
Recent Achievements in Total Synthesis for Integral Structural Revisions of Marine Natural Products


Abstract
:
1. Introduction
2. Examples of Total Synthesis of Marine Natural Products Resulting in Their Structural Revisions
2.1. Synthesis of Boholamide A
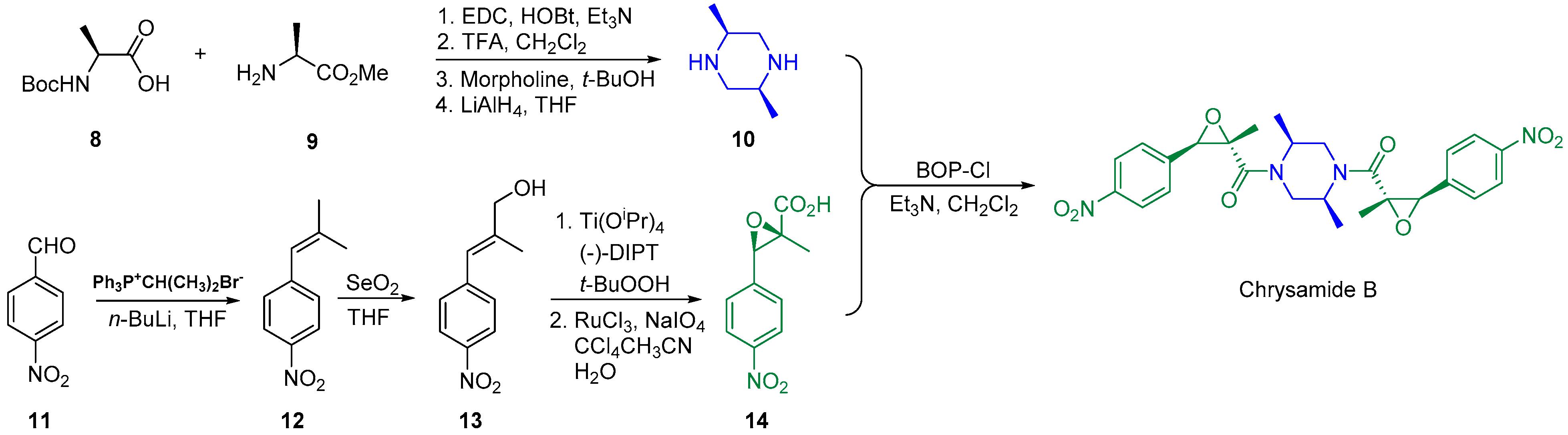
2.2. Synthesis of Chrysamide B
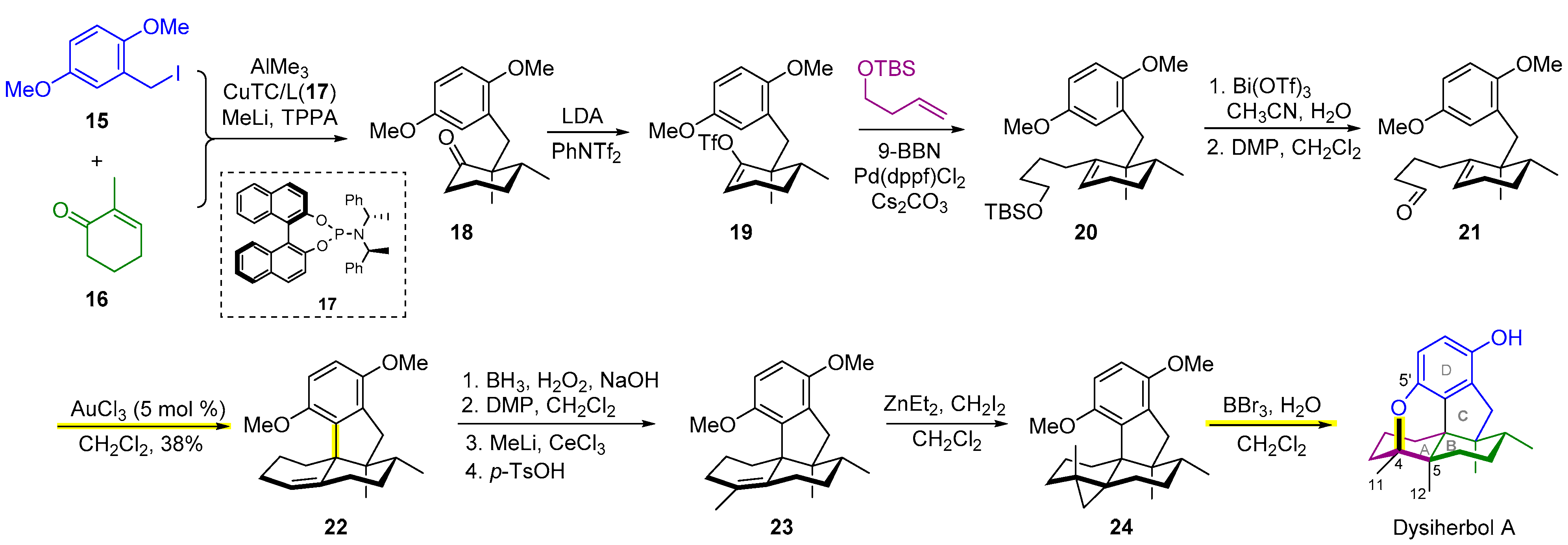
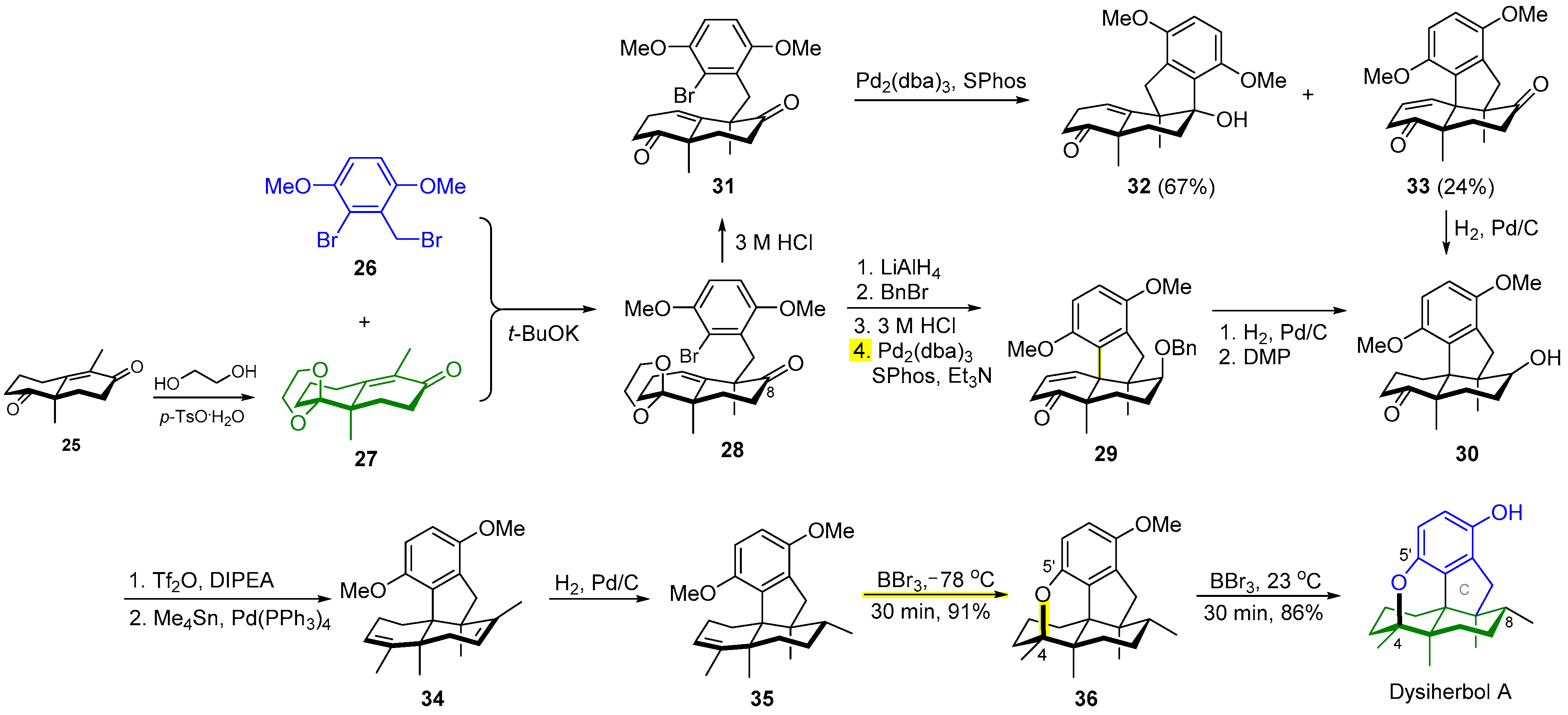
2.3. Synthesis of Dysiherbol A
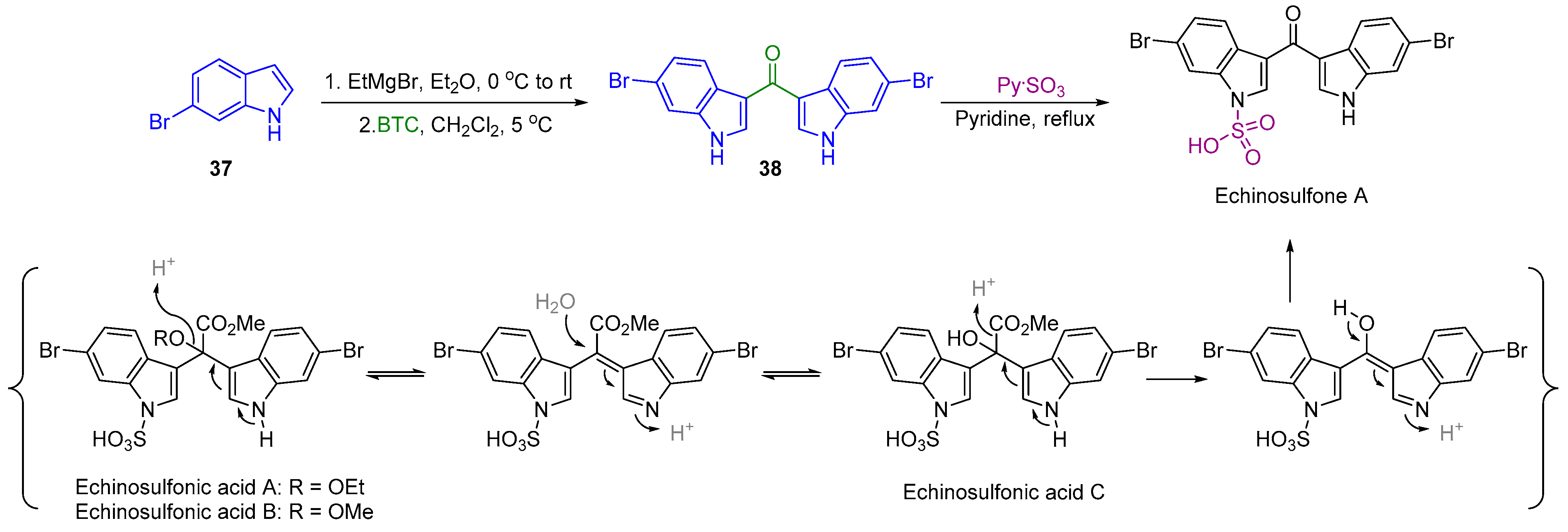
2.4. Synthesis of Echinosulfone A and Reconsideration of Structures of Echinosulfonic Acid A~D
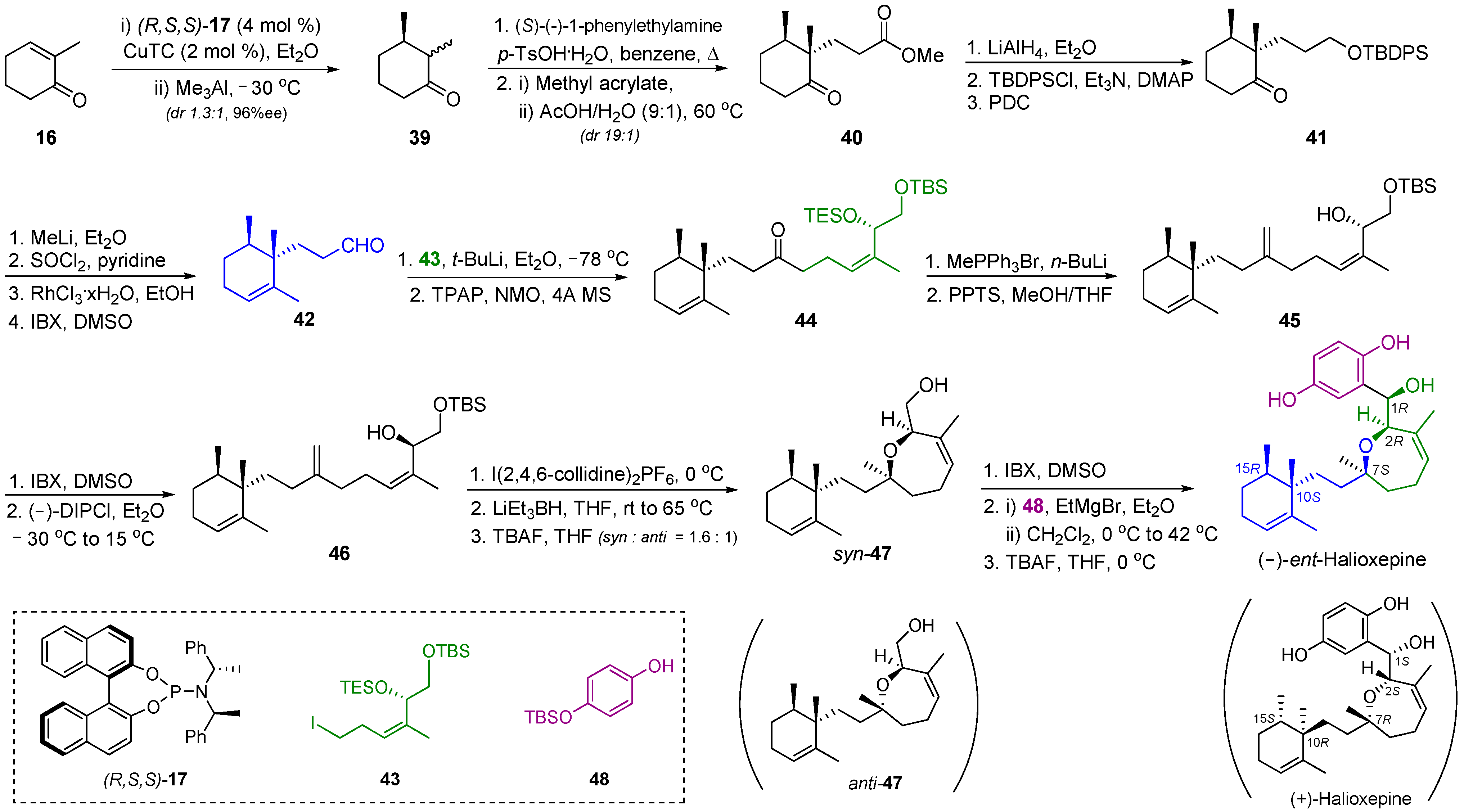
2.5. Synthesis of Halioxepine
2.6. Synthesis of Iriomoteolide-2a
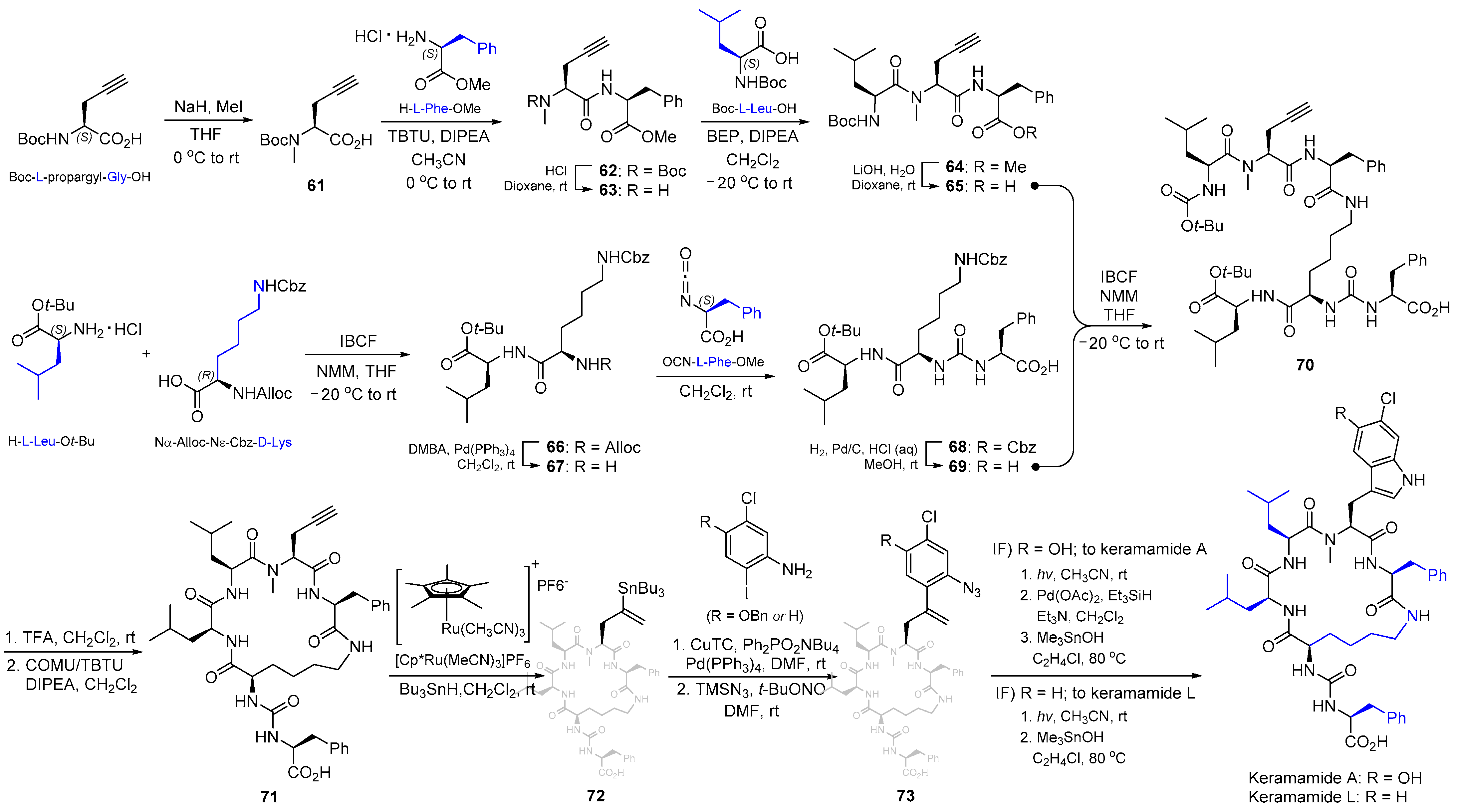
2.7. Synthesis of Keramamide A & L and Mozamamide A
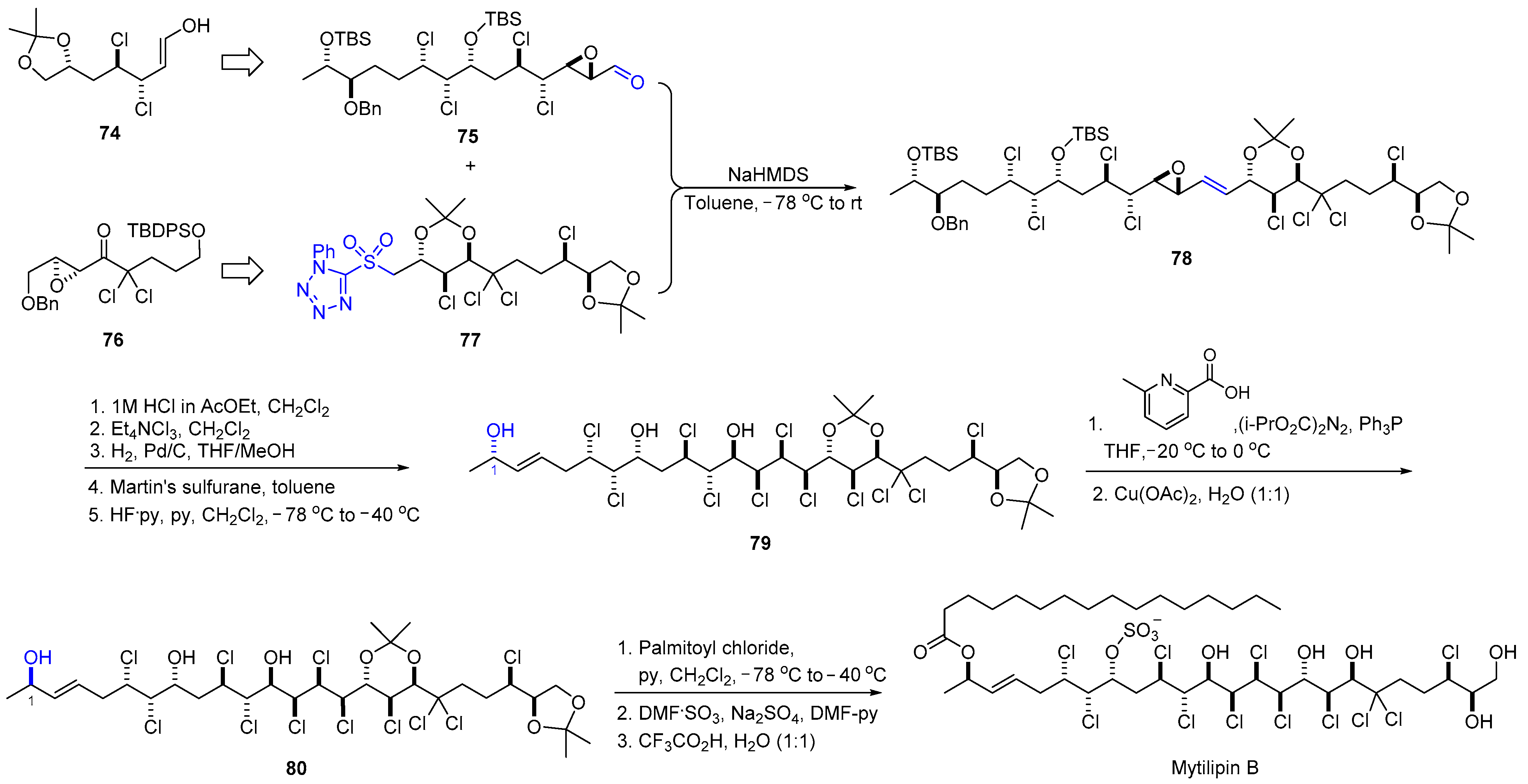
2.8. Synthesis of Mytilipin B
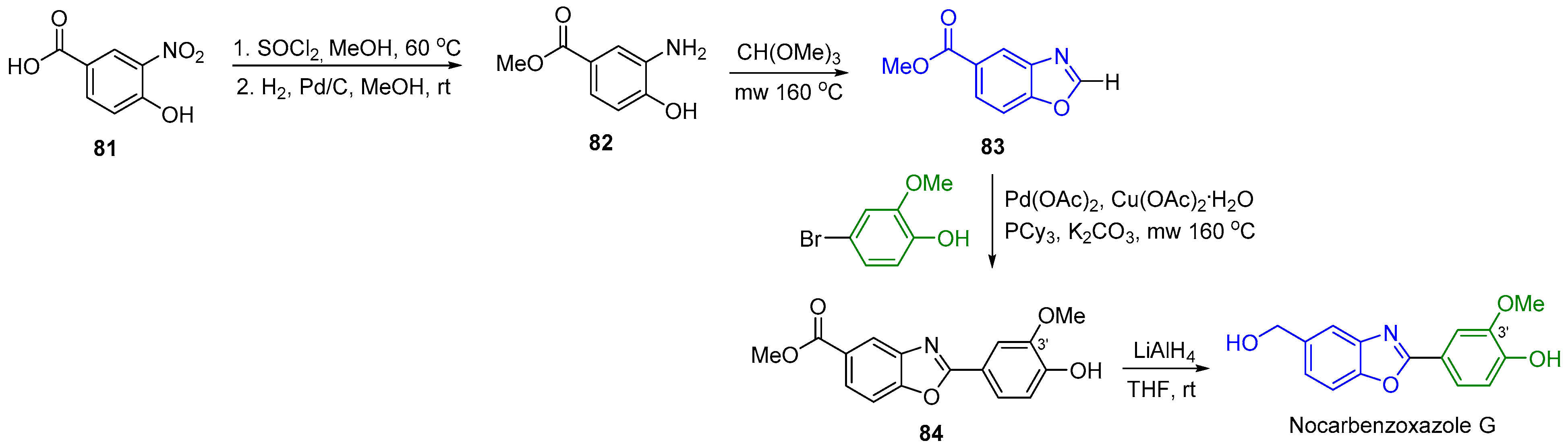
2.9. Synthesis of Nocarbenzoxazole G
2.10. Synthesis of Solomonamide A and B
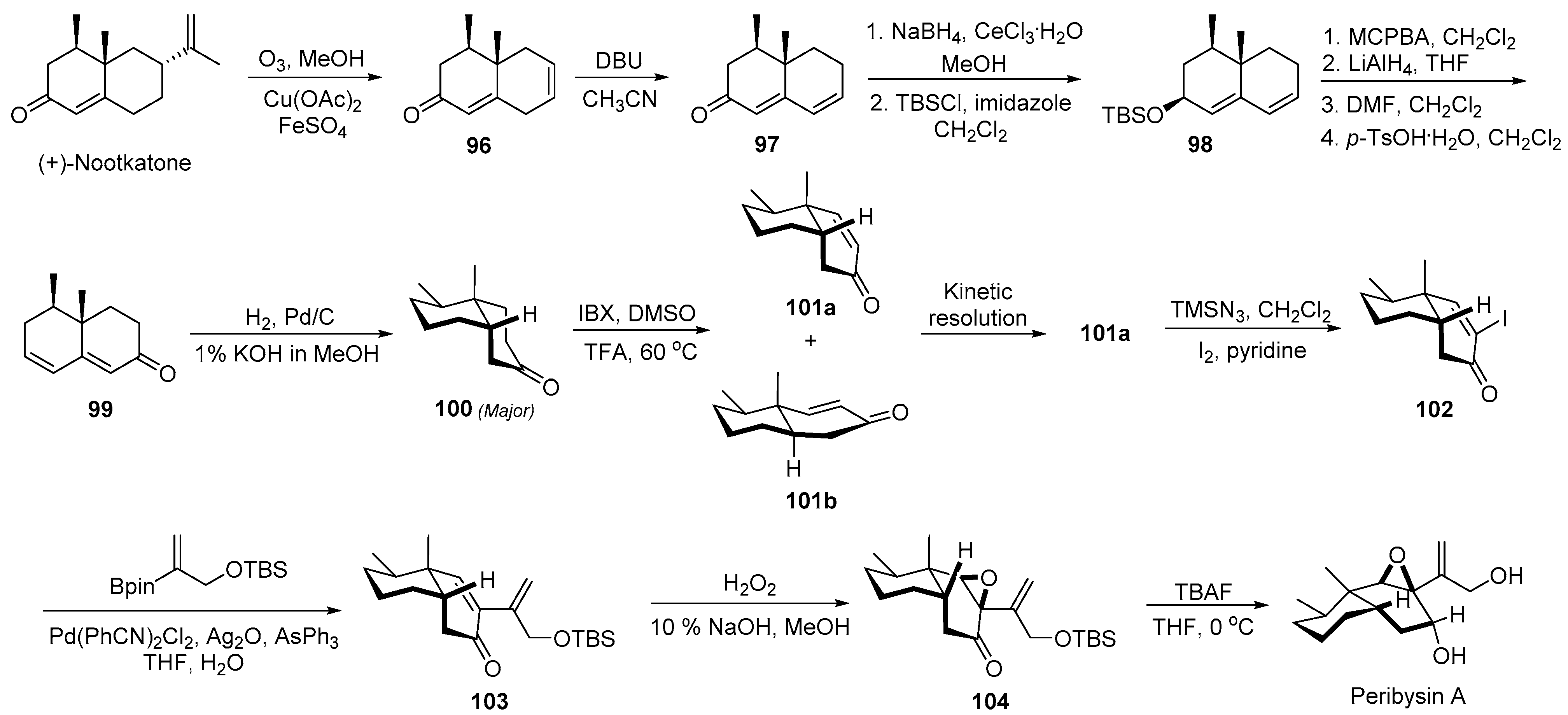
2.11. Synthesis of Peribysin A, B, C, F, and G
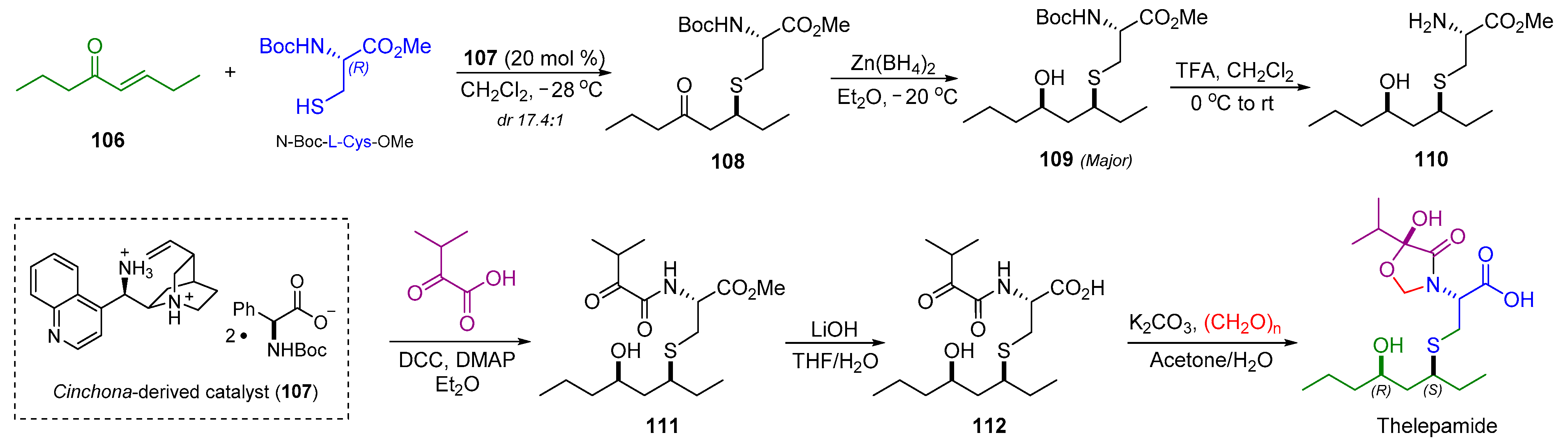
2.12. Synthesis of Thelepamide
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Colegate, S.M.; Molyneux, R.J. An introduction and overview. In Bioactive Natural Products: Detection, Isolation, and Structural Determination, 2nd ed.; Colegate, S.M., Molyneux, R.J., Eds.; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–11. [Google Scholar]
- Vuong, T.V. Natural products and their derivatives with antibacterial, antioxidant and anticancer activities. Antibiotics 2021, 10, 70. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural products as a foundation for drug discovery. Curr. Protoc. Pharmacol. 2009, 46, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Qian, P.Y.; Li, Z.; Xu, Y.; Li, Y.; Fusetani, N. Mini-review: Marine natural products and their synthetic analogs as antifouling compounds: 2009–2014. Biofouling 2015, 31, 101–122. [Google Scholar] [CrossRef]
- Brown, P.D.; Lawrence, A.L. The importance of asking “how and why?” in natural product structure elucidation. Nat. Prod. Rep. 2017, 34, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.; Aires-de-Sousa, J. Computational methodologies in the exploration of marine natural product leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Oh, J.; Lee, T.H. Structure elucidation of small organic molecules by contemporary computational chemistry methods. Arch. Pharmacal Res. 2020, 43, 1114–1127. [Google Scholar] [CrossRef]
- Yun, H.; Paek, S.M.; Jung, J.W.; Kim, N.J.; Kim, S.H.; Suh, Y.G. First total syntheses of (−)-macrosphelides J and K and elucidation of their absolute configuration. Chem. Commun. 2009, 18, 2463–2465. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem. Int. Ed. 2005, 44, 1012–1044. [Google Scholar] [CrossRef]
- Maier, M.E. Structural revisions of natural products by total synthesis. Nat. Prod. Rep. 2009, 26, 1105–1124. [Google Scholar] [CrossRef]
- Usami, Y. Recent synthetic studies leading to structural revisions of marine natural products. Mar. Drugs 2009, 7, 314–330. [Google Scholar] [CrossRef]
- Suyama, T.L.; Gerwick, W.H.; McPhail, K.L. Survey of marine natural product structure revisions: A synergy of spectroscopy and chemical synthesis. Bioorg. Med. Chem. 2011, 19, 6675–6701. [Google Scholar] [CrossRef] [Green Version]
- Chhetri, B.K.; Lavoie, S.; Sweeney-Jones, A.M.; Kubanek, J. Recent trends in the structural revision of natural products. Nat. Prod. Rep. 2018, 35, 514–531. [Google Scholar] [CrossRef]
- Han, F.; Liu, G.; Zhang, X.; Ding, Y.; Wang, L.; Wu, Y.; Chen, Y.; Zhang, Q. Total synthesis and structure revision of boholamide A. Org. Lett. 2021, 23, 4976–4980. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.; Zhu, L.; Peng, X.; Feng, Y.; Lu, Y.; Hu, X.; Liang, J.; Zhao, Q.; Wang, Z. Total synthesis and structure revision of chrysamide B. Org. Chem. Front. 2018, 5, 3402–3405. [Google Scholar] [CrossRef]
- Baars, J.; Grimm, I.; Blunk, D.; Neudörfl, J.M.; Schmalz, H.G. Enantioselective total synthesis and structural revision of dysiherbol A. Angew. Chem. Int. Ed. 2021, 60, 14915–14920. [Google Scholar] [CrossRef]
- Chong, C.; Lu, Z. Bioinspired total synthesis of marine anticancer meroterpenoids dysideanone B and dysiherbol A and structure revision of dysiherbol A. Synlett 2021, 32, 1777–1783. [Google Scholar]
- Chong, C.; Zhang, Q.; Ke, J.; Zhang, H.; Yang, X.; Wang, B.; Ding, W.; Lu, Z. Total synthesis of anti-cancer meroterpenoids dysideanone B and dysiherbol A and structural reassignment of dysiherbol A. Angew. Chem. Int. Ed. 2021, 60, 13807–13813. [Google Scholar] [CrossRef]
- Holland, D.C.; Kiefel, M.J.; Carroll, A.R. Structure revisions of the sponge-derived dibrominated bis-indole alkaloids, echinosulfone A and the echinosulfonic acids A to D. J. Org. Chem. 2020, 85, 3490–3496. [Google Scholar] [CrossRef]
- Neupane, P.; Salim, A.A.; Capon, R.J. Structure revision of the rare sponge metabolite echinosulfone A, and biosynthetically related echinosulfonic acids A–D. Tetrahedron Lett. 2020, 61, 151651–151653. [Google Scholar] [CrossRef]
- Liu, H.B.; Imler, G.H.; Baldridge, K.K.; O’Connor, R.D.; Siegel, J.S.; Deschamps, J.R.; Bewley, C.A. X-ray crystallography and unexpected chiroptical properties reassign the configuration of haliclonadiamine. J. Am. Chem. Soc. 2020, 142, 2755–2759. [Google Scholar] [CrossRef]
- Poock, C.; Kalesse, M. Total synthesis and structure revision of halioxepine. Chem. Eur. J. 2021, 27, 1615–1619. [Google Scholar] [CrossRef]
- Sakamoto, K.; Hakamata, A.; Tsuda, M.; Fuwa, H. Total synthesis and stereochemical revision of iriomocteolide-2a. Angew. Chem. Int. Ed. 2018, 57, 3801–3805. [Google Scholar] [CrossRef]
- Dong, Y.; Ding, W.; Sun, C.; Ji, X.; Ling, C.; Zhou, Z.; Chen, Z.; Chen, X.; Ju, J. Julichrome monomers from marine gastropod mollusk-associated Streptomyces and stereochemical revision of julichromes Q3.5 and Q3.3. Chem. Biodivers. 2020, 17, e2000057. [Google Scholar]
- Junk, L.; Kazmaier, U. Total synthesis of keramamides A and L from a common precursor by late-stage indole synthesis and configurational revision. Angew. Chem. Int. Ed. 2018, 57, 11432–11435. [Google Scholar] [CrossRef]
- Junk, L.; Kazmaier, U. Total synthesis and configurational revision of mozamide A, a hydroxy-brunsvicamide. J. Org. Chem. 2019, 84, 2489–2500. [Google Scholar] [CrossRef]
- Sondermann, P.; Carreira, E.M. Stereochemical revision, total synthesis, and solution state conformation of the complex chlorosulfolipid mytilipin B. J. Am. Chem. Soc. 2019, 141, 10510–10519. [Google Scholar] [CrossRef]
- Kim, T.; Lee, S.A.; Noh, T.; Choi, P.; Choi, S.-J.; Song, B.G.; Kim, Y.; Park, Y.-T.; Huh, G.; Kim, Y.-J.; et al. Synthesis, structure revision, and cytotoxicity of nocarbenzoxazole G. J. Nat. Prod. 2019, 82, 1325–1330. [Google Scholar] [CrossRef]
- Jachak, G.R.; Athawale, P.R.; Agarwal, H.; Barthwal, M.K.; Lauro, G.; Bifulco, G.; Reddy, D.S. Total synthesis of the potent anti-inflammatory natural product solomonamide A along with structural revision and biological activity evaluation. Org. Biomol. Chem. 2018, 16, 9138–9142. [Google Scholar] [CrossRef]
- Kashinath, K.; Jachak, G.R.; Athawale, P.R.; Marelli, U.K.; Gonnade, R.G.; Reddy, D.S. Total synthesis of the marine natural product solomonamide B necessitates stereochemical revision. Org. Lett. 2016, 18, 3178–3181. [Google Scholar] [CrossRef]
- Athawale, P.R.; Kalmode, H.P.; Motiwala, Z.; Kulkarni, K.A.; Reddy, D.S. Overturning the peribysin family natural products isolated from Periconia byssoides OUPS-N133: Synthesis and stereochemical revision of peribysins A, B, C, F, and G. Org. Lett. 2020, 22, 3104–3109. [Google Scholar] [CrossRef]
- Seitz, T.; Millán, R.E.; Lentz, D.; Jiménez, C.; Rodríguez, J.; Christmann, M. Synthesis of thelepamide via catalyst-controlled 1, 4-addition of cysteine derivatives and structure revision of thelepamide. Org. Lett. 2018, 20, 594–597. [Google Scholar] [CrossRef]
- Li, X.; Li, X.M.; Wang, B.G. Structural revision of wentiquinone C and related congeners from anthraquinones to xanthones using chemical derivatization and NMR analysis. Mar. Drugs 2019, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.P.; Lin, Z.; Fenton, D.S.; Leavitt, L.U.; Niu, C.; Lam, P.-Y.; Robes, J.M.; Peterson, R.T.; Concepcion, G.P.; Haygood, M.G.; et al. Boholamide A, an APD-class, hypoxia-selective cyclodepsipeptide. J. Nat. Prod. 2002, 83, 1249–1257. [Google Scholar] [CrossRef]
- Mcbrien, K.D.; Berry, R.L.; Lowe, S.E.; Neddermann, K.M.; Bursuker, I.; Huang, S.; Klohr, S.R.; Leet, J.E. Rakicidins, new cytotoxic lipopeptides from Micromonospora sp. fermentation, isolation and characterization. J. Antibiot. 1995, 48, 1446–1452. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wunderlich, D.; Sattler, I.; Feng, X.; Grabley, S.; Thiericke, R. Rakicidin C, a new cyclic depsipeptide from Streptomyces sp. Eur. J. Org. Chem. 2000, 2000, 3353–3356. [Google Scholar] [CrossRef]
- Igarashi, Y.; Shimasaki, R.; Miyanaga, S.; Oku, N.; Onaka, H.; Sakurai, H.; Saiki, I.; Kitani, S.; Nihira, T.; Wimonsiravude, W.; et al. Rakicidin D, an inhibitor of tumor cell invasion from marine-derived Streptomyces sp. J. Antibiot. 2010, 63, 563–565. [Google Scholar] [CrossRef]
- Oku, N.; Matoba, S.; Yamazaki, Y.M.; Shimasaki, R.; Miyanaga, S.; Igarashi, Y. Complete stereochemistry and preliminary structure activity relationship of rakicidin A, a hypoxia-selective cytotoxin from Micromonospora sp. J. Nat. Prod. 2014, 77, 2561–2565. [Google Scholar] [CrossRef]
- Kitani, S.; Ueguchi, T.; Igarashi, Y.; Leetanasaksakul, K.; Thamchaipenet, A.; Nihira, T. Rakicidin F, a new antibacterial cyclic depsipeptide from a marine sponge-derived Streptomyces sp. J. Antibiot. 2018, 71, 139–141. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, W.; Jiang, H.; Zhou, J.; Chen, X.; Lian, Y.; Jiang, H.; Lin, F. Rakicidins G–I, cyclic depsipeptides from marine Micromonospora chalcea FIM 02-523. Tetrahedron 2018, 74, 4151–4154. [Google Scholar] [CrossRef]
- Igarashi, M.; Shida, T.; Sasaki, Y.; Kinoshita, N.; Naganawa, H.; Hamada, M.; Takeuchi, T. Vinylamycin, a new depsipeptide antibiotic, from Streptomyces sp. J. Antibiot. 1999, 52, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Carr, G.; Poulsen, M.; Klassen, J.L.; Hou, Y.; Wyche, T.P.; Bugni, T.S.; Currie, C.R.; Clardy, J. Microtermolides A and B from termite-associated Streptomyces sp. and structural revision of vinylamycin. Org. Lett. 2012, 14, 2822–2825. [Google Scholar] [CrossRef]
- Nishioka, H.; Nakajima, S.; Nagashima, M.; Kojiri, K.; Suda, H. BE-43547 Series Substances, Their Manufacture with Streptomyces Species, and Their Use as Antitumor Agents. Patent JP10147594-A, 2 June 1998. [Google Scholar]
- Oppolzer, W.; Lienard, P. Efficient asymmetric synthesis of anti-aldols from bornanesultam derived boryl enolates. Tetrahedron Lett. 1993, 34, 4321–4324. [Google Scholar] [CrossRef]
- Decicco, C.P.; Grover, P. Total asymmetric synthesis of the potent immunosuppressive marine natural product microcolin A. J. Org. Chem. 1996, 61, 3534–3541. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Lin, X.; Zhao, B.; Wei, X.; Li, G.; Kaliaperumal, K.; Liao, S.; Yang, B.; Zhou, X.; et al. Chrysamides A-C, three dimeric nitrophenyl trans-epoxyamides produced by the deep-sea-derived fungus Penicillium chrysogenum SCSIO41001. Org. Lett. 2016, 18, 3650–3653. [Google Scholar] [CrossRef]
- Kojo, S.; Uchino, H.; Yoshimura, M.; Tanaka, K. Racemic d,l-asparagine causes enantiomeric excess of other coexisting racemic d,l-amino acids during recrystallization: A hypothesis accounting for the origin of l-amino acids in the biosphere. Chem. Commun. 2004, 2146–2147. [Google Scholar] [CrossRef]
- Rikken, G.L.J.A.; Raupach, E. Enantioselective magnetochiral photochemistry. Nature 2000, 405, 932–935. [Google Scholar] [CrossRef]
- Bérubé, C.; Carpentier, C.; Voyer, N. Total synthesis of chrysamide B. Tetrahedron Lett. 2017, 58, 2334–2336. [Google Scholar] [CrossRef]
- Fürstner, A.; Gastner, T. Total synthesis of cristatic acid. Org. Lett. 2000, 2, 2467–2470. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, P.; Zhang, D.; Baunach, M.; Hertweck, C.; Li, A. Bioinspired total synthesis of sespenine. Angew. Chem. Int. Ed. 2014, 53, 1–6. [Google Scholar] [CrossRef]
- Sun, C.; Lee, H.; Lee, D. Synthesis of the carbocyclic core of massadine. Org. Lett. 2015, 17, 5348–5351. [Google Scholar] [CrossRef]
- Jiao, W.H.; Shi, G.H.; Xu, T.T.; Chen, G.D.; Gu, B.B.; Wang, Z.; Peng, S.; Wang, S.P.; Li, J.; Han, B.N.; et al. Dysiherbols A–C and dysideanone E, cytotoxic and NF-κB inhibitory tetracyclic meroterpenes from a Dysidea sp. marine sponge. J. Nat. Prod. 2016, 79, 406–411. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-catalyzed organic reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Davies, P.W. Catalytic carbophilic activation: Catalysis by platinum and gold π acids. Angew. Chem. Int. Ed. 2007, 46, 3410–3449. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold (I)-catalyzed activation of alkynes for the construction of molecular complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [Green Version]
- Zi, W.; Toste, F.D. Recent advances in enantioselective gold catalysis. Chem. Soc. Rev. 2016, 45, 4567–4589. [Google Scholar] [CrossRef]
- Pflästerer, D.; Hashmi, A.S.K. Gold catalysis in total synthesis—Recent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef]
- Alexakis, A.; Albrow, V.; Biswas, K.; d’Augustin, M.; Prieto, O.; Woodward, S. Highly enantioselective copper (I)-phosphoramidite-catalysed additions of organoaluminium reagents to enones. Chem. Commun. 2005, 22, 2843–2845. [Google Scholar] [CrossRef]
- Kim, D.E.; Zhu, Y.; Newhouse, T.R. Vinylogous acyl triflates as an entry point to α, β-disubstituted cyclic enones via Suzuki–Miyaura cross-coupling. Org. Biomol. Chem. 2019, 17, 1796–1799. [Google Scholar] [CrossRef]
- Wildermuth, R.; Speck, K.; Magauer, T. Gold (i)-catalyzed enyne cyclizations: Studies toward the total synthesis of (+)-aureol. Synthesis 2016, 48, 1814–1824. [Google Scholar]
- Furukawa, J.; Kawabata, N.; Nishimura, J. Synthesis of cyclopropanes by the reaction of olefins with dialkylzinc and methylene iodide. Tetrahedron 1968, 24, 53–58. [Google Scholar] [CrossRef]
- Solé, D.; Vallverdú, L.; Solans, X.; Font-Bardía, M.; Bonjoch, J. Intramolecular Pd-mediated processes of amino-tethered aryl halides and ketones: Insight into the ketone α-arylation and carbonyl-addition dichotomy. A new class of four-membered azapalladacycles. J. Am. Chem. Soc. 2003, 125, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, S.; Yuan, Z.; Chen, P.; Xie, X.; Wang, X.; She, X. Palladium-promoted neutral 1,4-Brook rearrangement/intramolecular allylic cyclization cascade reaction: A strategy for the construction of vinyl cyclobutanols. Org. Lett. 2017, 19, 3478–3481. [Google Scholar] [CrossRef] [PubMed]
- Ovenden, S.P.B.; Capon, R.J. Echinosulfonic Acids A-C and echinosulfone A: Novel bromoindole sulfonic acids and a sulfone from a southern Australian marine sponge. J. Nat. Prod. 1999, 62, 1246–1249. [Google Scholar] [CrossRef]
- Heacock, R.A.; Kašpárek, S. The indole Grignard reagents. Adv. Heterocycl. Chem. 1969, 10, 43–112. [Google Scholar]
- Giannini, M.; Ornella Tinti, M.; Pisano, C.; Moretti, G.; Minetti, P.; Garattini, E.; Penco, S.G.M. Bis-Heterocyclic Compounds with Antitumour and Chemosensitising Activity. U.S. Patent 20060211759A1, 21 September 2006. [Google Scholar]
- Trianto, A.; Hermawan, I.; De Voogd, N.J.; Tanaka, J. Halioxepine, a new meroditerpene from an Indonesian sponge Haliclona sp. Chem. Pharm. Bull. 2011, 59, 1311–1313. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, G.; Benedit, G.; Fernández, R.; Pérez, M.; Rodriguez, J.; Jiménez, C.; Cuevas, C. Can stereoclusters separated by two methylene groups be related by DFT studies? The case of the cytotoxic meroditerpenes halioxepines. J. Nat. Prod. 2018, 81, 343–348. [Google Scholar] [CrossRef]
- Vuagnoux-d’Augustin, M.; Alexakis, A. Copper-catalyzed asymmetric conjugate addition of trialkylaluminium reagents to trisubstituted enones: Construction of chiral quaternary centers. Chem. Eur. J. 2007, 13, 9647–9662. [Google Scholar] [CrossRef]
- Tori, M.; Hisazumi, K.; Wada, T.; Sono, M.; Nakashima, K. Total synthesis of optically active liverwort sesquiterpenes, trifarienols A and B, using phenylethylamine as a chiral auxiliary. Tetrahedron Asymmetry 1999, 10, 961–971. [Google Scholar] [CrossRef]
- Stahl, P.; Kissau, L.; Mazitschek, R.; Huwe, A.; Furet, P.; Giannis, A.; Waldmann, H. Total synthesis and biological evaluation of the nakijiquinones. J. Am. Chem. Soc. 2001, 123, 11586–11593. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Kumagai, K.; Tsuda, M.; Masuda, A. Iriomoteolide-2a, a cytotoxic 23-membered macrolide from marine benthic dinoflagellate Amphidinium species. Heterocycles 2015, 91, 265–274. [Google Scholar]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon−proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1, 3-dimesityl-4, 5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
- Corey, E.J.; Bakshi, R.K.; Shibata, S. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc. 1987, 109, 5551–5553. [Google Scholar] [CrossRef]
- Williams, D.R.; Meyer, K.G. Total synthesis of (+)-amphidinolide K. J. Am. Chem. Soc. 2001, 123, 765–766. [Google Scholar] [CrossRef]
- Müller, S.; Liepold, B.; Roth, G.J.; Bestmann, H.J. An improved one-pot procedure for the synthesis of alkynes from aldehydes. Synlett 1996, 1996, 521–522. [Google Scholar] [CrossRef]
- Zurwerra, D.; Gertsch, J.; Altmann, K.H. Synthesis of (−)-dactylolide and 13-desmethylene-(−)-dactylolide and their effects on tubulin. Org. Lett. 2010, 12, 2302–2305. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.I.; Sato, M.; Ishibashi, M.; Shigemori, H.; Nakamura, T.; Ohizumi, Y. Keramamide A, a novel peptide from the Okinawan marine sponge Theonella sp. J. Chem. Soc. Perkin Trans. 1991, 1, 2609–2611. [Google Scholar] [CrossRef]
- Uemoto, H.; Yahiro, Y.; Shigemori, H.; Tsuda, M.; Takao, T.; Shimonishi, Y.; Kobayashi, J.I. Keramamides K and L, new cyclic peptides containing unusual tryptophan residue from theonella sponge. Tetrahedron 1996, 54, 6719–6724. [Google Scholar] [CrossRef]
- Junk, L.; Kazmaier, U. Synthesis of indoles and tryptophan derivatives via photoinduced nitrene C–H insertion. Org. Biomol. Chem. 2016, 14, 2916–2923. [Google Scholar] [CrossRef] [PubMed]
- Junk, L.; Kazmaier, U. A straightforward protocol for the synthesis of functionalized tryptophan peptides via Stille coupling, azidation and photoinduced nitrene insertion. Synlett 2016, 27, 1531–1536. [Google Scholar]
- Schmidt, E.W.; Harper, M.K.; Faulkner, D.J. Mozamides A and B, cyclic peptides from a theonellid sponge from Mozambique. J. Nat. Prod. 1997, 60, 779–782. [Google Scholar] [CrossRef]
- Elovson, J.; Vagelos, P.R. A new class of lipids: Chlorosulfolipids. Proc. Natl. Acad. Sci. USA 1969, 62, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Di Rosa, M.; Ianaro, A.; Poletti, R. Structure and stereochemistry of a new cytotoxic polychlorinated sulfolipid from adriatic shellfish. J. Am. Chem. Soc. 2002, 124, 13114–13120. [Google Scholar] [CrossRef]
- Blakemore, P.R.; Cole, W.J.; Kocienski, P.J.; Morley, A. A stereoselective synthesis of trans-1,2-disubstituted alkenes based on the condensation of aldehydes with metallated 1-phenyl-1H-tetrazol-5-yl sulfones. Synlett 1998, 1, 26–28. [Google Scholar] [CrossRef]
- Shibuya, G.M.; Kanady, J.S.; Vanderwal, C.D. Stereoselective dichlorination of allylic alcohol derivatives to access key stereochemical arrays of the chlorosulfolipids. J. Am. Chem. Soc. 2008, 130, 12514–12518. [Google Scholar] [CrossRef]
- Nilewski, C.; Deprez, N.R.; Fessard, T.C.; Li, D.B.; Geisser, R.W.; Carreira, E.M. Synthesis of undecachlorosulfolipid A: Re-evaluation of the nominal structure. Angew. Chem. Int. Ed. 2011, 50, 7940–7943. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, X.; Hao, H.; Li, W.; Lu, C. Nocarbenzoxazoles A–G, benzoxazoles produced by halophilic Nocardiopsis lucentensis DSM 44048. J. Nat. Prod. 2015, 78, 2123–2127. [Google Scholar] [CrossRef]
- Xu, D.; Chiaroni, A.; Fleury, M.B.; Largeron, M. Electrochemically induced cascade reaction for the assembly of libraries of biologically relevant 1,4-benzoxazine derivatives. J. Org. Chem. 2006, 71, 6374–6381. [Google Scholar] [CrossRef]
- Nakamura, H.; Yasui, Y.; Ban, H.S. Synthesis and biological evaluation of ortho-carborane containing benzoxazole as an inhibitor of hypoxia inducible factor (HIF)-1 transcriptional activity. J. Organomet. Chem. 2013, 747, 189–194. [Google Scholar] [CrossRef]
- Festa, C.; De Marino, S.; Sepe, V.; D’Auria, M.V.; Bifulco, G.; Debitus, C.; Bucci, M.; Vellecco, V.; Zampella, A. Solomonamides A and B, new anti-inflammatory peptides from Theonella swinhoei. Org. Lett. 2011, 13, 1532–1535. [Google Scholar] [CrossRef]
- Sheppard, G.S.; Wang, J.; Kawai, M.; BaMaung, N.Y.; Craig, R.A.; Erickson, S.A.; Lynch, L.; Patel, J.; Yang, F.; Searle, X.B.; et al. 3-amino-2-hydroxyamides and related compounds as inhibitors of methionine aminopeptidase-2. Bioorg. Med. Chem. Lett. 2004, 14, 865–868. [Google Scholar] [CrossRef]
- Kashinath, K.; Dhara, S.; Reddy, D.S. Breaking and making of olefins simultaneously using ozonolysis: Application to the synthesis of useful building blocks and macrocyclic core of solomonamides. Org. Lett. 2015, 17, 2090–2093. [Google Scholar] [CrossRef]
- Miller, D.G.; Wayner, D.D.M. Improved method for the Wacker oxidation of cyclic and internal olefins. J. Org. Chem. 1990, 55, 2924–2927. [Google Scholar] [CrossRef]
- Yamada, T.; Iritani, M.; Minoura, K.; Kawai, K.; Numata, A. Peribysins A–D, potent cell-adhesion inhibitors from a sea hare-derived culture of Periconia species. Org. Biomol. Chem. 2004, 2, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Doi, M.; Miura, A.; Harada, W.; Hiramura, M.; Minoura, K.; Tanaka, R.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, peribysins A, E, F and G, produced by a sea hare-derived Periconia sp. J. Antibiot. 2005, 58, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Angeles, A.R.; Dorn, D.C.; Kou, C.A.; Moore, M.A.S.; Danishefsky, S.J. Total synthesis of peribysin E necessitates revision of the assignment of its absolute configuration. Angew. Chem. Int. Ed. 2007, 46, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L.; Liew, W.-F. Iron/copper promoted fragmentation reactions of alpha-alkoxy hydroperoxides. The conversion of octalins into fourteen-membered ring macrolides. J. Am. Chem. Soc. 1985, 107, 2980–2982. [Google Scholar] [CrossRef]
- Čeković, Ž.; Green, M.M. Formation of remote double bonds by ferrous sulfate-cupric acetate promoted decomposition of alkyl hydroperoxides. J. Am. Chem. Soc. 1974, 96, 3000–3002. [Google Scholar] [CrossRef]
- Huang, D.; Schuppe, A.W.; Liang, M.Z.; Newhouse, T.R. Scalable procedure for the fragmentation of hydroperoxides mediated by copper and iron tetrafluoroborate salts. Org. Biomol. Chem. 2016, 14, 6197–6200. [Google Scholar] [CrossRef]
- Luche, J.L. Lanthanides in organic chemistry. 1. Selective 1, 2 reductions of conjugated ketones. J. Am. Chem. Soc. 1978, 100, 2226–2227. [Google Scholar] [CrossRef]
- Takagi, J.; Takahashi, K.; Ishiyama, T.; Miyaura, N. Palladium-catalyzed cross-coupling reaction of Bis(pinacolato)diboron with 1-alkenyl halides or triflates: Convenient synthesis of unsymmetrical 1,3-dienes via the borylation-coupling sequence. J. Am. Chem. Soc. 2002, 124, 8001–8006. [Google Scholar] [CrossRef]
- Rodriguez, J.; Nieto, R.M.; Blanco, M.; Valeriote, F.A.; Jimenez, C.; Crews, P. Thelepamide: An unprecedented ketide-amino acid from Thelepus crispus, a marine annelid worm. Org. Lett. 2014, 16, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Mahajan, S.; Enders, D. Organocatalytic carbon–sulfur bond-forming reactions. Chem. Rev. 2014, 114, 8807–8864. [Google Scholar] [CrossRef] [PubMed]
- Jouglet, B.; Oumoch, S.; Rousseau, G. An efficient hydroxymethylation of lactams. Synth. Commun. 1995, 25, 3869–3874. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Name | Initially Proposed Structure | Revised Structure | Discrepancy | Ref. |
|---|---|---|---|---|---|
| Molecular Class | Revision Clue | ||||
| 1 | Boholamide A | 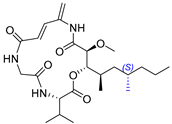 | 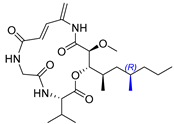 | Incorrect stereochemistry | [15] |
| Macrocyclic depsipeptide | Total synthesis | ||||
| 2 | Chrysamide B | 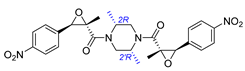 | 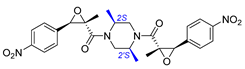 | Incorrect stereochemistry | [16] |
| Dimeric epoxyamide | Total synthesis | ||||
| 3 | Dysiherbol A | 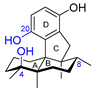 | 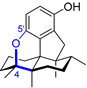 | Incorrect formula | [17,18,19] |
| Avarane meroterpene | Total synthesis | ||||
| 4 | Echinosulfone A | 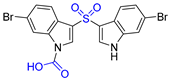 | 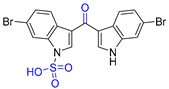 | Incorrect formula | [20] |
| bis-Indole alkaloid | Total synthesis | ||||
| 5 | Echinosulfonic acid A~D |  | 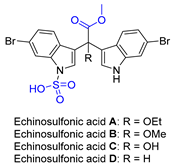 | Incorrect formula | [21] |
| bis-Indole alkaloid | Close inspection w/ plausible biosynthesis | ||||
| 6 | Haliclonadiamine | 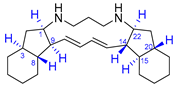 | 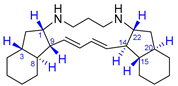 | Incorrect stereochemistry | [22] |
| bis-Indane | X-ray crystallography | ||||
| 7 | Halioxepine | 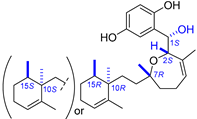 | 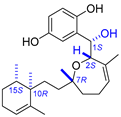 | Incorrect stereochemistry | [23] |
| Meroditerpene | Total synthesis | ||||
| 8 | Iriomoteolide-2a | 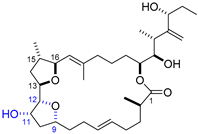 |  | Incorrect stereochemistry | [24] |
| 23-Membered macrocycle | Total synthesis | ||||
| 9 | Julichromes Q3.3 and Q3.5 | 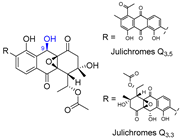 | 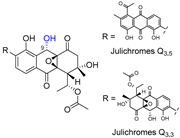 | Incorrect stereochemistry | [25] |
| Aromatic polyketide | X-ray crystallography | ||||
| 10 | Keramamide A and L | 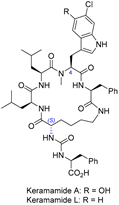 | 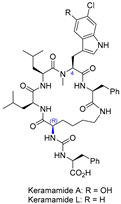 | Incorrect stereochemistry | [26] |
| Anabaenopeptin-type peptide | Total synthesis | ||||
| 11 | Mozamide A |  | 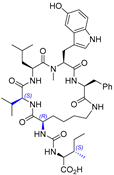 | Incorrect stereochemistry | [27] |
| Anabaenopeptin-type peptide | Total synthesis | ||||
| 12 | Mytilipin B | 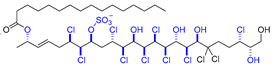 |  | Incorrect stereochemistry | [28] |
| Chlorosulfolipid | Total synthesis | ||||
| 13 | Nocarbenzoxazole G | 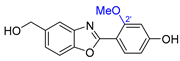 |  | Incorrect constitution | [29] |
| Nocarbobenzoxazole (2-arylbenzoxazole) | Total synthesis | ||||
| 14 | Solomonamide A and B | 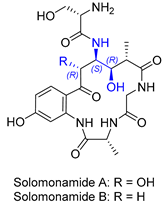 |  | Incorrect stereochemistry | [30,31] |
| Marocyclic peptide | Total synthesis | ||||
| 15 | Peribysins A, B, C, F, and G | 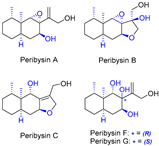 | 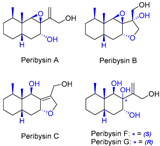 | Incorrect stereochemistry | [32] |
| Peribysin family | Total synthesis | ||||
| 16 | Thelepamide | 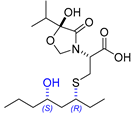 |  | Incorrect stereochemistry | [33] |
| Tetraketide -amino acid | Total synthesis | ||||
| 17 | Wentiquinone C | 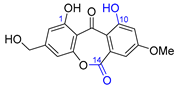 | 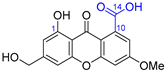 | Incorrect constitution | [34] |
| Xanthone | Chemical derivatization |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, M.W.; Kim, J.; Paek, S.-M. Recent Achievements in Total Synthesis for Integral Structural Revisions of Marine Natural Products. Mar. Drugs 2022, 20, 171. https://doi.org/10.3390/md20030171
Ha MW, Kim J, Paek S-M. Recent Achievements in Total Synthesis for Integral Structural Revisions of Marine Natural Products. Marine Drugs. 2022; 20(3):171. https://doi.org/10.3390/md20030171
Chicago/Turabian StyleHa, Min Woo, Jonghoon Kim, and Seung-Mann Paek. 2022. "Recent Achievements in Total Synthesis for Integral Structural Revisions of Marine Natural Products" Marine Drugs 20, no. 3: 171. https://doi.org/10.3390/md20030171
APA StyleHa, M. W., Kim, J., & Paek, S.-M. (2022). Recent Achievements in Total Synthesis for Integral Structural Revisions of Marine Natural Products. Marine Drugs, 20(3), 171. https://doi.org/10.3390/md20030171