

Astaxanthin Protection against Neuronal Excitotoxicity via Glutamate Receptor Inhibition and Improvement of Mitochondrial Function


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
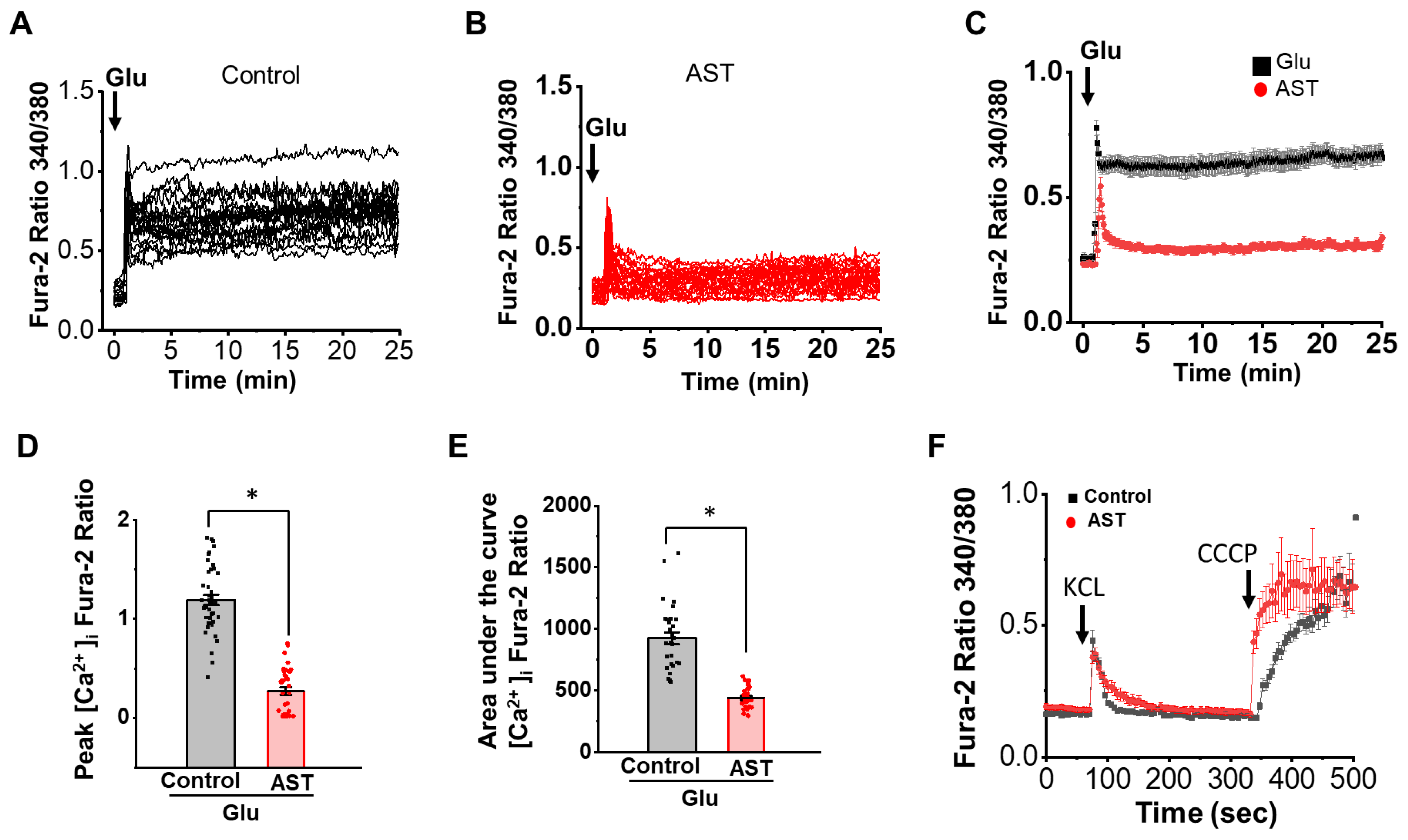
2.1. AST Modulates [Ca2+]i Dynamics in Excitotoxicity
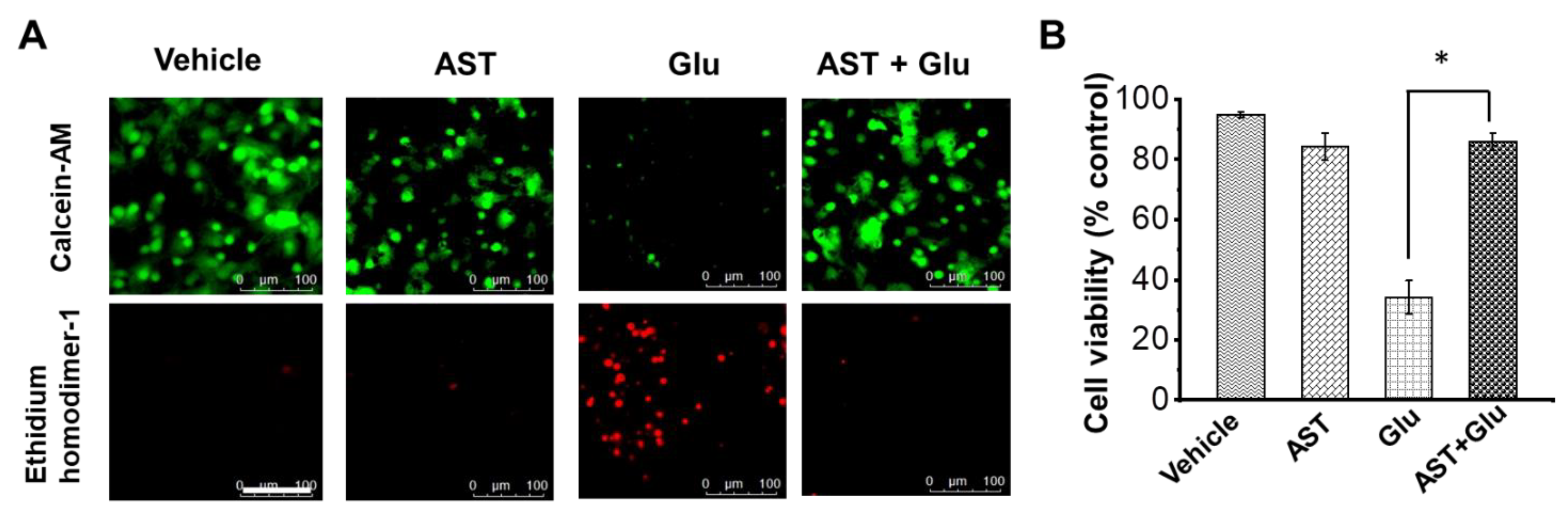
2.2. AST Attenuates Excitotoxic Neuronal Death
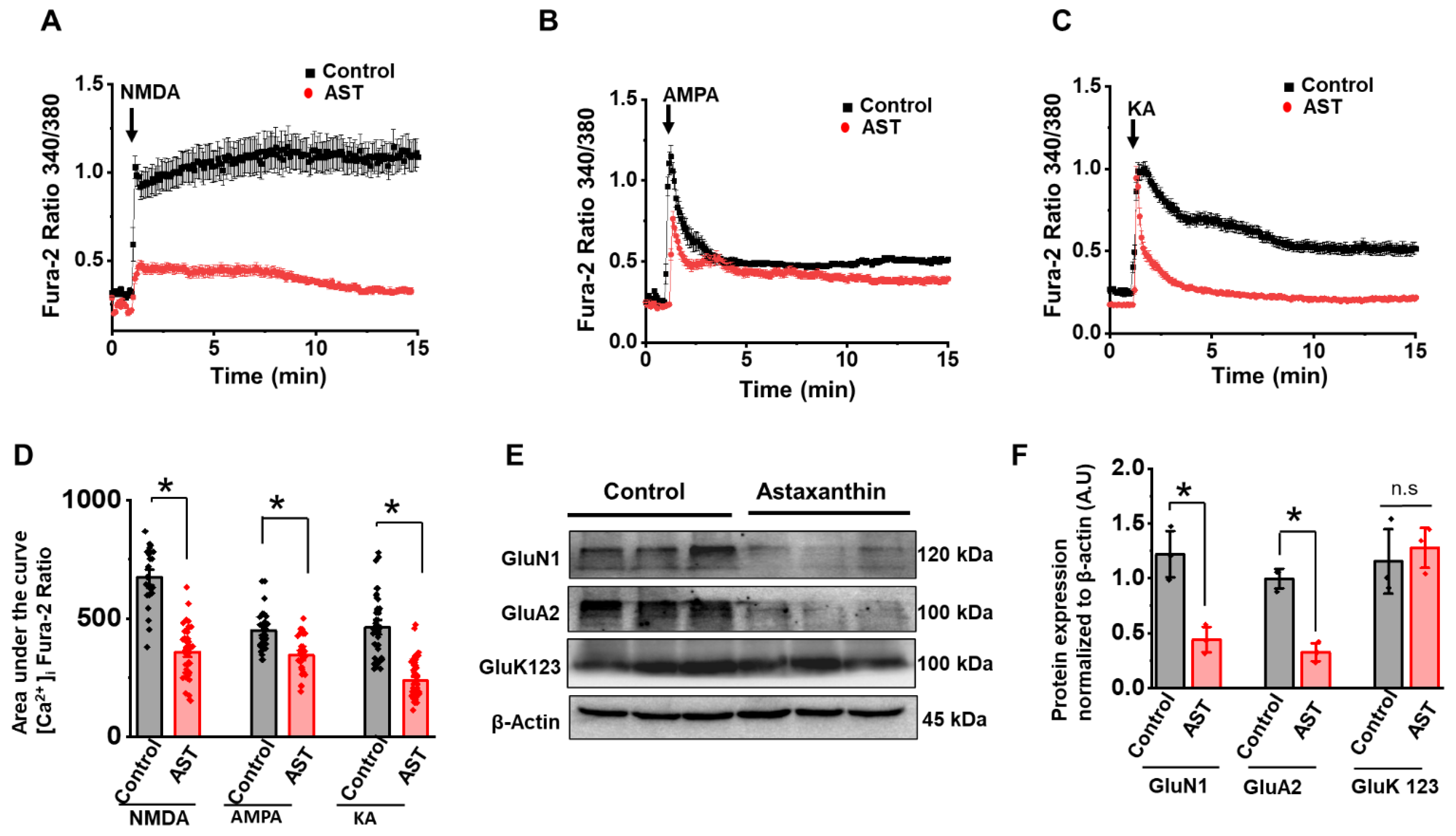
2.3. Chronic Treatment of AST Decreases NMDA, AMPA and KA Receptor-Mediated Ca2+ Influx and Their Protein Expression
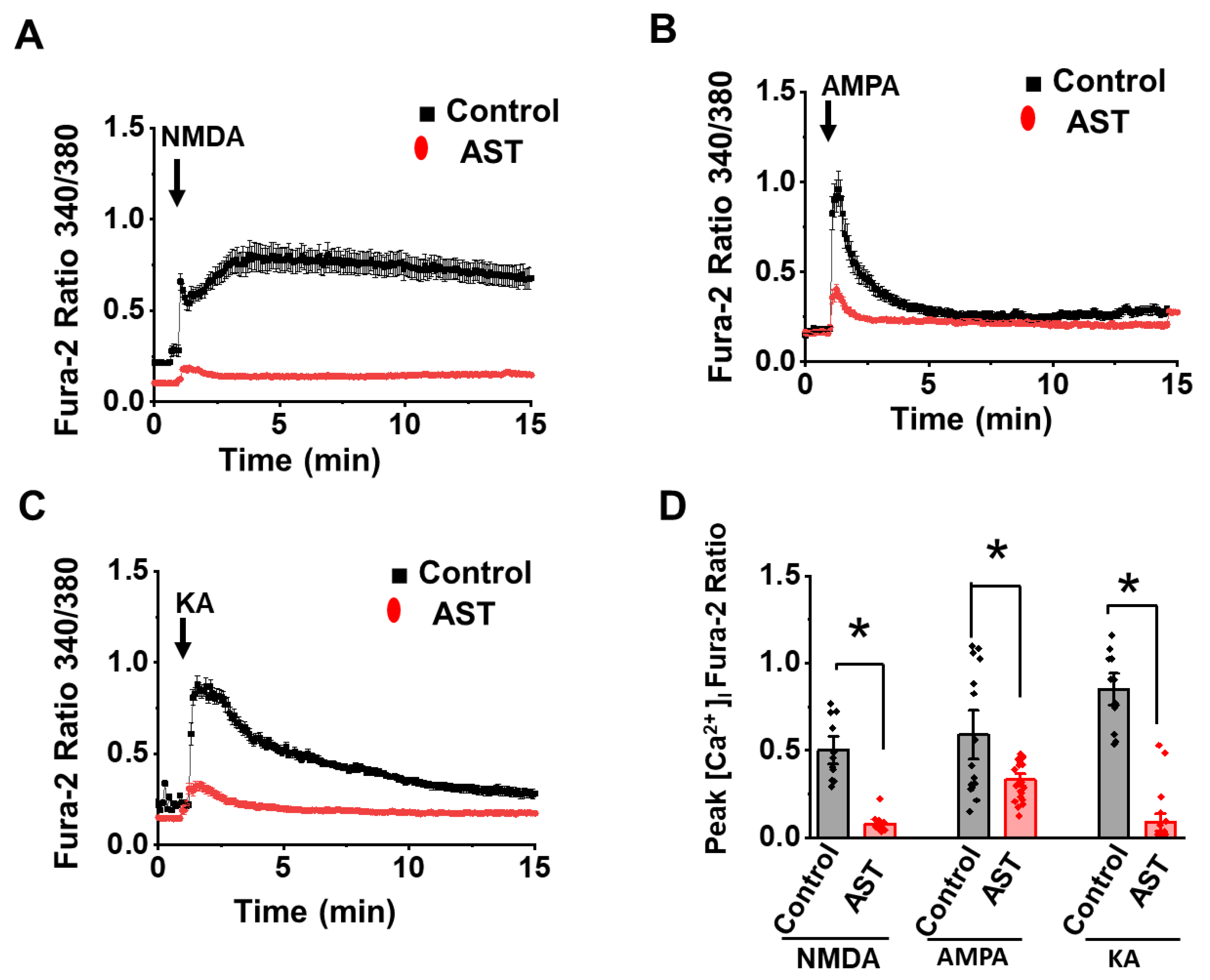
2.4. Acute Treatment of AST Inhibits NMDA, AMPA and KA Receptor [Ca2+] Response
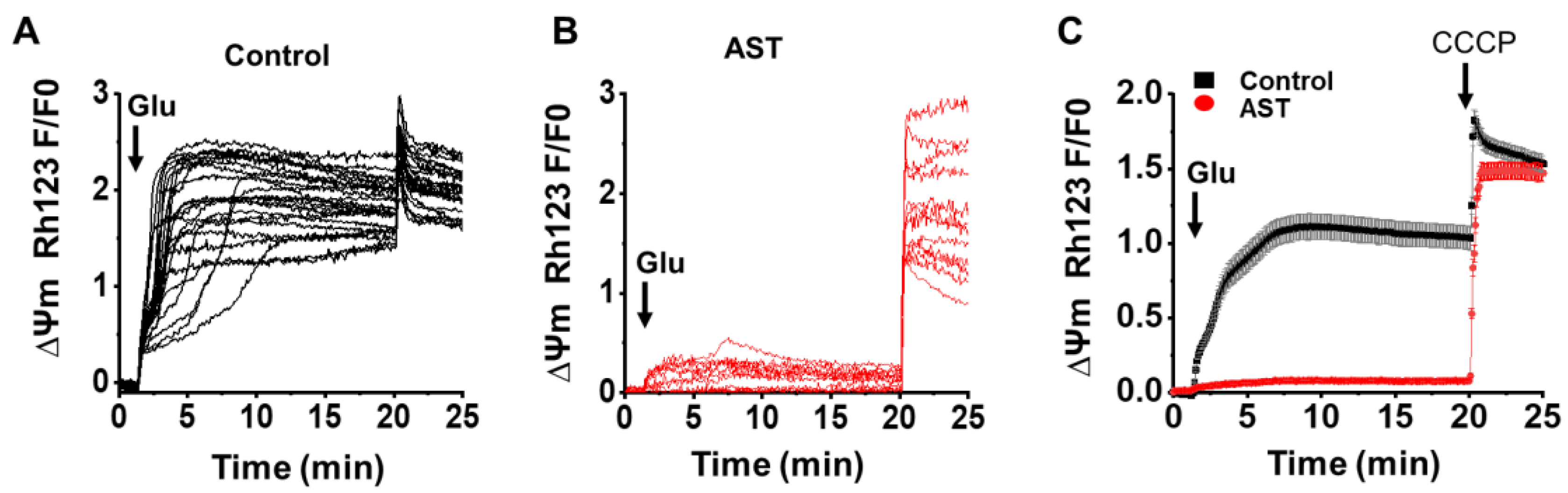
2.5. AST Abrogates Glutamate-Mediated Increase in Mitochondrial Membrane Potential (Ψm)
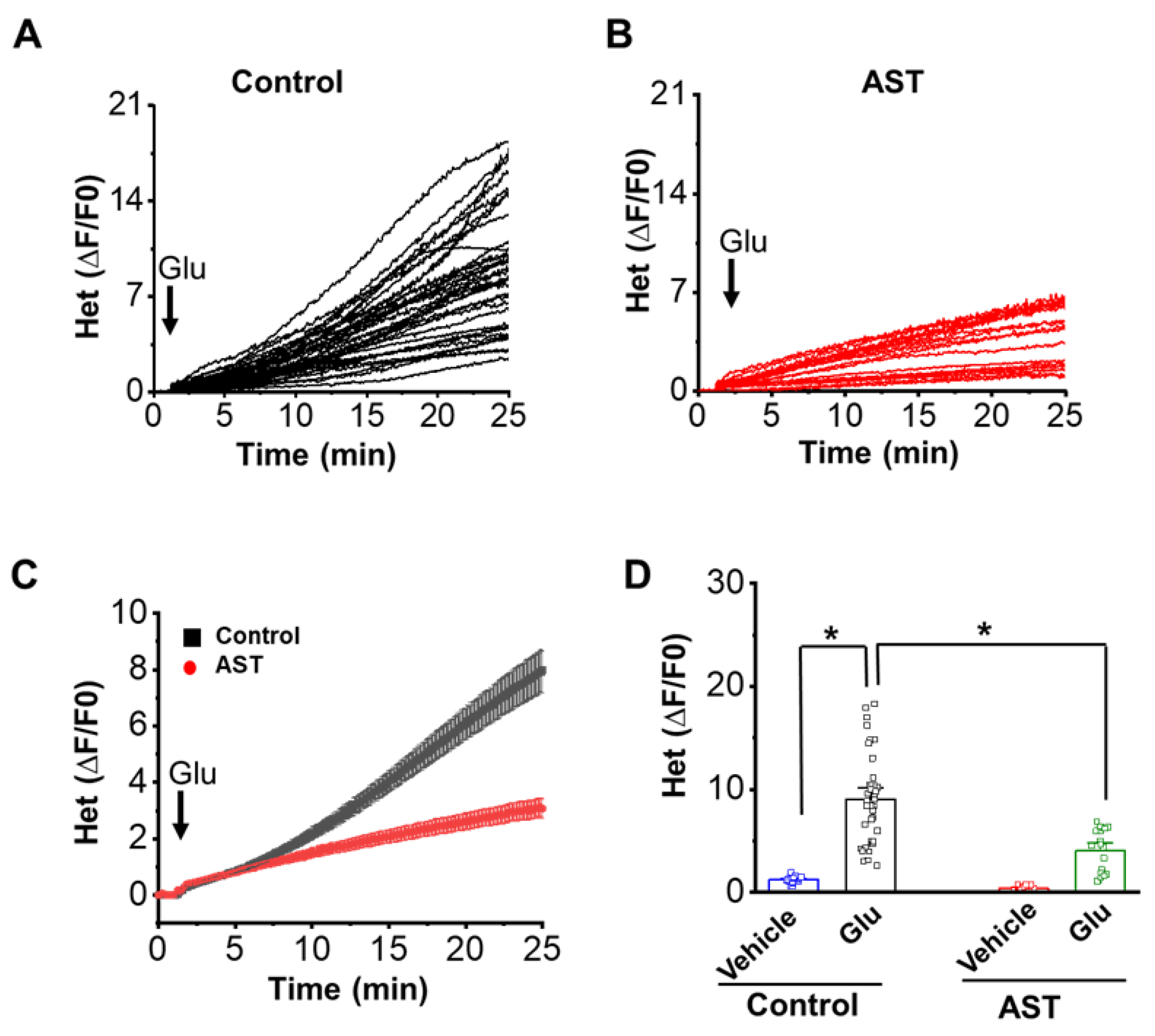
2.6. AST Inhibits Glutamate-Induced Reactive Oxygen Species (ROS)
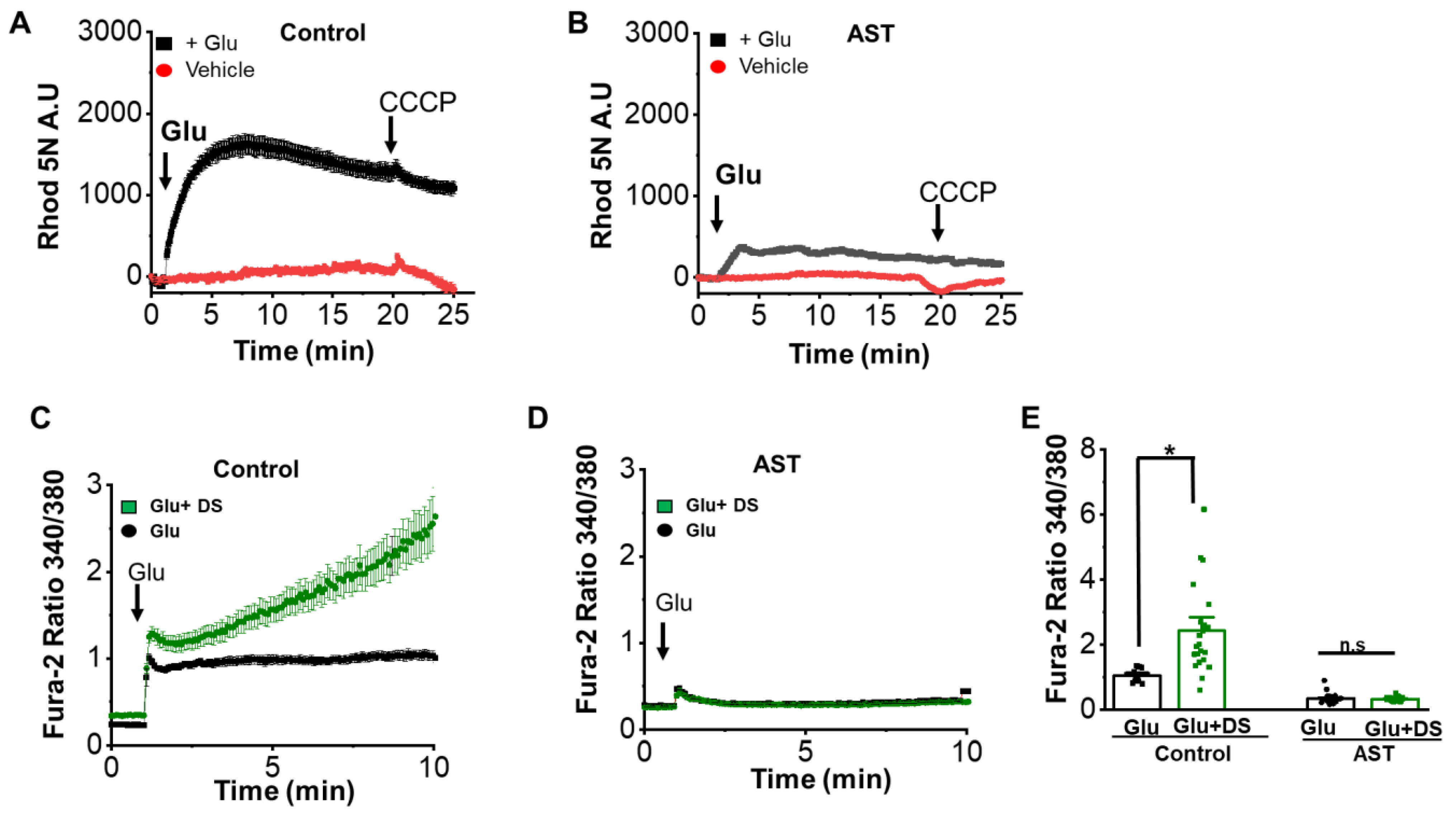
2.7. AST Affects Mitochondrial Calcium
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Experimental Animal
4.3. Preparation of Cortical Neuronal Culture
4.4. Measurement of Intracellular Calcium
4.5. Measurement of Mitochondrial Membrane Potential (Ψm) and Mitochondrial Calcium ([Ca2+]m)
4.6. Measurement of the Reactive Oxygen Species (ROS)
4.7. Neuronal Survival
4.8. Western Blot Analysis
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. AJNR Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar]
- Gasiorowska, A.; Wydrych, M.; Drapich, P.; Zadrozny, M.; Steczkowska, M.; Niewiadomski, W.; Niewiadomska, G. The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front. Aging Neurosci. 2021, 13, 654931. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, Y.Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.D.; Wilkinson, K.A.; Henley, J.M.; Mellor, J.R. Kainate receptors and synaptic plasticity. Neuropharmacology 2021, 196, 108540. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target Against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar]
- Olney, J.W.; Rhee, V.; Ho, O.L. Kainic acid: A powerful neurotoxic analogue of glutamate. Brain Res. 1974, 77, 507–512. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef]
- Tymianski, M.; Charlton, M.P.; Carlen, P.L.; Tator, C.H. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J. Neurosci. 1993, 13, 2085–2104. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.D.; Thayer, S.A. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [CrossRef] [PubMed]
- Budd, S.L.; Nicholls, D.G. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 1996, 67, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, S.; Reynolds, I.J. Glutamate-induced intracellular calcium changes and neurotoxicity in cortical neurons in vitro: Effect of chemical ischemia. Neuroscience 1994, 62, 667–679. [Google Scholar] [CrossRef]
- Alano, C.C.; Beutner, G.; Dirksen, R.T.; Gross, R.A.; Sheu, S.S. Mitochondrial permeability transition and calcium dynamics in striatal neurons upon intense NMDA receptor activation. J. Neurochem. 2002, 80, 531–538. [Google Scholar] [CrossRef]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Plotegher, N.; Filadi, R.; Pizzo, P.; Duchen, M.R. Excitotoxicity Revisited: Mitochondria on the Verge of a Nervous Breakdown. Trends Neurosci. 2021, 44, 342–351. [Google Scholar] [CrossRef]
- Khodorov, B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Vesce, S.; Kirk, L.; Chalmers, S. Interactions between mitochondrial bioenergetics and cytoplasmic calcium in cultured cerebellar granule cells. Cell. Calcium 2003, 34, 407–424. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Niu, H.; Shao, A.; Wu, C.; Dixon, B.J.; Zhang, J.; Yang, S.; Wang, Y. Astaxanthin as a Potential Neuroprotective Agent for Neurological Diseases. Mar. Drugs 2015, 13, 5750–5766. [Google Scholar] [CrossRef]
- Li, Y.; Gong, F.; Guo, S.; Yu, W.; Liu, J. Adonis amurensis Is a Promising Alternative to Haematococcus as a Resource for Natural Esterified (3S,3’S)-Astaxanthin Production. Plants 2021, 10, 1059. [Google Scholar] [CrossRef]
- Hayashi, M.; Ishibashi, T.; Maoka, T. Effect of astaxanthin-rich extract derived from Paracoccus carotinifaciens on cognitive function in middle-aged and older individuals. J. Clin. Biochem. Nutr. 2018, 62, 195–205. [Google Scholar] [CrossRef]
- Lobos, P.; Bruna, B.; Cordova, A.; Barattini, P.; Galaz, J.L.; Adasme, T.; Hidalgo, C.; Munoz, P.; Paula-Lima, A. Astaxanthin Protects Primary Hippocampal Neurons against Noxious Effects of Abeta-Oligomers. Neural Plast. 2016, 2016, 3456783. [Google Scholar] [CrossRef]
- Satoh, A.; Tsuji, S.; Okada, Y.; Murakami, N.; Urami, M.; Nakagawa, K.; Ishikura, M.; Katagiri, M.; Koga, Y.; Shirasawa, T. Preliminary Clinical Evaluation of Toxicity and Efficacy of a New Astaxanthin-rich Haematococcus pluvialis Extract. J. Clin. Biochem. Nutr. 2009, 44, 280–284. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction—A Mini-Review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef]
- Baburina, Y.; Krestinin, R.; Odinokova, I.; Sotnikova, L.; Kruglov, A.; Krestinina, O. Astaxanthin Inhibits Mitochondrial Permeability Transition Pore Opening in Rat Heart Mitochondria. Antioxidants 2019, 8, 576. [Google Scholar] [CrossRef]
- Brasil, F.B.; de Almeida, F.J.S.; Luckachaki, M.D.; Dall’Oglio, E.L.; de Oliveira, M.R. Astaxanthin prevents mitochondrial impairment in the dopaminergic SH-SY5Y cell line exposed to glutamate-mediated excitotoxicity: Role for the Nrf2/HO-1/CO-BR axis. Eur. J. Pharmacol. 2021, 908, 174336. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Nawaz, A.; Hecht, K.; Tobe, K. Astaxanthin as a Novel Mitochondrial Regulator: A New Aspect of Carotenoids, beyond Antioxidants. Nutrients 2021, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Afshari, A.R.; Fanoudi, S.; Rajabian, A.; Sadeghnia, H.R.; Mollazadeh, H.; Hosseini, A. Potential protective roles of phytochemicals on glutamate-induced neurotoxicity: A review. Iran J. Basic. Med. Sci. 2020, 23, 1113–1123. [Google Scholar]
- Sharma, K.; Sharma, D.; Sharma, M.; Sharma, N.; Bidve, P.; Prajapati, N.; Kalia, K.; Tiwari, V. Astaxanthin ameliorates behavioral and biochemical alterations in in-vitro and in-vivo model of neuropathic pain. Neurosci. Lett. 2018, 674, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Taheri, F.; Sattari, E.; Hormozi, M.; Ahmadvand, H.; Bigdeli, M.R.; Kordestani-Moghadam, P.; Anbari, K.; Milanizadeh, S.; Moghaddasi, M. Dose-Dependent Effects of Astaxanthin on Ischemia/Reperfusion Induced Brain Injury in MCAO Model Rat. Neurochem. Res. 2022, 47, 1736–1750. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, Y.; Li, S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur. J. Pharmacol. 2017, 806, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Garcia, F.; Lobos, P.; Ponce, A.; Cataldo, K.; Meza, D.; Farias, P.; Estay, C.; Oyarzun-Ampuero, F.; Herrera-Molina, R.; Paula-Lima, A.; et al. Astaxanthin Counteracts Excitotoxicity and Reduces the Ensuing Increases in Calcium Levels and Mitochondrial Reactive Oxygen Species Generation. Mar. Drugs 2020, 18, 335. [Google Scholar] [CrossRef]
- Li, S.; Gao, X.; Zhang, Q.; Zhang, X.; Lin, W.; Ding, W. Astaxanthin protects spinal cord tissues from apoptosis after spinal cord injury in rats. Ann. Transl. Med. 2021, 9, 1796. [Google Scholar] [CrossRef]
- Altunrende, M.E.; Gezen-Ak, D.; Atasoy, I.L.; Candas, E.; Dursun, E. The Role of Astaxanthin on Transcriptional Regulation of NMDA Receptors Voltage Sensitive Calcium Channels and Calcium Binding Proteins in Primary Cortical Neurons. Noro. Psikiyatr. Ars. 2018, 55, 295–300. [Google Scholar] [CrossRef]
- Krasil’nikova, I.; Surin, A.; Sorokina, E.; Fisenko, A.; Boyarkin, D.; Balyasin, M.; Demchenko, A.; Pomytkin, I.; Pinelis, V. Insulin Protects Cortical Neurons Against Glutamate Excitotoxicity. Front. Neurosci. 2019, 13, 1027. [Google Scholar] [CrossRef]
- Peng, T.I.; Jou, M.J.; Sheu, S.S.; Greenamyre, J.T. Visualization of NMDA receptor-induced mitochondrial calcium accumulation in striatal neurons. Exp. Neurol. 1998, 149, 1–12. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Sharipov, R.R.; Boyarkin, D.P.; Belosludtseva, N.V.; Dubinin, M.V.; Krasilnikova, I.A.; Bakaeva, Z.V.; Zgodova, A.E.; Pinelis, V.G.; Surin, A.M. The effect of DS16570511, a new inhibitor of mitochondrial calcium uniporter, on calcium homeostasis, metabolism, and functional state of cultured cortical neurons and isolated brain mitochondria. Biochim. Biophys. Acta. Gen. Subj. 2021, 1865, 129847. [Google Scholar] [CrossRef] [PubMed]
- Taksima, T.; Chonpathompikunlert, P.; Sroyraya, M.; Hutamekalin, P.; Limpawattana, M.; Klaypradit, W. Effects of Astaxanthin from Shrimp Shell on Oxidative Stress and Behavior in Animal Model of Alzheimer’s Disease. Mar. Drugs 2019, 17, 628. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef]
- Lu, Y.; Xie, T.; He, X.X.; Mao, Z.F.; Jia, L.J.; Wang, W.P.; Zhen, J.L.; Liu, L.M. Astaxanthin rescues neuron loss and attenuates oxidative stress induced by amygdala kindling in adult rat hippocampus. Neurosci. Lett. 2015, 597, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chen, K.C.; Liaw, K.C.; Peng, C.C.; Peng, R.Y. Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity. Molecules 2020, 25, 214. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Chang, Y.; Lu, C.W.; Chen, Y.J.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Astaxanthin protects against kainic acid-induced seizures and pathological consequences. Neurochem. Int. 2018, 116, 85–94. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Brand, M.D.; Gerencser, A.A. Mitochondrial bioenergetics and neuronal survival modelled in primary neuronal culture and isolated nerve terminals. J. Bioenerg. Biomembr. 2015, 47, 63–74. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ward, M.W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 2000, 23, 166–174. [Google Scholar] [CrossRef]
- Atlante, A.; Calissano, P.; Bobba, A.; Giannattasio, S.; Marra, E.; Passarella, S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001, 497, 1–5. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Gagliardi, S.; Minervini, G.M.; Ciotti, M.T.; Marra, E.; Calissano, P. Glutamate neurotoxicity in rat cerebellar granule cells: A major role for xanthine oxidase in oxygen radical formation. J. Neurochem. 1997, 68, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Budd, S.L.; Castilho, R.F.; Nicholls, D.G. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997, 415, 21–24. [Google Scholar] [CrossRef]
- Sengpiel, B.; Preis, E.; Krieglstein, J.; Prehn, J.H. NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: Role of mitochondria. Eur. J. Neurosci. 1998, 10, 1903–1910. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Pan, L.; Zhou, Y.; Li, X.F.; Wan, Q.J.; Yu, L.H. Preventive treatment of astaxanthin provides neuroprotection through suppression of reactive oxygen species and activation of antioxidant defense pathway after stroke in rats. Brain Res. Bull. 2017, 130, 211–220. [Google Scholar] [CrossRef]
- Angelova, P.R.; Vinogradova, D.; Neganova, M.E.; Serkova, T.P.; Sokolov, V.V.; Bachurin, S.O.; Shevtsova, E.F.; Abramov, A.Y. Pharmacological Sequestration of Mitochondrial Calcium Uptake Protects Neurons Against Glutamate Excitotoxicity. Mol. Neurobiol. 2019, 56, 2244–2255. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandy, S.K.; Nimonkar, M.M.; Dash, S.S.; Mehta, B.; Markandeya, Y.S. Astaxanthin Protection against Neuronal Excitotoxicity via Glutamate Receptor Inhibition and Improvement of Mitochondrial Function. Mar. Drugs 2022, 20, 645. https://doi.org/10.3390/md20100645
Kandy SK, Nimonkar MM, Dash SS, Mehta B, Markandeya YS. Astaxanthin Protection against Neuronal Excitotoxicity via Glutamate Receptor Inhibition and Improvement of Mitochondrial Function. Marine Drugs. 2022; 20(10):645. https://doi.org/10.3390/md20100645
Chicago/Turabian StyleKandy, Swapna Kannothum, Madhura Milind Nimonkar, Suravi Sasmita Dash, Bhupesh Mehta, and Yogananda S. Markandeya. 2022. "Astaxanthin Protection against Neuronal Excitotoxicity via Glutamate Receptor Inhibition and Improvement of Mitochondrial Function" Marine Drugs 20, no. 10: 645. https://doi.org/10.3390/md20100645
APA StyleKandy, S. K., Nimonkar, M. M., Dash, S. S., Mehta, B., & Markandeya, Y. S. (2022). Astaxanthin Protection against Neuronal Excitotoxicity via Glutamate Receptor Inhibition and Improvement of Mitochondrial Function. Marine Drugs, 20(10), 645. https://doi.org/10.3390/md20100645