Abstract
A rapid and sensitive method using ultra-high performance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC-QTOF-MS/MS) was applied for the analysis of the metabolic profile of acarviostatin-containing aminooligosaccharides derived from Streptomyces sp. HO1518. A total of ninety-eight aminooligosaccharides, including eighty potential new compounds, were detected mainly based on the characteristic fragment ions originating from quinovosidic bond cleavages in their molecules. Following an LC-MS-guided separation technique, seven new aminooligosaccharides (10–16) along with four known related compounds (17–20) were obtained directly from the crude extract of strain HO1518. Compounds 10–13 represent the first examples of aminooligosaccharides with a rare acarviostatin II02-type structure. In addition, all isolates displayed considerable inhibitory effects on three digestive enzymes, which revealed that the number of the pseudo-trisaccharide core(s), the feasible length of the oligosaccharides, and acyl side chain exerted a crucial influence on their bioactivities. These results demonstrated that the UPLC-QTOF-MS/MS-based metabolomics approach could be applied for the rapid identification of aminooligosaccharides and other similar structures in complex samples. Furthermore, this study highlights the potential of acylated aminooligosaccharides with conspicuous α-glucosidase and lipase inhibition for the future development of multi-target anti-diabetic drugs.
1. Introduction
Type 2 diabetes mellitus (T2DM) is the most prevalent metabolic syndrome characterized by prolonged high levels of blood glucose, reflected by 537 million patients and 6.7 million deaths in 2021. The number of cases of diabetes is estimated to increase further to 783 million by 2045, which placed immense economic and social pressures on patients [1,2,3,4,5]. Currently, α-glucosidases (mainly α-amylases and disaccharidases), secreted from the intestinal chorionic epithelium capable of converting dietary carbohydrates into glucose, are still recognized as an important pharmacological target for anti-diabetic drug development. Acarbose, a typical anti-diabetes drug functioning as an α-glucosidases inhibitor, potently inhibits the α-glucosidases in vivo to retard carbohydrate digestion and avoid blood glucose elevation [6,7]. However, the specific kinase Mak1 derived from the human microbiome selectively phosphorylates acarbose at the C6-OH of C7N cyclohexitol ring in acarbose, leading to its inactivation [8]. Therefore, an urgent demand for the discovery of new α-glucosidase inhibitors with high efficacy has declared a public-health imperative for the treatment of T2DM.
Natural products, characteristic of enormous structural diversity and complexity, have been recognized as an attractive source of leading compounds and therapeutic agents attributable to their remarkable pharmacological activities [9]. Traditionally, the bioactivity-based approach remains the most commonly employed screening method to isolate natural products; however, an increasing number of compounds already described are repeatedly isolated during bioassay-guided purification [10,11]. To avoid the rediscovery of known molecules and screen new chemical entities, several dereplication strategies, including ultraviolet-visible spectroscopy (UV-Vis), nuclear magnetic resonance spectroscopy (NMR), or mass spectrometry (MS) have been developed [12,13]. Among them, MS-based dereplication has the advantage of high sensitivity and versatility, enabling users to obtain multiple types of data in a single experiment, which generally serves as the first choice of a structure-based pipeline for the discovery of unknown secondary metabolites [14]. Recently, time-of-flight mass spectrometry (TOF-MS), especially the quadrupole time-of-flight mass spectrometry (QTOF-MS), has become one of the most powerful tools for untargeted analysis of complex mixtures derived from plants and microorganisms and is attributable to their capability of providing accurate mass data and structural information [15]. Because the ultra-high performance liquid chromatography (UPLC) can shorten the analysis time of multi-component extract and increase sensitivity and reproducibility in comparison to conventional HPLC, UPLC coupled to QTOF-MS has become the crucial platform for analyzing the metabolite profiling of certain plants or microbe [16,17].
Marine Streptomyces, capable of producing structurally novel and biologically active secondary metabolites, has been recognized as a highly prolific resource of pharmaceutically and industrially meaningful small molecules [18,19]. In our continuing efforts to search for new anti-diabetic lead compounds from the Streptomyces species, we have recently discovered a series of rare acylated aminooligosaccharides with intriguing inhibitory activities against α-glucosidases and pancreatic lipase (PL) from the Streptomyces sp. HO1518 [20,21]. Structurally, these aminooligosaccharides possess a single or repeated pseudo-trisaccharide unit(s), which are combined with d-glucopyranose groups attached to the reducing and non-reducing terminus through α-(1→4) glycosidic bond. Pseudo-trisaccharide is composed of an acarviosine moiety and a d-glucopyranose group through α-(1→4) quinovosidic bond, and acarviosine is comprised of an unsaturated C7N cyclohexitol residue and a 4-amino-4,6-dideoxy-d-glucopyranose unit via α-(1→4) pseudo-glycosidic bond. Based on their structure feature, this class of naturally occurring oligosaccharide is referred to as acarviostatins followed by a Roman numeral and two digits, such as acarviostatin I01 (acarbose). The Roman numeral represents the number of the pseudo-trisaccharide cores, the middle digit denotes the number of glucose residues at the non-reducing end, and the last digit corresponds to the number of glucose units at the reducing end [22,23,24].
Since this family of oligosaccharides displayed conspicuous inhibitory activities against α-glucosidases and PL [20,21], this inspires our great interest to decipher the whole metabolic profiling of aminooligosaccharides in strain HO1518, which may contribute to exploring their structure-activity relationships and screening the optimal antidiabetic candidate molecules. To this end, a rapid and sensitive UPLC-QTOF-MS/MS method was developed to determine aminooligosaccharides secreted by strain HO1518 based on the MS and MS2 fragmentation patterns of nine reference standards (1–9) (Figure 1). This analytical approach resulted in the identification of ninety-eight oligosaccharides, including eighty new ones. Then, guided by the UPLC-QTOF-MS/MS, further study of the fermentation broth of strain HO1518 led to the isolation of seven new aminooligosaccharides (10–16) and four known congeners (17–20), among which 10–13 represent a new type of pseudo-octasaccharide. Compound 9 was the most potent α-amylase inhibitor with the IC50 value of 0.03 μM and was 282-fold more effective than that of acarbose (8.51 μM), while 19 exhibited the strongest activity against lipase with the IC50 value of 1.00 μM, and was almost equal to that of the anti-obesity orlistat (0.34 μM).
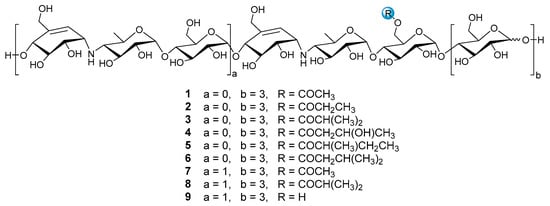
Figure 1.
The structures of compounds 1–9.
2. Results and Discussion
Nine reference compounds D6-O-acetyl-acarviostatin I03 (Ac-Aca I03, 1), D6-O-propionyl-acarviostatin I03 (Pr-Aca I03, 2), D6-O-isobutyryl-acarviostatin I03 (isoBu-Aca I03, 3), D6-O-β-hydroxybutyryl-acarviostatin I03 (Hbu-Aca I03, 4), D6-O-2-methyl-butyryl-acarviostatin I03 (Mbu-Aca I03, 5), D6-O-isovaleryl-acarviostatin I03 (isoVa-Aca I03, 6), D6-O-acetyl-acarviostatin II03 (Ac-Aca II03, 7), D6-O-isobutyryl-acarviostatin II03 (isoBu-Aca II03, 8) and acarviostatin II03 (Aca II03, 9), previously isolated from Streptomyces sp. HO1518 by our group, can be grouped into two types, namely acarviostatin I03-type (1–6) and acarviostatin II03-type (7–9), based on the number of pseudo-trisaccharide units. Since the amine residues of aminooligosaccharides are readily protonated [25], the positive-ion mode HRMS/MS analysis of nine references was performed. Two series of nomenclatures bi and yj, with respect to the fragmentation of glycoconjugates in the FAB-MS/MS spectra, have been adopted in this study [26]. The bi represents fragments containing the sugar moiety counted from the non-reducing end, while the yj refers to ions possessing the aglycone at the reducing end. These fragments can provide multidimensional MS information, including retention times, molecular formulas, base peaks, sugar constituents, as well as the relative abundance of ions.
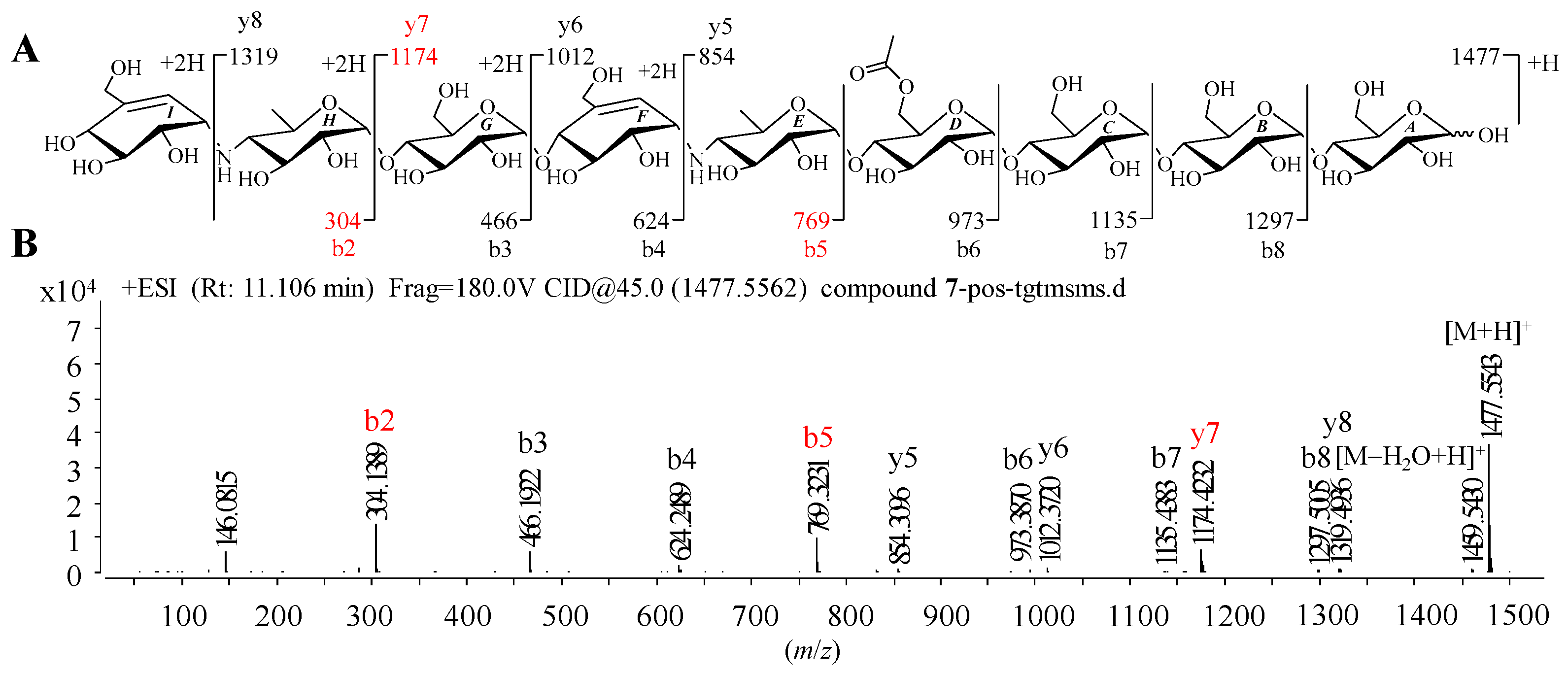
Given that 1–9 showed similar HRMS/MS fragmentation patterns (Figure S1), the representative D6-O-acetyl-acarviostatin II03 (Ac-Aca II03, 7) harboring two pseudo-trisaccharide cores is taken as an example of how to take advantage of MS/MS data to identify the structure of acarviostatins. In the positive HRESIMS/MS spectrum of 7, a strong protonated molecular ion at m/z 1477 was observed (Figure 2B). The high-intensity peaks in 7 were m/z 304 (b2), 769 (b5) and 1174 (y7), which resulted from the cleavages of two quinovosidic bonds. The peak intensity of some fragments with secondary amine residues, such as 146, was relatively high, which is conducive for the structure identification of aminooligosaccharides. In addition, the crucial ions at m/z 973 (b6), 1135 (b7), 1298 (b8), 854 (y5), 1012 (y6), 1174 (y7) and 1398 (y8) in 7, were 42 mass units more than those of its deacyl product 9, revealing that the location of the acetyl group of 7 was assigned at C-D6. Therefore, the structure of Ac-Aca II03 was established.
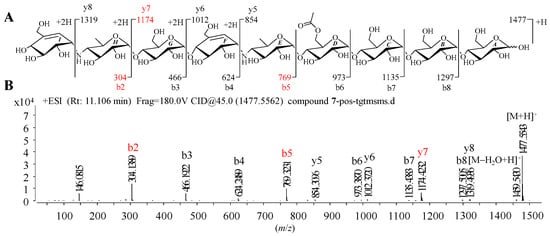
Figure 2.
Positive HRESIMS/MS fragmentation and spectrum of 7. (A) Positive-ion HRESIMS/MS fragmentation pattern of 7; (B) HRESIMS/MS spectra of 7.
It is worth noting that these most abundant fragments in 7 were produced by the rupture of the quinovosidic bonds between the quinovopyranose and glucose units. Similarly, the relatively high fragment ions in the other eight standards also originated from the dissociation of the quinovosidic bond, suggesting that the cleavage of this bond was easy to achieve when compared with those of pseudo-glycosidic, glycosidic and acyl bonds (Figure S1). The resultant fragments were thus regarded as characteristic fragment ions, as outlined in Table 1. In brief, the standards 1–6 sharing one pseudo-trisaccharide have the same base peak at m/z 304, whereas the other references 7–9 possessing two pseudo-trisaccharides have the mutual fragment ions at m/z 304 and 769.

Table 1.
Information of reference aminooligosaccharides 1–9.
On the basis of the features of mass spectrometry data of reference, aminooligosaccharides, potential fragmentation rules of oligosaccharides were summarized. First, the glycosidic, pseudo-glycosidic and quinovosidic bonds in acarviostatins could be dissociated to some extent, and the quinovosidic bond was more fragile than two other ordinary bonds. Therefore, the most abundant signals in the positive HRMS/MS spectra were produced by the quinovosidic bond cleavages, which played pivotal roles in the structural determination of undescribed acarviostatins. Second, the fragments harboring a single or repeated amine-containing moiety (moieties) tended to display higher intensity, largely attributable to the considerably strong basicity of secondary amine residues that readily formed protonated molecules, which offered important information for the structure identification of new oligosaccharides. Third, some diagnostic product ions bi and yj in the acylated aminooligosaccharides could be applied for the assignment of the location of the acyloxy side chain.
After the establishment of the fragmentation rules of aminooligosaccharides, the UPLC-QTOF-MS/MS data of the crude extract of Streptomyces sp. HO1518 was analyzed. The result showed that, except for those of nine reference acarviostatins, a considerable number of newly appeared protonated molecular ions at m/z 812, 1115, 1132, 1273, 1597, 1759, etc., were detected. Further analysis of the quasi-molecular signals implied that the predicted aminooligosaccharides in strain HO1518 possess 0–3 repeating pseudo-trisaccharide moieties accompanied with a 0–1 glucose unit attached to the non-reducing end and 0–5 glucose residues on the reducing termini. In most cases, the hydroxy group at C6 of the glucose unit in pseudo-trisaccharide moiety at the proximal of the reducing terminus was acylated by an acyl group with 2–6 carbon chain. The assembly of the repeating pseudo-trisaccharide units with different numbers of glucose residues at the reducing and/or non-reducing end, together with the diversity of acyl side chain led to the identification of ninety-eight aminooligosaccharides in strain HO1518 (Figures S6–S8), among which eighty are new compounds, including seventy-three acylated aminooligosaccharides and seven precursors. The structure of each oligosaccharide in the extract was determined on the basis of the molecular ion peak, characteristic fragments mainly corresponded to quinovosidic bond cleavages, as well as the qualitative retention time (Tables S10–S12).
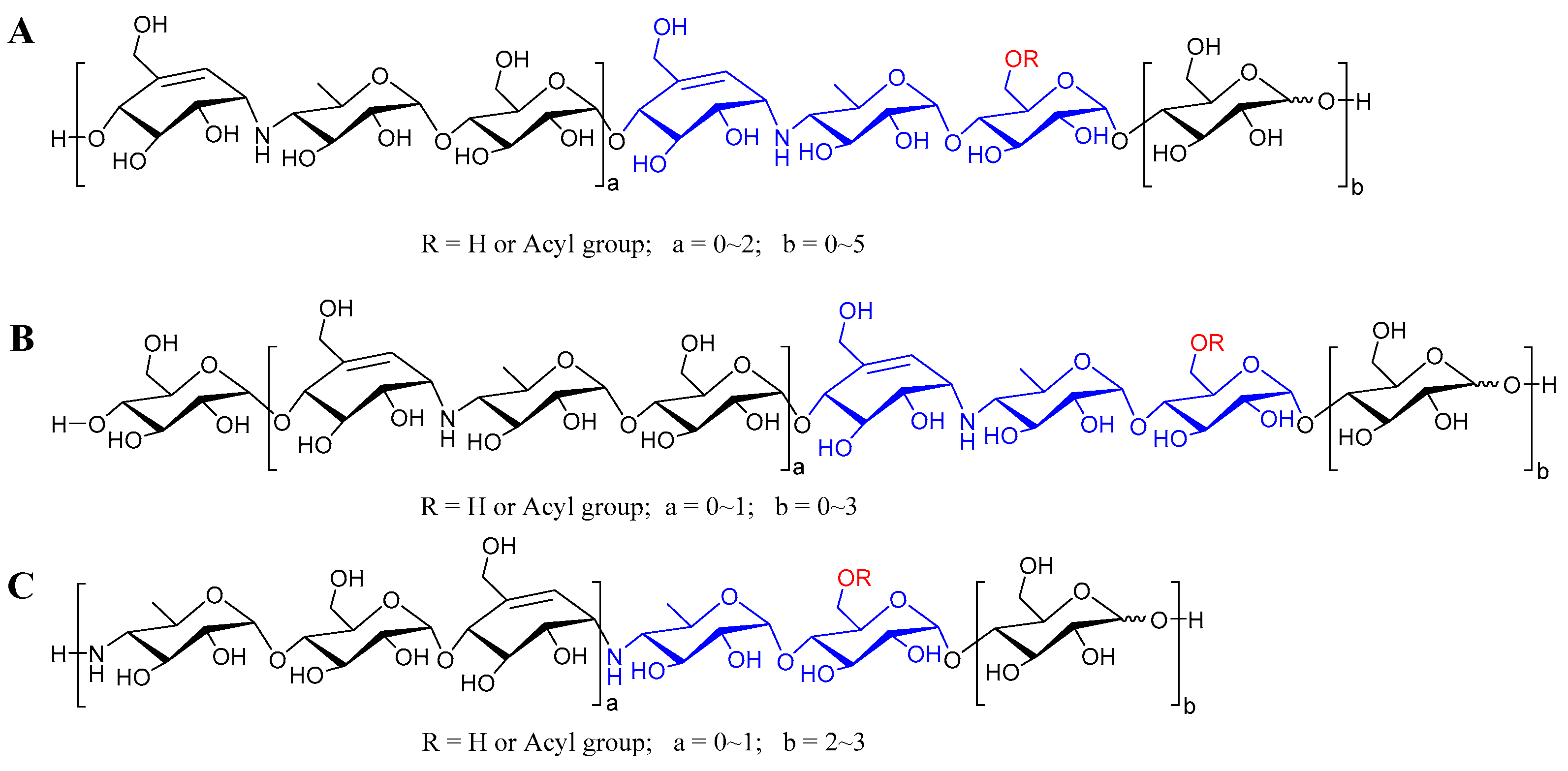
According to the abundant fragment ion peaks (m/z 304, 466, 146) arising from the cleavage of the first quinovosidic bond numbered at the non-reducing terminus, all the aminooligosaccharides are directly divided into three groups, namely acarviostatins with glucose(s) at the reducing end (Aca-glu), acarviostatins with glucose(s) at both ends (glu-Aca-glu), and acarviostatins with an incomplete pseudo-trisaccharide at the non-reducing end (incAca-glu) (Figure 3). The structures of sixty-three oligosaccharides can be categorized as group Aca-glu, which accounts for a major portion (more than 64%) of the metabolic profiling of strain HO1518. Amongst them, acarviostatin II05 (Aca II05) contains up to five glucose units at the reducing end, while D6-O-propionyl-acarviostatin III03 (Pr-Aca III03) possesses three pseudo-trisaccharides. The abundant MS2 fragment ion at m/z 304, produced by the loss of the acarviosine moiety at the non-reducing end, is the basic and characteristic peak for group Aca-glu. The structures of fifteen aminooligosaccharides are assigned as group glu-Aca-glu. Due to the presence of an additional glucose unit appended to the non-reducing terminus in group glu-Aca-glu compared with those in group Aca-glu, the typical peak ion of aminooligosaccharides is m/z 466. Group incAca-glu contains twenty aminooligosaccharides. The rare absence of an unsaturated cyclohexitol unit in the partial pseudo-trisaccharide core at the non-reducing termini results in a high-intensity characteristic MS/MS fragment at m/z 146, corresponding to the loss of the 4-amino-4,6-dideoxy-d-glucopyranose unit in the non-reducing end. Moreover, when discriminating aminooligosaccharides harbor more than one pseudo-trisaccharide core, the above-mentioned characteristic peak in each group combined with the second typical fragment ion peaks at m/z 769 for Aca-glu, 931 for glu-Aca-glu, and 611 for incAca-glu and would be greatly helpful for the judgment of their structures.
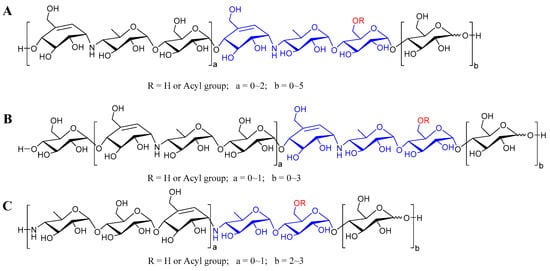
Figure 3.
The general structures of aminooligosaccharides from Streptomyces sp. HO1518. (A) The general structures of acarviostatins with glucoses at the reducing terminus; (B) The general structures of acarviostatins with glucoses at the reducing and non-reducing terminus; (C) The general structures of acarviostatins with an incomplete pseudo-trisaccharide at the non-reducing terminus.
Some aminooligosaccharides, in particular the acylated acarviostatins, share identical m/z values and molecular formulas, suggesting that these compounds should be structural isomers. The reasons for this were attributed to the different assembly sequences of the same number of monosaccharides or the isomerism of the acyl side chains. For example, the protonated molecular ion at m/z 1012 in the extracted ion chromatograms (EIC) shows two peaks (Figure S9A). The minor peak with a retention time of 10.19 min was assigned as D6-O-acetyl-acarviostatin I12 (Ac-Aca I12) attributable to the most abundant fragment at m/z 466 (Figure S9B), while the major peak appearing at a retention time of 11.19 min was inferred as D6-O-acetyl-acarviostatin I03 (Ac-Aca I03) due to the most abundant ion at m/z 304 (Figure S9C). Therefore, these abundant signals produced by the cleavage of quinovosidic bonds could confirm the number of the d-glucopyranose attached to the reducing and/or non-reducing terminus. In addition, it is worth mentioning that twenty aminooligosaccharides belonging to group incAca-glu represent a new type of oligosaccharides. To the best of our knowledge, this is the first report of acylated aminooligosaccharides directly ending with 4-amino-4-deoxy-D-quinovopyranose unit at the non-reducing end.
Motivated by the metabolic profile of aminooligosaccharides, the LC-MS guided fractionation procedure was performed to acquire new oligosaccharides. The extract of the strain HO1518 was prepared and subjected to C18 column chromatography to yield six fractions. After careful analysis of these fractions using LC-HRMS/MS, the molecular weights related to various aminooligosaccharides were found to enrich the fractions F1 and F2. Then, several new ion peaks at m/z 1273, 1329, 1343, 1491, and 1519 in group Aca-glu were selected as target compounds (Figure S5). In addition, many newly appeared quasi-molecular ion peaks of acarviostatins, especially those in group incAca-glu, also inspired our great interest to further perform the chemical search of strain HO1518 for the discovery of new anti-diabetic agents. Nevertheless, to our regret, we failed to acquire these novel compounds due to the trace amount of these two groups of oligomers.
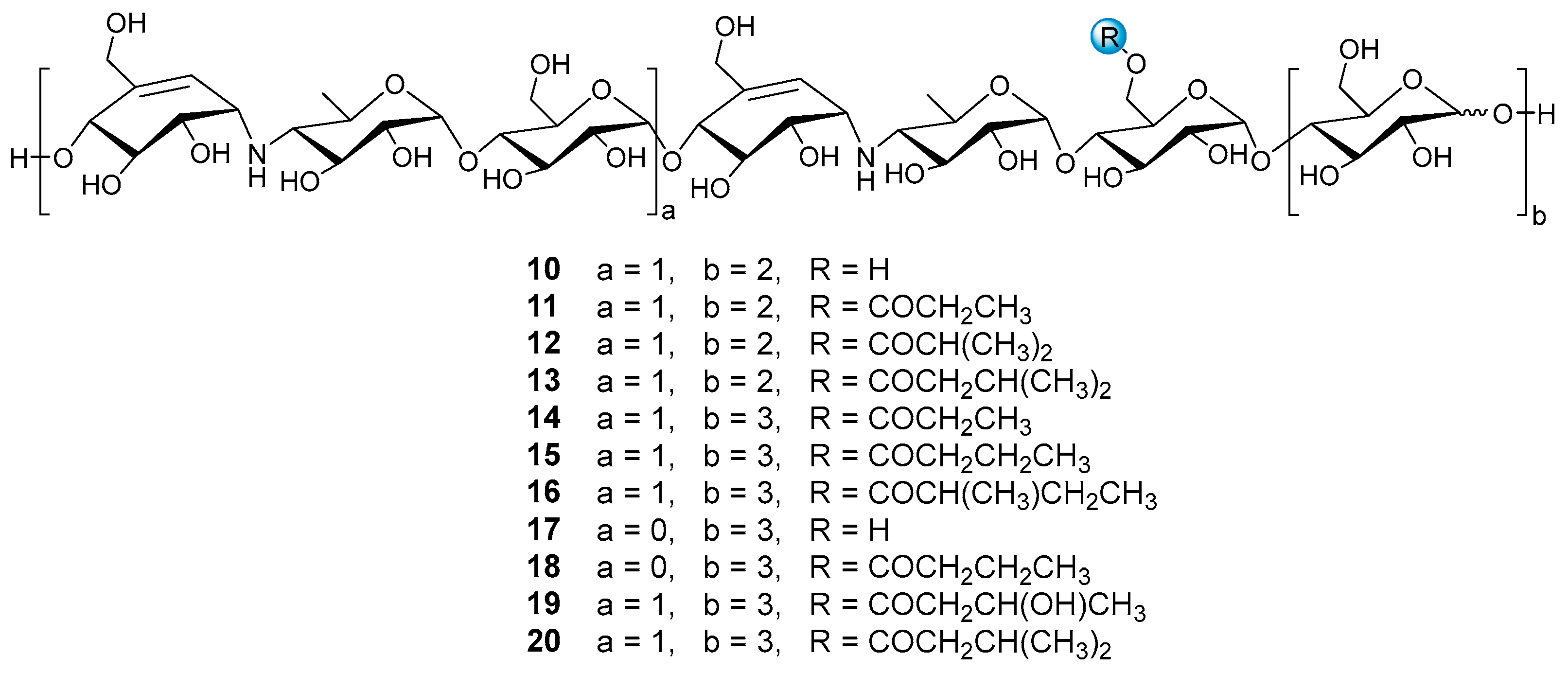
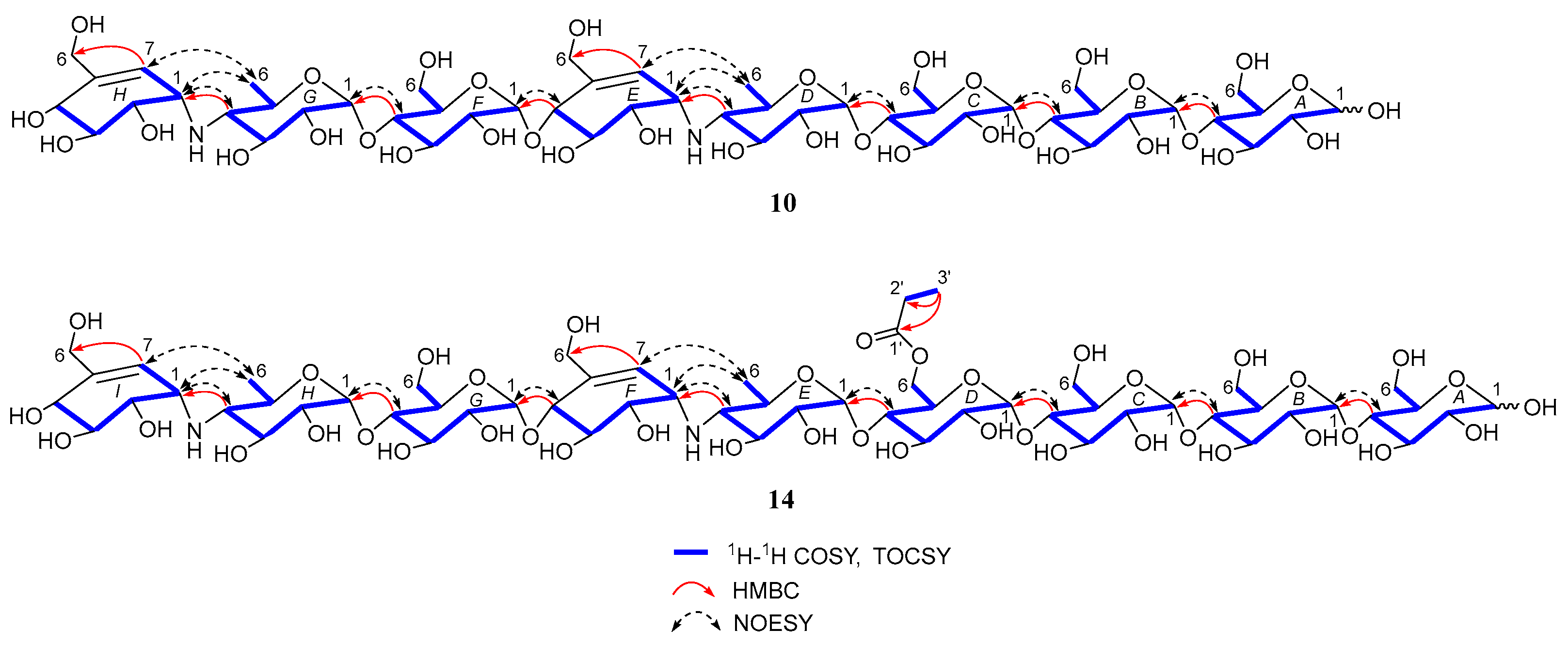
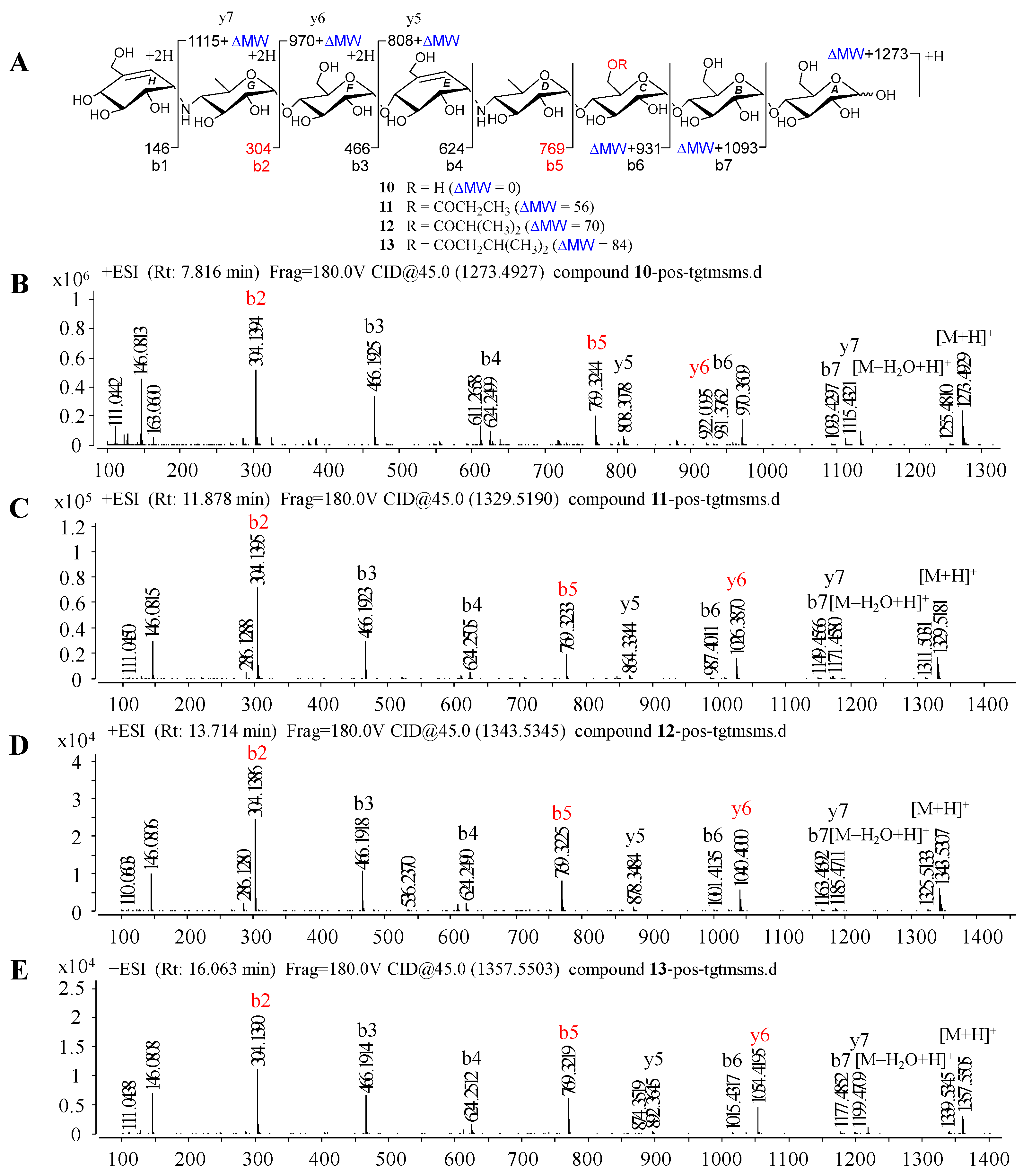
Under the guidance of the aforementioned quasi-molecular peaks, seven new aminooligosaccharide congeners (10–16) and four known related compounds (17–20) were isolated (Figure 4). Compound 10, white amorphous powder, was assigned a HRESIMS ion peak at m/z 1273.4938 ([M + H]+, calcd for 1273.4927), which matched a molecular formula of C50H84N2O35 with 10 degrees of hydrogen deficiency (Figure S27). The 1D and 2D NMR spectra, especially the 1D-selective TOCSY, 2D-TOCSY, HSQC, HMBC, and HSQC-TOCSY, allowed the construction of the gross structure of 10 (Figure 5). This deduction was further supported by several crucial fragments at m/z 304 (b2), 769 (b5), and 970 (y6) observed in the HRESIMS/MS spectrum of 10, corresponding to the fission of the quinovosidic bond (Figure 6). The molecular formula of 11 was determined as C53H88N2O36 (m/z 1329.5234 ([M + H]+, calcd for 1329.5190) by HRESIMS, suggesting that 11 was an acetylated derivative of 10. A careful analysis of 1H and 13C NMR spectra between 10 and 11 revealed that the hydroxyl group at C-D6 was acetylated in 11, which was further verified by the 1H-1H COSY cross peak of H-2′/H-3′ and the HMBC correlations from H-2′ (δH 2.48) and H-3′ (δH 1.13) to C-1′ (δC 180.8) as well as H-3′ to C-2′ (δC 30.6) (Figure 5). Similarly, the carbon signals (δC 180.1, 33.9, 18.3 and 18.2 for 12, and δC 176.2, 42.9, 25.5, 21.7 and 21.7 for 13) indicated 12 and 13 to be isobutyryl and isovaleryl substituted analogs of 10, respectively.
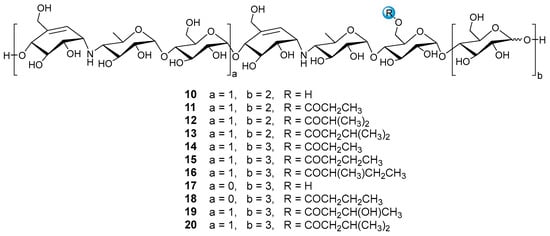
Figure 4.
The structures of compounds 10–20.
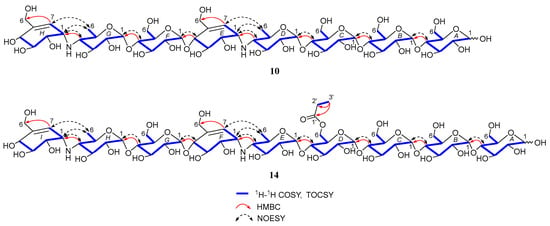
Figure 5.
Key 2D NMR correlations of compounds 10 and 14.
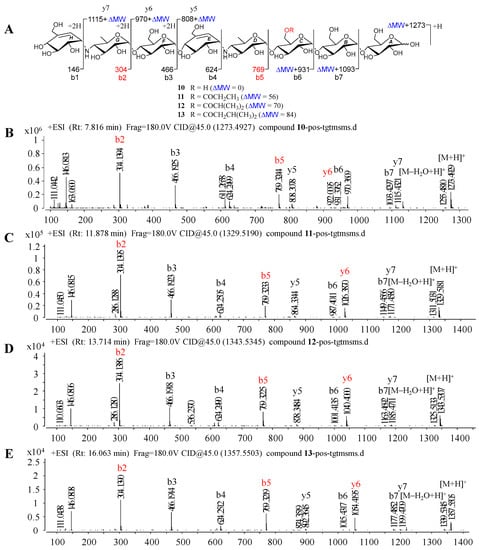
Figure 6.
Positive HRESIMS/MS fragmentation and spectra of 10–13. (A) Positive-ion HRESIMS/MS fragmentation patterns of 10–13; (B–E) HRESIMS/MS spectra of 10–13.
The molecular formula of 14 was assigned as C59H98N2O41 by HRESIMS ion peak at m/z 1491.5715 ([M + H]+, calcd for 1491.5718), which was 162 mass units more than that of 11. Detailed inspection of the NMR spectroscopic data of 11 and 14 suggested that they should feature an analogous planar structure (Figure 4), with the major difference being the presence of an additional glucose unit at the reducing end in 14 (δC 102.3, 79.9, 76.1, 74.3, 74.1 and 63.5). After a careful comparison of 1H and 13C NMR data of 15 and 16 (Table S2) with those of 14, three compounds were found to share the identical acarviostatin II03-type core skeleton, but the side chain differentiated. The propionyl functionality in 14 was replaced by the butyryl group in 15 (δH 2.45, 1.65, 1.18 and 0.94) or the 2-methyl-butyryl group in 16 (δH 2.52, 1.63, 1.51, 1.13 and 0.88), which could be supported by the 1H-1H COSY cross-peaks (H-2′/H-3′/H-4′ in 15, and H-5′/H-2′/H-3′/H-4′ in 16) and the HRMS/MS spectra (Figure 5 and Figure S2).
T2DM is one of the most serious chronic diseases worldwide, which is closely linked to disturbances of glucose and lipid metabolism. Inhibiting α-glucosidase and lipase involved in the breakdown of carbohydrates and fats can reduce glucose and free fatty acid absorption in the gastrointestinal tract, which contributes to avoiding postprandial hyperglycemia and restoring normal levels of insulin secretion of pancreatic β-cells in diabetic patients [27,28,29]. Thus, compounds 9–20 were evaluated for their inhibitory activities against PPA, sucrase and PL, as presented in Table 2.
Table 2.
The inhibitory activities of 9–20 against PPA, sucrase and PL.
All the compounds showed inhibition of three metabolic enzymes under the assay conditions. Twelve isolates (9–20) showed remarkable inhibition of PPA, with the IC50 values ranging from 0.03 to 0.40 μM (Figure S3). Compounds 9–16, 19 and 20 possessing the repeated pseudo-trisaccharide moieties exhibited more potential α-amylase inhibitory effects than those with a single pseudo-trisaccharide (17 and 18), among which 9 (IC50 = 0.03 μM) was 282-fold more effective than that of acarbose (IC50 = 8.51 μM). Nevertheless, the more pseudo-trisaccharide cores might pose an unfavorable effect for sucrase inhibition by comparison with the suppressing activities of 9, 10, and 17. When the hydroxyl group occurred at C-6 in 10 (IC50 = 13.05 μM) was acylated, 11–13 presented stronger inhibitory activity against sucrase with IC50 values of 4.34, 6.79 and 7.06 μM, respectively, which implied that the acyl group shows a positive contribution to their inhibitory potency toward sucrase. Furthermore, twelve compounds displayed considerable inhibitory ability against PL with IC50 values in a range of 1.00–31.56 μM, while acarbose was inactive with an IC50 value of 191.00 μM. Of these compounds, 13, 16, 19 and 20, sharing an acarviostatin II03-type structure, showed potent inhibitory effects against lipase with IC50 values of 1.56, 1.34, 1.00 and 1.43 μM, respectively, which was nearly equal to the positive control orlistat (IC50 = 0.34 μM). It is noteworthy, that increasing the acyl chain length in aminooligosaccharides contributes to enhancing their lipase inhibitory activities, as referred to 9–20. These biological results highlight the potential of acylated aminooligosaccharides with prominent α-glucosidase and lipase inhibition for the future development of multi-target anti-diabetic drugs.
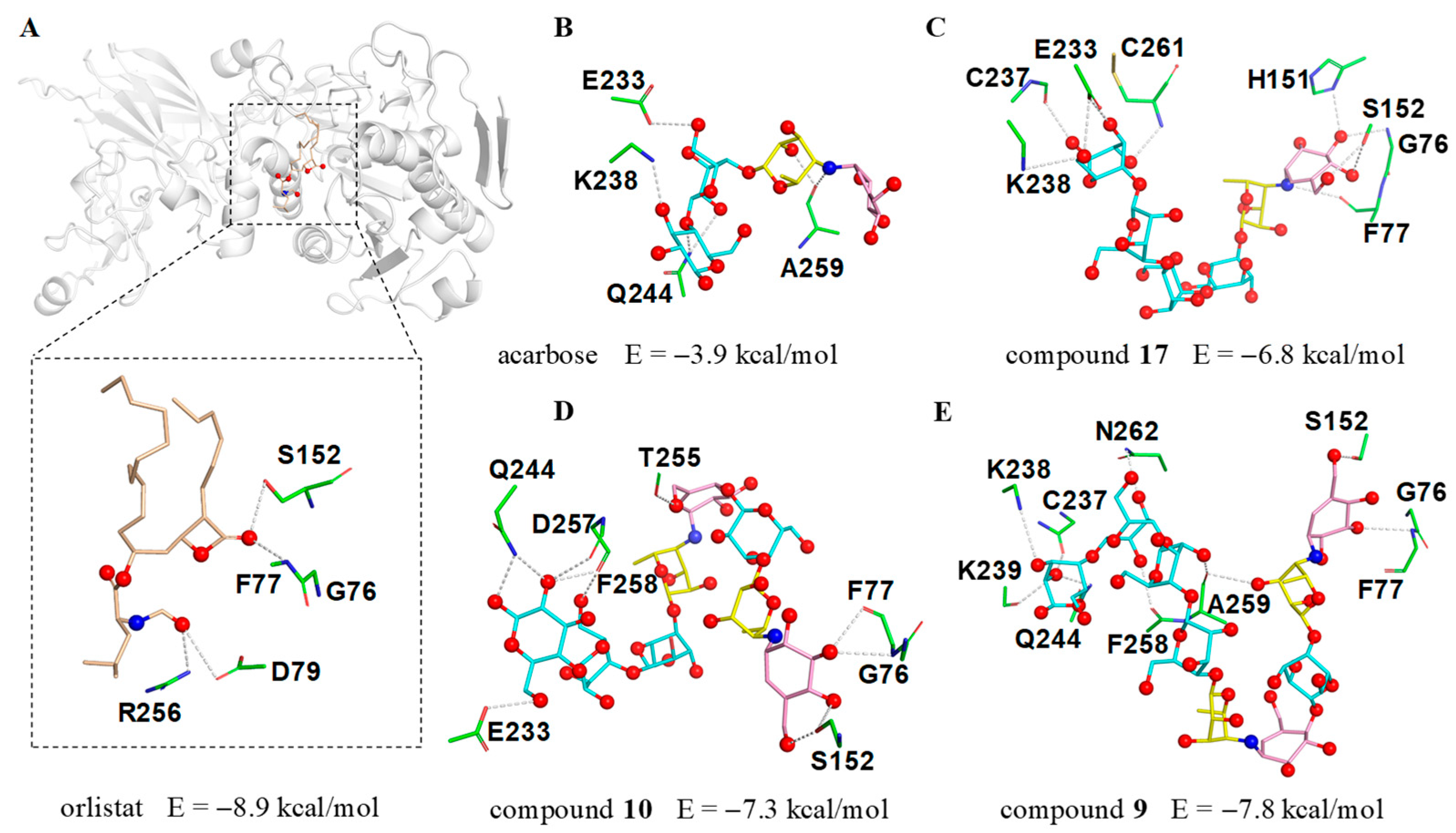
The significant PL inhibitory activity for aminooligosaccharides prompted us to further investigate the potential molecular recognition mechanism between this class of compounds and PL, thus the molecular docking simulations were implemented using a previously reported crystal structure of human PL (PDB ID: 1LPB) [30,31]. To better understand the binding mode of different aminooligosaccharides, the anti-obesity drug orlistat was firstly docked into the same domain for comparative purposes. As shown in Figure 7A, orlistat abrogated the activity of human PL by occupying the substrate binding canyon of PL and stabilized itself via strong interactions with a series of key residues (G76, F77, D79, S152 and R256) in the catalytic active site. Based on this, three characteristic aminooligosaccharides 9 (IC50 = 7.64 μM), 10 (IC50 = 12.66 μM), 17 (IC50 = 31.56 μM) as well as acarbose (negative control) belonging to acarviostatins II03, II02, I03 and I01 type, respectively, were selected as ligands for further detailed study.
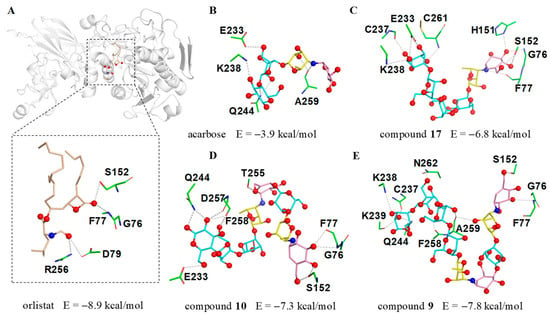
Figure 7.
The docking results of human PL (PDB ID: 1LPB) with inhibitors. (A) The possible interactions between 1LPB and orlistat; (B) acarbose; (C) acarviostatin I03; (D) acarviostatin II02; (E) acarviostatin II03. Wheat: orlistat; pink: C7N cyclohexitol; yellow: 4-amino-4,6-dideoxy-d-glucopyranose; cyan: d-glucopyranose.
As we anticipated, the cyclohexitol ring of the pseudo-trisaccharide unit for 9, 10 and 17 could be perfectly docked into the catalytic cavity of PL by forming three hydrogen bonds with G76, F77 and S152, whereas the pseudo-tetrasaccharide acarbose, only located outside the catalytic pocket with no bonding site observed in the catalytic active center, indicates that the chain length and the pseudo-trisaccharide core(s) of aminooligosaccharides were crucial in hinting PL activity (Figure 7B–E). Besides the common residues mentioned above, 17 also interacted with four residues E233, C237, K238, and C261, while 10 showed interactions with the five residues E233, Q244, T255, D257, and F258. Notably, compound 9 (calculated binding energy = −7.8 kcal/mol), possessing an extra seven polar contacts with PL, exhibits a higher potency toward lipase than 10 and 17 (calculated binding energy = −7.3 and −6.8 kcal/mol), suggesting that the total strength of individual contact between ligand and PL is a definitive factor to forming a stable substrate-enzyme complex. These results were consistent with the aforementioned biological results and demonstrated the pseudo-trisaccharide unit(s) along with the glucose residues of aminooligosaccharides played a crucial role in their lipase inhibitory activity.
3. Materials and Methods
3.1. General Experimental Procedures
The optical rotations were performed on an Anton Paar MCP-500 spectropolarimeter (Anton Paar, Graz, Austria) at 20 °C. UV spectra were recorded on a JASCO V-550 UV/VIS spectrophotometer (Jasco Corporation, Tokyo, Japan). IR data were measured using a FT-IR Vertex 70 v spectrometer (Bruker, Fällanden, Switzerland). The 1D and 2D NMR spectra were acquired using a Bruker Avance 500 MHz spectrometer with TMS as an internal standard (Bruker, Fällanden, Switzerland). HRESIMS data were collected on a Thermo Q Exactive high-resolution mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). HRMS/MS data were recorded on an Agilent Q-TOF 6545 mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with an electrospray ionization source (ESI). MCI gel CHP20/P120 (Mitsubishi Chemical Corporation, Tokyo, Japan) and SiliaSphere C18 (50 µm, Silicycle, QuébecK, QC, Canada) were used for column chromatography. UPLC analysis was performed using an Agilent 1200Series LC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a binary pump, an online degasser, an autoplate-sampler, and a thermostatically controlled column compartment. The Thermo ultimate 3000 (Thermo Fisher Scientific, Waltham, MA, USA), equipped with an Alltech 3300 ELSD detector and VWD detector was used for HPLC. Preparative HPLC was performed using a SilGreen C18 column (250 × 20 mm, 5 μm, 12 nm, Greenherbs CO., Ltd., Beijing, China), while semi-preparative HPLC was performed utilizing a TSK-gel 100 V C18 column (250 × 10 mm, 5 μm, 12 nm, Tosoh Corporation, Tokyo, Japan).
3.2. Reagents
The HPLC-grade methanol and acetonitrile were purchased from CINC High Purity Solvents (Shanghai) Co., Ltd. (Shanghai, China). The other solvents were of analytical grade (Sinopharm Chemical Reagent Co., Ltd., Beijing, China). Porcine pancreatic α-amylase (PPA) and PL were purchased from Sigma Aldrich Co. (St Louis, MO, USA). Sucrase and acarbose were obtained from Shanghai yuanye Bio-Technology Co., Ltd. (Shanghai, China). Orlistat was bought from Shanghai xushuo Bio-Technology Co., Ltd. (Shanghai, China). Standards 1–9 were previously isolated from Streptomyces sp. HO1518 by our group.
3.3. Bacterial Material
The Streptomyces sp. HO1518 was isolated from a sediment sample collected from the Rizhao coastal area, Shandong Province of China, in summer 2010. This strain (Voucher Specimen No. M2018176) is preserved at the China Center for Type Culture Collection (CCTCC), Wuhan University.
3.4. UPLC Analysis
The ethanol extract of strain HO1518 was separated using an XBridge C18 column (4.6 × 150 mm, 3.5 μm; Waters, Milford, MA, USA). The mobile phase was composed of A (0.01% aqueous ammonia) and B (acetonitrile) with a flow rate of 0.3 mL/min. The column temperature was maintained at 40 °C, and the injection volume was 1 μL. The elution program was as follows: 0–1 min, 5% B; 1–21 min, 5–35% B; 21–26 min, 35–65% B; 26–27 min, 65–100% B; 27–28.5 min, 100% B; 28.5–29 min, 100–5% B; 29–30 min, 5% B.
3.5. QTOF-MS/MS Analysis
HRMS/MS spectra were performed on an Agilent Q-TOF 6545 mass spectrometer equipped with an electrospray ionization source (ESI). The operating parameters were set as follows: drying gas (nitrogen, N2) flow rate, 6.0 L/min; drying gas temperature, 320 °C; nebulizer, 45 psig; sheath gas temperature, 350 °C; sheath gas flow, 12 L/min; capillary, 3500 V; skimmer, 65 V; OCT RF V, 750 V; and fragmentor voltage, 180 V. For MS/MS experiments, the collision energy was adjusted from 10V to 45V to optimize signals and obtain maximal structural information from the ions of interest. The system was operated under the Masshunter workstation software, version B.02.00 (Agilent Technologies, Santa Clara, CA, USA). Each sample was analyzed in positive-ion mode to provide sufficient information for structural identification. The mass range was set at m/z 50–2000.
3.6. Fermentation, Extraction and Isolation
The 70 L fermented broth of strain HO1518 was filtered to remove mycelia and the supernatant was subjected to XAD-16 resin by column chromatography, eluting with anhydrous ethanol to obtain the crude extract. The ethanol extract (9.2 g) was separated into 6 fractions (Frs. 1–6) on a C18 reverse-phase (RP) silica gel column by step gradient elution with MeOH/H2O (5–100%, v/v). Since the majority of aminooligosaccharide derivatives were present in Frs. 1 and 2, the MS-guided fractionation was carried out.
Fr. 1 (4.2 g) was subjected to reversed-phase C18 silica gel using the gradient elution with MeOH/H2O (5–100%, v/v) to obtain six subfractions (Frs. 1-1–1-6). Fr. 1-1 (0.2 g) was chromatographed over the MCI column and further purified by a preparative RP HPLC system equipped with a preparative SilGreen C18 column (MeCN/H2O, 8 mL/min, 6:94) to produce 10 (1.6 mg, tR 9.6 min). Fr. 1-3 (0.3 g) was fractionated by HPLC using an isocratic mobile phase of 10% MeCN/H2O to obtain 19 (4.5 mg, tR 15.1 min), whereas Fr. 1-4 (0.4 g) was successively separated by MCI and HPLC with an isocratic mobile phase of 10% MeCN/H2O to acquire 14 (13.2 mg, tR 40.5 min). Fr. 1-5 (0.8 g) was repeatedly purified by HPLC (MeCN/H2O, 8 mL/min, 16:84) to yield 18 (10.2 mg, tR 14.3 min). Fr. 1-6 (0.8 g) was fractionated by the MCI column, which was purified by HPLC on a preparative SilGreen C18 column (MeCN/H2O, 8 mL/min, 18:82) to yield 16 (4.2 mg, tR 12.2 min) and 20 (6.2 mg, tR 12.8 min). Fr. 2 (3.9 g) was subjected to the MCI column, eluting with MeOH/H2O (5–100%, v/v) to afford four subfractions (Frs. 2-1–2-4). Fr. 2-1 (0.2 g) was purified by HPLC with an isocratic phase of 18% MeOH/H2O to yield 17. Fr. 2-2 (0.5 g) was separated by the reversed-phase C18 silica gel column, and then purified by HPLC (MeCN/H2O, 8 mL/min, 16:84) to obtain 12 (1.9 mg, tR 10.4 min) and 15 (5.9 mg, tR 11.9 min). Fr. 2-3 (0.3 g) was purified by HPLC using 10% MeCN/H2O to produce 11 (2.2 mg, tR 17.0 min), while Fr. 2-4 (0.2 g) was separated by the RP HPLC system (MeCN/H2O, 8 mL/min, 6:94) to afford 13 (2.0 mg, tR 44.3 min).
Acarviostatin II02 (Aca II02, 10): White amorphous powder, +127.2 (c 1.05, H2O). UV (H2O) end absorption; IR νmax 3319, 1663, 1396, 1149, 1031 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S1; positive ESIMS: m/z 1273 [M + H]+; HRESIMS: m/z 1273.4938 [M + H]+ (calcd for C50H84N2O35, 1273.4927).
D6-O-Propionyl-acarviostatin II02 (Pr-Aca II02, 11): White amorphous powder, +142.4 (c 1.01, H2O). UV (H2O) end absorption; IR νmax 3337, 1726, 1407, 1149, 1026 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S1; positive ESIMS: m/z 1329 [M + H]+; HRESIMS: m/z 1329.5234 [M + H]+ (calcd for C53H88N2O36, 1329.5190).
D6-O-Isobutyryl-acarviostatin II02 (isoBu-Aca II02, 12): White amorphous powder, +136.1 (c 1.02, H2O). UV (H2O) end absorption; IR νmax 3329, 1721, 1365, 1147, 1014 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S1; positive ESIMS: m/z 1343 [M + H]+; HRESIMS: m/z 1343.5365 [M + H]+ (calcd for C54H90N2O36, 1343.5346).
D6-O-Isovaleryl-acarviostatin II02 (isoVa-Aca II02, 13): White amorphous powder, +143.0 (c 0.99, H2O). UV (H2O) end absorption; IR νmax 3305, 1647, 1407, 1150, 1013 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S1; positive ESIMS: m/z 1357 [M + H]+; HRESIMS: m/z 1357.5520 [M + H]+ (calcd for C55H92N2O36, 1357.5503).
D6-O-Propionyl-acarviostatin II03 (Pr-Aca II03, 14): White amorphous powder, +136.3 (c 0.90, H2O). UV (H2O) end absorption; IR νmax 3304, 1734, 1402, 1148, 1023 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S2; positive ESIMS: m/z 1491 [M + H]+; HRESIMS: m/z 1491.5727 [M + H]+ (calcd for C59H98N2O41, 1491.5718).
D6-O-Butyryl-acarviostatin II03 (Bu-Aca II03, 15): White amorphous powder, +150.3 (c 0.53, H2O). UV (H2O) end absorption; IR νmax 3324, 1729, 1568, 1149, 1024 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S2; positive ESIMS: m/z 1505 [M + H]+; HRESIMS: m/z 1505.5872 [M + H]+ (calcd for C59H98N2O41, 1505.5874).
D6-O-2-Methyl-butyryl-acarviostatin II03 (Mbu-Aca II03, 16): White amorphous powder, +130.1 (c 1.03, H2O). UV (H2O) end absorption; IR νmax 3303, 1645, 1406, 1149, 1014 cm−1. 1H (500 MHz) and 13C (125 MHz) NMR spectroscopic data, see Table S2; positive ESIMS: m/z 1519 [M + H]+; HRESIMS: m/z 1519.6039 [M + H]+ (calcd for C61H102N2O41, 1519.6031).
3.7. PPA Inhibition Assay
The PPA inhibitory activities of compounds 9–20 were conducted based on a previously reported method. Commercial α-amylase inhibitor acarbose was used as the positive control [20].
3.8. Sucrase Inhibition Assay
The sucrase inhibition assay of compounds 9–20 was evaluated according to the previously reported method. Acarbose was also used as the positive control [21].
3.9. PL Inhibition Assay
The lipase inhibition assay of compounds 9–20 was performed according to the method outlined by McDougall et al. with slight modification [32]. Orlistat was measured as a positive control.
3.10. Molecular Docking
The molecular docking simulations were performed by AutoDock Vina software, version 1.5.7 [33]. The crystal structure of human PL was downloaded from the RSCB Protein Data Bank (PDB ID: 1LPB). The binding site parameters (x: 4.342 Å; y: 24.299 Å; z: 47.471 Å) and the grid box dimensions (30 × 30 × 30 Å) were set. The results of molecular docking were evaluated on the basis of the binding energy, criteria of binding structure, and possible interactions between ligand and the critical catalytic triad of protein 1LPB.
4. Conclusions
In summary, the hyphenated system UPLC-QTOF-MS/MS that could provide qualitative retention time and reliable mass spectrometry information was utilized to reveal the metabolic profiling of aminooligosaccharides secreted by Streptomyces sp. HO1518. A total of ninety-eight aminooligosaccharides, including eighty new compounds, were detected and characterized from the extract of stain HO1518. Among them, twenty structural intriguing oligomers that ended with the 4-amino-4-deoxy-D-quinovopyranose unit at the non-reducing terminus were reported for the first time. The subsequent MS-guided fractionation method resulted in the isolation of seven new oligosaccharides (10–16) and four known analogs (17–20). Notably, compounds 10–13 are the first reported examples of oligosaccharides with a rarely occurring acarviostatin II02-type structure. All the compounds exhibited significant inhibitory activities against three digestive enzymes, among which compounds 9–16, 19 and 20 sharing two pseudo-trisaccharides were the most effective inhibitors of α-amylase and lipase. Furthermore, primary structure-activity relationships of 9–20 revealed that the number of the pseudo-trisaccharide core and acyl side chain play pivotal roles in their biological activity, which was evidenced by molecular docking analysis. These results of this study highlighted the advantages of UPLC-QTOF-MS/MS for the rapid structural identification of oligosaccharides, and this strategy could be extended to other investigations for high-throughput analysis of natural products with similar structures. More importantly, this study not only provided new lead compounds for further scientific research towards anti-diabetic drug discovery, but also shed light on the structural optimization of aminooligosaccharides analogs for medicinal scientists.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md20030189/s1, Figures S1–S9: HRMS data for an extract of strain HO1518, Figures S10–S154: HRESIMS, IR, UV, and 1D and 2D NMR spectra of compounds 10–16, Tables S1–S9: NMR data of 10–16, Tables S10–S12: information of 98 acarviostatins [20,21,34,35,36,37,38,39,40,41].
Author Contributions
Y.W. designed and coordinated this study and reviewed the manuscript; H.L. and Y.R. reviewed and edited the manuscript; J.X., Z.L. and Z.F. performed MS data analysis, fermentation, isolation and biological evaluations experiments; Z.L. performed the docking analysis; J.X., H.L. and Z.L. elucidated structures and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Key R&D Program of China (No. 2018YFA0900600), the National Natural Science Foundation of China (Nos. 41876084, 31670099), the Strategic Priority Research Program “Molecular mechanism of Plant Growth and Development” of the Chinese Academy of Sciences (Nos. XDB27020202, XDB27020103), the Construction of the Registry and Database of Bioparts for Synthetic Biology of the Chinese Academy of Sciences (No. ZSYS-016), the Program of Shanghai Academic Research Leader (No. 20XD1404400), the International Partnership Program of the Chinese Academy of Sciences (No. 153D31KYSB20170121), the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (No. TSBICIP-KJGG-002-15), and the National Key Laboratory of Plant Molecular Genetics, SIPPE, CAS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to Shizheng Bu and Yining Liu for NMR, HRESIMS and ESIMS/MS data acquisitions (the Core Facility Center of the Institute of the CAS Centre for Excellence in Molecular Plant Science, Chinese Academy of Sciences).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xie, K.B.; Zhang, X.L.; Sui, S.Y.; Ye, F.; Dai, J.G. Exploring and applying the substrate promiscuity of a C-glycosyltransferase in the chemo-enzymatic synthesis of bioactive C-glycosides. Nat. Commun. 2020, 11, 5162. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus-mechanisms and treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Heni, M.; Tabák, A.G.; Machann, J.; Schick, F.; Randrianarisoa, E.; de Angelis, M.H.; Birkenfeld, A.L.; Stefan, N.; Peter, A.; et al. Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat. Med. 2021, 27, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Hjorth, M.F.; Astrup, A. Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Gao, H.; Tang, W.; Nie, S.P. Plant non-starch polysaccharides that inhibit key enzymes linked to type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2017, 1401, 28–36. [Google Scholar] [CrossRef]
- Bailey, C.J.; Day, C. The future of new drugs for diabetes management. Diabetes Res. Clin. Pract. 2019, 155, 107785. [Google Scholar] [CrossRef]
- Balaich, J.; Estrella, M.; Wu, G.J.; Jeffrey, P.D.; Biswas, A.; Zhao, L.P.; Korennykh, A.; Donia, M.S. The human microbiome encodes resistance to the antidiabetic drug acarbose. Nature 2021, 600, 110–115. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Wolfender, J.L.; Litaudon, M.; Touboul, D.; Queiroz, E.F. Innovative omics-based approaches for prioritisation and targeted isolation of natural products—New strategies for drug discovery. Nat. Prod. Rep. 2019, 36, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Chen, Y.C.; Liu, Z.M.; Li, S.N.; Wang, Y.; Ren, Y.H.; Liu, H.X.; Zhang, W.M. Lithocarpins E−G, potent anti-tumor tenellone-macrolides from the deep-sea fungus Phomopsis lithocarpus FS508. Chin. J. Chem. 2021, 39, 1104–1112. [Google Scholar] [CrossRef]
- Covington, B.C.; McLean, J.A.; Bachmann, B.O. Comparative mass spectrometry-based metabolomics strategies for the investigation of microbial secondary metabolites. Nat. Prod. Rep. 2017, 34, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.X.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-based molecular networking for the discovery of drug leads in natural product bioassay-guided fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Henke, M.T.; Kelleher, N.L. Modern mass spectrometry for synthetic biology and structure-based discovery of natural products. Nat. Prod. Rep. 2016, 33, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Rivera, G.; Ballesteros-Vivas, D.; Parada-Alfonso, F.; Ibañez, E.; Cifuentes, A. Recent applications of high resolution mass spectrometry for the characterization of plant natural products. Trends Anal. Chem. 2019, 112, 87–101. [Google Scholar] [CrossRef]
- Bozicevic, A.; Dobrzynski, M.; De Bie, H.; Gafner, F.; Garo, E.; Hamburger, M. Automated comparative metabolite profiling of large LC-ESIMS data sets in an ACD/MS workbook suite add-in, and data clustering on a new open-source web platform FreeClust. Anal. Chem. 2017, 89, 12682–12689. [Google Scholar] [CrossRef]
- Panusa, A.; Petrucci, R.; Marrosu, G.; Multari, G.; Gallo, F.R. UHPLC-PDA-ESI-TOF/MS metabolic profiling of Arctostaphylos pungens and Arctostaphylos uva-ursi. A comparative study of phenolic compounds from leaf methanolic extracts. Phytochemistry 2015, 115, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Robertsen, H.L.; Musiol-Kroll, E.M. Actinomycete-derived polyketides as a source of antibiotics and lead structures for the development of new antimicrobial drugs. Antibiotics 2019, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.Q. Genomics-driven natural product discovery in actinomycetes. Trends Biotechnol. 2018, 36, 238–241. [Google Scholar] [CrossRef]
- Liu, H.-L.; E, H.-C.; Xie, D.-A.; Cheng, W.-B.; Tao, W.-Q.; Wang, Y. Acylated aminooligosaccharides with inhibitory effects against α-amylase from Streptomyces sp. HO1518. Mar. Drugs 2018, 16, 403. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Liu, H.L.; Liu, Z.F.; Ren, Y.H.; Wang, Y. Acylated aminooligosaccharides from the yellow sea Streptomyces sp. HO1518 as both α-glucosidase and lipase inhibitors. Mar. Drugs 2020, 18, 576. [Google Scholar] [CrossRef] [PubMed]
- McCranie, E.K.; Bachmann, B.O. Bioactive oligosaccharide natural products. Nat. Prod. Rep. 2014, 31, 1026–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.L.; Liu, Z.F.; Zhang, X.W.; Liu, H.L.; Wang, Y. Microbial oligosaccharides with biomedical applications. Mar. Drugs 2021, 19, 350. [Google Scholar] [CrossRef] [PubMed]
- Truscheit, E.; Frommer, W.; Junge, B.; Müller, L.; Schmidt, D.D.; Wingender, W. Chemistry and biochemistry of microbial α-glucosidase inhibitors. Angew. Chem. Int. Ed. 1981, 20, 744–761. [Google Scholar] [CrossRef]
- Lang, Y.Z.; Zhao, X.; Liu, L.L.; Yu, G.L. Applications of mass spectrometry to structural analysis of marine oligosaccharides. Mar. Drugs 2014, 12, 4005–4030. [Google Scholar] [CrossRef] [Green Version]
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Padhi, S.; Nayak, A.K.; Behera, A. Type II diabetes mellitus: A review on recent drug based therapeutics. Biomed. Pharmacother. 2020, 131, 110708. [Google Scholar] [CrossRef]
- Al-Mrabeh, A.; Zhyzhneuskaya, S.V.; Peters, C.; Barnes, A.C.; Melhem, S.; Jesuthasan, A.; Aribisala, B.; Hollingsworth, K.G.; Lietz, G.; Mathers, J.C.; et al. Hepatic lipoprotein export and remission of human type 2 diabetes after weight loss. Cell Metab. 2020, 31, 233–249. [Google Scholar] [CrossRef]
- Bray, G.A.; Heisel, W.E.; Afshin, A.; Jensen, M.D.; Dietz, W.H.; Long, M.; Kushner, R.F.; Daniels, S.R.; Wadden, T.A.; Tsai, A.G.; et al. The science of obesity management: An endocrine society scientific statement. Endocr. Rev. 2018, 39, 79–132. [Google Scholar] [CrossRef] [Green Version]
- Egloff, M.P.; Marguet, F.; Buono, G.; Verger, R.; Cambillau, C.; van Tilbeurgh, H. The 2.46 Å resolution structure of the pancreatic lipase-colipase complex inhibited by a C11 alkyl phosphonate. Biochemistry 1995, 34, 2751–2762. [Google Scholar] [CrossRef]
- Van Tilbeurgh, H.; Egloff, M.P.; Martinez, C.; Rugani, N.; Verger, R.; Cambillau, C. Interfacial activation of the lipase-procolipase complex by mixed micelles revealed by X-ray crystallography. Nature 1993, 362, 814–820. [Google Scholar] [CrossRef] [PubMed]
- McDougall, G.J.; Kulkarni, N.N.; Stewart, D. Berry polyphenols inhibit pancreatic lipase activity in vitro. Food Chem. 2009, 115, 193–199. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.D.; Frommer, W.; Junge, B.; Müller, L.; Wingender, W.; Truscheit, E.; Schäfer, D. α-Glucosidase inhibitors. New complex oligosaccharides of microbial origin. Naturwissenschaften 1977, 64, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, K.; Murai, H.; Murao, S. Isolation and structure-activity relationship of some amylostatins (F-lb Fraction) produced by Streptomyces diastaticus subsp amylostaticus No. 9410. Agric. Biol. Chem. 1982, 46, 1941–1945. [Google Scholar]
- Geng, P.; Qiu, F.; Zhu, Y.Y.; Bai, G. Four acarviosin-containing oligosaccharides identified from Streptomyces coelicoflavus ZG0656 are potent inhibitors of α-amylase. Carbohydr. Res. 2008, 343, 882–892. [Google Scholar] [CrossRef]
- Qian, M.X.; Nahoum, V.; Bonicel, J.; Bischoff, H.; Henrissat, B.; Payan, F. Enzyme-catalyzed condensation reaction in a mammalian α-amylase. High resolution structural analysis of an enzymeinhibitor complex. Biochemistry 2001, 40, 7700–7709. [Google Scholar] [CrossRef]
- Weiss, S.C.; Skerra, A.; Schiefner, A. Structural basis for the interconversion of maltodextrins by MalQ, the amylomaltase of Escherichia coli. J. Biol. Chem. 2015, 290, 21352–21364. [Google Scholar] [CrossRef] [Green Version]
- Si, D.Y.; Zhong, D.F.; Xu, Q.M. Two butylated aminooligosaccharides isolated from the culture filtrate of Streptomyces luteogriseus. Carbohydr. Res. 2001, 335, 127–132. [Google Scholar] [CrossRef]
- Zhong, D.F.; Si, D.Y.; He, W.Y.; Zhao, L.M.; Xu, Q.M. Structural revision of isovalertatins M03, M13, and M23 isolated from the culture of Streptomyces luteogriseus. Carbohydr. Res. 2001, 331, 69–75. [Google Scholar] [CrossRef]
- Si, D.Y.; Zhong, D.F.; He, W.Y.; Zhao, L.M. Structural revision of isovalertatins D03 and D23 isolated from the culture filtrate of Streptomyces luteogriseus. Chin. Chem. Lett. 2001, 12, 327–330. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).