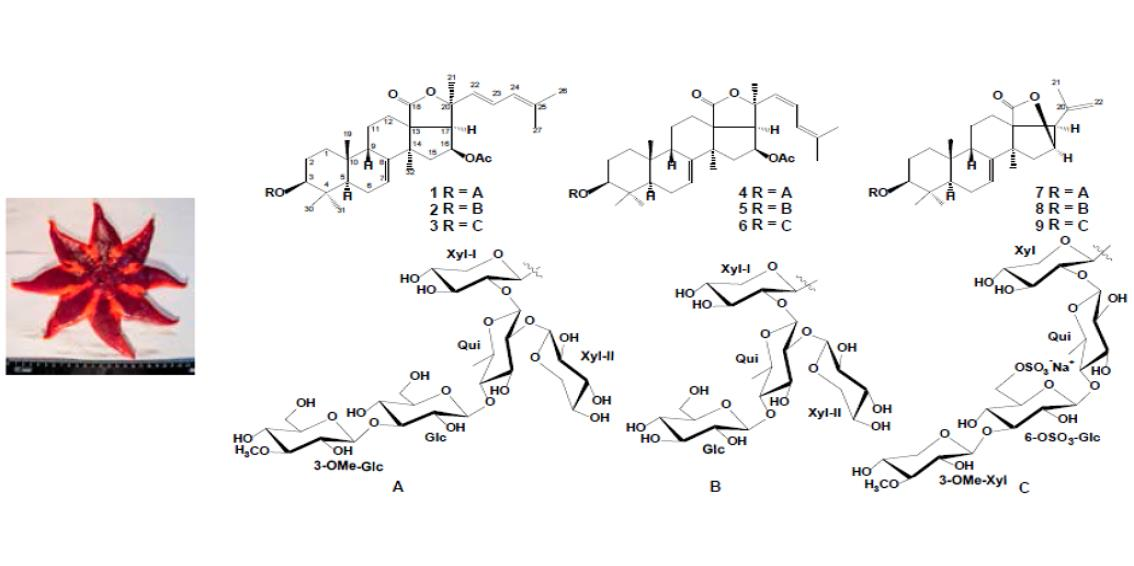
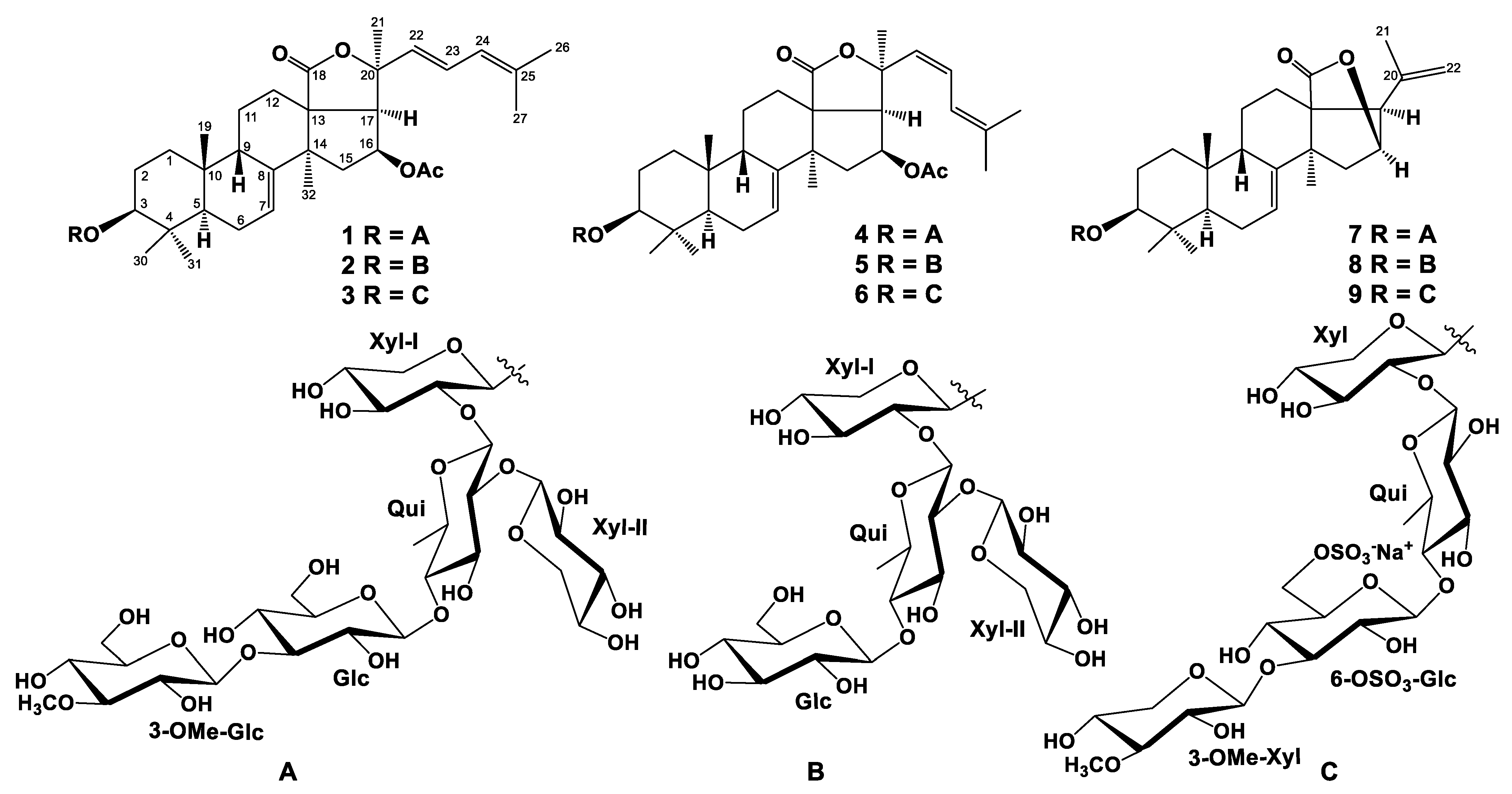
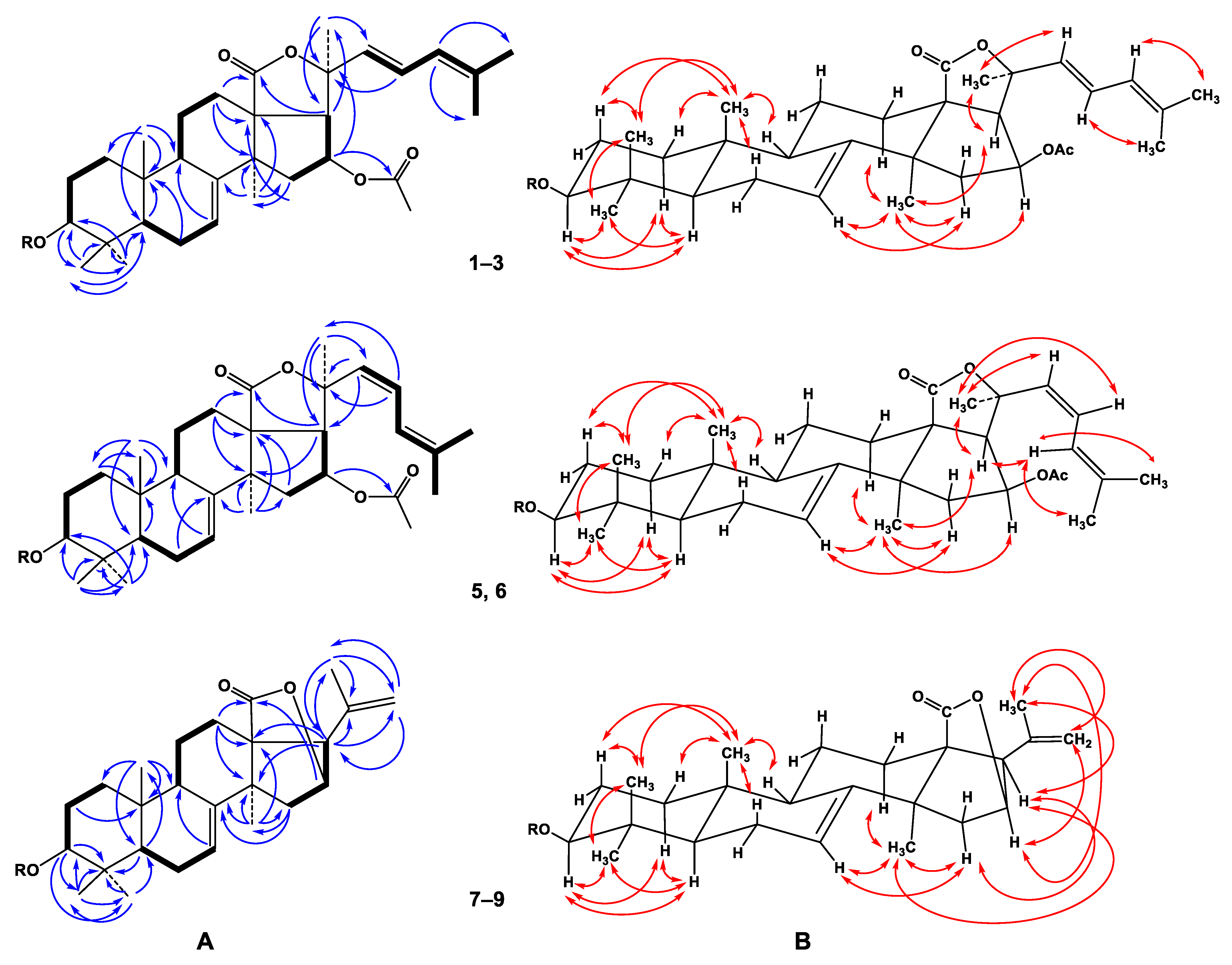
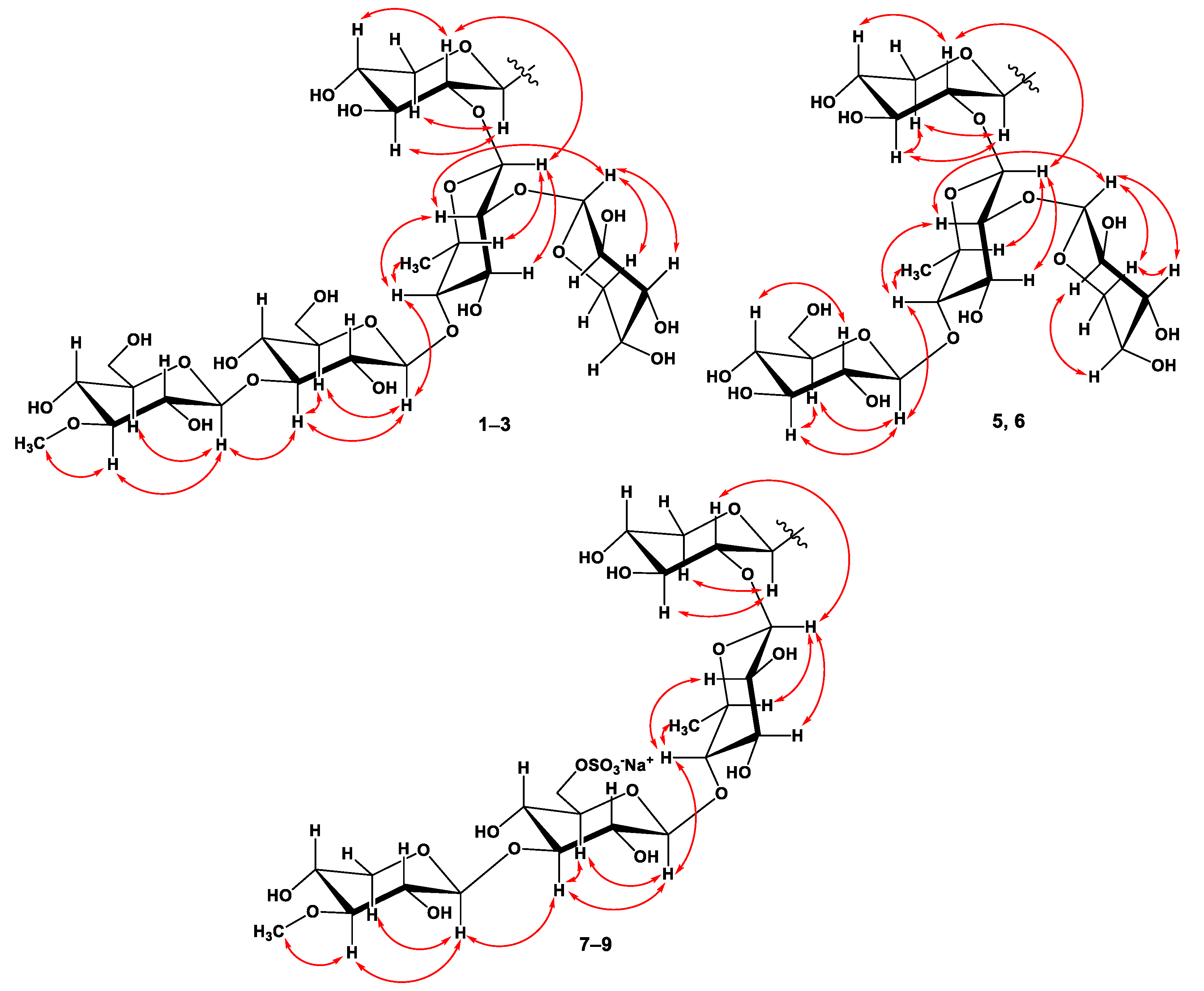
2.1. The Isolation and Structure Elucidation of Compounds 1–9 from S. pacificus
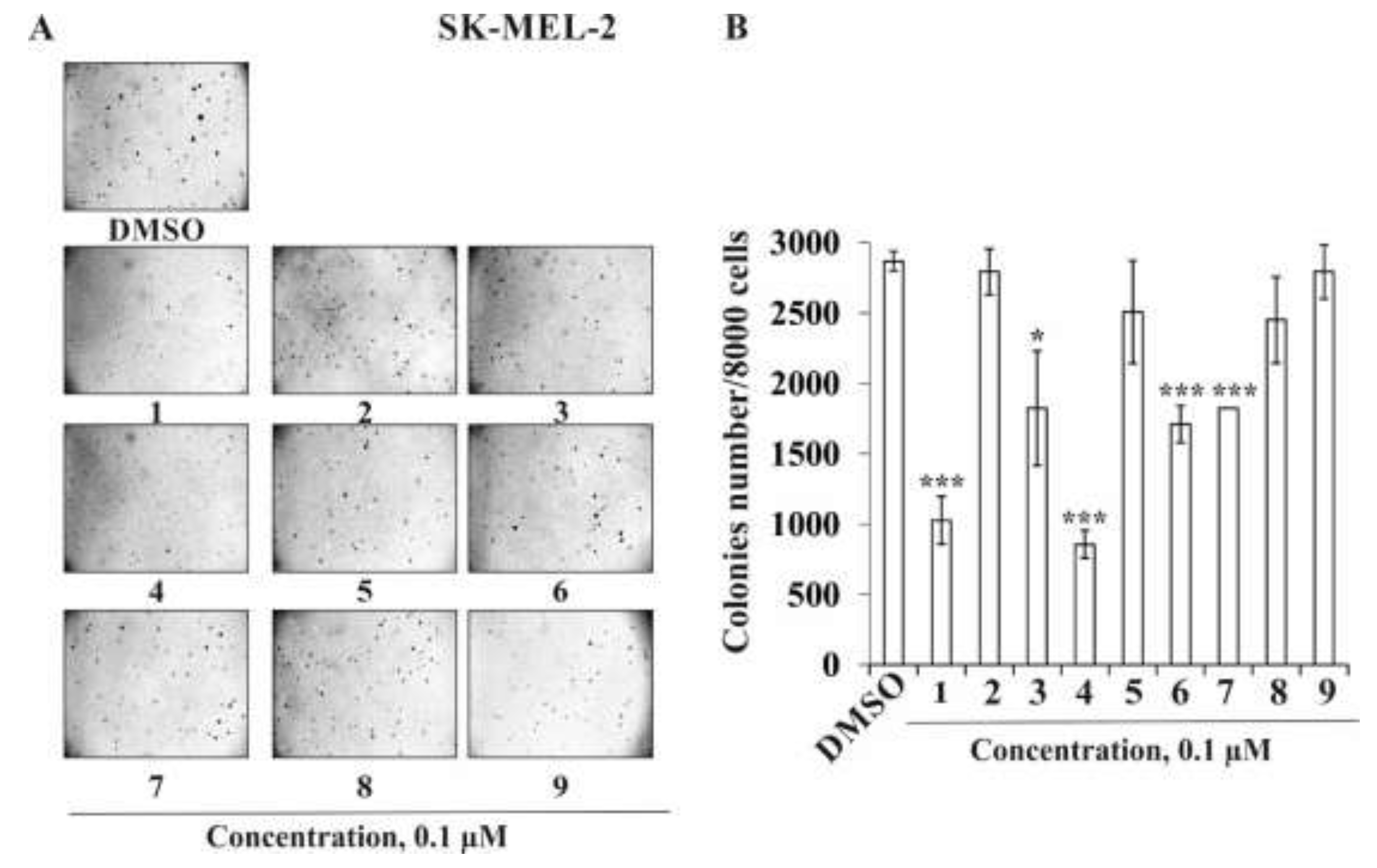
2.2. The Biological Activity Investigation
2.2.1. The Effect of the Triterpene Glycosides on Cell Viability
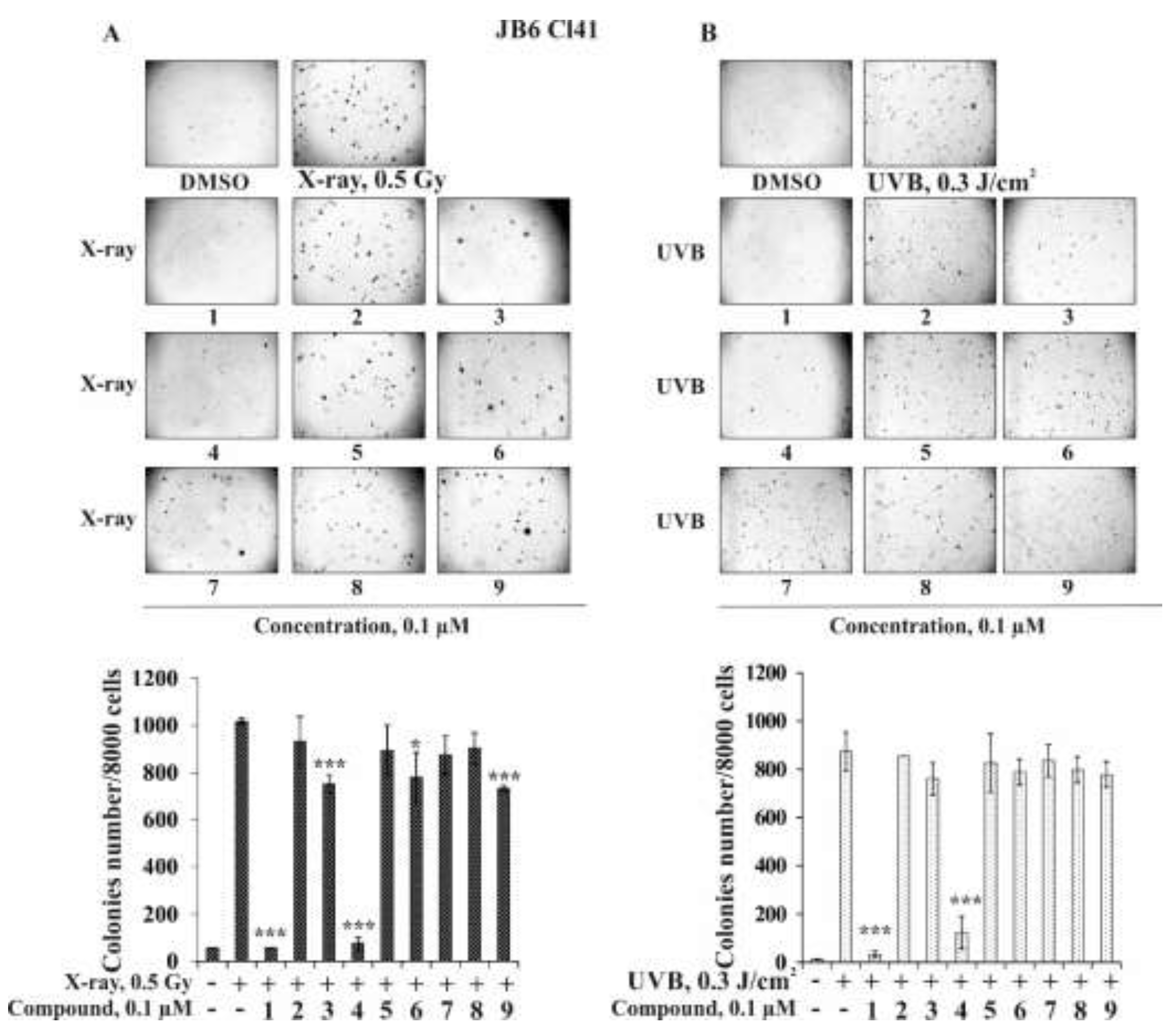
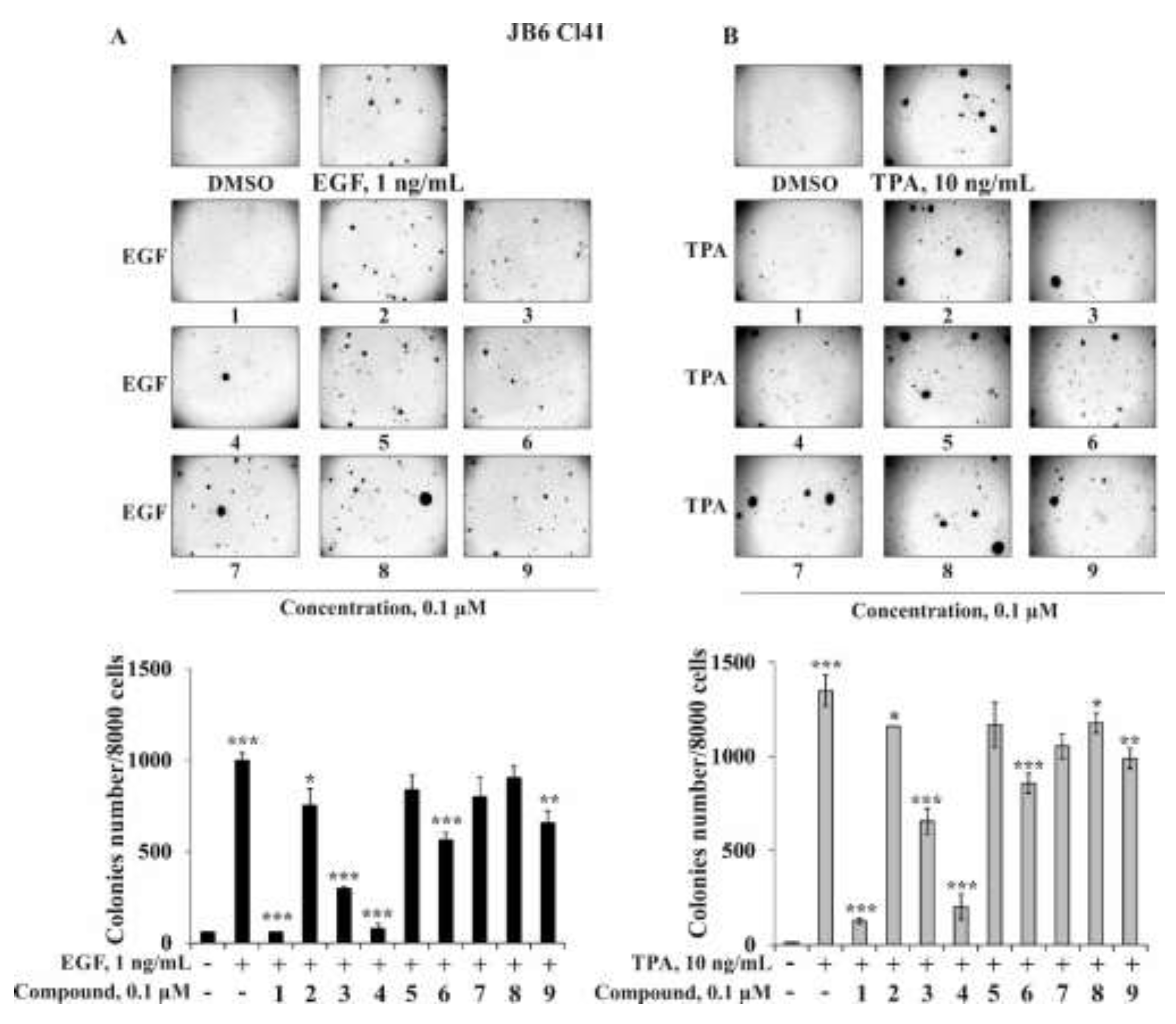
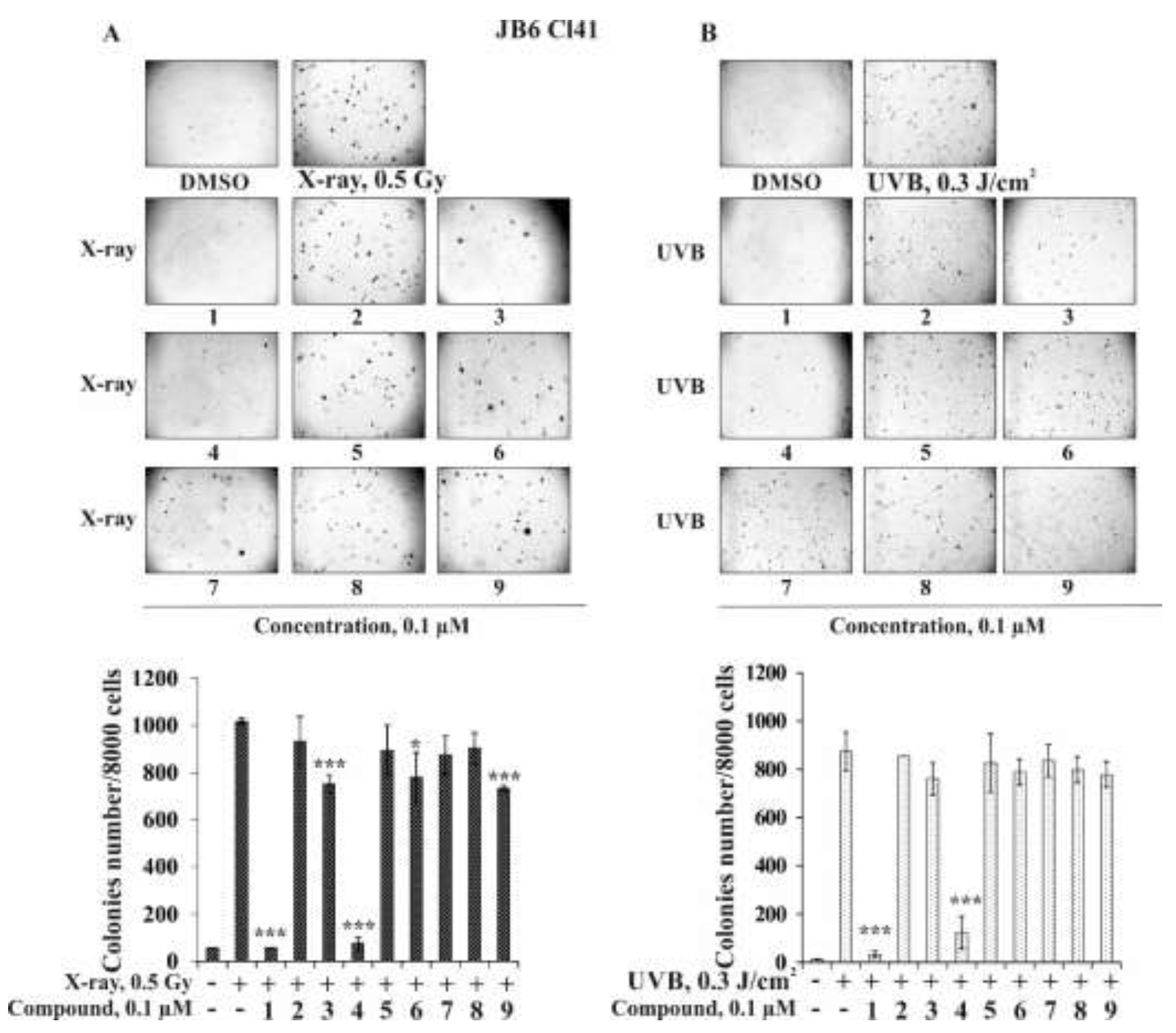
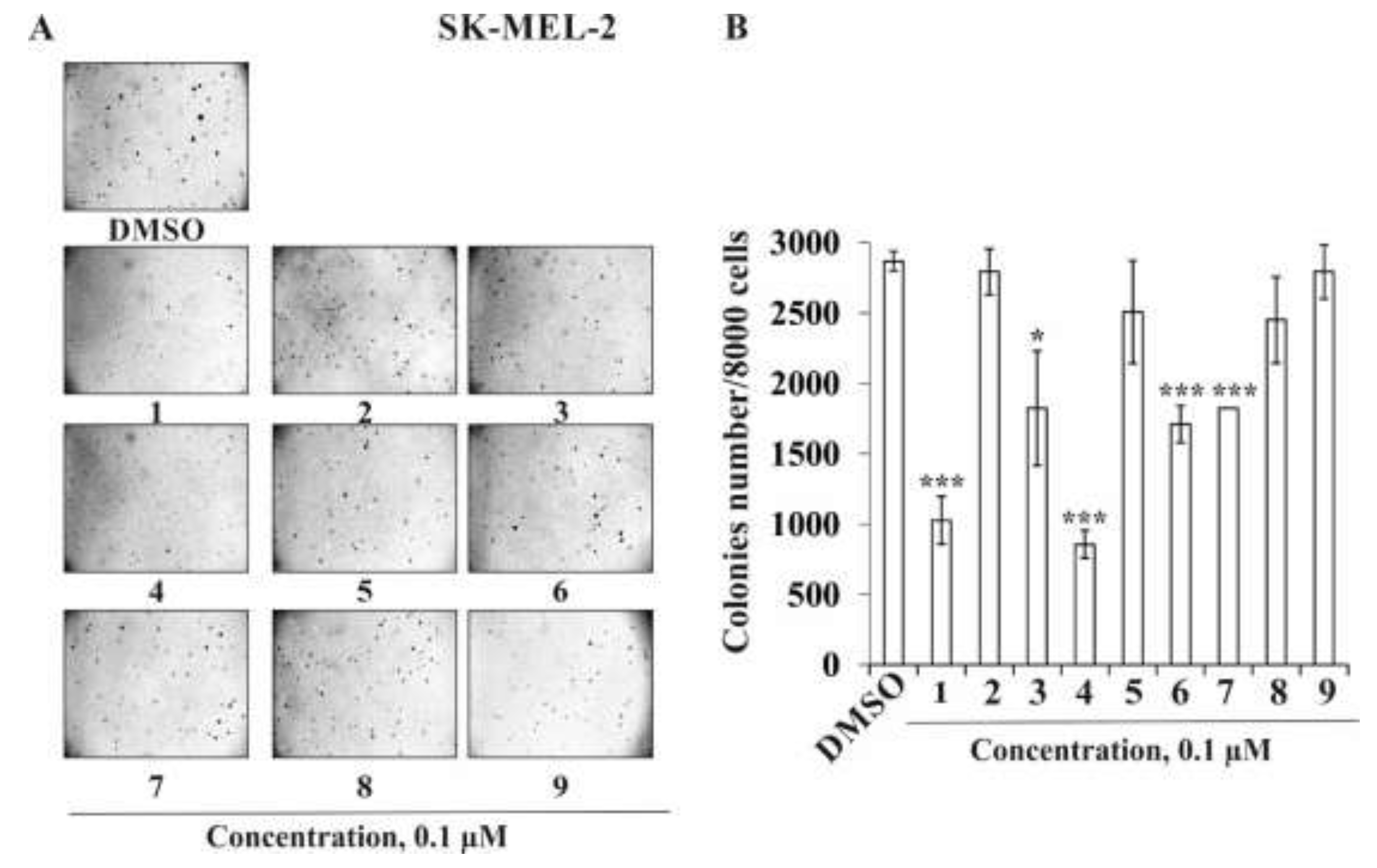
2.2.2. The Effect of the Triterpene Glycosides on Neoplastic Cell Transformation Induced by Carcinogenic Factors
3. Materials and Methods
3.1. General Methods
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Spectral Data of New Compounds
3.5. Acid Hydrolysis and Determination of Absolute Configurations of Monosaccharides
3.6. Cell Lines and Culture Conditions
3.7. Cell Viability Assay
3.8. Hemolysis Assay
3.9. The Soft Agar Assay
3.9.1. Neoplastic Cell Transformation of Normal Cells Induced by Carcinogenic Factors
3.9.2. Colony Formation of Cancer Cells
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baselga, J.; Bhardwaj, N.; Cantley, L.C.; DeMatteo, R.; DuBois, R.N.; Foti, M.; Gapstur, S.M.; Hahn, W.C.; Helman, L.J.; Jensen, R.A.; et al. AACR Cancer Progress Report 2015. Clin. Cancer Res. 2015, 21, S1–S128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, A.; Carpi, A.; Ferrari, P.; Biava, P.M.; Rossi, G. Immunotherapy and hormone-therapy in metastatic breast cancer: A review and an update. Curr. Drug Targets 2016, 17, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. Causes of cancer, including hazardous circumstances. In World Cancer Report: Cancer Research for Cancer Prevention, 1st ed.; WHO: Lyon, France, 2014; pp. 49–137. [Google Scholar]
- Maru, G.B.; Hudlikar, R.R.; Kumar, G.; Gandhi, K.; Mahimkar, M.B. Understanding the molecular mechanisms of cancer prevention by dietary phytochemicals: From experimental models to clinical trials. World J. Biol. Chem. 2016, 7, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Signal transduction pathways in cancer development and as targets for cancer prevention. Prog. Nucleic Acid Res. Mol. Biol. 2005, 79, 237–297. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res. 1976, 36, 2699–2702. [Google Scholar] [PubMed]
- Wattenberg, L.W. Chemoprevention of cancer. Cancer Res. 1985, 45, 1–8. [Google Scholar] [CrossRef]
- Lee, K.W.; Bode, A.M.; Dong, Z. Molecular targets of phytochemicals for cancer prevention. Nat. Rev. Cancer 2011, 11, 211–218. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munroa, M.H.G.; Prinsepd, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [Green Version]
- Kalinin, V.I.; Aminin, D.L.; Avilov, S.A.; Silchenko, A.S.; Stonik, V.A. Triterpene glycosides from sea cucumbers (Holothurioidea, Echinodermata), biological activities and functions. Stud. Nat. Prod. Chem. 2008, 35, 135–196. [Google Scholar]
- Careaga, V.P.; Maier, M.S. Cytotoxic triterpene glycosides from sea cucumbers. In Handbook of Anticancer Drugs from Marine Origin; Kim, S.-K., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 515–528. [Google Scholar]
- Chludil, H.D.; Murray, A.P.; Seldes, A.M.; Maier, M.S. Biologically active triterpene glycosides from sea cucumbers (Holothuroidea, Echinodermata). In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier Science Publisher: Amsterdam, The Netherlands, 2003; Volume 28, pp. 587–615. [Google Scholar]
- Kim, S.K.; Himaya, S.W.A. Chapter 20-Triterpene glycosides from sea cucumbers and their biological activities. Adv. Food Nutr. Res. 2012, 63, 297–319. [Google Scholar] [CrossRef]
- Maier, M.S.; Roccatagliata, A.J.; Kuriss, A.; Chludil, H.; Seldes, A.M.; Pujol, C.A.; Damonte, E.B. Two new cytotoxic and virucidal trisulfated triterpene glycosides from the Antarctic sea cucumber Staurocucumis liouvillei. J. Nat. Prod. 2001, 64, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Minale, L.; Riccio, R.; Zollo, F. Steroidal oligoglycosides and polyhydroxysteroids from Echinoderms. Fortschr. Chem. Org. Nat. 1993, 62, 75–308. [Google Scholar] [CrossRef]
- Stonik, V.A. Marine polar steroids. Russ. Chem. Rev. 2001, 70, 673–715. [Google Scholar] [CrossRef]
- Iorizzi, M.; De Marino, S.; Zollo, F. Steroidal oligoglycosides from the Asteroidea. Curr. Org. Chem. 2001, 5, 951–973. [Google Scholar] [CrossRef]
- Stonik, V.A.; Ivanchina, N.V.; Kicha, A.A. New polar steroids from starfish. Nat. Prod. Commun. 2008, 3, 1587–1610. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Xu, T.H.; Yang, B.; Lin, X.P.; Zhou, X.F.; Yang, X.W.; Liu, Y.H. Chemical constituents and bioactivities of starfish. Chem. Biodivers. 2011, 8, 740–791. [Google Scholar] [CrossRef] [PubMed]
- Ivanchina, N.V.; Kicha, A.A.; Stonik, V.A. Steroid glycosides from marine organisms. Steroids 2011, 76, 425–454. [Google Scholar] [CrossRef] [PubMed]
- Ivanchina, N.V.; Kicha, A.A.; Malyarenko, T.V.; Stonik, V.A. Advances in Natural Products Discovery; Gomes, A.R., Rocha-Santos, T., Duarte, A., Eds.; Nova Science Publishers: New York, NY, USA, 2017; Volume 6, pp. 191–224. [Google Scholar]
- Stonik, V.A.; Kicha, A.A.; Malyarenko, T.V.; Ivanchina, N.V. Asterosaponins: Structures, taxonomic distribution, biogenesis and biological activities. Mar. Drugs 2020, 18, 584. [Google Scholar] [CrossRef]
- Xia, J.M.; Miao, Z.; Xie, C.L.; Zhang, J.W.; Yang, X.W. Chemical constituents and bioactivities of starfish: An update. Chem. Biodivers. 2020, 17, e1900638. [Google Scholar] [CrossRef] [Green Version]
- Aminin, D.L.; Pislyagin, E.A.; Menchinskaya, E.S.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Immunomodulatory and anticancer activity of sea cucumber triterpene glycosides. Stud. Nat. Prod. Chem. 2014, 41, 75–94. [Google Scholar]
- Mondol, M.A.M.; Shin, H.J.; Rahman, M.A.; Islam, M.T. Sea cucumber glycosides: Chemical structures, producing species and important biological properties. Mar. Drugs 2017, 15, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.C.; Sun, Y.; Li, W.; Lin, Y.; Sha, Y.; Pei, Y.H. A new triterpene glycoside from Asterias rollentoni. J. Asian Nat. Prod. Res. 2006, 8, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Ivanchina, N.V.; Kalinovsky, A.I.; Malyarenko, T.V.; Kicha, A.A.; Dmitrenok, P.S. A holothurian triterpene glycoside holothurin A2 (= echinoside A) isolated from the starfish Choriaster granulatus. Nat. Prod. Commun. 2019, 14, 1–3. [Google Scholar] [CrossRef]
- Malyarenko, T.V.; Kicha, A.A.; Kalinovsky, A.I.; Dmitrenok, P.S.; Malyarenko, O.S.; Kuzmich, A.S.; Stonik, V.A.; Ivanchina, N.V. New triterpene glycosides from the Far Eastern starfish Solaster pacificus and their biological activity. Biomolecules 2021, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Popov, R.S.; Kalinin, V.I.; Andrijaschenko, P.V.; Dmitrenok, P.S.; Yurchenko, E.A. Triterpene glycosides from the sea cucumber Eupentacta fraudatrix. Structure and cytotoxic action of cucumarioside D with a terminal 3-O-Me-glucose residue unique for this species. Nat. Prod. Commun. 2018, 13, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.D.; Schupp, P.J.; Schrör, J.-P.; Engemann, A.; Rohde, S.; Kelman, D.; de Voogd, N.; Carroll, A.; Motti, C.A. Twilight zne sponges from Guam yield theonellin isocyanate and psammaplysins I and J. J. Nat. Prod. 2012, 75, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Sharma, U.; Schulz, T.C.; McLean, A.B.; Robins, A.J.; West, L.M. Bicyclic C21 terpenoids from the marine sponge Clathria compressa. J. Nat. Prod. 2012, 75, 1223–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afiyatullov, S.S.; Kalinovsky, A.I.; Stonik, V.A. Structures of cucumariosides C1 and C2 – two new triterpene glycosides from the holothurian Eupentacta fraudatrix. Chem. Nat. Comp. 1987, 23, 691–696. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Andrijaschenko, P.V.; Dmitrenok, P.S.; Martyyas, E.A.; Kalinin, V.I. Triterpene glycosides from the sea cucumber Eupentacta fraudatrix. Structure and cytotoxic action of cucumariosides A2, A7, A9, A10, A11, A13 and A14, seven new minor non-sulfated tetraosides and an aglycone with an uncommon 18-hydroxy group. Nat. Prod. Commun. 2012, 7, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Popov, R.S.; Ivanchina, N.V.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I.; Dolmatov, I.Y.; Stonik, V.A.; Dmitrenok, P.S. Metabolite profiling of triterpene glycosides of the Far Eastern sea cucumber Eupentacta fraudatrix and their distribution in various body components using LC-ESI QTOF-MS. Mar. Drugs 2017, 15, 302. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Lazaro, M. How many times should we screen a chemical library to discover an anticancer drug. Drug Discov. Today 2015, 20, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.D.; Gonzalez, F.J.; Perdew, G.H.; Peters, J.M. Molecular regulation of carcinogenesis: Friend and foe. Toxicol. Sci. 2018, 165, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M.; Gonzalez, F.J. The evolution of carcinogenesis. Toxicol. Sci. 2018, 165, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Rundhaug, J.E.; Fischer, S.M. Molecular mechanisms of mouse skin tumor promotion. Cancers 2010, 2, 436–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AAT Bioquest. Available online: https://www.aatbio.com/tools/ic50-calculator (accessed on 10 November 2020).
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of in-vitro bioassay methods: Application in herbal drug research. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1–3 | 5, 6 | 7–9 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 1.51 m | 36.0 | 1.49 m | 36.1 | 1.49 m | 35.9 |
| 2 | 2.22 m 1.97 m | 27.1 | 2.20 m 1.95 m | 27.0 | 2.20 dd (13.4, 4.0) 1.90 m | 27.1 |
| 3 | 3.34 dd (12.0, 4.1) | 89.2 | 3.36 dd (11.6, 3.8) | 88.9 | 3.33 dd (11.7, 4.5) | 88.8 |
| 4 | 39.5 | 39.4 | 39.3 | |||
| 5 | 1.09 m | 48.0 | 1.07 dd (9.5, 5.7) | 47.8 | 1.01 dd (11.7, 3.5) | 47.5 |
| 6 | 2.09 m 2.05 m | 23.2 | 2.07 m | 23.2 | 2.06 m 1.98 m | 23.2 |
| 7 | 5.71 m | 120.5 | 5.67 m | 120.5 | 5.65 brd (7.1) | 122.6 |
| 8 | 145.5 | 145.8 | 147.4 | |||
| 9 | 3.52 m | 47.2 | 3.49 brd (14.1) | 47.3 | 3.02 brd (14.3) | 46.4 |
| 10 | 35.5 | 35.5 | 35.5 | |||
| 11 | 1.85 m 1.59 m | 22.4 | 1.84 m 1.52 m | 22.5 | 2.00 m 1.47 m | 21.7 |
| 12 | 2.18 m 2.01 m | 30.9 | 2.17 m 2.03 m | 30.7 | 2.37 ddd (14.2, 10.0, 8.1) 1.88 m | 20.0 |
| 13 | 58.9 | 58.3 | 56.7 | |||
| 14 | 47.6 | 48.0 | 46.0 | |||
| 15 | 2.45 dd (12.4, 7.7) 1.84 m | 43.0 | 2.50 dd (12.7, 7.9) 1.78 dd (12.7, 7.9) | 43.6 | 2.17 d (13.7) 1.97 dd (13.7, 2.4) | 43.8 |
| 16 | 5.94 q (8.2) | 72.9 | 6.01 q (7.9) | 72.6 | 4.75 brd (2.4) | 80.4 |
| 17 | 2.83 d (8.8) | 56.1 | 3.11 d (7.9) | 57.4 | 2.95 s | 59.0 |
| 18 | 179.2 | 179.1 | 180.6 | |||
| 19 | 1.24 s | 23.9 | 1.24 s | 23.8 | 1.04 s | 23.8 |
| 20 | 83.1 | 83.8 | 139.9 | |||
| 21 | 1.57 s | 30.2 | 1.71 s | 29.0 | 1.76 s | 23.0 |
| 22 | 5.92 d (15.7) | 134.0 | 5.79 d (12.2) | 132.1 | 5.07 s 4.98 s | 113.9 |
| 23 | 6.56 dd (15.7, 11.0) | 122.4 | 6.13 t (12.2) | 120.3 | ||
| 24 | 5.86 brd (11.0) | 125.2 | 6.49 d (12.2) | 121.1 | ||
| 25 | 134.6 | 136.8 | ||||
| 26 | 1.61 s | 17.9 | 1.74 s | 17.5 | ||
| 27 | 1.69 s | 25.6 | 1.70 s | 26.0 | ||
| 30 | 1.14 s | 17.2 | 1.17 s | 17.3 | 1.09 s | 17.0 |
| 31 | 1.34 s | 28.6 | 1.33 s | 28.7 | 1.31 s | 28.5 |
| 32 | 1.18 s | 32.1 | 1.12 s | 32.3 | 1.36 s | 33.9 |
| CO | 170.3 | 169.6 | ||||
| CH3-CO | 1.98 s | 21.2 | 1.98 s | 21.2 | ||
| Position | 1, 7 | 2, 5, 8 | 3, 6, 9 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| δH | δC | HMBC | δH | δC | HMBC | δH | δC | HMBC | |
| Xyl-I | Xyl-I | Xyl (= Xyl-I) | |||||||
| 1 | 4.87 d (7.0) | 105.1 | C3-agl | 4.87 d (7.2) | 105.1 | C3-agl | 4.76 d (7.3) | 105.5 | C3-agl |
| 2 | 3.93 dd (8.7, 7.0) | 83.2 | C1, C3-Xyl-I, C1-Qui | 3.94 dd (8.8, 7.2) | 83.3 | C1, C3-Xyl-I, C1-Qui | 4.06 m | 83.0 | |
| 3 | 4.20 t (8.7) | 77.8 | C2-Xyl-I | 4.21 t (8.8) | 77.8 | C2, C4-Xyl-I | 4.11 m | 77.3 | C4-Xyl-I |
| 4 | 4.13 m | 70.4 | 4.13 m | 70.2 | 4.13 m | 70.9 | |||
| 5 | 4.34 dd (11.3, 5.2) 3.71 dd (11.3, 9.7) | 66.5 | C1, C4-Xyl-I | 4.33 dd (11.4, 5.3) 3.72 dd (11.4, 9.1) | 66.5 | C1, C3, C4-Xyl-I C1-Xyl-I | 4.28 m 3.61 m | 66.5 | C4-Xyl-I C1-Xyl-I |
| Qui | Qui | Qui | |||||||
| 1 | 5.20 d (7.8) | 103.0 | C2-Xyl-I | 5.21 d (7.2) | 103.0 | C2-Xyl-I | 5.06 d (7.6) | 104.9 | C2-Xyl-I |
| 2 | 4.13 m | 82.6 | C1, C3-Qui | 4.15 m | 82.7 | C1, C3-Qui | 3.96 dd (9.4, 7.6) | 76.1 | C3-Qui |
| 3 | 4.10 t (9.0) | 75.7 | C2-Qui | 4.13 m | 75.7 | C2-Qui | 4.01 dd (9.4, 8.6) | 74.7 | C2-Qui |
| 4 | 3.60 t (9.0) | 86.6 | C3, C5, C6-Qui, C1-Glc-I | 3.62 m | 86.6 | C3, C5-Qui | 3.54 t (8.6) | 88.3 | C6-Qui, C1-Glc |
| 5 | 3.67 m | 71.0 | 3.68 m | 71.0 | 3.76 m | 71.5 | |||
| 6 | 1.70 d (6.7) | 17.9 | C4, C5-Qui | 1.71 d (6.4) | 18.0 | C4, C5-Qui | 1.69 d (6.1) | 17.9 | C4, C5-Qui |
| Glc (=Glc-I) | Glc | 6-OSO3-Glc (=Glc) | |||||||
| 1 | 4.92 d (7.8) | 104.7 | C4-Qui | 4.96 d (8.0) | 105.3 | C4-Qui | 4.85 d (8.1) | 104.9 | C4-Qui |
| 2 | 4.00 dd (9.0, 7.8) | 73.5 | C3-Glc-I | 4.01 dd (9.0, 8.0) | 74.7 | C1, C3-Glc | 3.98 t (8.8) | 73.7 | |
| 3 | 4.22 t (9.0) | 88.0 | C2, C4-Glc-I, C1-Glc-II | 4.23 t (9.0) | 78.1 | C2, C4-Glc | 4.18 t (8.8) | 86.5 | C4-Glc, C1-Xyl-II |
| 4 | 4.05 t (9.0) | 69.6 | C3, C5, C6-Glc-I | 4.18 t (9.0) | 71.5 | C3, C6-Glc | 3.81 t (9.1) | 70.1 | C3, C5, C6-Glc |
| 5 | 3.98 m | 77.7 | 4.06 m | 78.2 | 4.29 m | 75.2 | C4-Glc | ||
| 6 | 4.45 dd (11.9, 2.2) 4.18 dd (11.9, 6.5) | 61.9 | 4.55 dd (11.4, 2.1) 4.29 dd (11.4, 6.2) | 62.3 | 5.25 dd (10.4, 2.7) 4.82 dd (10.4, 9.4) | 67.6 | C5-Glc | ||
| 3-OMe-Glc (=Glc-II) | Xyl-II | 3-OMe-Xyl (=Xyl-II) | |||||||
| 1 | 5.27 d (8.0) | 105.5 | C3-Glc-I | 5.39 d (6.9) | 105.9 | C2-Qui | 5.21 d (7.5) | 105.9 | C3-Glc |
| 2 | 3.99 dd (9.0, 8.0) | 74.9 | C1-Glc-II | 4.07 m | 75.6 | C1, C3-Xyl-II | 3.92 t (8.3) | 74.5 | C1, C2-Xyl-II |
| 3 | 3.71 t (9.0) | 87.8 | OMe, C2-Glc-II | 4.11 m | 77.0 | C2, C4-Xyl-II | 3.58 t (8.9) | 87.6 | C2, C4-Xyl-II, OMe |
| 4 | 4.14 t (9.0) | 70.2 | C5, C6-Glc-II | 4.13 m | 70.4 | 4.07 m | 69.9 | C5-Xyl-II | |
| 5 | 3.96 m | 78.2 | 4.34 dd (11.4, 4.5) 3.66 dd (11.4, 9.2) | 66.9 | C1, C3, C4-Xyl-II C3, C4-Xyl-II | 4.20 dd (11.3, 5.6) 3.61 dd (11.3, 10.7) | 66.9 | C1, C3, C4-Xyl-II C1, C3, C4-Xyl-II | |
| 6 | 4.46 dd (11.5, 2.4) 4.27 dd (11.5, 4.9) | 62.1 | C5-Glc-II | ||||||
| 3-OMe | 3.87 s | 60.5 | C3-Glc-II | 3.85 s | 60.4 | C3-Xyl-II | |||
| Xyl-II | |||||||||
| 1 | 5.39 d (7.0) | 105.8 | C2-Qui | ||||||
| 2 | 4.05 dd (8.2, 7.0) | 75.5 | C1, C3-Xyl-II | ||||||
| 3 | 4.10 t (8.2) | 77.0 | C2, C4-Xyl-II | ||||||
| 4 | 4.13 m | 70.5 | |||||||
| 5 | 4.33 dd (11.5, 5.1) 3.66 dd (11.5, 9.7) | 66.9 | C1, C3, C4-Xyl-II C1, C3, C4-Xyl-II | ||||||
| Compound | JB6 Cl41 | SK-MEL-2 | SK-MEL-28 | RPMI-7951 | |||
|---|---|---|---|---|---|---|---|
| IC50, µM | IC50, µM | SI | IC50, µM | SI | IC50, µM | SI | |
| 1 | 6.4 ± 0.05 | 0.7 ± 0.07 | 9.1 | 8.4 ± 0.07 | 0.8 | 26.7 ± 0.1 | 0.24 |
| 2 | 31.5 ± 4.1 | 37.6 ± 0.2 | 0.8 | 29.0 ± 0.02 | 1.0 | 36.4 ± 0.2 | 0.87 |
| 3 | 6.0 ± 0.1 | 0.68 ± 0.06 | 8.8 | 14.8 ± 0.03 | 0.4 | 27.9 ± 0.09 | 0.22 |
| 4 | 6.1 ± 0.4 | 0.67 ± 0.04 | 9.1 | 8.3 ± 0.1 | 1.4 | 29.2 ± 0.04 | 0.2 |
| 5 | 8.7 ± 0.2 | 37.3 ± 0.01 | 0.2 | 25.3 ± 0.09 | 0.3 | 38.0 ± 0.3 | 0.23 |
| 6 | 6.6 ± 0.3 | 0.69 ± 0.03 | 9.5 | 8.2 ± 0.2 | 0.8 | 32.4 ± 0.5 | 0.2 |
| 7 | 8.7 ± 0.07 | 5.6 ± 0.1 | 1.4 | 23.0 ± 0.07 | 0.3 | 31.4 ± 0.1 | 0.25 |
| 8 | 32.8 ± 2.8 | >50 | n.d. | 42.0 ± 0.4 | 0.8 | >50 | n.d. |
| 9 | 7.4 ± 0.8 | 0.75 ± 0.03 | 9.9 | 23.3 ± 0.6 | 0.32 | 32.4 ± 0.08 | 0.23 |
| Compound | Human erythrocytes (O(I)+) |
|---|---|
| ED50, µM | |
| Cucumarioside A2-2 | 0.95 ± 0.04 |
| 1 | 2.03 ± 0.19 |
| 2 | 4.08 ± 0.36 |
| 3 | 0.72 ± 0.05 |
| 4 | 2.48 ± 0.05 |
| 5 | 7.02 ± 0.46 |
| 6 | 1.68 ± 0.14 |
| 7 | 4.07 ± 0.10 |
| 8 | 6.16 ± 0.05 |
| 9 | 2.85 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malyarenko, T.V.; Malyarenko, O.S.; Kicha, A.A.; Kalinovsky, A.I.; Dmitrenok, P.S.; Ivanchina, N.V. In Vitro Anticancer and Cancer-Preventive Activity of New Triterpene Glycosides from the Far Eastern Starfish Solaster pacificus. Mar. Drugs 2022, 20, 216. https://doi.org/10.3390/md20030216
Malyarenko TV, Malyarenko OS, Kicha AA, Kalinovsky AI, Dmitrenok PS, Ivanchina NV. In Vitro Anticancer and Cancer-Preventive Activity of New Triterpene Glycosides from the Far Eastern Starfish Solaster pacificus. Marine Drugs. 2022; 20(3):216. https://doi.org/10.3390/md20030216
Chicago/Turabian StyleMalyarenko, Timofey V., Olesya S. Malyarenko, Alla A. Kicha, Anatoly I. Kalinovsky, Pavel S. Dmitrenok, and Natalia V. Ivanchina. 2022. "In Vitro Anticancer and Cancer-Preventive Activity of New Triterpene Glycosides from the Far Eastern Starfish Solaster pacificus" Marine Drugs 20, no. 3: 216. https://doi.org/10.3390/md20030216
APA StyleMalyarenko, T. V., Malyarenko, O. S., Kicha, A. A., Kalinovsky, A. I., Dmitrenok, P. S., & Ivanchina, N. V. (2022). In Vitro Anticancer and Cancer-Preventive Activity of New Triterpene Glycosides from the Far Eastern Starfish Solaster pacificus. Marine Drugs, 20(3), 216. https://doi.org/10.3390/md20030216