
Evaluation of Magnesium-Phosphate Particle Incorporation into Co-Electrospun Chitosan-Elastin Membranes for Skin Wound Healing

 , , ,
, , , 
 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Characterization
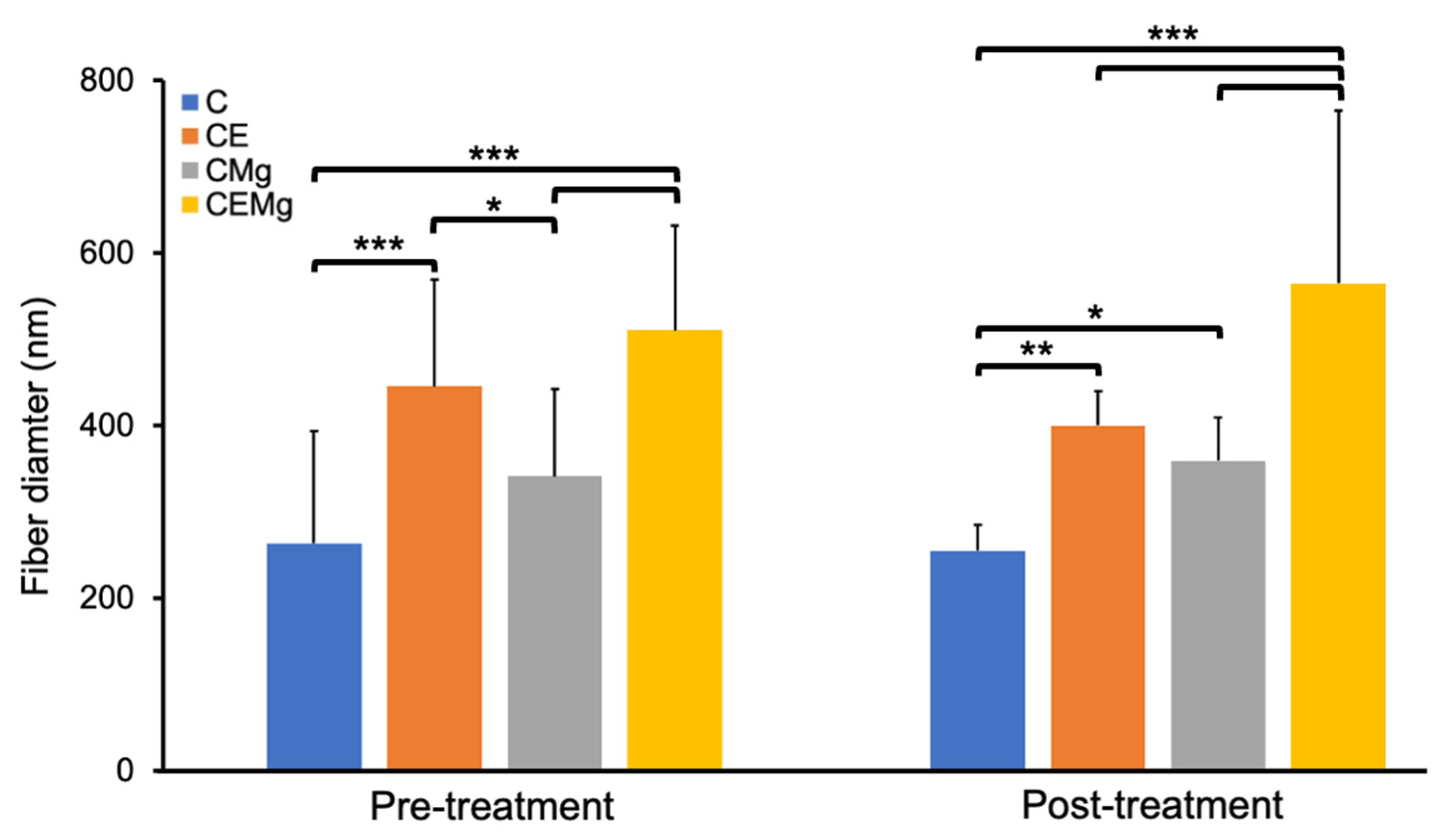
2.1.1. Fiber Morphology and Fiber Diameter Analysis
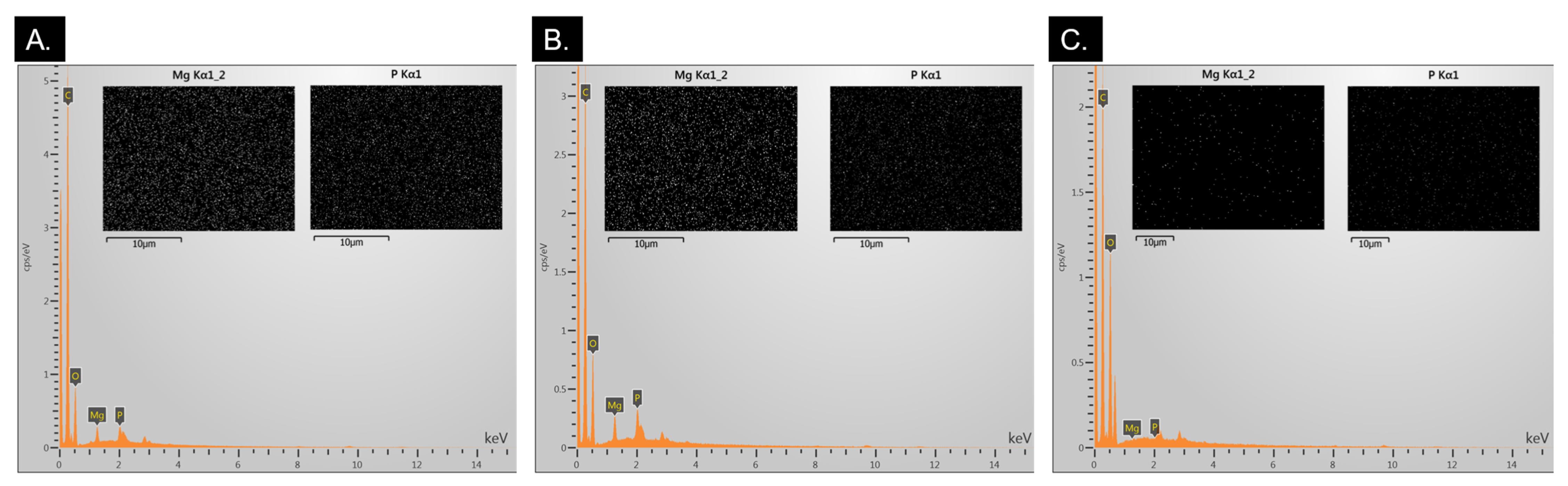
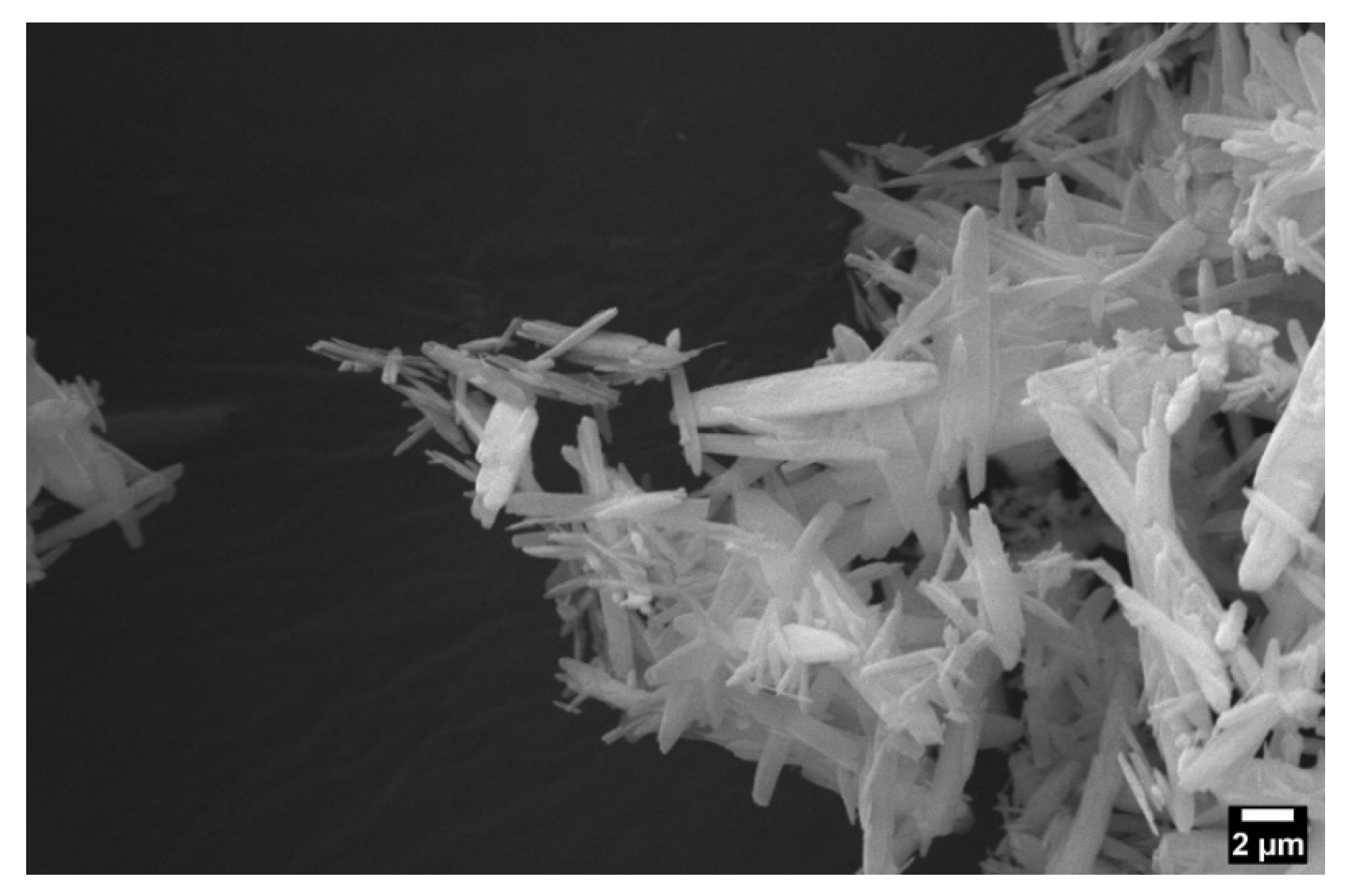
2.1.2. EDS Analysis for MgP Incorporation Verification
2.1.3. FTIR Analysis
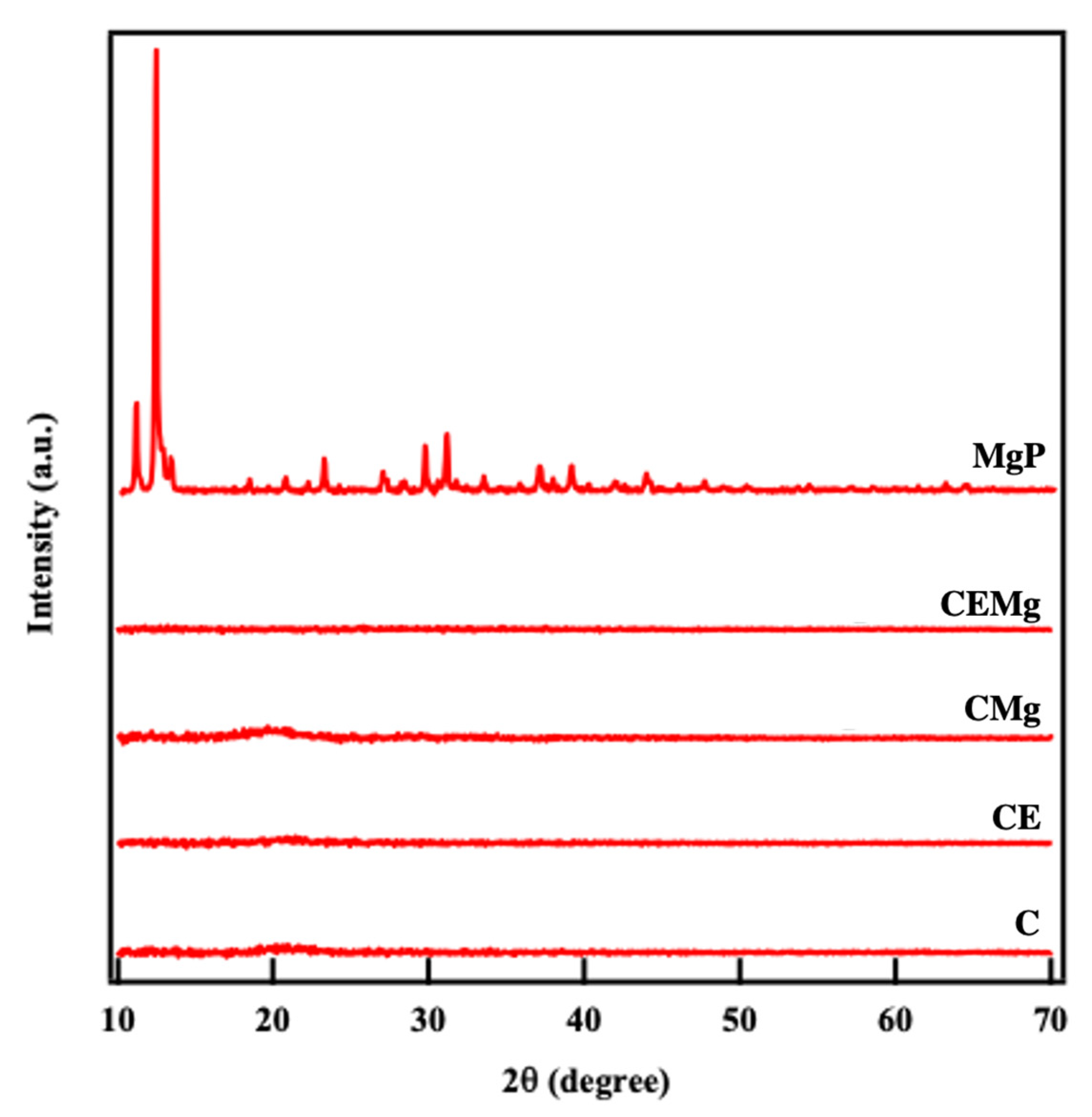
2.1.4. X-Ray Diffraction (XRD) Crystallography Analysis
2.1.5. Water Contact Angle Analysis
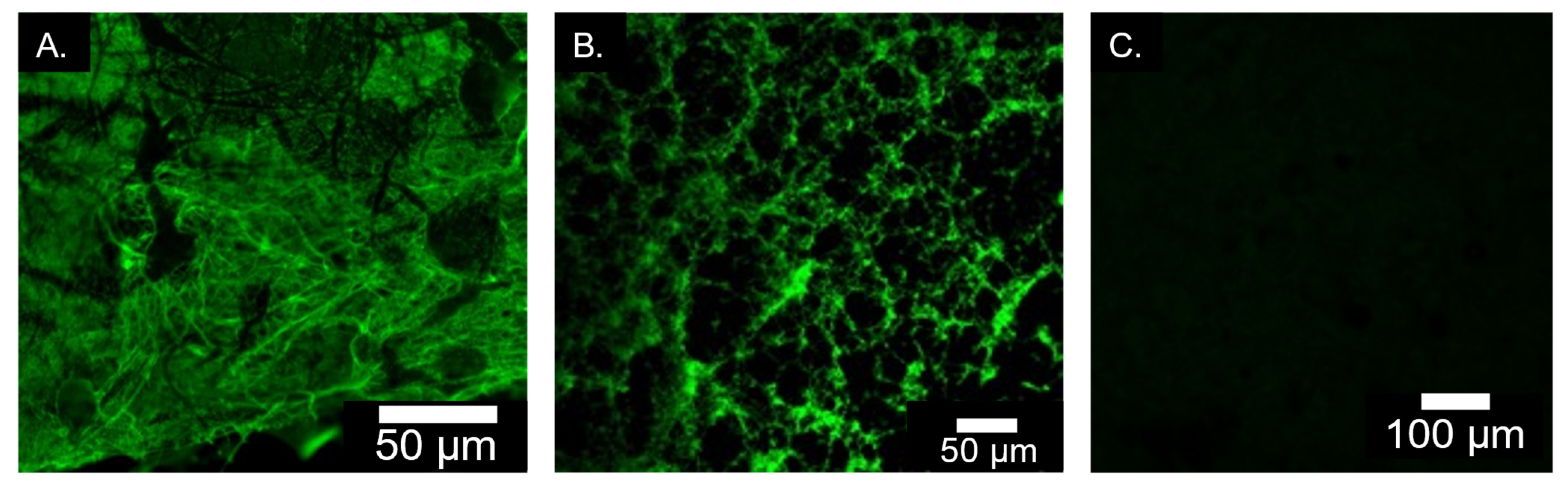
2.1.6. Immunofluorescence Staining for Elastin Incorporation
2.1.7. MgP Size and Zeta Potential Analysis
2.1.8. Combustion Analysis
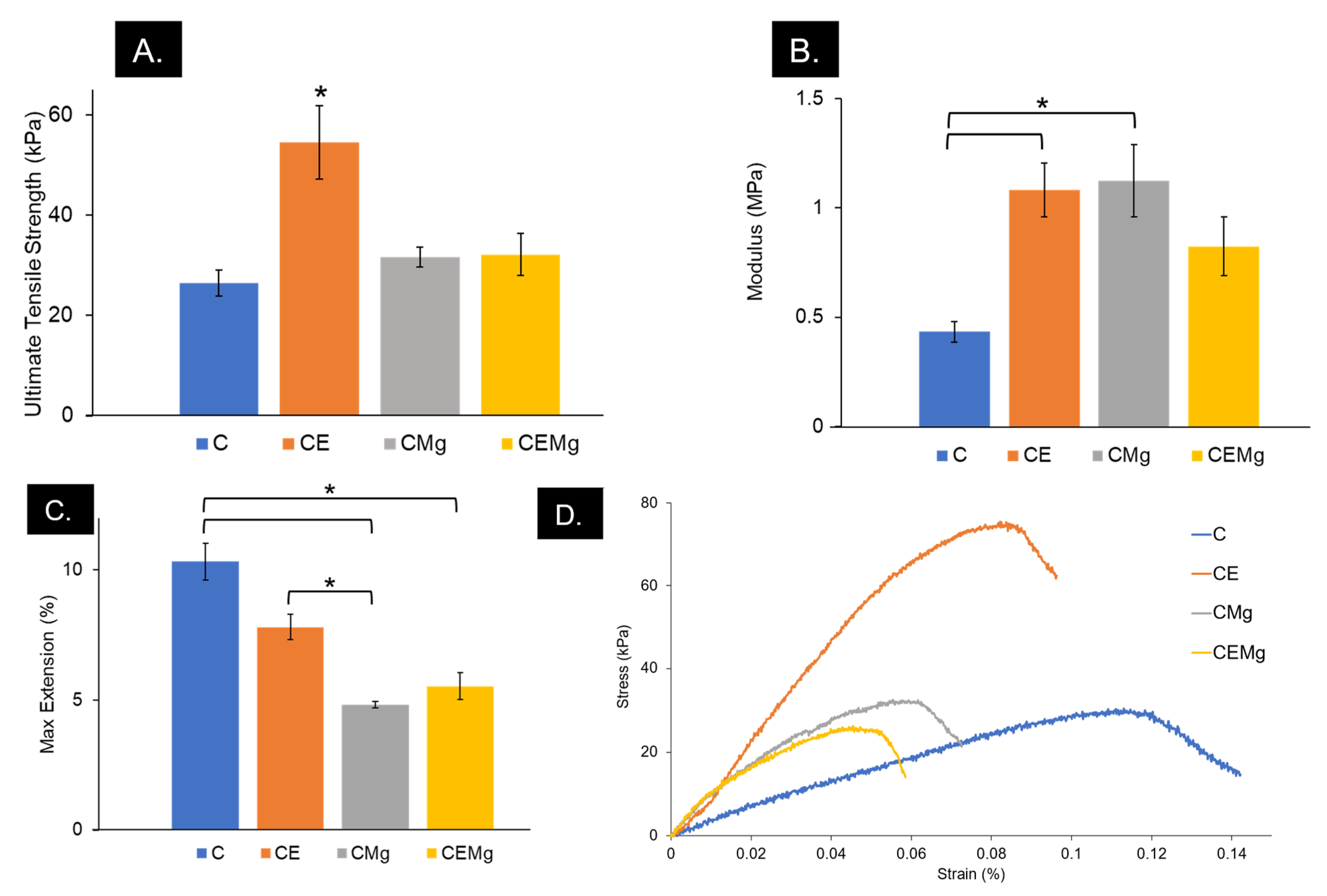
2.1.9. Tensile Testing
2.2. In Vitro Analysis
2.2.1. In Vitro Magnesium Release
2.2.2. In Vitro Degradation Profiles of Membranes
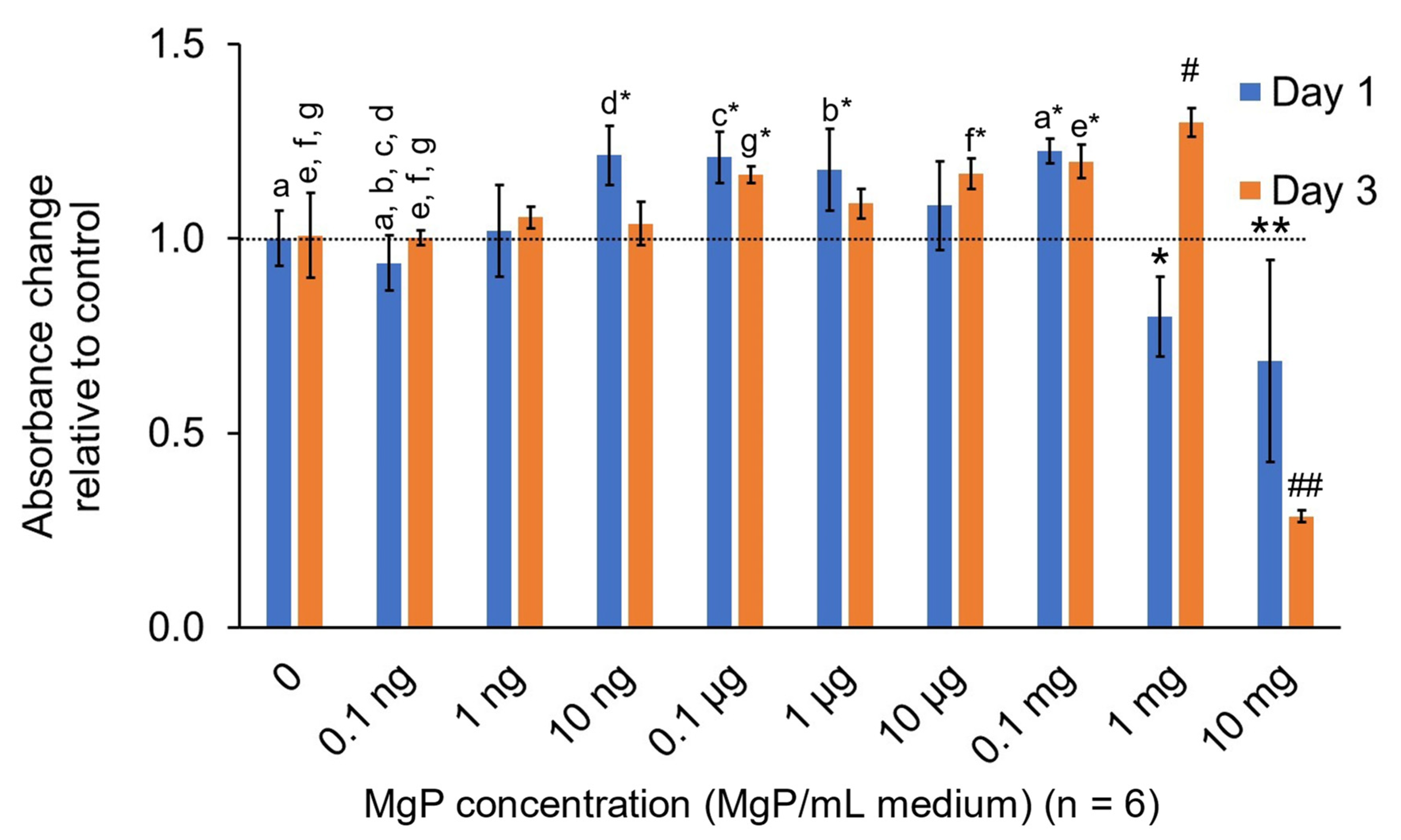
2.2.3. In Vitro MgP Cytotoxicity
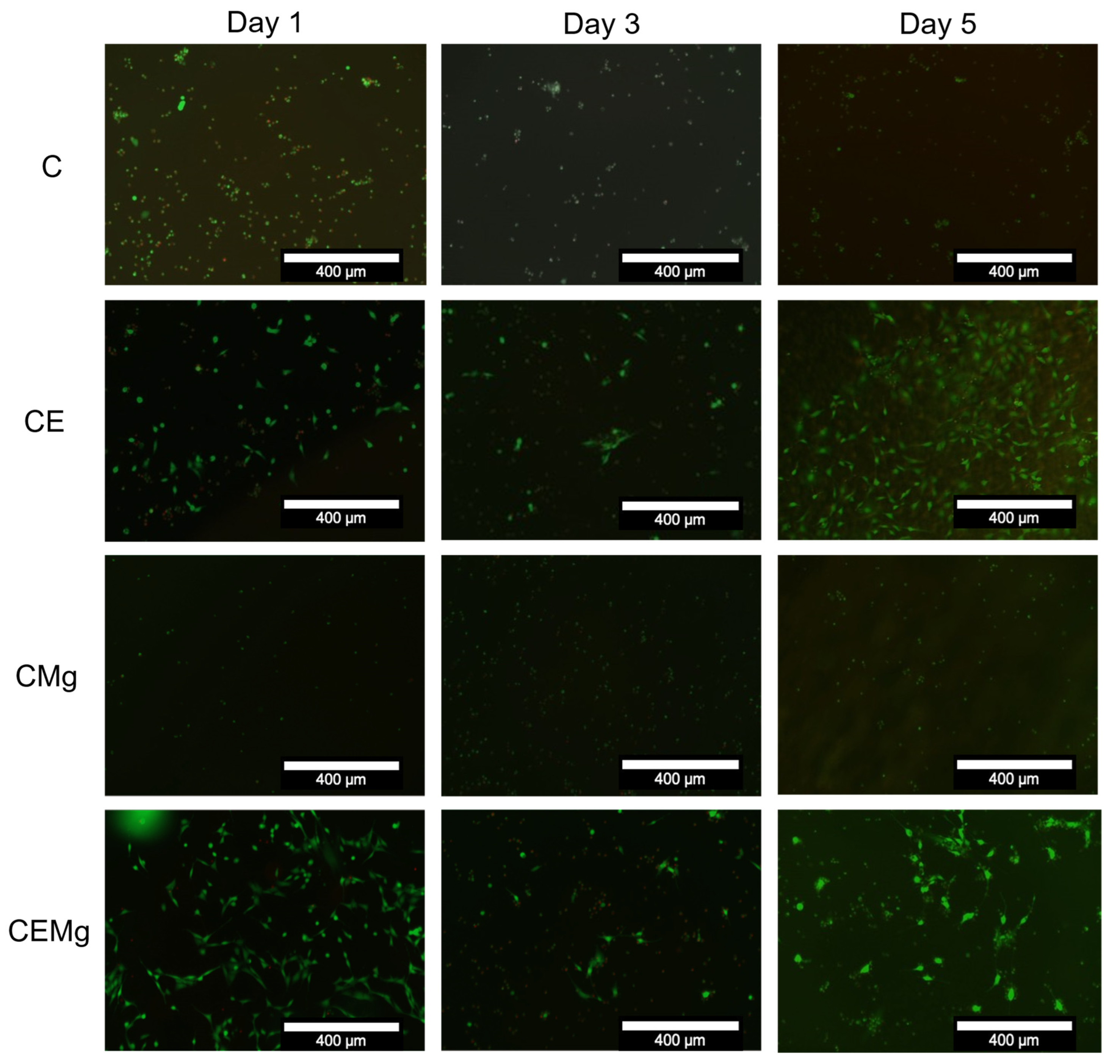
2.2.4. In Vitro Cytocompatibility of Membranes with NIH3T3 Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Membrane Fabrication
4.2. Magnesium-Phopshpate Particle (MgP) Synthesis
4.3. Characterization
4.3.1. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray Spectroscopy (EDS) Analysis
4.3.2. FTIR Analysis
4.3.3. X-Ray Diffraction (XRD) Crystallography Analysis
4.3.4. Water Contact Angle Analysis
4.3.5. Immunofluorescence Staining for Elastin Incorporation
4.3.6. MgP Size and Zeta Potential Analysis
4.3.7. Combustion Analysis
4.3.8. Tensile Testing
4.4. In Vitro Analysis
4.4.1. In Vitro Magnesium Release
4.4.2. In Vitro Degradation Study
4.4.3. In Vitro MgP Cytotoxicity
4.4.4. In Vitro Cytocompatibility of Membranes with NIH3T3 Fibroblasts
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Center for Disease Control and Prevention. National Hospital Ambulatory Medical Care Survey: 2018 Emergency Department Summary Tables. Available online: https://www.cdc.gov/nchs/ahcd/web_tables.htm (accessed on 18 October 2021).
- American Burn Association. Burn Injury Fact Sheet. Natl. Burn Aware. Week 2008, 4–10, 1–2. Available online: http://ameriburn.org/wp-content/uploads/2017/12/nbaw-factsheet_121417-1.pdf (accessed on 3 December 2020).
- Phillips, T.; Stanton, B.; Provan, A.; Lew, R. A study of the impact of leg ulcers on quality of life: Financial, social, and psychologic implications. J. Am. Acad. Dermatol. 1994, 31, 49–53. [Google Scholar] [CrossRef]
- Vig, K.; Chaudhari, A.; Tripathi, S.; Dixit, S.; Sahu, R.; Pillai, S.; Dennis, V.A.; Singh, S.R. Advances in Skin Regeneration Using Tissue Engineering. Int. J. Mol. Sci. 2017, 18, 789. [Google Scholar] [CrossRef]
- Ozhathil, D.; Tay, M.; Wolf, S.; Branski, L. A Narrative Review of the History of Skin Grafting in Burn Care. Medicina 2021, 57, 380. [Google Scholar] [CrossRef] [PubMed]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn Wound Infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.E.; Logan, B.; Wu, J.; Alousi, A.M.; Ho, V.; Bolaños-Meade, J.; Weisdorf, D. Graft-versus-Host Disease Treatment: Predictors of Survival. Biol. Blood Marrow Transplant. 2010, 16, 1693–1699. [Google Scholar] [CrossRef]
- Dixit, S.; Baganizi, D.R.; Sahu, R.; Dosunmu, E.; Chaudhari, A.; Vig, K.; Pillai, S.R.; Singh, S.R.; Dennis, V.A. Immunological challenges associated with artificial skin grafts: Available solutions and stem cells in future design of synthetic skin. J. Biol. Eng. 2017, 11, 49. [Google Scholar] [CrossRef]
- Supp, D.M.; Boyce, S.T. Engineered skin substitutes: Practices and potentials. Clin. Dermatol. 2005, 23, 403–412. [Google Scholar] [CrossRef]
- MacNeil, S. Progress and opportunities for tissue-engineered skin. Nature 2007, 445, 874–880. [Google Scholar] [CrossRef]
- Kozusko, S.D.; Riccio, C.; Goulart, M.; Bumgardner, J.; Jing, X.L.; Konofaos, P. Chitosan as a Bone Scaffold Biomaterial. J. Craniofacial Surg. 2018, 29, 1788–1793. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.L.; Voigt, D.W.; Wiebelhaus, P.; Paul, C.N. Using Skin Replacement Products to Treat Burns and Wounds. Adv. Ski. Wound Care 2001, 14, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.E.; Clausen, J.; Kavanagh, S. Experience with Biobrane: Uses and Caveats for Success. Eplasty 2009, 9, e25. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19633707 (accessed on 17 December 2020). [PubMed]
- Metcalfe, A.; Ferguson, M.W.J. Tissue engineering of replacement skin: The crossroads of biomaterials, wound healing, embryonic development, stem cells and regeneration. J. R. Soc. Interface 2006, 4, 413–437. [Google Scholar] [CrossRef]
- Rybarczyk, M.M.; Schafer, J.; Elm, C.M.; Sarvepalli, S.; Vaswani, P.A.; Balhara, K.S.; Carlson, L.C.; Jacquet, G.A. A systematic review of burn injuries in low- and middle-income countries: Epidemiology in the WHO-defined African Region. Afr. J. Emerg. Med. 2017, 7, 30–37. [Google Scholar] [CrossRef]
- Dai, T.; Tanaka, M.; Huang, Y.-Y.; Hamblin, M.R. Chitosan preparations for wounds and burns: Antimicrobial and wound-healing effects. Expert Rev. Anti. Infect. Ther. 2011, 9, 857–879. [Google Scholar] [CrossRef]
- Pandey, A.R.; Singh, U.S.; Momin, M.; Bhavsar, C. Chitosan: Application in tissue engineering and skin grafting. J. Polym. Res. 2017, 24, 125. [Google Scholar] [CrossRef]
- Xu, C.; Lei, C.; Meng, L.; Wang, C.; Song, Y. Chitosan as a barrier membrane material in periodontal tissue regeneration. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100, 1435–1443. [Google Scholar] [CrossRef]
- Luna-Hernández, E.; Cruz-Soto, M.; Padilla-Vaca, F.; Mauricio-Sánchez, R.; Ramirez-Wong, D.; Muñoz, R.; Granados-López, L.; Ovalle-Flores, L.; Menchaca-Arredondo, J.; Hernández-Rangel, A.; et al. Combined antibacterial/tissue regeneration response in thermal burns promoted by functional chitosan/silver nanocomposites. Int. J. Biol. Macromol. 2017, 105, 1241–1249. [Google Scholar] [CrossRef]
- Ahmed, R.; Tariq, M.; Ali, I.; Asghar, R.; Khanam, P.N.; Augustine, R.; Hasan, A. Novel electrospun chitosan/polyvinyl alcohol/zinc oxide nanofibrous mats with antibacterial and antioxidant properties for diabetic wound healing. Int. J. Biol. Macromol. 2018, 120, 385–393. [Google Scholar] [CrossRef]
- Deng, A.; Yang, Y.; Du, S.; Yang, S. Electrospinning of in situ crosslinked recombinant human collagen peptide/chitosan nanofibers for wound healing. Biomater. Sci. 2018, 6, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Yadav, S.K.; Patel, R.R.; Kumar, N.; Bansal, M.; Mishra, B. Tinidazole functionalized homogeneous electrospun chitosan/poly (ε-caprolactone) hybrid nanofiber membrane: Development, optimization and its clinical implications. Int. J. Biol. Macromol. 2017, 103, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Rodrigues, G.; Martins, G.; Roberto, M.; Mafra, M.; Henriques, C.; Silva, J.C. In vitro and in vivo evaluation of electrospun nanofibers of PCL, chitosan and gelatin: A comparative study. Mater. Sci. Eng. C 2015, 46, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Boucard, N.; Viton, C.; Agay, D.; Mari, E.; Roger, T.; Chancerelle, Y.; Domard, A. The use of physical hydrogels of chitosan for skin regeneration following third-degree burns. Biomaterials 2007, 28, 3478–3488. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, H.; Yang, X.; Zhang, W.; Jiang, M.; Wen, T.; Wang, J.; Guo, R.; Liu, H. Preparation and Application of Quaternized Chitosan- and AgNPs-Base Synergistic Antibacterial Hydrogel for Burn Wound Healing. Molecules 2021, 26, 4037. [Google Scholar] [CrossRef]
- Murali, V.P.; Fujiwara, T.; Gallop, C.; Wang, Y.; Wilson, J.A.; Atwill, M.T.; Kurakula, M.; Bumgardner, J.D. Modified electrospun chitosan membranes for controlled release of simvastatin. Int. J. Pharm. 2020, 584, 119438. [Google Scholar] [CrossRef] [PubMed]
- Murali, V.P.; Guerra, F.D.; Ghadri, N.; Christian, J.M.; Stein, S.H.; Jennings, J.A.; Smith, R.A.; Bumgardner, J.D. Simvastatin loaded chitosan guided bone regeneration membranes stimulate bone healing. J. Periodontal Res. 2021, 56, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Ghadri, N.; Anderson, K.M.; Adatrow, P.; Stein, S.H.; Su, H.; Garcia-Godoy, F.; Karydis, A.; Bumgardner, J.D. Evaluation of Bone Regeneration of Simvastatin Loaded Chitosan Nanofiber Membranes in Rodent Calvarial Defects. J. Biomater. Nanobiotechnol. 2018, 09, 210–231. [Google Scholar] [CrossRef]
- Daamen, W.F.; Veerkamp, J.H.; Van Hest, J.C.M.; Van Kuppevelt, T.H. Elastin as a biomaterial for tissue engineering. Biomaterials 2007, 28, 4378–4398. [Google Scholar] [CrossRef]
- Sell, S.A.; Wolfe, P.S.; Garg, K.; McCool, J.M.; Rodriguez, I.A.; Bowlin, G.L. The Use of Natural Polymers in Tissue Engineering: A Focus on Electrospun Extracellular Matrix Analogues. Polymers 2010, 2, 522–553. [Google Scholar] [CrossRef]
- Alimohammad, A.; Mohammadali, M.; Mahmod, K.; Khadijeh, S. A Study of The Effect of Magnesium Hydroxide on The Wound Healing Process in Rats. Med. J. Islamic Acad. Sci. 2006, 16, 165–170. Available online: https://medicaljournal-ias.org/jvi.aspx?pdir=ias&plng=eng&un=IAS-63626 (accessed on 25 February 2022).
- Al Alawi, A.M.; Majoni, S.W.; Falhammar, H. Magnesium and Human Health: Perspectives and Research Directions. Int. J. Endocrinol. 2018, 2018, 9041694. [Google Scholar] [CrossRef]
- Tang, X.; Wang, X.; Sun, Y.; Zhao, L.; Li, D.; Zhang, J.; Sun, H.; Yang, B. Magnesium Oxide-Assisted Dual-Cross-Linking Bio-Multifunctional Hydrogels for Wound Repair during Full-Thickness Skin Injuries. Adv. Funct. Mater. 2021, 31, 2105718. [Google Scholar] [CrossRef]
- Daniela, B.; Bernardini, D.; Nasulewic, A.; Mazur, A.; Maier, J.A.M. Magnesium and microvascular endothelial cells: A role in inflammation and angiogenesis. Front. Biosci. 2005, 10, 1177–1182. [Google Scholar] [CrossRef]
- Lapidos, K.A.; Woodhouse, E.C.; Kohn, E.C.; Masiero, L. Mg(++)-induced endothelial cell migration: Substratum selectivity and receptor-involvement. Angiogenesis 2001, 4, 21–28. [Google Scholar] [CrossRef]
- Zhou, H.; Luchini, T.J.F.; Bhaduri, S.B. Microwave assisted synthesis of amorphous magnesium phosphate nanospheres. J. Mater. Sci. Mater. Med. 2012, 23, 2831–2837. [Google Scholar] [CrossRef]
- Gillen, G.; Joel, B.; Jegdish, B.; Jessica, J.; Warren, H.; Hitesh, A.; Diego, P.; Timothy, M.; Vishnu, M.; Sanjay, M. Use of calcium phosphate- silver nanoparticles in chitosan coatings on titanium and for drug delivery. Front. Bioeng. Biotechnol. 2016, 4. [Google Scholar] [CrossRef]
- Nayak, R.; Padhye, R.; Kyratzis, I.L.; Truong, Y.B.; Arnold, L. Effect of viscosity and electrical conductivity on the morphology and fiber diameter in melt electrospinning of polypropylene. Text. Res. J. 2012, 83, 606–617. [Google Scholar] [CrossRef]
- Cramariuc, B.; Cramariuc, R.; Scarlet, R.; Manea, L.R.; Lupu, I.G.; Cramariuc, O. Fiber diameter in electrospinning process. J. Electrost. 2013, 71, 189–198. [Google Scholar] [CrossRef]
- Feng, J.J. The stretching of an electrified non-Newtonian jet: A model for electrospinning. Phys. Fluids 2002, 14, 3912–3926. [Google Scholar] [CrossRef]
- Malhotra, B.D.; Kaushik, A. Metal oxide–chitosan based nanocomposite for cholesterol biosensor. Thin Solid Films 2009, 518, 614–620. [Google Scholar] [CrossRef]
- Feng, K.-J.; Yang, Y.-H.; Wang, Z.-J.; Jiang, J.-H.; Shen, G.-L.; Yu, R.-Q. A nano-porous CeO2/Chitosan composite film as the immobilization matrix for colorectal cancer DNA sequence-selective electrochemical biosensor. Talanta 2006, 70, 561–565. [Google Scholar] [CrossRef]
- Sencadas, V.; Correia, D.; Areias, A.; Botelho, G.; Fonseca, A.; Neves, I.; Ribelles, J.G.; Mendez, S.L. Determination of the parameters affecting electrospun chitosan fiber size distribution and morphology. Carbohydr. Polym. 2011, 87, 1295–1301. [Google Scholar] [CrossRef]
- Elsabee, M.Z.; Naguib, H.F.; Morsi, R. Chitosan based nanofibers, review. Mater. Sci. Eng. C 2012, 32, 1711–1726. [Google Scholar] [CrossRef]
- Sapkota, S.; Chou, S.-F. Electrospun Chitosan-based Fibers for Wound Healing Applications. J. Biomater. 2020, 4, 51. [Google Scholar] [CrossRef]
- Su, H.; Liu, K.-Y.; Karydis, A.; Abebe, D.G.; Wu, C.; Anderson, K.M.; Ghadri, N.; Adatrow, P.; Fujiwara, T.; Bumgardner, J.D. In vitro and in vivo evaluations of a novel post-electrospinning treatment to improve the fibrous structure of chitosan membranes for guided bone regeneration. Biomed. Mater. 2016, 12, 015003. [Google Scholar] [CrossRef]
- Dos Santos, L.A.L. Natural Polymeric Biomaterials: Processing and Properties. Ref. Modul. Mater. Sci. Mater. Eng. 2017, 3–4. [Google Scholar] [CrossRef]
- Swindle-Reilly, K.E.; Paranjape, C.S.; Miller, C.A. Electrospun poly(caprolactone)-elastin scaffolds for peripheral nerve regeneration. Prog. Biomater. 2014, 3, 20. [Google Scholar] [CrossRef]
- Nitti, P.; Gallo, N.; Natta, L.; Scalera, F.; Palazzo, B.; Sannino, A.; Gervaso, F. Influence of Nanofiber Orientation on Morphological and Mechanical Properties of Electrospun Chitosan Mats. J. Heal. Eng. 2018, 2018, 3651480. [Google Scholar] [CrossRef]
- Vondran, J.L.; Sun, W.; Schauer, C.L. Crosslinked, electrospun chitosan–poly(ethylene oxide) nanofiber mats. J. Appl. Polym. Sci. 2008, 109, 968–975. [Google Scholar] [CrossRef]
- Pan, F.; Giovannini, G.; Zhang, S.; Altenried, S.; Zuber, F.; Chen, Q.; Boesel, L.F.; Ren, Q. pH-responsive silica nanoparticles for the treatment of skin wound infections. Acta Biomater. 2022, 145, 172–184. [Google Scholar] [CrossRef]
- Shen, Z.; Cai, N.; Xue, Y.; Chan, V.; Yu, B.; Wang, J.; Song, H.; Deng, H.; Yu, F. Engineering Sustainable Antimicrobial Release in Silica-Cellulose Membrane with CaCO3-Aided Processing for Wound Dressing Application. Polymers 2019, 11, 808. [Google Scholar] [CrossRef]
- Nqakala, Z.B.; Sibuyi, N.R.S.; Fadaka, A.O.; Meyer, M.; Onani, M.O.; Madiehe, A.M. Advances in Nanotechnology towards Development of Silver Nanoparticle-Based Wound-Healing Agents. Int. J. Mol. Sci. 2021, 22, 11272. [Google Scholar] [CrossRef]
- Kim, K.; Yu, M.; Zong, X.; Chiu, J.; Fang, D.; Seo, Y.-S.; Hsiao, B.S.; Chu, B.; Hadjiargyrou, M. Control of degradation rate and hydrophilicity in electrospun non-woven poly(d,l-lactide) nanofiber scaffolds for biomedical applications. Biomaterials 2003, 24, 4977–4985. [Google Scholar] [CrossRef]
- Sousa, I.; Mendes, A.; Pereira, R.F.; Bártolo, P.J. Collagen surface modified poly(ε-caprolactone) scaffolds with improved hydrophilicity and cell adhesion properties. Mater. Lett. 2014, 134, 263–267. [Google Scholar] [CrossRef]
- Perez-Puyana, V.; Villanueva, P.; Jiménez-Rosado, M.; de la Portilla, F.; Romero, A. Incorporation of Elastin to Improve Polycaprolactone-Based Scaffolds for Skeletal Muscle via Electrospinning. Polymers 2021, 13, 1501. [Google Scholar] [CrossRef]
- Barenghi, R.; Beke, S.; Romano, I.; Gavazzo, P.; Farkas, B.; Vassalli, M.; Brandi, F.; Scaglione, S. Elastin-Coated Biodegradable Photopolymer Scaffolds for Tissue Engineering Applications. BioMed Res. Int. 2014, 2014, 624645. [Google Scholar] [CrossRef]
- Sionkowska, A. Current research on the blends of natural and synthetic polymers as new biomaterials: Review. Prog. Polym. Sci. 2011, 36, 1254–1276. [Google Scholar] [CrossRef]
- Costa, R.R.; Castro, E.; Arias, F.J.; Rodríguez-Cabello, J.C.; Mano, J.F. Multifunctional Compartmentalized Capsules with a Hierarchical Organization from the Nano to the Macro Scales. Biomacromolecules 2013, 14, 2403–2410. [Google Scholar] [CrossRef]
- Su, H.; Fujiwara, T.; Bumgardner, J. A Study of Combining Elastin in the Chitosan Electrospinning to Increase the Mechanical Strength and Bioactivity. Mar. Drugs 2021, 19, 169. [Google Scholar] [CrossRef]
- Hunter, R.J. Zeta Potential in Colloidal Science; Academic Press: Cambridge, MA, USA, 1988; Volume 2, p. 98. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zeta Potential (mV) | Diameter (nm) |
|---|---|
| −11.8 ± 0.11 | 1660 ± 140 |
| Membrane Type | Ash Content (%) |
|---|---|
| C | 3.64 |
| CE | 2.95 |
| CMg | 6.92 |
| CEMg | 8.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryan, A.; Wales, E.; Vedante, S.; Blanquer, A.; Neupane, D.; Mishra, S.; Bačáková, L.; Fujiwara, T.; Jennings, J.A.; Bumgardner, J.D. Evaluation of Magnesium-Phosphate Particle Incorporation into Co-Electrospun Chitosan-Elastin Membranes for Skin Wound Healing. Mar. Drugs 2022, 20, 615. https://doi.org/10.3390/md20100615
Bryan A, Wales E, Vedante S, Blanquer A, Neupane D, Mishra S, Bačáková L, Fujiwara T, Jennings JA, Bumgardner JD. Evaluation of Magnesium-Phosphate Particle Incorporation into Co-Electrospun Chitosan-Elastin Membranes for Skin Wound Healing. Marine Drugs. 2022; 20(10):615. https://doi.org/10.3390/md20100615
Chicago/Turabian StyleBryan, Alex, Ethan Wales, Samarth Vedante, Andreu Blanquer, Dipesh Neupane, Sanjay Mishra, Lucie Bačáková, Tomoko Fujiwara, Jessica Amber Jennings, and Joel D. Bumgardner. 2022. "Evaluation of Magnesium-Phosphate Particle Incorporation into Co-Electrospun Chitosan-Elastin Membranes for Skin Wound Healing" Marine Drugs 20, no. 10: 615. https://doi.org/10.3390/md20100615
APA StyleBryan, A., Wales, E., Vedante, S., Blanquer, A., Neupane, D., Mishra, S., Bačáková, L., Fujiwara, T., Jennings, J. A., & Bumgardner, J. D. (2022). Evaluation of Magnesium-Phosphate Particle Incorporation into Co-Electrospun Chitosan-Elastin Membranes for Skin Wound Healing. Marine Drugs, 20(10), 615. https://doi.org/10.3390/md20100615