
Regioselective Synthesis of 6-O-Acetyl Dieckol and Its Selective Cytotoxicity against Non-Small-Cell Lung Cancer Cells

 , ,
, ,
Abstract

1. Introduction
2. Results
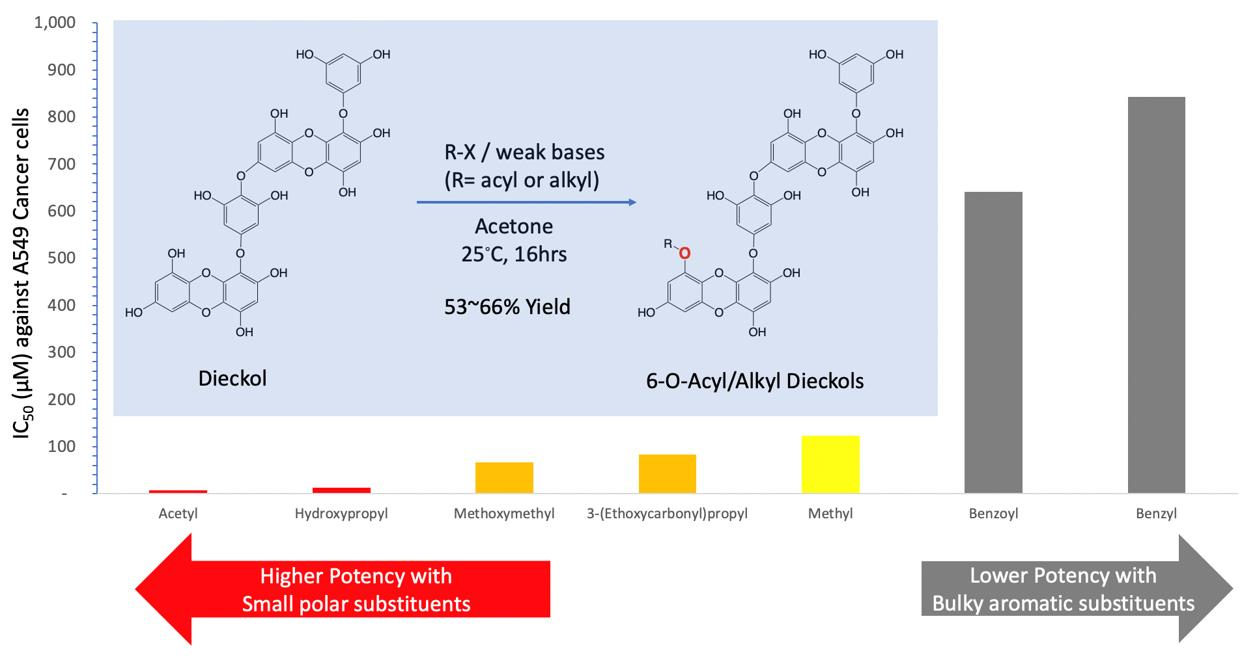
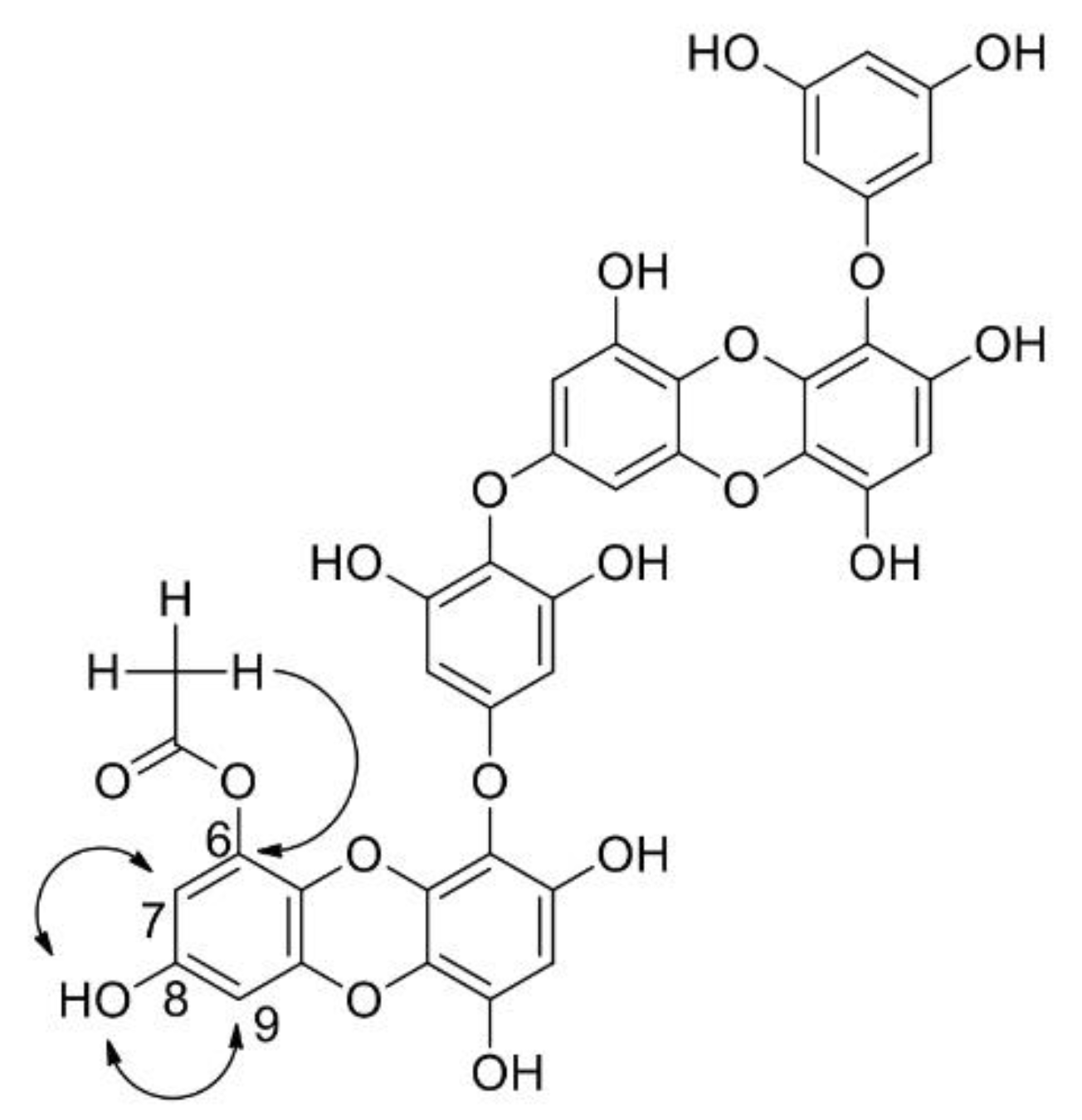
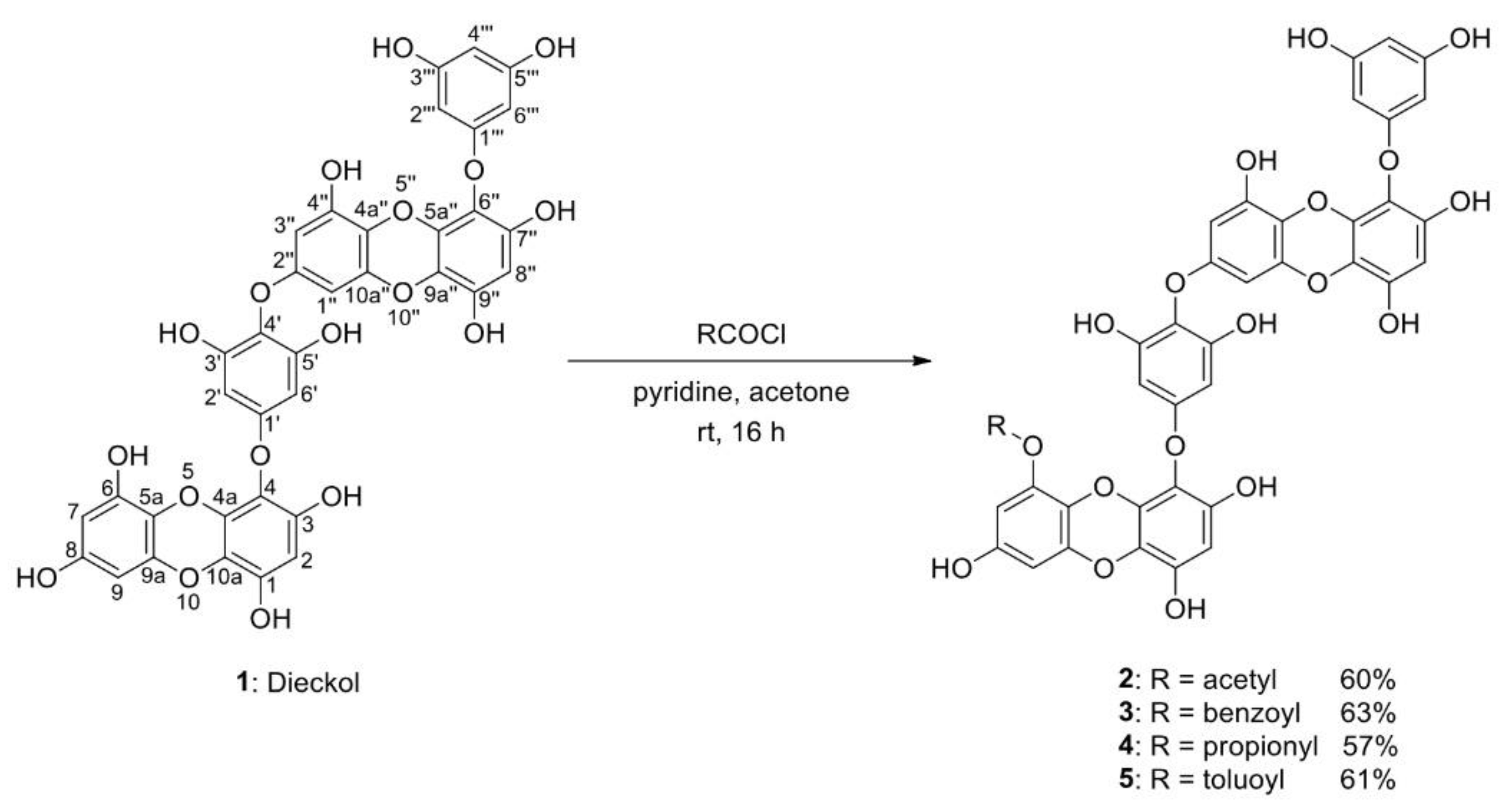
2.1. Acylation Reactions of Dieckol (1)
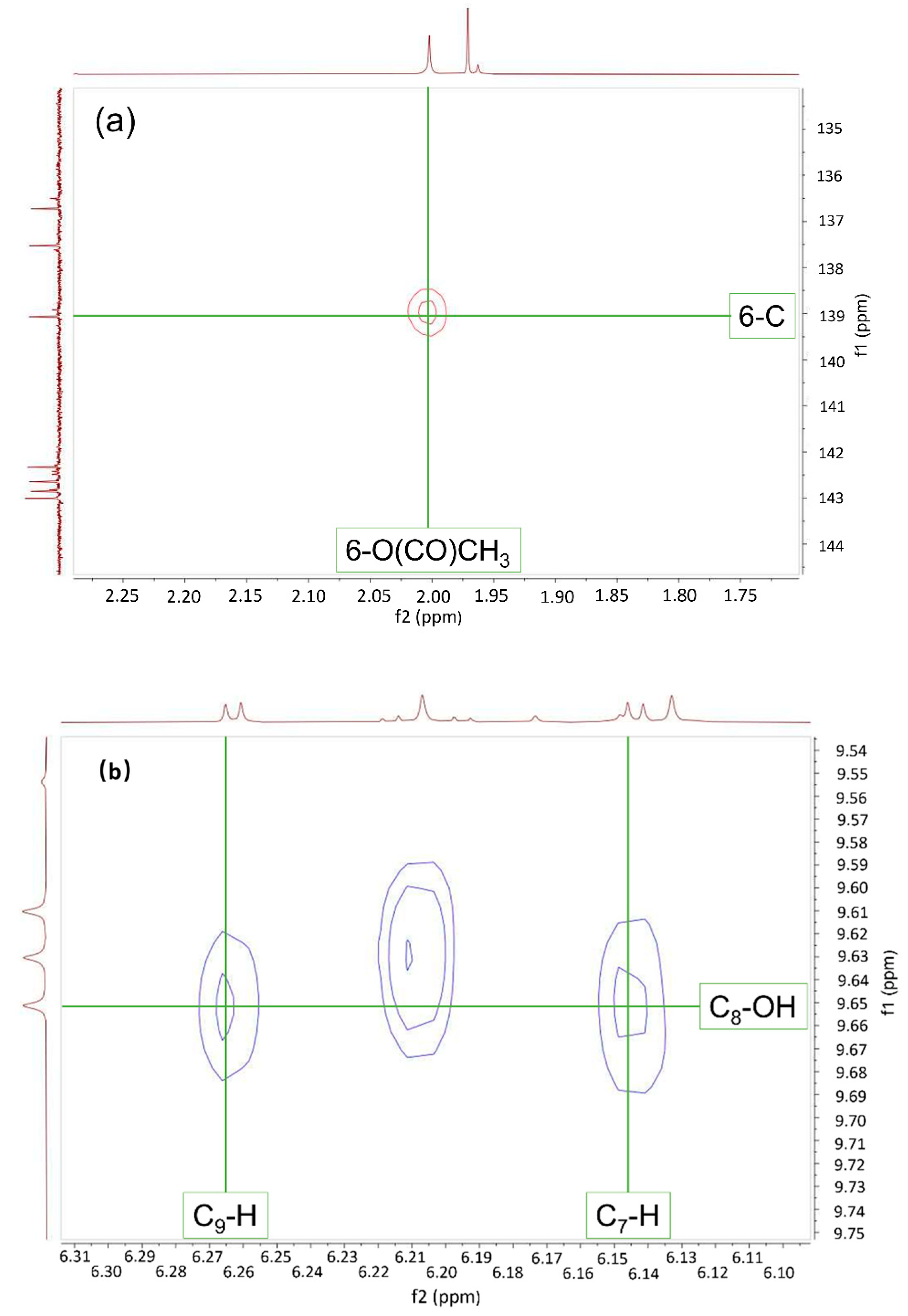
2.2. Spectroscopic Determination of Exact Structures of O-Acyl Dieckols
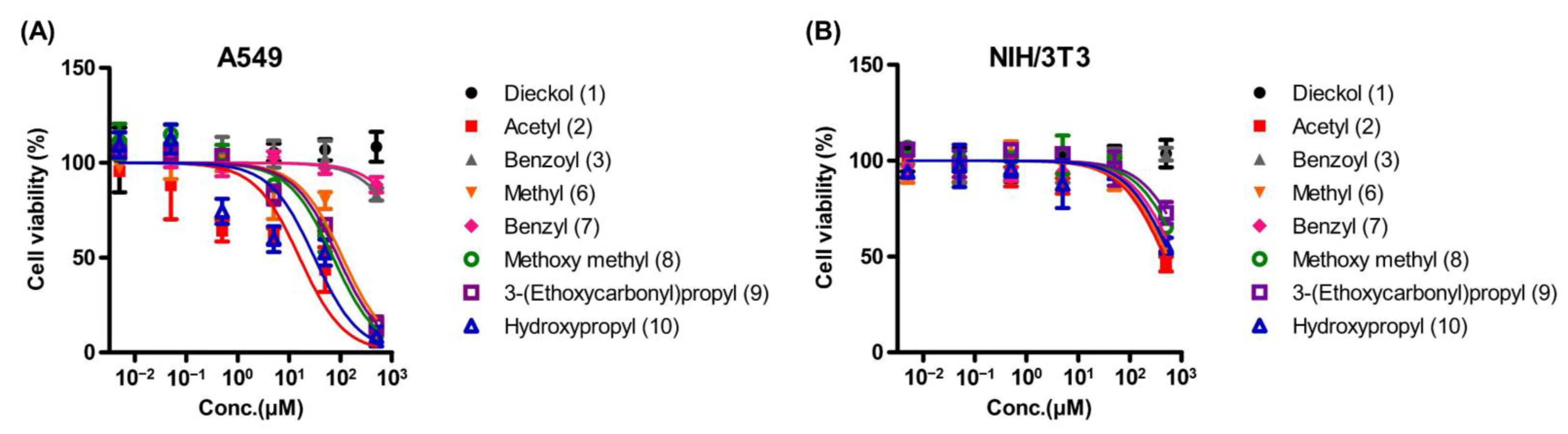
2.3. Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Analytical Methods
4.3. Preparation of Mono O-Acyl DKs
4.3.1. General Method of Preparation
4.3.2. Acetyl DK (2)
4.3.3. Benzoyl DK (3)
4.3.4. Propionyl DK (4)
4.3.5. Toluoyl DK (5)
4.4. Preparation of Mono-O-Alkyl DKs
4.5. Cell Lines and Culture
4.6. Cytotoxicity Assay
4.7. Selectivity Index
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of Anticancer Drug Toxicities: Paradigm Shift in Adverse Effect Profile. Life 2021, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Chemotherapy to Treat Cancer. National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/side-effects (accessed on 20 October 2022).
- Chemotherapy Side Effects, American Cancer Society. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/chemotherapy/chemotherapy-side-effects.html (accessed on 20 October 2022).
- Singh, I.P.; Bharate, S.B. Phloroglucinol compounds of natural origin. Nat. Prod. Rep. 2006, 23, 558–591. [Google Scholar] [CrossRef]
- Koirala, P.; Jung, H.A.; Choi, J.S. Recent advances in pharmacological research on Ecklonia species: A review. Arch. Pharmacal Res. 2017, 40, 981–1005. [Google Scholar] [CrossRef] [PubMed]
- Phloronol Inc. (25 September 2013–8 December 2014). Phase I Study of PH100 (Ecklonia Cava Phlorotannins). Identifier. NCT04335045. Available online: https://clinicaltrials.gov/ct2/show/NCT04335045 (accessed on 20 October 2022).
- Bota Bio Co., Ltd. (4 March 2016–4 May 2018). Phase 2a Study to Evaluate the Safety and Efficacy of PH100 Tablet in T2DM Patients with Recent Cardiovascular (PH100_IIa). Identifier NCT04141241. Available online: https://clinicaltrials.gov/ct2/show/NCT04141241 (accessed on 20 October 2022).
- Hwang, H.; Chen, T.; Nines, R.G.; Shin, H.-C.; Stoner, G.D. Photochemoprevention of UVB-induced skin carcinogenesis in SKH-1 mice by brown algae polyphenols. Int. J. Cancer 2006, 119, 2742–2749. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-I.; Ahn, J.-H.; Choi, Y.S.; Choi, J.-H. Brown algae phlorotannins enhance the tumoricidal effect of cisplatin and ameliorate cisplatin nephrotoxicity. Gynecol. Oncol. 2014, 136, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Sadeeshkumar, V.; Duraikannu, A.; Ravichandran, S.; Kodisundaram, P.; Fredrick, W.S.; Gobalakrishnan, R. Modulatory efficacy of dieckol on xenobiotic-metabolizing enzymes, cell proliferation, apoptosis, invasion and angiogenesis during NDEA-induced rat hepatocarcinogenesis. Mol. Cell. Biochem. 2017, 433, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, Y.T.; Jeon, Y.J. Antioxidant dieckol downregulates the Rac1/ROS signaling pathway and inhibits Wiskott-Aldrich syndrome protein (WASP)-family verprolin-homologous protein 2 (WAVE2)-mediated invasive migration of B16 mouse melanoma cells. Mol. Cells 2012, 33, 363–369. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.; Qian, Z.-J.; Lee, S.-H.; Kim, S.-K. Dieckol from Ecklonia cava Regulates Invasion of Human Fibrosarcoma Cells and Modulates MMP-2 and MMP-9 Expression via NF-κB Pathway. Evid. Based. Complement. Altern. Med. 2011, 2011, 140462. [Google Scholar] [CrossRef]
- Park, S.J.; Jeon, Y.J. Dieckol from Ecklonia cava suppresses the migration and invasion of HT1080 cells by inhibiting the focal adhesion kinase pathway downstream of Rac1-ROS signaling. Mol. Cells 2012, 33, 141–149. [Google Scholar] [CrossRef]
- Hyun, K.-H.; Yoon, C.-H.; Kim, R.-K.; Lim, E.-J.; An, S.; Park, M.-J.; Hyun, J.-W.; Suh, Y.; Kim, M.-J.; Lee, S.-J. Eckol suppresses maintenance of stemness and malignancies in glioma stem-like cells. Toxicol. Appl. Pharmacol. 2011, 254, 32–40. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Yang, Y.-I.; Lee, K.-T.; Choi, J.-H. Dieckol, isolated from the edible brown algae Ecklonia cava, induces apoptosis of ovarian cancer cells and inhibits tumor xenograft growth. J. Cancer Res. Clin. Oncol. 2014, 141, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; He, Y.; Yoon, B.; Koo, S.; Yang, Z.; Kang, E.J.; Lee, B.H.; Han, S.-Y.; Yoo, Y.C.; Lee, K.B.; et al. Synthesis of rhodamine-labelled dieckol: Its unique intracellular localization and potent anti-inflammatory activity. Chem. Commun. 2014, 50, 13045–13048. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Yang, Z.; Yoon, B.; He, Y.; Uhm, S.; Shin, H.-C.; Lee, B.H.; Yoo, Y.C.; Lee, K.B.; Han, S.-Y.; et al. Blood-brain barrier-permeable fluorone-labeled dieckols acting as neuronal ER stress signaling inhibitors. Biomaterials 2015, 61, 52–60. [Google Scholar] [CrossRef]
- Kim, Y.; Shin, J.; Kang, S.M.; Song, J.; Shin, H.-C.; Keum, Y.-S.; Hwang, H.J.; Park, K. Highly Regioselective Preparation and Characterization of New 6-O-Substituted Dieckol Derivatives. J. Ind. Eng. Chem. 2020, 91, 285–295. [Google Scholar] [CrossRef]
- Glombitza, K.-W.; Gerstberger, G. Phlorotannins with dibenzodioxin structural elements from the brown alga Eisenia arborea. Phytochemistry 1985, 24, 543–551. [Google Scholar] [CrossRef]
- Glombitza, K.-W.; Vogels, H.P. Antibiotics from Algae. XXXV. Phlorotannins from Ecklonia maxima. Planta Med. 1985, 51, 308–312. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, C.; Shen, L.; Shin, H.-C.; Lee, K.B.; Ji, B. Comparative analysis of oxidative mechanisms of phloroglucinol and dieckol by electrochemical, spectroscopic, cellular and computational methods. RSC Adv. 2018, 8, 1963–1972. [Google Scholar] [CrossRef]
- Lung Cancer Facts 2022. Lung Cancer Research Foundation: New York, NY, USA. 2022. Available online: https://www.lungcancerresearchfoundation.org/lung-cancer-facts/?gclid=Cj0KCQjwj7CZBhDHARIsAPPWv3e0skZT68gIHJYE7y9s9O57VZ8efgbRWpLflxrAQ9fz6DH7ZS8VpAgaApqHEALw_wcB (accessed on 20 October 2022).
- Basumallik, N.; Agarwal, M. Small Cell Lung Cancer. In StatPearls; StatPearls Publishing: Tempa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482458/#:~:text=Lung%20cancer%20is%20histologically%20divided,while%20NSCLC%20comprises%20approximately%2085%25 (accessed on 20 October 2022).
- Lee, J.G.; Hwang, H.J.; Lee, Y.S.; Shin, H. Conformational Isomerism for Eckol and its Skeleton: Theoretical Study. Bull. Korean Chem. Soc. 2019, 40, 935–936. [Google Scholar] [CrossRef]
- Rudyk, R.; Molina, M.; Yurquina, A.; Gómez, M.; Blanco, S.; Ferretti, F. A theoretical and experimental study on the structure and dipole moment of phloroglucinol in ethanol. J. Mol. Struct. 2004, 673, 231–238. [Google Scholar] [CrossRef]
- Chang, M.-C.; Wu, J.-Y.; Liao, H.-F.; Chen, Y.-J.; Kuo, C.-D. Comparative assessment of therapeutic safety of norcantharidin, N-farnesyloxy-norcantharimide, and N-farnesyl-norcantharimide against Jurkat T cells relative to human normal lymphoblast: A quantitative pilot study. Medicine 2016, 95, e4467. [Google Scholar] [CrossRef]
- Calderón-Montaño, J.M.; Martínez-Sánchez, S.M.; Jiménez-González, V.; Burgos-Morón, E.; Guillén-Mancina, E.; Jiménez-Alonso, J.J.; Díaz-Ortega, P.; García, F.; Aparicio, A.; López-Lázaro, M. Screening for Selective Anticancer Activity of 65 Extracts of Plants Collected in Western Andalusia, Spain. Plants 2021, 10, 2193. [Google Scholar] [CrossRef] [PubMed]
- Indrayanto, G.; Putra, G.-S.; Suhud, F. Chapter 6: Validation of in-vitro bioassay methods: Application in herbal drug research. In Profiles of Drug Substances, Excipients and Related Methodology; Al-Majed, A.-A., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 46, pp. 273–307. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
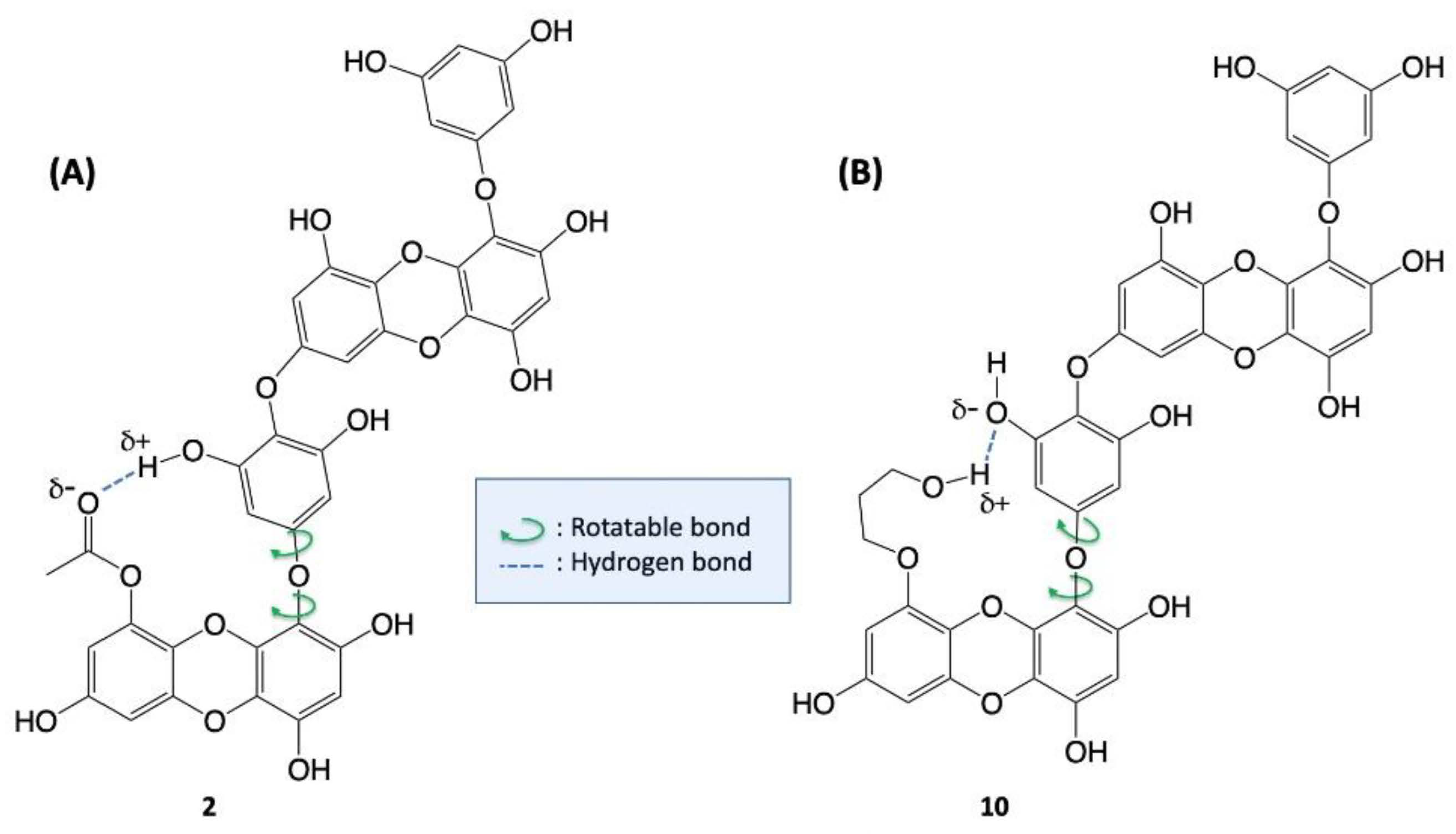{kind=link}
{kind=link}
| No. | δC | δH | (J in Hz) | HMBC (H → C) | NOESY |
|---|---|---|---|---|---|
| 1 | 142.61 | 9.65 | C2, C1, C10a | C2−H | |
| 2 | 99.04 | 6.22 | C4, C3, C1, C10a | C3−H, C1−H | |
| 3 | 146.85 | 9.38 | C4, C3, C2 | C2−H | |
| 4 | 122.30 | ||||
| 4a | 136.71 | ||||
| 5a | 126.36 | ||||
| 6 | 139.05 | ||||
| 7 | 104.91 | 6.16 | d (2.74) | C9, C8, C6, C5a | C8−H |
| 8 | 153.52 | 9.67 | C9, C8, C7 | C9−H, C7−H | |
| 9 | 101.01 | 6.28 | d (2.74) | C9a, C8, C7, C5a | C8−H |
| 9a | 142.99 | ||||
| 10a | 123.56 | ||||
| 1′ | 156.12 | ||||
| 2′ | 94.32 | 5.92 | C1′, C3′, C4′, C5′, C6′ | C3′−H | |
| 3′ | 151.66 | 9.31 | C2′, C3′, C4′, C5′, C6′ | C2′−H | |
| 4′ | 124.63 | ||||
| 5′ | 151.66 | 9.31 | C2′, C3′, C4′, C5′, C6′ | C6′−H | |
| 6′ | 94.32 | 5.92 | C1′, C2′, C3′, C4′, C5′ | C5′−H | |
| 1″ | 93.72 | 5.81 | d (2.86) | C10a″, C2″, C3″, C4a″ | |
| 2″ | 154.68 | - | |||
| 3″ | 98.81 | 6.06 | d (2.86) | C1″, C2″, C4″, C4a″ | C4"−H |
| 4″ | 146.33 | 9.63 | C3″, C4″, C4a″ | C3″−H | |
| 4a″ | 124.52 | ||||
| 5a″ | 137.51 | ||||
| 6″ | 122.61 | ||||
| 7″ | 146.46 | 9.19 | C6″, C7″, C8″ | C8″−H | |
| 8″ | 98.73 | 6.15 | C6″, C7″, C9″, C9a″ | C7″−H, C9″−H | |
| 9″ | 142.32 | 9.42 | C8″, C9″, C9a″ | C8″−H | |
| 9a″ | 123.58 | ||||
| 10a″ | 142.84 | ||||
| 1‴ | 160.70 | ||||
| 2‴ | 94.07 | 5.73 | d ( 2.08 ) | C1‴, C3‴, C4‴, C5‴, C6‴ | C3‴−H |
| 3‴ | 159.20 | 9.12 | C2‴, C3‴, C4‴, C5‴, C6‴ | C2‴−H, C4‴−H | |
| 4‴ | 96.63 | 5.80 | t ( 2.08 ) | C2‴, C3‴, C5‴, C6‴ | C3‴−H, C5‴−H |
| 5‴ | 159.20 | 9.12 | C2‴, C3‴, C4‴, C5‴, C6‴ | C4‴−H, C6‴−H | |
| 6‴ | 94.07 | 5.73 | d ( 2.08 ) | C1‴, C2‴, C3‴, C4‴, C5‴ | C5‴−H |
| 6-O(CO)CH3 | 168.11 | ||||
| 6-O(CO)CH3 | 20.18 | 2.02 | C6, C6-O(CO)CH3 | C2′−H, C6′−H |
| No. | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 142.37 | 9.47 | 142.61 | 9.65 | 142.64 | 9.65 | 142.65 | 9.67 | 142.66 | 9.65 |
| 2 | 98.63 | 6.16 | 99.04 | 6.22 | 99.10 | 6.22 | 99.21 | 6.23 | 99.19 | 6.23 |
| 3 | 146.31 | 9.25 | 146.85 | 9.38 | 146.80 | 9.37 | 146.76 | 9.34 | 146.78 | 9.34 |
| 4 | 122.68 | - | 122.30 | - | 122.23 | - | 122.14 | - | 122.17 | - |
| 4a | 137.63 | - | 136.71 | - | 136.81 | - | 137.05 | - | 137.07 | - |
| 5a | 122.99 | - | 126.36 | - | 126.37 | - | 126.47 | - | 126.51 | - |
| 6 | 146.45 | 9.57 | 139.05 | - | 139.16 | 139.07 | - | 139.19 | - | |
| 7 | 98.90 | 5.99 | 104.91 | 6.16 | 104.95 | 6.16 | 105.03 | 6.38 | 105.07 | 6.37 |
| 8 | 153.47 | 9.19 | 153.52 | 9.67 | 153.53 | 9.66 | 153.55 | 9.76 | 153.53 | 9.73 |
| 9 | 94.27 | 5.81 | 101.01 | 6.28 | 100.96 | 6.28 | 101.13 | 6.35 | 101.04 | 6.34 |
| 9a | 142.99 | - | 142.99 | - | 143.00 | - | 143.16 | - | 143.17 | - |
| 10a | 123.63 | - | 123.56 | - | 123.53 | - | 123.52 | - | 123.58 | - |
| 1′ | 156.30 | - | 156.12 | - | 156.04 | - | 155.79 | - | 155.81 | - |
| 2′ | 94.90 | 5.95 | 94.32 | 5.92 | 94.22 | 5.90 | 94.20 | 5.70 | 94.21 | 5.69 |
| 3′ | 151.55 | 9.32 | 151.66 | 9.31 | 151.65 | 9.30 | 151.41 | 9.17 | 151.43 | 9.16 |
| 4′ | 124.62 | - | 124.63 | - | 124.62 | - | 124.61 | - | 124.68 | - |
| 5′ | 151.55 | 9.32 | 151.66 | 9.31 | 151.65 | 9.30 | 151.41 | 9.17 | 151.43 | 9.16 |
| 6′ | 94.90 | 5.95 | 94.32 | 5.92 | 94.22 | 5.90 | 94.20 | 5.70 | 94.21 | 5.69 |
| 1″ | 93.94 | 5.82 | 93.72 | 5.81 | 93.73 | 5.81 | 93.83 | 5.78 | 93.93 | 5.79 |
| 2″ | 154.63 | - | 154.68 | - | 154.67 | - | 154.66 | - | 154.70 | - |
| 3″ | 98.46 | 6.02 | 98.81 | 6.06 | 98.83 | 6.05 | 98.85 | 6.06 | 98.92 | 6.07 |
| 4″ | 146.36 | 9.67 | 146.33 | 9.63 | 146.34 | 9.64 | 146.33 | 9.65 | 146.34 | 9.65 |
| 4a″ | 124.42 | - | 124.52 | - | 124.52 | - | 124.50 | - | 124.54 | - |
| 5a″ | 137.46 | - | 137.51 | - | 137.52 | - | 137.48 | - | 137.51 | - |
| 6″ | 122.61 | - | 122.61 | - | 122.63 | - | 122.60 | - | 122.64 | - |
| 7″ | 146.49 | 9.20 | 146.46 | 9.19 | 146.47 | 9.18 | 146.46 | 9.19 | 146.48 | 9.19 |
| 8″ | 98.75 | 6.14 | 98.73 | 6.15 | 98.74 | 6.15 | 98.73 | 6.15 | 98.75 | 6.15 |
| 9″ | 142.27 | 9.42 | 142.32 | 9.42 | 142.33 | 9.44 | 142.31 | 9.41 | 142.34 | 9.41 |
| 9a″ | 123.55 | - | 123.58 | - | 123.60 | - | 123.58 | - | 123.62 | - |
| 10a″ | 142.80 | - | 142.84 | - | 142.84 | - | 142.80 | - | 142.82 | - |
| 1‴ | 160.69 | - | 160.70 | - | 160.72 | - | 160.70 | - | 160.73 | - |
| 2‴ | 94.05 | 5.72 | 94.07 | 5.73 | 94.09 | 5.73 | 94.07 | 5.73 | 94.10 | 5.73 |
| 3‴ | 159.20 | 9.13 | 159.20 | 9.12 | 159.21 | 9.12 | 159.20 | 9.12 | 159.23 | 9.12 |
| 4‴ | 96.63 | 5.80 | 96.63 | 5.80 | 96.63 | 5.80 | 96.62 | 5.80 | 96.65 | 5.81 |
| 5‴ | 159.20 | 9.13 | 159.20 | 9.12 | 159.21 | 9.12 | 159.20 | 9.12 | 159.23 | 9.12 |
| 6‴ | 94.05 | 5.72 | 94.07 | 5.73 | 94.09 | 5.73 | 94.07 | 5.73 | 94.10 | 5.73 |
| Substituent | Cytotoxicity b (IC50 a, μM) | SI Value c | |
|---|---|---|---|
| A549 | NIH/3T3 | ||
| Intact DK (1) | N/D | N/D | N/A |
| Acetyl (2) | 7.02 ± 1.53 | 481.42 + 18.61 | 68.58 |
| Benzoyl (3) | 640.92 ± 17.86 | N/D | N/A |
| Methyl (6) | 123.02 ± 1.85 | 575.79 ± 22.32 | 4.68 |
| Benzyl (7) | 842.26 ± 10.80 | 563.42 ± 34.02 | 0.67 |
| Methoxymethyl (8) | 66.10 ± 8.47 | 653.45 ± 27.46 | 9.89 |
| 3-(Ethoxycarbonyl)propyl (9) | 82.45 ± 6.49 | 719.30 ± 54.58 | 8.72 |
| Hydroxypropyl (10) | 13.07 ± 5.06 | 562.90 ± 51.77 | 43.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.-C.; Kim, Y.; Choi, J.; Kang, H.B.; Han, S.-Y.; Park, K.; Hwang, H.J. Regioselective Synthesis of 6-O-Acetyl Dieckol and Its Selective Cytotoxicity against Non-Small-Cell Lung Cancer Cells. Mar. Drugs 2022, 20, 683. https://doi.org/10.3390/md20110683
Shin H-C, Kim Y, Choi J, Kang HB, Han S-Y, Park K, Hwang HJ. Regioselective Synthesis of 6-O-Acetyl Dieckol and Its Selective Cytotoxicity against Non-Small-Cell Lung Cancer Cells. Marine Drugs. 2022; 20(11):683. https://doi.org/10.3390/md20110683
Chicago/Turabian StyleShin, Hyeon-Cheol, Yongkyun Kim, Jaeyeong Choi, Hyun Bae Kang, Seung-Yun Han, Kwangyong Park, and Hye Jeong Hwang. 2022. "Regioselective Synthesis of 6-O-Acetyl Dieckol and Its Selective Cytotoxicity against Non-Small-Cell Lung Cancer Cells" Marine Drugs 20, no. 11: 683. https://doi.org/10.3390/md20110683
APA StyleShin, H.-C., Kim, Y., Choi, J., Kang, H. B., Han, S.-Y., Park, K., & Hwang, H. J. (2022). Regioselective Synthesis of 6-O-Acetyl Dieckol and Its Selective Cytotoxicity against Non-Small-Cell Lung Cancer Cells. Marine Drugs, 20(11), 683. https://doi.org/10.3390/md20110683