
Rare β-Resorcylic Acid Derivatives from a Halophyte-Associated Fungus Colletotrichum gloeosporioides JS0419 and Their Antifungal Activities

 , and
, and
Abstract
:1. Introduction
2. Results
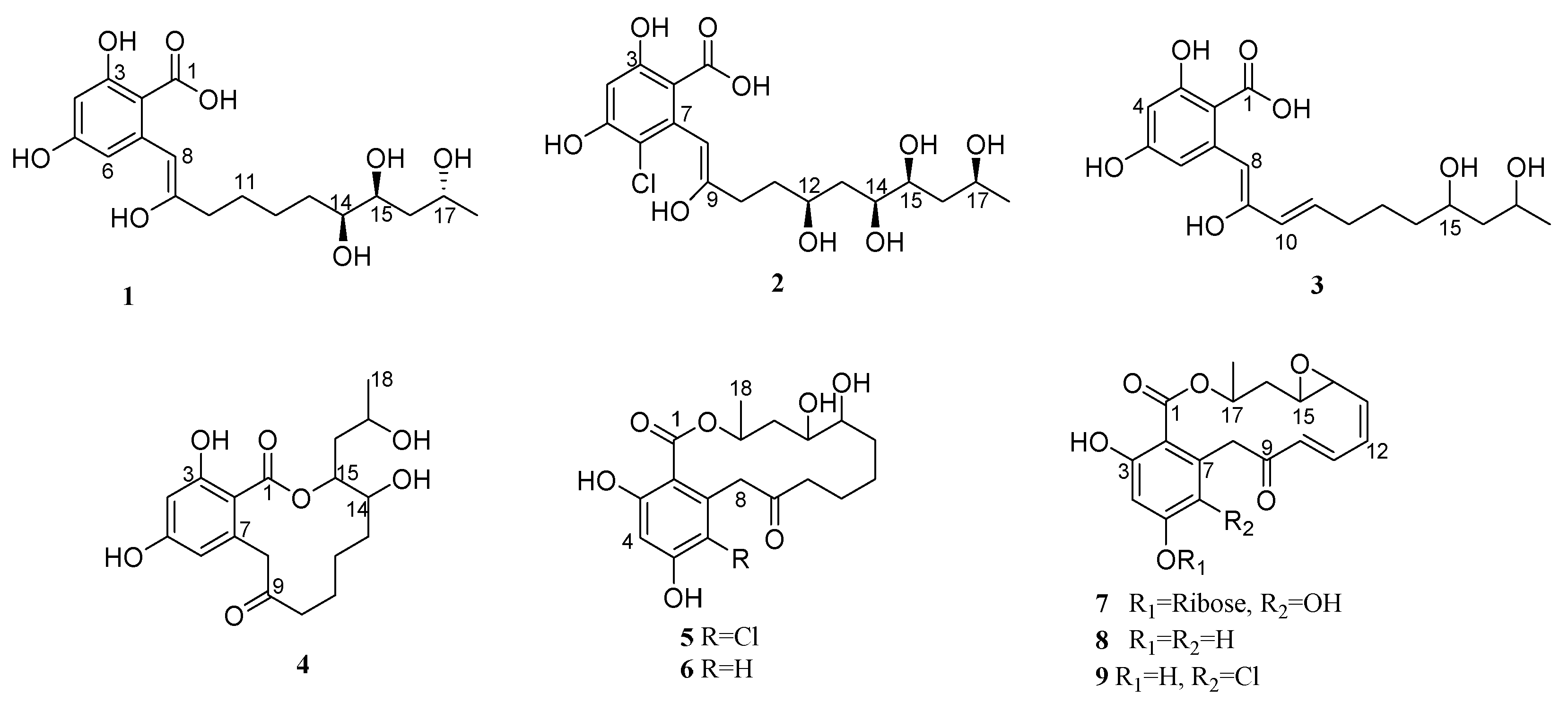
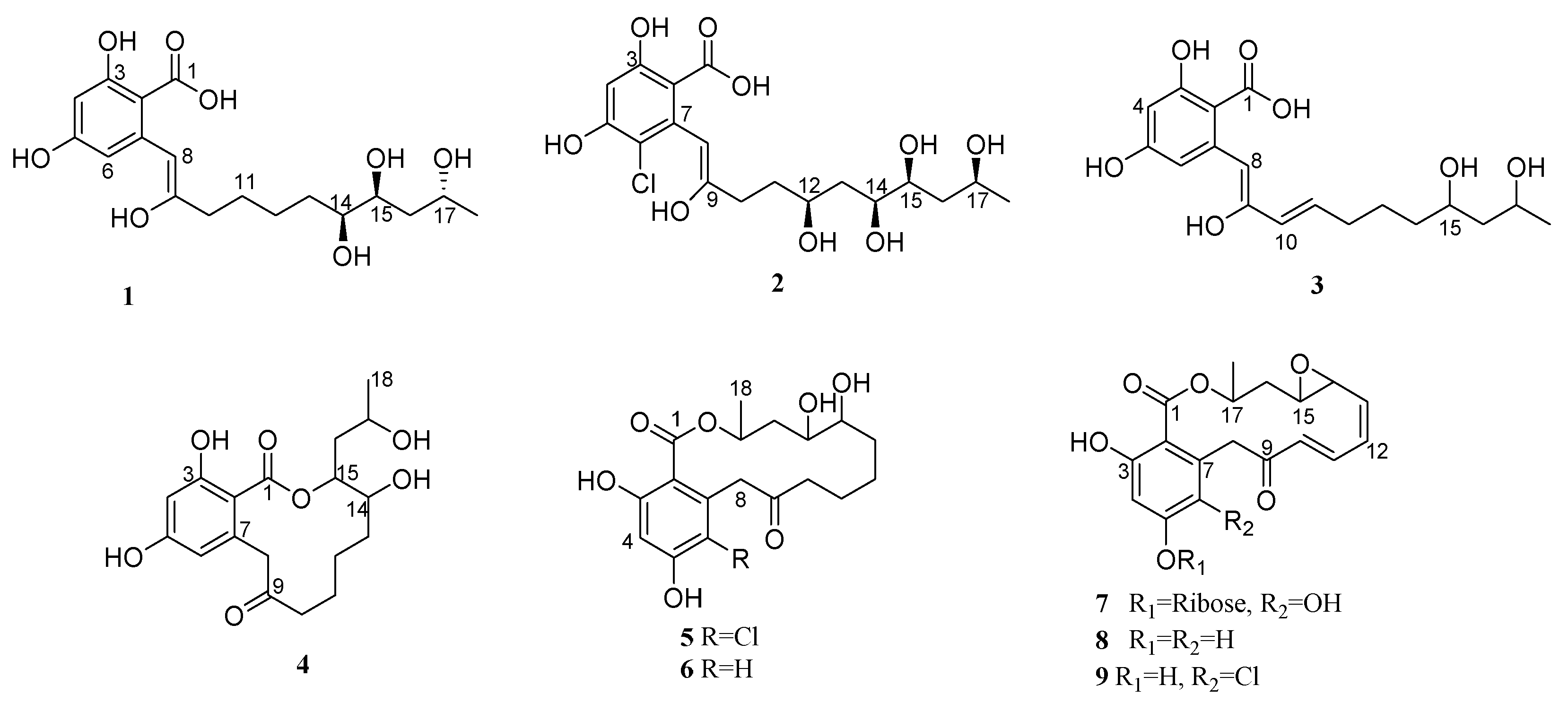
2.1. Structure Determination of the Compounds
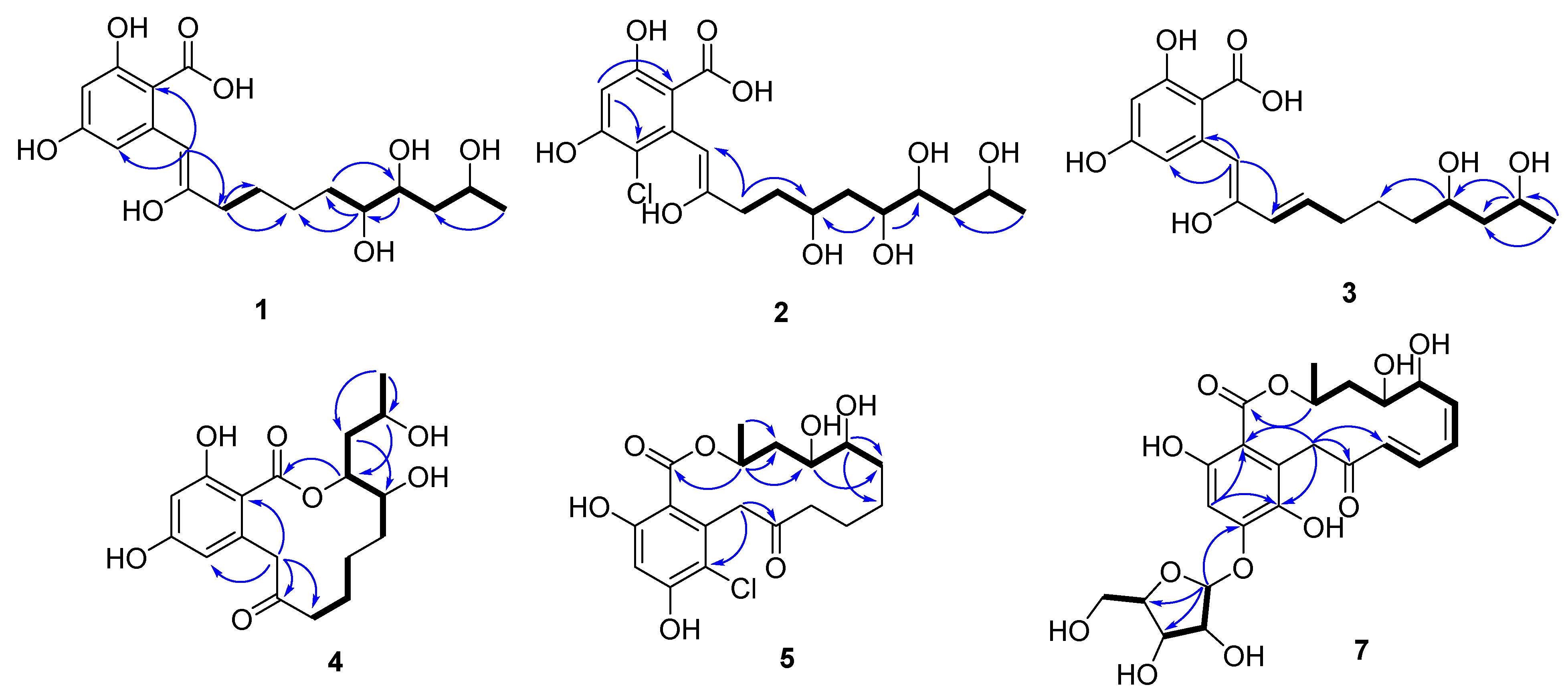
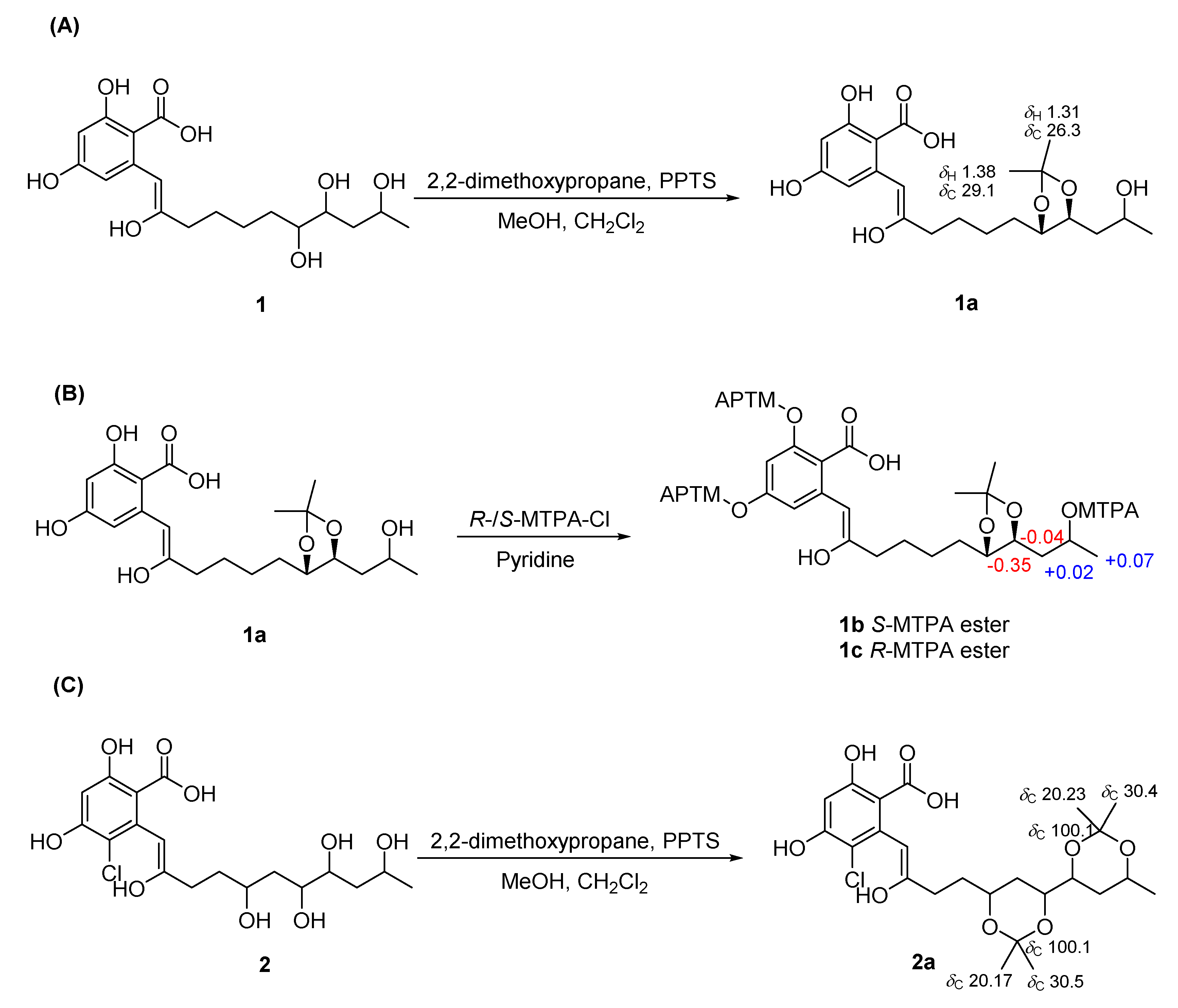
2.2. Establishment of Stereochemistry
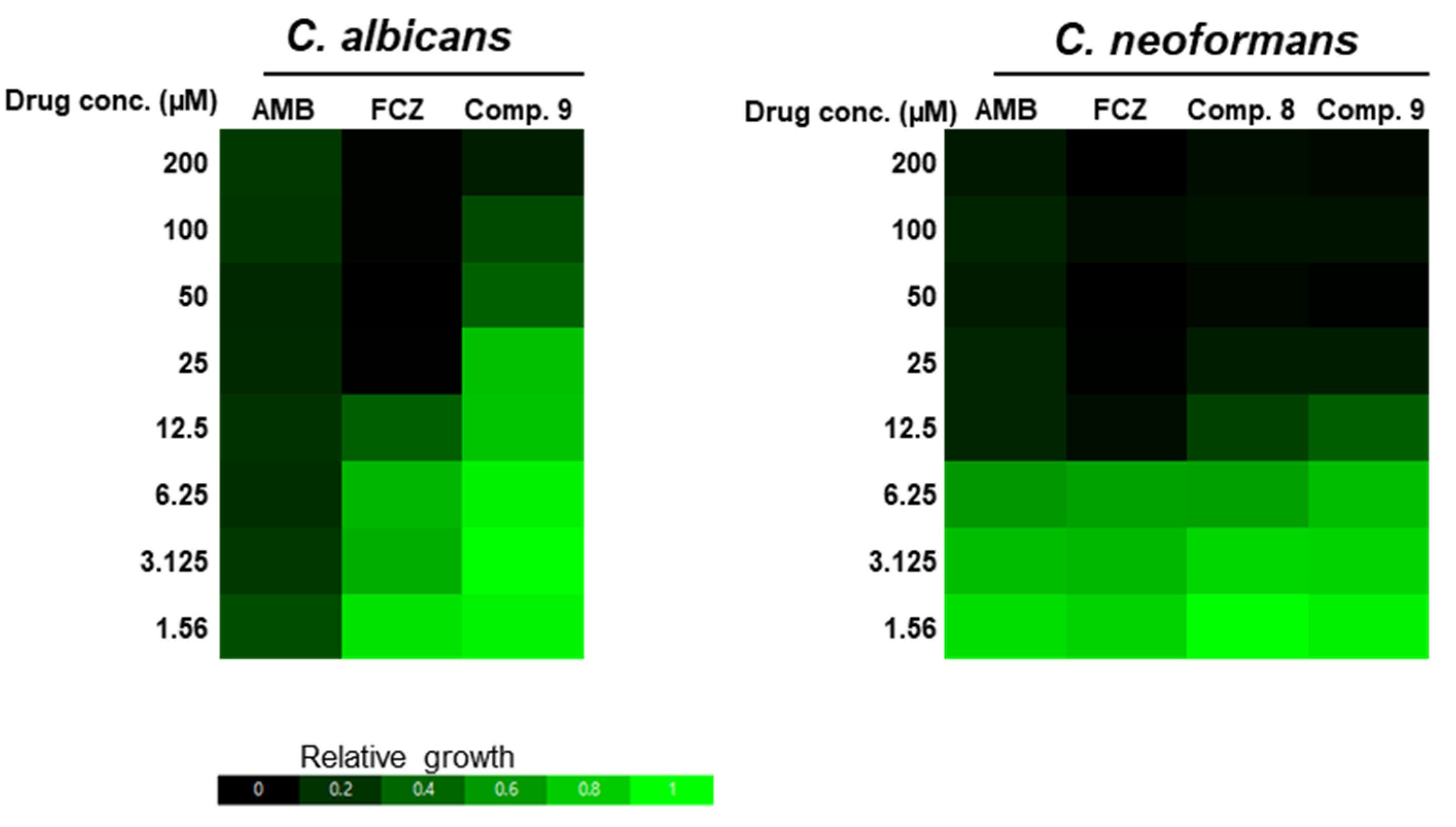
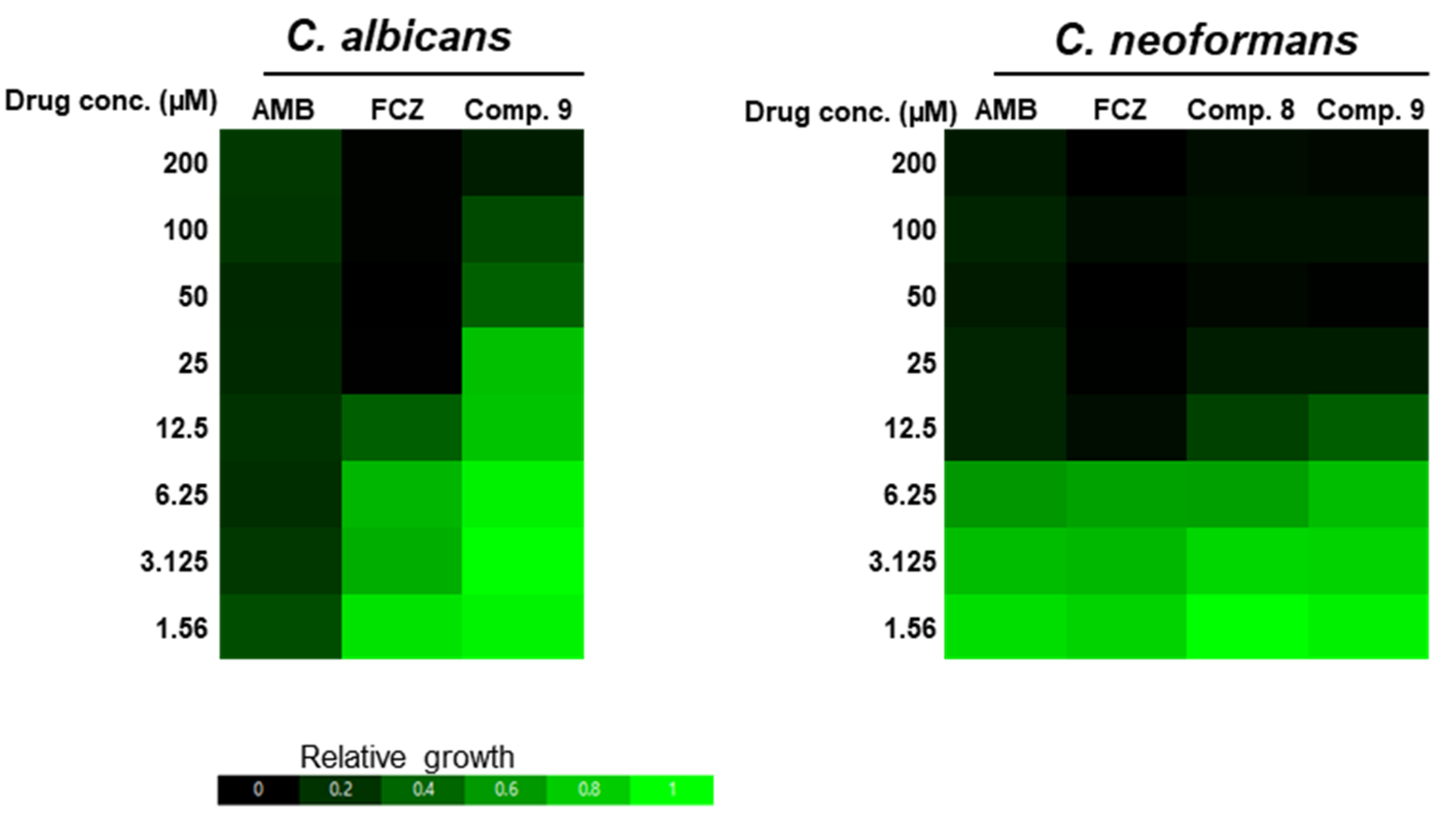
2.3. Antifungal Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Strain
3.3. Cultivation and Extraction of the Fungal Strain
3.4. Isolation of Compounds
3.5. Preparation of Acetonide Derivatives of 1 (1a) and 2 (2a)
3.6. Preparation of (R)-and (S)-α-Methoxy-α-trifluoromethylphenylacetic Acid (MTPA) Ester Derivatives of 1 (1b/1c)
3.7. Antifungal Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sandor, G.; Attila, K.; Tamas, E.; Gyula, B.; Gabor, V. Filamentous fungi from Plantago lanceolata L. leaves: Contribution to the pattern and stability of bioactive metabolites. Phytochemistry 2013, 86, 127–136. [Google Scholar]
- Zhao, N.; Wang, L.; Tian, C. Halophyte-endophyte coupling: A promising bioremediation system for oil-contaminated soil in Northwest China. Environ. Sci. Technol. 2013, 47, 11938–11939. [Google Scholar] [CrossRef] [PubMed]
- Firakova, S.; Sturdikova, M.; Muckova, M. Bioactive secondary metabolites produced by microorganisms associated with plants. Biologia 2007, 62, 251–257. [Google Scholar] [CrossRef]
- Winssinger, N.; Barluenga, S. Chemistry and biology of resorcyclic acid lactones. Chem. Commun. 2007, 1, 22–36. [Google Scholar] [CrossRef]
- Takehana, K.; Sato, S.; Kobayasi, T.; Maeda, T. A radicicol-related macrocyclic nonaketide compound, antibiotic LL-Z1640-2, inhibits the JNK/p38 pathways in signal-specific manner. Biochem. Biophys. Res. Commun. 1999, 257, 19–23. [Google Scholar] [CrossRef]
- Stob, M.; Baldwin, R.S.; Tuite, J.; Andrews, F.N.; Gillette, K.G. Isolation of an anabolic, uterotrophic compound from corn infected with Gibberella zeae. Nature 1962, 196, 1318. [Google Scholar] [CrossRef]
- Ellestad, G.A.; Lovell, G.A.; Perkinson, N.A.; Hargreaves, R.T.; McGahren, W.J. New zearalenone related macrolides and isocoumarins from an unidentified fungus. J. Org. Chem. 1978, 43, 2339–2343. [Google Scholar] [CrossRef]
- Nair, M.S.R.; Carey, S.T.; James, J.C. Metabolites of pyrenomycetes. XIV: Structure and partial stereochemistry of the antibiotic macrolides hypothemycin and dihydrohypothemycin. Tetrahedron Lett. 1980, 21, 2445–2449. [Google Scholar] [CrossRef]
- Aver, W.A.; Peña-Rodriguez, L. Minor metabolites of Monocillium nordinii. Phytochemistry 1987, 26, 1353–1355. [Google Scholar] [CrossRef]
- Sugawara, F.; Kim, K.-W.; Kobayashi, K.; Uzawa, J.; Yoshida, S.; Murfushi, N.; Takahashi, N.; Strobel, G.A. Zearalenone derivatives produced by the fungus Drechslera portulacae. Phytochemistry 1992, 31, 1987–1990. [Google Scholar] [CrossRef]
- Isaka, M.; Suyarnsestakorn, C.; Tanticharoen, M.; Kongsaeree, P.; Thebtaranonth, Y. Aigialomycins A−E, new resorcylic macrolides from the marine mangrove fungus Aigialus parvus. J. Org. Chem. 2002, 67, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, V.; Mayer-Bartschmid, A.; Muller, H.; Greif, G.; Kleymann, G.; Zitzmann, W.; Tichy, H.-V.; Stadler, M. Pochonin A-F, new antiviral and antiparasitic resorcyclic acid lactones from Pochonia chlamydosporia var. catenulata. J. Nat. Prod. 2003, 66, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; He, Z.; Xue, J.; Chen, X.; Wei, X. β-resorcylic acid lactones from a Paecilomyces Fungus. J. Nat. Prod. 2010, 73, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xue, J.; Zou, Y.; He, S.; Wei, X. Three new β-resorcylic acid lactones from Paecilomyces sp. SC0924. Chin. J. Chem. 2012, 30, 1273–1277. [Google Scholar] [CrossRef]
- Wicklow, D.T.; Jordan, A.M.; Gloer, J.B. Antifugal metabolites (monorden, monocillin I, II, III) from Colletotrichum graminicola, a systemic vascular pathogen of maize. Mycol. Res. 2009, 113, 1433–1442. [Google Scholar] [CrossRef]
- Wee, J.C.; Sundermann, K.; Licari, P.; Galazzo, J. Cytotoxic hypothemycin analogues from Hypomyces subiculosus. J. Nat. Prod. 2006, 69, 1456–1459. [Google Scholar] [CrossRef]
- Hoshino, Y.; Ivanova, V.B.; Yazawa, K.; Ando, A.; Mikami, Y. Queenslandon, a new antifungal compound produced by Chrysosporium queenslandicum: Production, isolation and structure elucidation. J. Antibiot. 2002, 55, 516–519. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Agatsuma, T.; Nakano, H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 1998, 16, 2639–2645. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.D.; Morimoto, R.I. Molecular chaperones and the stress of oncogenesis. Oncogene 2004, 23, 2907–2918. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Wedge, D.E.; Nagle, D.G. A new 2D-TLC bioautography method for the discovery of novel antifungal agents to control plant pathogens. J. Nat. Prod. 2000, 63, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Chapla, V.M.; Zeraik, M.L.; Leptokarydis, I.H.; Silva, G.H.; Bolzani, V.S.; Young, M.C.; Pfenning, L.H.; Araújo, A.R. Antifungal compounds produced by Colletotrichum gloeosporioides, an endophytic fungus from Michelia champaca. Molecules 2014, 19, 19243–19252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Agarie, S. Physiological roles of betacyanin in a halophyte, Suaeda japonica Makino. Plant Prod. Sci. 2010, 13, 351–359. [Google Scholar] [CrossRef]
- Kim, J.W.; Shim, S.H. The fungus Colletotrichum as a source for bioactive secondary metabolites. Arch. Pharm. Res. 2019, 42, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Lee, C.; Kim, S.; Song, J.H.; Kang, K.S.; Deyrup, S.T.; Nam, S.-J.; Xia, X.; Shim, S.H. Neuroprotective glycosylated cyclic lipodepsipeptides, Colletotrichamides A−E, from a halophyte-associated fungus, Colletotrichum gloeosporioides JS419. J. Org. Chem. 2019, 84, 10999–11006. [Google Scholar] [CrossRef] [PubMed]
- Shinonaga, H.; Kawamura, Y.; Ikeda, A.; Aoki, M.; Sakai, N.; Fujimoto, N.; Kawashima, A. Pochonins K-P: New radicicol analogues from Pochonia chlamydosporia var. chlamydosporia and their WNT-5A expression inhibitory activities. Tetrahedron 2009, 65, 3446–3453. [Google Scholar] [CrossRef]
- Garbaccio, R.M.; Stachel, S.J.; Baeschlin, D.K.; Danishefsky, S.J. Concise asymmetric syntheses of radicicol and monocillin I. J. Am. Chem. Soc. 2001, 123, 10903–10908. [Google Scholar] [CrossRef]
- Pospisil, S.; Sedmera, P.; Halada, P.; Petricek, M. Extracellular carbohydrate metabolites from Streptomyces coelicolor A3(2). J. Nat. Prod. 2007, 70, 768–771. [Google Scholar] [CrossRef]
- Nagle, D.G.; Gerwick, W.H. Structure and stereochemistry of constanolactones A-G, lactonized cyclopropyl oxylipins from the red marine alga Constantinea simplex. J. Org. Chem. 1994, 59, 7227–7237. [Google Scholar] [CrossRef]
- Beignet, J.; Cox, L.R. Using a temporary silicon connection in stereoselective allylation with allylsilanes: Application to the synthesis of stereodefined 1,2,4-triols. Org. Lett. 2003, 5, 4231–4234. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon−proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Fidanze, S.; Song, F.; Szlosek-Pinaud, M.; Small, P.L.C.; Kishi, Y. Complete structure of the mycolactones. J. Am. Chem. Soc. 2001, 123, 10117–10118. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Rogers, B.N.; Richardson, T.I. Configurational assignment of polyene macrolide antibiotics using the [13C]acetonide analysis. Acc. Chem. Res. 1998, 31, 9–17. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Castanheira, M.; Diekema, D.J.; Messer, S.A.; Moet, G.J.; Jones, R.N. Comparison of european committee on antimicrobial susceptibility testing (EUCAST) and etest methods with the CLSI broth microdilution method for echinocandin susceptibility testing of Candida species. J. Clin. Microbiol. 2010, 48, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhang, S.; Zhou, T.; Zhan, J. Three new resorcylic acid derivatives from Sporotrichum laxum. Bioorg. Med. Chem. Lett. 2013, 23, 5806–5809. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Meng, L.; Mándi, A.; Li, X.; Kurtán, T.; Wang, B. Structure, absolute configuration, and conformational study of resorcylic acid derivatives and related congeners from the fungus Penicillium brocae. RSC Adv. 2015, 5, 39870–39877. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
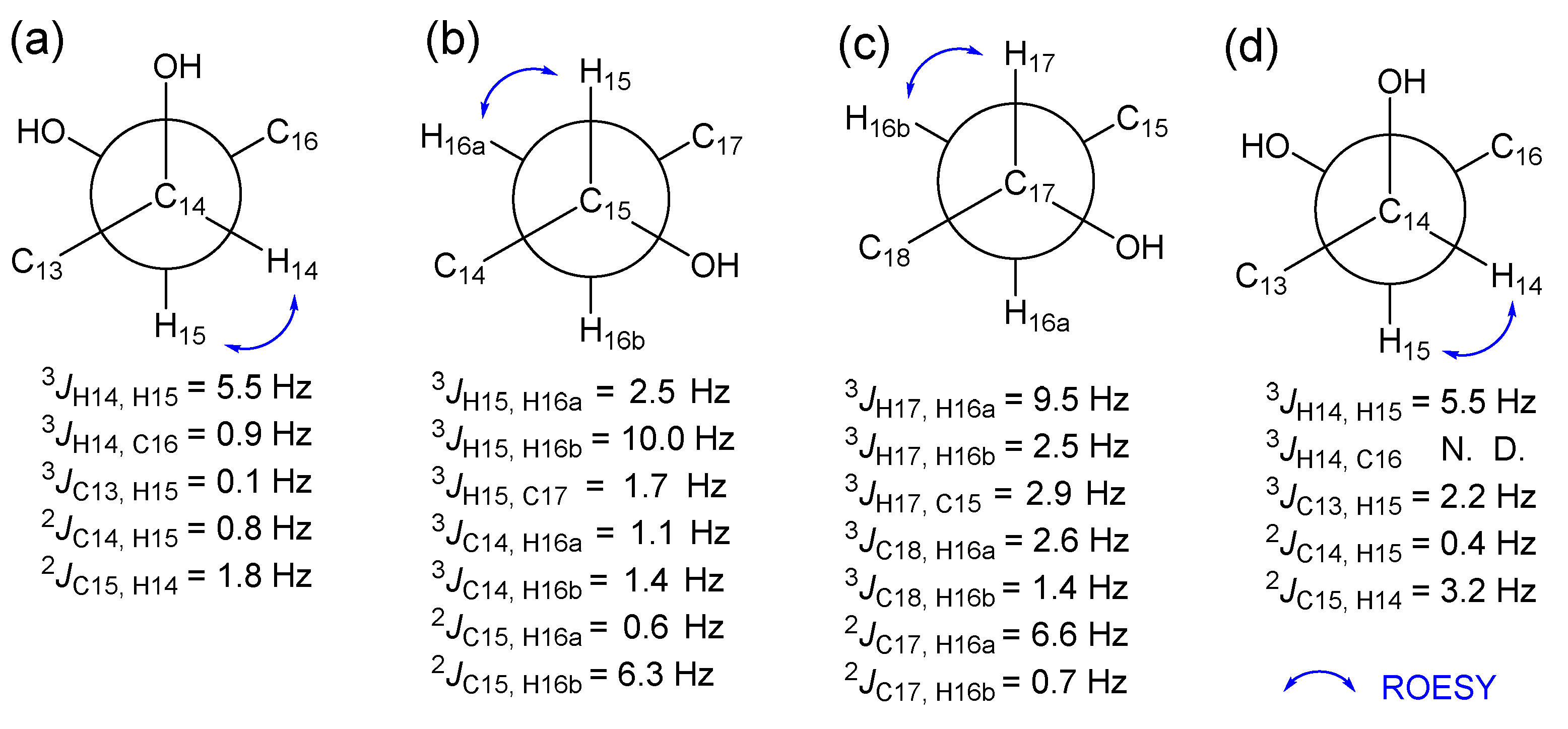{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC, Type b | δH (J in Hz) a | δC, Type b | δH (J in Hz) a | δC, Type d | δH (J in Hz) c | |
| 1 | 168.1, C | 167.6, C | 167.6, C | |||
| 2 | 99.6, C | 99.0, C | 100.0, C | |||
| 3 | 164.8, C | 165.6, C | 167.3, C | |||
| 4 | 102.8, CH | 6.29, d (2.0) | 103.8, CH | 6.42, s | 103.0, CH | 6.31, d (2.0) |
| 5 | 167.7, C | 163.3, C | 164.9, C | |||
| 6 | 103.9, CH | 6.28, d (2.0) | 108.3, C | 104.6, CH | 6.34, d (2.0) | |
| 7 | 141.5, C | 137.5, C | 141.5, C | |||
| 8 | 105.4, CH | 6.31, s | 101.9, CH | 6.72, s | 106.0, CH | 6.37, s |
| 9 | 159.0, C | 159.9, C | 153.5, C | |||
| 10 | 34.1, CH2 | 2.52, t (7.5) | 30.8, CH2 | 2.75, ddd (15.0, 9.5, 5.0) 2.67, dd (9.5, 7.0) | 123.4, CH | 6.16, d (15.5) |
| 11 | 28.1, CH2 | 1.71, m | 35.5, CH2 | 1.96, m 1.64, m | 137.4, CH | 6.54, dt (15.5, 2.5) |
| 12 | 26.5, CH2 | 1.63, m 1.44, m | 70.4, CH | 3.91, dq (7.0, 3.5) | 33.8, CH2 | 2.28, q (7.0) |
| 13 | 33.4, CH2 | 1.65, m 1.42, m | 40.3, CH2 | 1.81, dt (8.5, 3.0) 1.64, m | 26.1, CH2 | 1.65, m 1.54, m |
| 14 | 76.1, CH | 3.42, ddd (8.0, 5.5, 2.5) | 74.8, CH | 3.61, m | 38.4, CH2 | 1.51, m |
| 15 | 72.9, CH | 3.66, ddd (9.8, 5.5, 2.5) | 75.0, CH | 3.61, m | 71.3, CH | 3.75, m |
| 16 | 42.4, CH2 | 1.61, m 1.48, ddd (14, 9.8, 2.8) | 42.0, CH2 | 1.69, ddd (14.5, 5.5, 3.0) 1.60, td (7.0, 2.5) | 46.9, CH2 | 1.60, m 1.56, m |
| 17 | 65.6, CH | 4.00, dqd (9.5, 6.0, 2.8) | 65.6, CH | 4.00, dq (12.5, 6.0) | 67.7, CH | 3.95, m |
| 18 | 24.6, CH3 | 1.20, d (6.0) | 23.6, CH3 | 1.19, d (6.0) | 23.8, CH3 | 1.18, d (6.0) |
| No. | 4 | 5 | 7 | |||
|---|---|---|---|---|---|---|
| δC, Type b | δH (J in Hz) a | δC, Type b | δH (J in Hz) a | δC, Type b | δH (J in Hz) a | |
| 1 | 171.9, C | 172.5, C | 174.8, C | |||
| 2 | 107.5, C | 107.1, C | 117.1, C | |||
| 3 | 165.5, C | 163.5, C | 168.5, C | |||
| 4 | 103.0, CH | 6.26 (d, 2.5) | 104.2, C | 105.3, CH | 6.87 (s) | |
| 5 | 163.9, C | 160.9, C | 156.7, C | |||
| 6 | 113.9, CH | 6.14 (d, 2.5) | 117.2, CH | 6.41 (s) | 157.3, C | |
| 7 | 140.3, C | 137.6, C | 134.3, C | |||
| 8 | 51.4, CH2 | 4.48 (d, 18.5) 3.82 (d, 18.5) | 48.3, CH2 | 4.84 (d, 18.5) 4.25 (d, 18.5) | 46.4, CH2 | 4.01 (d, 16.0) 3.93 (d, 16.0) |
| 9 | 212.5, C | 208.7, C | 199.2, C | |||
| 10 | 42.5, CH2 | 2.70 (ddd, 16.0, 9.5, 2.0) 2.38 (ddd, 16.0, 9.5, 2.3) | 39.2, CH2 | 2.98 (ddd, 18.5, 12.0, 3.5) 2.55 (dt, 19.0, 3.5) | 131.8, CH | 6.08 (d, 16.0) |
| 11 | 23.1, CH2 | 1.98 (m) 1.82 (m) | 22.6, CH2 | 1.93 (m) 1.33 (m) | 140.8, CH | 7.59 (dd, 16.0, 10.0) |
| 12 | 25.5, CH2 | 1.58 (m) | 23.1, CH2 | 1.53 (m) 1.21 (dd, 11.5, 6.0) | 131.1, CH | 6.22 (t, 10.0) |
| 13 | 32.4, CH2 | 1.54 (m) 1.45 (m) | 31.4, CH2 | 1.53 (m) 1.37 (m) | 137.4, CH | 5.77 (dd, 10.0, 4.5) |
| 14 | 73.1, CH | 3.73 (ddd, 9.0, 7.0, 2.5) | 76.4, CH | 3.59 (dt, 11.0, 2.5) | 56.6, CH | 3.36 (m) |
| 15 | 77.6, CH | 4.97 (ddd, 9.0, 7.5, 4.0) | 69.3, CH | 3.49 (br d, 10.5) | 56.9, CH | 3.08 (dt, 8.5, 3.0) |
| 16 | 42.4, CH2 | 2.01 (m) 1.77 (ddd, 14.5, 7.0, 3.0) | 36.6, CH2 | 1.98 (dd, 15.5, 11.0) 1.72 (dd, 15.5, 11.0) | 38.1, CH2 | 2.44 (dt, 14.5, 3.0) 1.66 (ddd, 14.0, 9.0, 4.5) |
| 17 | 65.4, CH | 3.90 (dtd, 9.5, 6.5, 3.0) | 71.7, CH | 5.43 (m) | 72.4, CH | 5.38 (m) |
| 18 | 24.3, CH3 | 1.18 (d, 6.5) | 21.7, CH3 | 1.41 (d, 6.0) | 18.6, CH3 | 1.53 (d, 6.5) |
| 1′ | 103.4, CH | 5.65 (d, 4.5) | ||||
| 2′ | 73.7, CH | 4.22 (dd, 6.5, 4.5) | ||||
| 3′ | 71.1, CH | 4.09 (dd, 6.5, 2.5) | ||||
| 4′ | 88.3, CH | 4.18 (dd, 6.5, 3.0) | ||||
| 5′ | 63.2, CH2 | 3.69 (dd, 12.5, 3.5) 3.65 (dd, 12.0, 3.0) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bang, S.; Kim, J.; Oh, J.; Kim, J.-S.; Yu, S.-R.; Deyrup, S.; Bahn, Y.-S.; Shim, S.H. Rare β-Resorcylic Acid Derivatives from a Halophyte-Associated Fungus Colletotrichum gloeosporioides JS0419 and Their Antifungal Activities. Mar. Drugs 2022, 20, 195. https://doi.org/10.3390/md20030195
Bang S, Kim J, Oh J, Kim J-S, Yu S-R, Deyrup S, Bahn Y-S, Shim SH. Rare β-Resorcylic Acid Derivatives from a Halophyte-Associated Fungus Colletotrichum gloeosporioides JS0419 and Their Antifungal Activities. Marine Drugs. 2022; 20(3):195. https://doi.org/10.3390/md20030195
Chicago/Turabian StyleBang, Sunghee, Jaekyeong Kim, Jiwon Oh, Ji-Seok Kim, Seong-Ryong Yu, Stephen Deyrup, Yong-Sun Bahn, and Sang Hee Shim. 2022. "Rare β-Resorcylic Acid Derivatives from a Halophyte-Associated Fungus Colletotrichum gloeosporioides JS0419 and Their Antifungal Activities" Marine Drugs 20, no. 3: 195. https://doi.org/10.3390/md20030195
APA StyleBang, S., Kim, J., Oh, J., Kim, J.-S., Yu, S.-R., Deyrup, S., Bahn, Y.-S., & Shim, S. H. (2022). Rare β-Resorcylic Acid Derivatives from a Halophyte-Associated Fungus Colletotrichum gloeosporioides JS0419 and Their Antifungal Activities. Marine Drugs, 20(3), 195. https://doi.org/10.3390/md20030195