
Sesquiterpene and Sorbicillinoid Glycosides from the Endophytic Fungus Trichoderma longibrachiatum EN-586 Derived from the Marine Red Alga Laurencia obtusa

 ,
,  , and
, and
Abstract
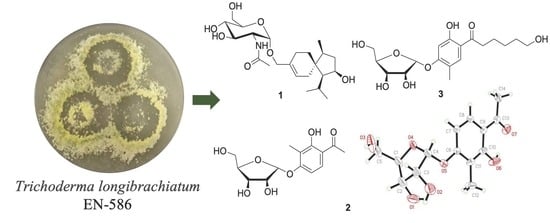
:
1. Introduction
2. Results and Discussion
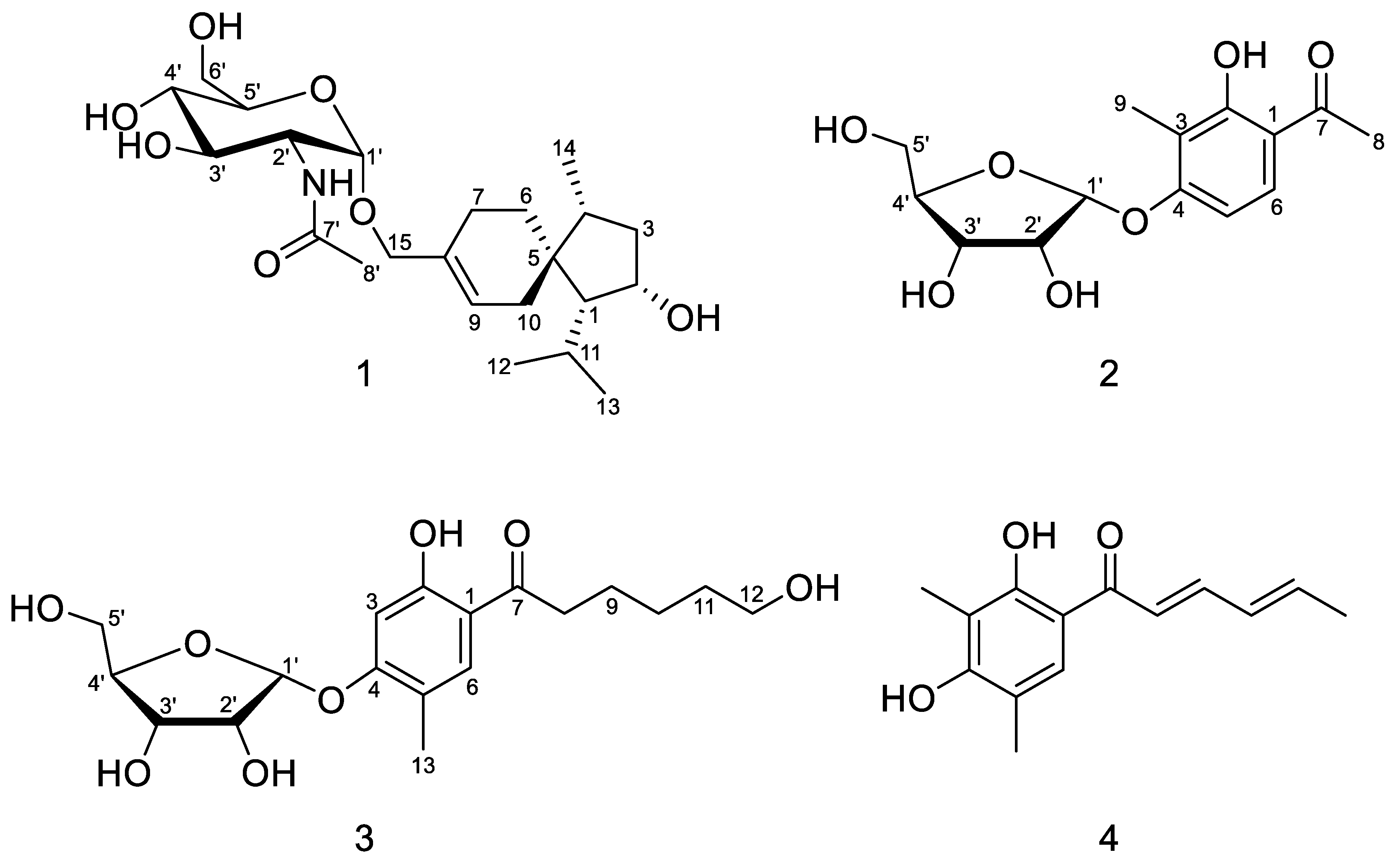
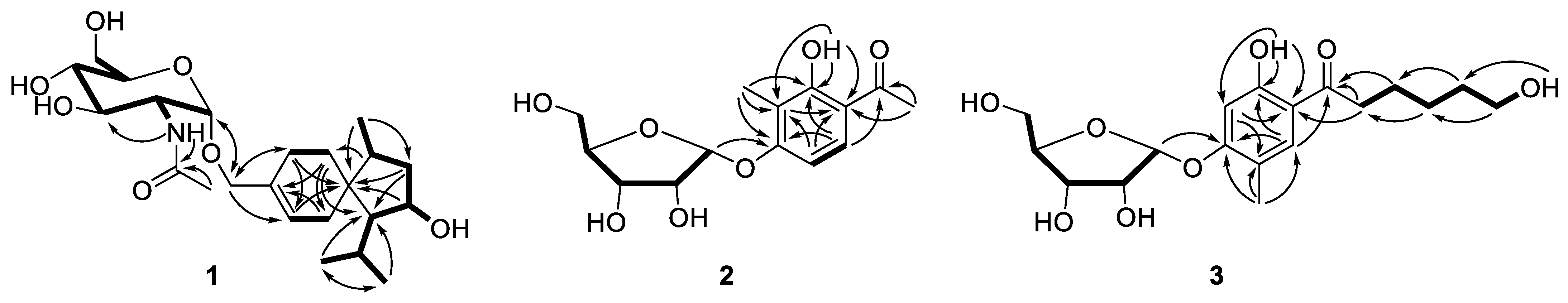
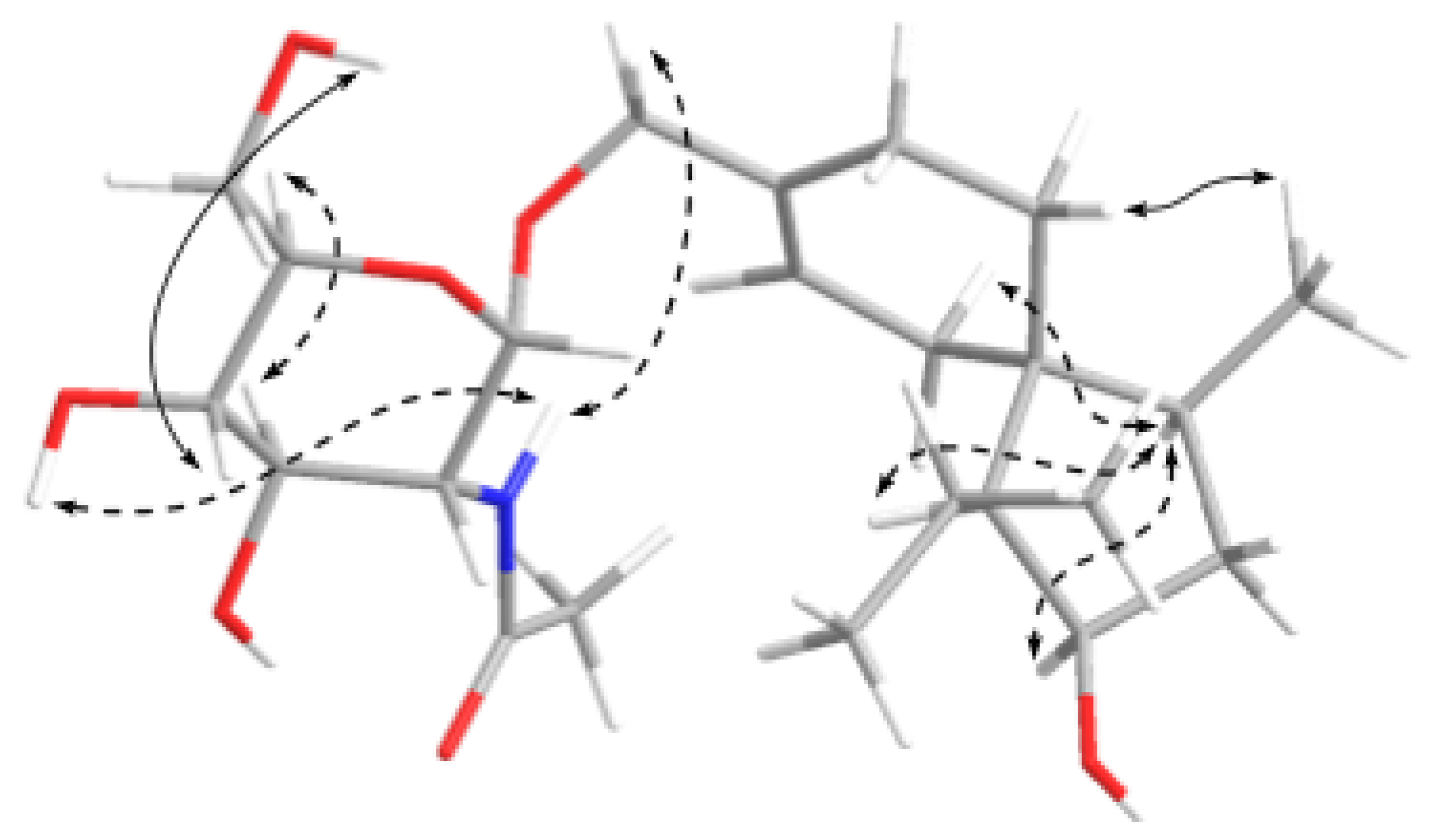
2.1. Structure Elucidation
2.2. Antimicrobial Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. X-ray Crystallographic Analysis of Compound 2
3.5. Acid Hydrolysis and Derivatization of Compound 1
3.6. Acid Hydrolysis and Derivatization of Compound 3
3.7. Computational NMR Chemical Shift Calculation and DP4+ Analysis of Compound 1
3.8. Antimicrobial Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baquero, F. Threats of antibiotic resistance: An obliged reappraisal. Int. Microbiol. 2021, 24, 499–506. [Google Scholar] [CrossRef]
- André, A.; Wojtowicz, N.; Touré, K.; Stien, D.; Eparvier, V. New acorane sesquiterpenes isolated from the endophytic fungus Colletotrichum gloeosporioides SNB-GSS07. Tetrahedron Lett. 2017, 58, 1269–1272. [Google Scholar] [CrossRef]
- Li, G.H.; Yang, Z.S.; Zhao, P.J.; Zheng, X.; Luo, S.L.; Sun, R.; Niu, X.M.; Zhang, K.Q. Three new acorane sesquiterpenes from Trichoderma sp. YMF1.02647. Phytochem. Lett. 2011, 4, 86–88. [Google Scholar] [CrossRef]
- Wu, S.H.; Zhao, L.X.; Chen, Y.W.; Huang, R.; Miao, C.P.; Wang, J. Sesquiterpenoids from the endophytic fungus Trichoderma sp. PR-35 of Paeonia delavayi. Chem. Biodivers. 2011, 8, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.P.; Deng, Y.L.; Lin, X.J.; Chen, B.; Li, J.; Liu, H.J.; Chen, S.H.; Liu, L. Anti-inflammatory mono- and dimeric sorbicillinoids from the marine-derived Fungus Trichoderma reesei 4670. J. Nat. Prod. 2019, 82, 947–957. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Ji, N.Y.; Wang, B.G. Mycochemistry of marine algicolous fungi. Fungal Divers. 2016, 80, 301–342. [Google Scholar] [CrossRef]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef]
- González, A.G.; Martín, J.D.; Norte, M.; Rivera, P.; Ruano, J.Z. Two new Cl5 acetylenes from the marine red alga Laurencia obtusa. Tetrahedron 1984, 40, 3443–3447. [Google Scholar] [CrossRef]
- Swamy, M.L.A. Marine algal sources for treating bacterial diseases. Adv. Food Nutr. Res. 2011, 64, 72–81. [Google Scholar]
- Li, H.L.; Yang, S.Q.; Li, X.M.; Li, X.; Wang, B.G. Structurally diverse alkaloids produced by Aspergillus creber EN-602, an endophytic fungus obtained from the marine red alga Rhodomela confervoides. Bioorg. Chem. 2021, 110, 104822. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Li, X.M.; Yang, S.Q.; Meng, L.H.; Li, X.; Wang, B.G. Prenylated phenol and benzofuran derivatives from Aspergillus terreus EN-539, an endophytic fungus derived from marine red alga Laurencia okamurai. Mar. Drugs 2019, 17, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.H.; Li, X.M.; Zhang, F.Z.; Wang, Y.N.; Wang, B.G. Talascortenes A−G, highly oxygenated diterpenoid acids from the sea-anemone-derived endozoic fungus Talaromyces scorteus AS-242. J. Nat. Prod. 2020, 83, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Y.; Wang, C.Y.; Li, X.M.; Yang, S.Q.; Li, X.; Wang, B.G.; Si, S.Y.; Meng, L.H. Cytochalasin derivatives from the endozoic Curvularia verruculosa CS-129, a fungus isolated from the deep-sea squat lobster Shinkaia crosnieri living in the cold seep environment. J. Nat. Prod. 2021, 84, 3122–3130. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Park, S.H.; Kim, B.Y.; Jo, S.I.; Lee, S.K.; Shin, J.; Oh, D.C. Deinococcucins A−D, aminoglycolipids from Deinococcus sp., a gut bacterium of the carpenter Ant Camponotus japonicus. J. Nat. Prod. 2017, 80, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Xuan, L.J.; Xu, Y.M.; Zhang, J.S. Seven new sesquiterpene glycosides from the root bark of Dictamnus dasycarpus. J. Nat. Prod. 2001, 64, 935–938. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the dtereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Rebollar-Ramos, D.; Macías-Ruvalcaba, M.L.; Figueroa, M.; Raja, H.A.; González-Andrade, M.; Mata, R. Additional α-glucosidase inhibitors from Malbranchea flavorosea (Leotiomycetes, Ascomycota). J. Antibiot. 2018, 71, 862–871. [Google Scholar] [CrossRef]
- May, D.S.; Kang, H.S.; Santarsiero, B.D.; Krunic, A.; Shen, Q.; Burdette, J.E.; Swanson, S.M.; Orjala, J. Ribocyclophanes A−E, glycosylated cyclophanes with antiproliferative activity from two cultured terrestrial cyanobacteria. J. Nat. Prod. 2018, 81, 572–578. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.; Chae, H.S.; Lee, C.; Choi, H.G.; Ryu, J.; Li, W.; Lee, H.; Jeong, G.S.; Chin, Y.W.; Shim, S.H. New aromatic compounds from the fruiting body of Sparassis crispa (wulf.) and their inhibitory activities on proprotein convertase subtilisin/kexin type 9 mRNA expression. J. Agric. Food Chem. 2017, 65, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.J.; Zhao, Y.; Xie, Z.L.; Liang, L.Z.; Shao, W.Y.; Zhu, L.P.; Yang, D.P.; Zhu, X.F.; Li, H.J. Novel sorbicillin analogues from the marine fungus Trichoderma sp. associated with the Seastar Acanthaster planci. Nat. Prod. Commun. 2012, 7, 1337–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, X.M.; Teuscher, F.; Li, D.L.; Diesel, A.; Ebel, R.; Proksch, P.; Wang, B.G. Chaetopyranin, a benzaldehyde derivative, and other related metabolites from Chaetomium globosum, an endophytic fungus derived from the marine red alga Polysiphonia urceolata. J. Nat. Prod. 2006, 69, 1622–1625. [Google Scholar] [CrossRef]
- Crystallographic Data of Compound 2 Have Been Deposited in the Cambridge Crystallographic Data Centre as CCDC 2131452. Available online: http://www.ccdc.cam.ac.uk/datarequest/cif (accessed on 29 December 2021).
- Sheldrick, G.M. SADABS, Software for Empirical Absorption Correction; University of Gottingen: Gottingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL, Structure Determination Software Programs; Bruker Analytical X-ray system Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL, Program for the Refinement of Crystal Structures; University of Gottingen: Gottingen, Germany, 2014. [Google Scholar]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, B69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Lee, H.S.; Woo, L.; Rho, J.R.; Seo, Y.; Cho, K.W.; Sim, C.J. New triterpenoid saponins from the sponge Erylus nobilis. J. Nat. Prod. 2001, 64, 767–771. [Google Scholar] [CrossRef]
- Lee, S.R.; Lee, D.; Park, M.; Lee, J.C.; Park, H.; Kang, K.S.; Kim, C.; Beemelmanns, C.; Kim, K.H. Absolute configuration and corrected NMR assignment of 17-hydroxycyclooctatin, a fused 5-8-5 tricyclic diterpene. J. Nat. Prod. 2020, 83, 354–361. [Google Scholar] [CrossRef]
- Kawazoe, R.; Matsuo, Y.; Saito, Y.; Tanaka, T. Computationally assisted structural revision of flavoalkaloids with a seven-membered ring: Aquiledine, isoaquiledine, and cheliensisine. J. Nat. Prod. 2020, 83, 3347–3353. [Google Scholar] [CrossRef]
- Chi, L.P.; Li, X.M.; Wan, Y.P.; Li, X.; Wang, B.G. Ophiobolin sesterterpenoids and farnesylated phthalide derivatives from the deep sea cold-seep-derived fungus Aspergillus insuetus SD-512. J. Nat. Prod. 2020, 83, 3652–3660. [Google Scholar] [CrossRef]
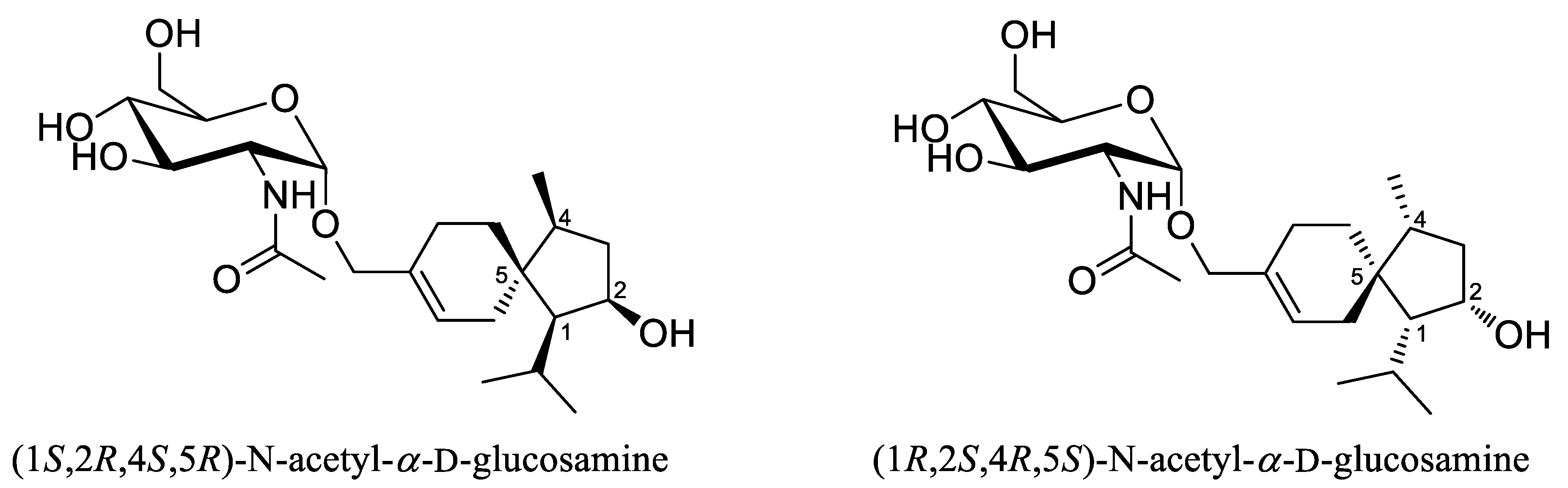
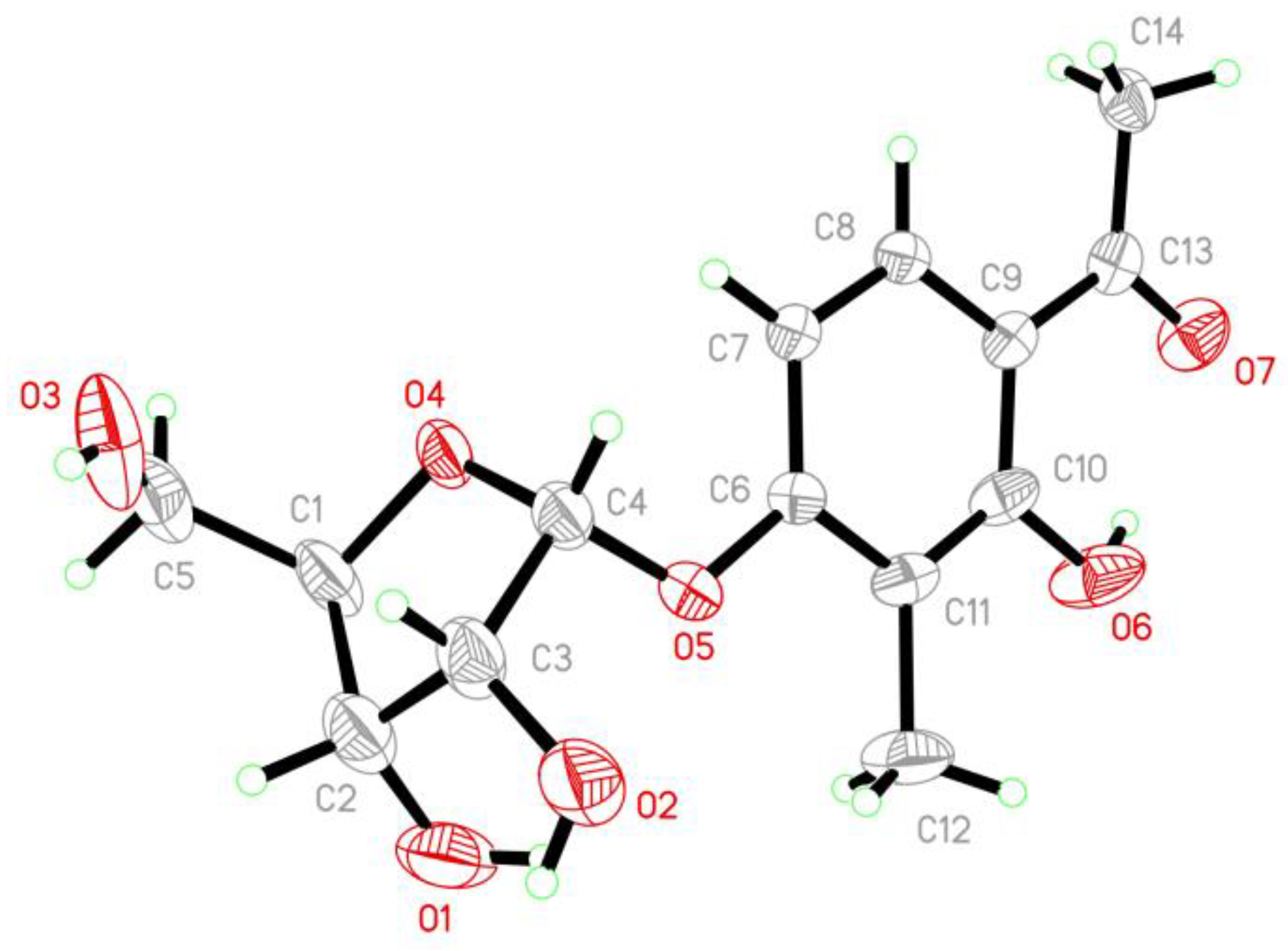

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 1.24, overlap | 59.3, CH | 113.9, C | 113.2, C | ||
| 2 | 4.22, brs | 64.6, CH | 161.2, C | 162.0, C | ||
| 3 | α 1.12, dd (11.4, 3.7) β 1.67, m | 28.5, CH2 | 113.9, C | 6.59, s | 102.1, CH | |
| 4 | 1.55, m | 46.2, CH | 161.0, C | 161.3, C | ||
| 5 | 44.2, C | 6.75, d (9.0) | 106.2, CH | 118.5, C | ||
| 6 | α 1.19, m β 1.63, m | 31.8, CH2 | 7.77, d (9.0) | 130.2, CH | 7.71, s | 131.8, CH |
| 7 | α 1.38, m β 1.71, overlap | 26.2, CH2 | 203.9, C | 205.3, C | ||
| 8 | 137.3, C | 2.58, s | 26.4, CH3 | 2.97, t (7.3) | 37.6, CH2 | |
| 9 | 5.69, brs | 124.1, CH | 2.05, s | 7.8, CH3 | 1.62, m | 24.1, CH2 |
| 10 | α 1.79, overlap β 2.07, dt (18.9, 3.0) | 34.5, CH2 | 1.35, m | 25.2, CH2 | ||
| 11 | 1.61, m | 29.8, CH | 1.45, m | 32.3, CH2 | ||
| 12 | 0.88, d (6.5) | 23.3, CH3 | 3.39, dt (6.3, 11.5) | 60.6, CH2 | ||
| 13 | 0.83, d (6.5) | 22.8, CH3 | 2.13, s | 15.3, CH3 | ||
| 14 | 0.80, d (6.7) | 14.2, CH3 | ||||
| 15 | α 4.08, d (13.0) β 4.01, d (13.0) | 66.6, CH2 | ||||
| 2-OH | 4.54, d (6.0) | 12.84, s | 12.51, s | |||
| 12-OH | 4.33, t (5.1) | |||||
| 1′ | 4.65, d (3.4) | 96.1, CH | 5.75, d (4.4) | 99.9, CH | 5.71, d (4.4) | 99.7, CH |
| 2′ | 3.65, td (8.1, 3.4) | 53.9, CH | 4.11, m | 71.4, CH | 4.11, m | 71.4, CH |
| 3′ | 3.45, dd (10.9, 8.1) | 70.8, CH | 3.96, overlap | 69.1, CH | 3.96, overlap | 69.1, CH |
| 4′ | 3.15, d (9.0) | 70.6, CH | 3.98, overlap | 86.5, CH | 3.96, overlap | 86.5, CH |
| 5′ | 3.39, ddd (10.0, 5.4, 2.3) | 72.9, CH | 3.47, d (3.6) | 61.4, CH2 | 3.47, dd (3.6, 4.9) | 61.4, CH2 |
| 6′ | α 3.49, overlapped β 3.60, m | 60.8, CH2 | ||||
| 7′ | 169.3, C | |||||
| 8′ | 1.82, s | 22.6, CH3 | ||||
| 2′-OH | 4.61, brs | 4.56, d (8.7) | ||||
| 2′-NH | 7.60, d (8.1) | |||||
| 3′-OH | 4.80, brs | 4.95, brs | 4.92, d (4.6) | |||
| 4′-OH | 5.03, brs | |||||
| 5′-OH | 4.80, brs | 4.81, t (4.9) | ||||
| 6′-OH | 4.46, dd (11.8, 3.5) | |||||
| Strains | Compounds | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | Positive Control | |
| A. brassicaeb | 32 | - | - | 16 | 0.5 |
| C. cornigerumb | 64 | - | - | 64 | 0.5 |
| C. gloeosporioidesb | 16 | 64 | 32 | - | 0.5 |
| C. gloeosporioides Penzb | 16 | 32 | 32 | 2 | 0.5 |
| C. spiciferab | 8 | 16 | 8 | 2 | 0.25 |
| F. graminearumb | - | - | - | 32 | 0.5 |
| F. oxysporumb | 32 | 32 | 32 | 1 | 0.5 |
| F. oxysporum f. sp. radicis lycopersici b | 32 | - | 64 | 32 | 0.5 |
| F. proliferatumb | 32 | 64 | 64 | 2 | 0.5 |
| P. digitatumb | 64 | 32 | 32 | 16 | 0.5 |
| P. piricola Noseb | 32 | - | 64 | 2 | 0.5 |
| A. hydrophiliac | 64 | 4 | 4 | 8 | 2 |
| E. colic | - | 64 | 16 | 16 | 1 |
| methicillin-resistant S. aureus c | 64 | - | - | 16 | 8 |
| P. aeruginosac | - | - | - | 16 | 1 |
| V. harveyic | 4 | 16 | 16 | 16 | 0.5 |
| V. parahaemolyticusc | - | - | - | 4 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, X.-M.; Yang, S.-Q.; Zhang, F.-Z.; Wang, B.-G.; Li, H.-L.; Meng, L.-H. Sesquiterpene and Sorbicillinoid Glycosides from the Endophytic Fungus Trichoderma longibrachiatum EN-586 Derived from the Marine Red Alga Laurencia obtusa. Mar. Drugs 2022, 20, 177. https://doi.org/10.3390/md20030177
Wang Y, Li X-M, Yang S-Q, Zhang F-Z, Wang B-G, Li H-L, Meng L-H. Sesquiterpene and Sorbicillinoid Glycosides from the Endophytic Fungus Trichoderma longibrachiatum EN-586 Derived from the Marine Red Alga Laurencia obtusa. Marine Drugs. 2022; 20(3):177. https://doi.org/10.3390/md20030177
Chicago/Turabian StyleWang, Ying, Xiao-Ming Li, Sui-Qun Yang, Fan-Zhong Zhang, Bin-Gui Wang, Hong-Lei Li, and Ling-Hong Meng. 2022. "Sesquiterpene and Sorbicillinoid Glycosides from the Endophytic Fungus Trichoderma longibrachiatum EN-586 Derived from the Marine Red Alga Laurencia obtusa" Marine Drugs 20, no. 3: 177. https://doi.org/10.3390/md20030177
APA StyleWang, Y., Li, X.-M., Yang, S.-Q., Zhang, F.-Z., Wang, B.-G., Li, H.-L., & Meng, L.-H. (2022). Sesquiterpene and Sorbicillinoid Glycosides from the Endophytic Fungus Trichoderma longibrachiatum EN-586 Derived from the Marine Red Alga Laurencia obtusa. Marine Drugs, 20(3), 177. https://doi.org/10.3390/md20030177