3. Materials and Methods
With regard to the general techniques used in this study, all moisture sensitive reactions were carried out under argon atmosphere. Anhydrous solvents were obtained as follows: THF and DME distilled from sodium and benzophenone; dichloromethane, toluene, triethylamine and diisopropylamine, distilled from CaH2. Column chromatography was performed with 230–400 mesh silica gel under low pressure of 5–10 psi. TLC was carried out with silica gel 60-F-254 plates, visualized under UV light and stained with phosphomolybdic acid. In addition, 1H NMR and 13C NMR spectra were recorded on Bruker Avance ARX- 400 (400 and 100 MHz), or Bruker DRX500 (500 and 125 MHz) spectrometers. High and low resolution mass spectra were carried out by the Mass Spectroscopy Center at Purdue University. HPLC analysis and preparative HPLC were performed on Agilent 1100 Series instruments (Agilent Technologies, Santa Clara, CA, USA, Agilent 1200 Series Autosampler used for analytical work).
(S)-4-(1-phenyl-1H-tetrazol-5-ylthio)butane-1,2-diol (13): To a stirred solution of alcohol 10 (1.505 g, 10.3 mmol), 1-(4-Hydroxyphenyl)-1H-tetrazole-5-thiol (3.67 g, 20.6 mmol) and triphenylphosphine (4.05 g, 15.5 mmol) in THF (30 mL), we added DIAD (3.6 mL, 18.5 mmol) at 0 °C. The reaction mixture was warmed up to rt and stirred overnight, before it was poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography (20% EtOAc/hexanes) provided the corresponding sulfide as colorless oil (2.68 g, 85%). Rf value (EtOAc/hexane 1:1): 0.75; [α]D20 = +12.5 (c = 1.0, CHCl3); IR (film, cm−1) 3384, 2936, 1644, 1596, 1499, 1462, 1388, 1318, 1280, 1074, 760; 1H NMR (400 MHz, CDCl3): 7.58 (brs, 5 H), 4.13–3.85 (m, 2 H), 3.81–3.68 (m, 1 H), 3.68–3.56 (m, 2 H), 3.55–3.42 (m, 1 H), 2.55 (brs, 1 H), 2.09–1.90 (m, 2 H); 13C NMR (100 MHz, CDCl3): 155.1, 133.5, 130.4, 129.9, 124.0, 69.5, 66.4, 33.7, 29.8.
To a stirred solution of the above acetonide (2.60 g, 8.49 mmol) in MeOH (100 mL), we added p-TsOH (320 mg, 1.70 mmol) at rt and stirred for 24 h. Et3N (2 mL) was added at 0 °C to quench the reaction. The mixture was concentrated in vacuo. Flash chromatography on silica gel (5% MeOH/CHCl3) resulted in diol 13 (2.06 g, 91%) as a white solid. Rf value (EtOAc/hexane/MeOH 80:20:6): 0.5; [α]D20 = +10.7 (c = 0.5, CHCl3); IR (film, cm−1) 3411, 3384, 2936, 1644, 1596, 1499, 1462, 1388, 1318, 1280, 1075, 759; 1H NMR (400 MHz, CDCl3): 7.58 (brs, 5 H), 4.13–3.85 (m, 2 H), 3.81–3.68 (m, 1 H), 3.68–3.56 (m, 2 H), 3.55–3.42 (m, 1 H), 2.55 (brs, 1 H), 2.09–1.90 (m, 2 H); 13C NMR (100 MHz, CDCl3): 155.1, 133.5, 130.4, 129.9, 124, 69.5, 66.4, 33.7, 29.8.
(S)-5-(3-(oxiran-2-yl)propyl)-1-phenyl-1H-tetrazole (14): To a stirred solution of diol 13 (2.06 g, 7.74 mmol) in DCM (60 mL), Bu2SnO (3.85 g, 15.5 mmol), triethylamine (1.3 mL, 9.29 mmol) and tosyl chloride (1.58 g, 8.12 mmol) were added at 0 °C. The reaction mixture was stirred for 6 h, followed by dilution with water (10 mL). The organic layer was separated and the aqueous layer was extracted with DCM. The combined organic extracts were dried over anhydrous MgSO4 and concentrated. The crude product was purified by flash column chromatography on silica gel (ethyl acetate–hexanes 1:1) to yield the corresponding tosylate (2.52 g, 81%). Rf value (EtOAc/hexane 1:1): 0.4; [α]D20 = +12.0 (c = 0.65, CHCl3); IR (film, cm−1): 3400, 3060, 2946, 1597, 1499, 1387, 1357, 1243; 1H NMR (400 MHz, CDCl3): 7.75 (d, J = 8.2 Hz, 2 H), 7.53 (s, 5 H), 7.30 (d, J = 8.2 Hz, 2 H), 4.03–3.92 (m, 4 H), 3.50–3.43 (m, 2 H), 2.03–1.87 (m, 2 H), 2.39 (s, 3 H); 13C NMR (100 MHz, CDCl3): 154.4, 144.9, 133.3, 132.3, 130.1, 129.8, 129.7, 127.7, 123.6, 73.1, 66.9, 32.7, 29.2, 21.4.
To a stirred solution of the above tosylate (3.54 g, 8.79 mmol) in CH3OH–DCM (9:1, 90 mL), K2CO3 (1.58 g, 11.4 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 1 h, concentrated, and then diluted with dichloromethane (30 mL) and water (10 mL). The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic extracts were dried over anhydrous MgSO4 and concentrated to provide 14 (1.58 g, 82%) as a colorless oil. Rf value (EtOAc/hexane 1:1): 0.65; [α]D20 = −15.5 (c = 1, CHCl3); IR (film, cm−1): 3056, 2991, 2924, 1596, 1500, 1461; 1H NMR (400 MHz, CDCl3): 7.56–7.49 (m, 5 H), 3.54–3.45 (m, 2 H), 3.06–3.01 (m, 1 H), 2.78–2.74 (m, 1 H), 2.52 (dd, J = 4.6, 2.7 Hz, 1 H) 2.29–2.21 (t, J = 4.6, 1 H),1.92 (td, J = 14.2, 6.9 Hz, 1 H); 13C NMR (100 MHz, CDCl3): 153.9, 133.5, 130.1, 29.8, 129.8, 123.7, 50.6, 46.8, 32.0.
(R)-5-(3-(tert-butyldimethylsilyloxy)-5-methylhex-5-enylthio)-1-phenyl-1H-tetrazole (8): To a stirred solution of epoxide 14 (1.49 g, 6 mmol) and CuCN (54 mg, 0.6 mmol) in THF (40 mL) at −78 °C, we added isopropenylmagnesium bromide (3.6 mL, 0.9 mmol). The resulting suspension was warmed up to 0 °C and stirred for 30 min. The reaction mixture was cooled again to −78 °C and more vinylmagnesium bromide (12 mL, 6 mmol) was added dropwise. The reaction mixture was warmed up to 0 °C and stirred for 1 h, before 20 mL of saturated NH4Cl and 10 mL of NH4OH were added to quench the reaction. The layers were separated and the aqueous layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with brine and dried over anhydrous magnesium sulfate. Filtration and concentration under reduced pressure gave a crude product. Flash chromatography on silica gel (20% EtOAc/hexanes) afforded the corresponding alcohol as a colorless oil (1.6 g, 92%). Rf value (EtOAc/hexane 1:2) 0.25; [α]D20 = +15.8 (c = 2, CHCl3); IR (film, cm−1): 3414, 2932, 1596, 1500, 1388, 1074, 761; 1H NMR (400 MHz, CDCl3) δ 7.63–7.48 (m, 1H), 4.85 (s, 1H), 4.77 (s, 1H), 3.89 (ddq, J = 12.8, 6.5, 3.3 Hz, 1H), 3.58–3.48 (m, 1H), 2.75 (s, 1H), 2.20 (d, J = 6.5 Hz, 1H), 2.09–1.97 (m, 1H), 1.89 (ddd, J = 21.4, 8.0, 6.2 Hz, 1H), 1.73 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 154.7, 142.1, 133.5, 130.1, 129.7, 123.7, 113.6, 66.8, 45.8, 36.6, 29.9, 22.4.
To a stirred solution of the above alcohol (942mg, 3.24 mmol) in DMF (6 mL), we added imidazole (353 mg, 5.18 mmol) and TBSCl (538 mg, 3.57 mmol), respectively, at 0 °C. The reaction mixture was stirred at 23 °C for 12 h. A solution of saturated NaHCO3 (aq) was added and the aqueous layer was extracted by diethyl ether. The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography (4% EtOAc/hexanes) gave the silyl ether 8 (1.25 g, 95%) as a colorless oil. Rf value (EtOAc/hexane 1:10) 0.45; [α]D20 = +12.5 (c = 1, CHCl3); IR (film, cm−1): 2953, 2929, 2857, 1598, 1500, 1387, 1074, 821, 775; 1H NMR (400 MHz, CDCl3) δ 7.81–7.37 (m, 5H), 4.77 (s, 1H), 4.70 (s, 1H), 3.96 (tdd, J = 7.5, 5.4, 4.0 Hz, 1H), 3.55–3.34 (m, 2H), 2.27 (dd, J = 13.6, 5.3 Hz, 1H), 2.17 (dd, J = 13.6, 7.6 Hz, 1H), 2.09–1.95 (m, 1H), 1.93–1.79 (m, 1H), 1.71 (s, 3H), 0.88 (s, 9H), 0.06 (d, J = 1.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 154.3, 141.8, 133.6, 129.9, 129.7, 123.7, 113.5, 69.3, 45.8, 35.6, 29.5, 25.8, 22.8, 17.9, −4.4, −4.7. MS (ESI, m/z) [M+Na]+ 427.2.
Acetonide 15: To a stirred solution of olefin 8 (1.23 g, 3 mmol) and aldehyde 7 (645 mg, 3.3 mmol) in DCM (30 mL), we added SnCl4 (4.5 mL, 1 M soln in DCM, 4.5 mmol) at −78 °C; the reaction mixture was warmed up to 0 °C over 1 h and stirred for 4 h. The reaction mixture was then poured into sat NaHCO3 (20 mL) with crushed ice. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:3) to give the corresponding diol (1.4 g, 78% yield). To a stirred solution of the diol (1.4 g, 2.34 mmol) and 2-methoxypropene (0.66 mL, 7 mmol) in DCM (40 mL), we added PPTS (50 mg, 0.2 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h before Et3N (1.0 mL) was added. The solvents were removed in vacuo and the crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the acetonide 12 (1.47 g, 98%) as a colorless oil. Rf value (EtOAc/hexane 1:4): 0.55; [α]D20 = −1.8 (c = 1, CHCl3); IR (film, cm−1) 3068, 2886, 1500, 1410, 1097; 1H NMR (400 MHz, CDCl3) δ7.56–7.53 (m, 5H), 7.32–7.25 (m, 5H), 4.90 (s, 1H), 4.85 (s, 1H), 4.55 (AB, JAB = 12.0 Hz, ΔυAB = 20.7 Hz, 2H), 4.13–4.10 (m, 1H), 4.0–3.97 (m 1H), 3.52–3.40 (m, 2H), 3.43–3.38 (m, 2H), 2.50–2.45 (m, 1H), 2.22–2.13 (m, 3H), 2.02–1.99 (m, 1H), 1.86–1.81 (m, 1H), 1.46 (s, 3H), 1.35 (s, 3H), 1.11 (s, 3H), 0.88 (s, 9H), 0.06 (s, 6H); 13C NMR (400 MHz, CDCl3) δ 154.3, 143.0, 138.2, 133..6, 130.0, 129.7, 128.2, 127.5, 127.3, 123.7, 114.8, 107.2, 81.6, 78.7, 74.8, 73.5, 69.3, 44.2, 36.4, 35.4, 29.4, 28.6, 26.5, 25.8, 19.2, 18.0, −4.3, −4.8; MS (ESI, m/z) [M + Na]+ 661.3.
Sulfone 5: To a stirred solution of thus obtained sulfide 15 (502 mg, 0.79 mmol) in ethanol (13 mL), we added a soln of ammonium molybdate (320 mg, 0.26 mmol) in hydrogen peroxide (1.6 mL) and water (0.8 mL) at rt. The reaction mixture was stirred for 12 h and poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the sulfone 5 as a colorless oil (519 mg, 98% yield). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = −2.6 (c = 1, CHCl3); IR (film, cm−1) 3038, 2887, 1512, 1215, 1097; 1H NMR (400 MHz, CDCl3) δ7.69–7.58 (m, 5H), 7.33–7.27 (m, 5H), 4.94 (s, 1H), 4.87 (s, 1H), 4.56 (AB, JAB = 12.0 Hz, ΔυAB = 20.7 Hz, 2H), 4.12–4.05 (m, 1H), 4.07–4.03 (m 1H), 3.90–3.73 (m, 2H), 3.47–3.41 (m, 2H), 2.54–2.50 (m, 1H), 2.22–2.17 (m, 2H), 2.15–2.10 (m, 2H), 2.0–1.92 (m, 1H), 1.46 (s, 3H), 1.36 (s, 3H), 1.14 (s, 3H), 0.90 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.3, 142.8, 138.1, 133, 131.3, 129.6, 128.3, 127.5, 127.4, 125, 115.2, 107.3, 81.5, 79.1, 77.4, 76.7, 75, 73.5, 68.2, 52.3, 43.9, 36.4, 28.6, 28, 26.5, 25.8, 19.2, 17.9, −4.4, −4.9; MS (ESI, m/z) [M + Na]+ 693.3. HRMS (ESI) [M + Na]+ calcd for C34H50N4O6SSiNa 693.3118, found 693.3111.
(2R,3S)-3-(methoxymethoxy)-2-methylpent-4-en-1-ol (20): To a stirred solution of racemic alcohol 19 (15.2 g, 88.4 mmol) in vinyl acetate (60 mL) and pentane (120 mL), we added immobilized lipase PS 30 (20% on celite, 15.2 g); the suspension was stirred at 23 °C for 30 h, monitored by 1H NMR, until a half conversion was obtained. Suction filtration furnished a crude product, which was purified by flash chromatography (EtOAc/hexane 1:1) to give the (+)-alcohol as a colorless oil (7.70 g, 44%), along with corresponding acetate (9.3 g, 49%). Rf value (EtOAc/hexane 1:5) 0.45; [α]D20 = +8.8 (c = 1.7, CHCl3); IR (film, cm−1): 3422, 2981, 1744, 1372, 1236, 1026, 947; 1H NMR (400 MHz, CDCl3) δ 5.90–5.68 (m, 1H), 5.22 (d, J = 17.3 Hz, 1H), 5.11–4.95 (m, 1H), 4.40 (s, 1H), 3.38 (s, 1H), 2.54–2.18 (m, 2H), 1.38 (d, J = 1.5 Hz, 9H); 13C NMR (100 MHz, CDCl3) δ 171.4, 139.0, 114.9, 81.1, 68.9, 42.1, 27.9.
To a stirred solution of diisopropylamine (15.8 mL, 0.112 mol) in THF (80 mL) at −78 °C, we added n-BuLi (1.6 M in hexane, 71.8 mL, 0.107 mol) dropwise. The mixture was kept at −78 °C for 20 min before a solution of the above alcohol (7.4 g, 42.9 mmol) was added dropwise. After another 20 min of stirring, MeI (6.7 mL, 0.107 mol) was added to the reaction mixture in a dropwise manner. The reaction was stirred at −10 °C for 4 h before 50 mL sat NH4Cl was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 50 mL). The combined organic solution was washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (10% EtOAc/hexanes) afforded the anti-methyl alcohol (5.84 g, 73%) as a colorless oil. Rf value (EtOAc/hexane 1:5) 0.5; [α]D20 = −9.9 (c = 1.26, CHCl3); IR (film, cm−1): 3414, 2982, 2936, 1744, 1394, 1370, 1235, 1024, 991; 1H NMR (400 MHz, CDCl3) δ 5.79 (ddd, J = 17.0, 10.4, 6.2 Hz, 1H), 5.24 (dd, J = 17.2, 1.0 Hz, 1H), 5.13 (dd, J = 10.4, 1.1 Hz, 1H), 4.10 (q, J = 6.5 Hz, 1H), 3.01 (d, J = 5.9 Hz, 1H), 2.41 (p, J = 7.1 Hz, 1H), 1.40 (s, 9H), 1.09 (dd, J = 7.2, 1.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.7, 138.2, 116.2, 80.9, 74.6, 45.7, 27.9.
To a stirred solution of the above alcohol (5.41 g, 29.1 mmol) in DMF (20 mL), we added DIPEA (12.6 mL, 72.6 mmol), MOMCl (4.4 mL, 58.2 mmol), and the catalytic amount of TBAI at 0 °C, respectively. The reaction mixture was stirred for 12 h before poured into saturated NaHCO3 (aq) and the aqueous layer was extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) afforded the MOM ether (6.42 g, 96%) as a colorless oil. Rf value (EtOAc/hexane 1:5) 0.65; [α]D20 = +70 (c = 1.34, CHCl3); IR (film, cm−1): 2946, 2936, 1745, 1394, 1371, 1291, 1160, 1026, 991, 845; 1H NMR (400 MHz, CDCl3) δ 5.79 (ddd, J = 17.0, 10.4, 6.2 Hz, 1H), 5.24 (dd, J = 17.2, 1.0 Hz, 1H), 5.13 (dd, J = 10.4, 1.1 Hz, 1H), 4.65 (d, J = 6.7 Hz, 1H), 4.47 (d, J = 6.7 Hz, 1H), 4.10 (q, J = 6.5 Hz, 1H), 3.34 (s, 3H), 3.01 (d, J = 5.9 Hz, 1H), 2.41 (p, J = 7.1 Hz, 1H), 1.40 (s, 9H), 1.09 (dd, J = 7.2, 1.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.7, 138.2, 116.2, 94.5, 80.9, 74.6, 55.6, 45.7, 27.9.
To a stirred suspension of LiAlH4 (1.54 g, 38.5 mmol) in Et2O (40 mL), a solution of the above ester (4.44 g, 19.3 mmol) in Et2O (10 mL) was transferred in at 0 °C. The reaction mixture was stirred at 0 °C for 1 h before being quenched by adding 20 mL saturated NH4Cl (aq) and 20 mL 25% Rochelle salt solution. The mixture was stirred at rt for 4 h and extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% Et2O/hexanes) afforded alcohol 20 (3.06 g, 99%). Rf value (EtOAc/hexane 1:2) 0.4; [α]D20 = +150 (c = 2.04, CHCl3); IR (film, cm−1): 3440, 3090, 1612, 1394, 1291, 1156, 1025; 1H NMR (400 MHz, CDCl3) δ 5.75 (ddd, J = 17.0, 10.7, 7.3 Hz, 1H), 5.29–5.19 (m, 2H), 4.66 (d, J = 6.6 Hz, 1H), 4.54 (d, J = 6.7 Hz, 1H), 4.19–4.11 (m, 1H), 3.69–3.59 (m, 1H), 3.55–3.48 (m, 1H), 3.37 (s, 3H), 2.57 (s, 1H), 2.00–1.87 (m, 1H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 135.5, 118, 94.2, 79.6, 65.2, 55.6, 39.6, 11.7. MS (ESI, m/z) [M + Na]+ 183.1.
(4R,5S)-methyl 5-(methoxymethoxy)-4-methylhept-6-en-2-ynoate (21): To a stirred solution of DMSO (3.7 mL, 52.3 mmol) in DCM (50 mL), we added (COCl)2 (2.7 mL, 31.4 mmol) at −78 °C. The mixture was stirred for 5 min, before a solution of alcohol 14 (3.35 g, 20.9 mmol) in DCM (10 mL) was added dropwise. The resulting suspension was stirred at −78 °C for 30 min. Et3N (14.6 mL, 0.105 mol) was added slowly. The reaction mixture was stirred at −78 °C for 1 h and allowed to warm up to room temperature and stirred for 30 min, before pouring it into 1M NaHSO4 solution. The organic layer was separated and the aqueous layer was extracted with DCM. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give crude aldehyde (3.35 g), which was used in the next step without further purification. Rf value (EtOAc/hexane 1:2) 0.7.
To a stirred solution of the CBr4 (13.86 g, 41.8 mmol) in DCM (50 mL) at 0 °C, we added PPh3 (21.93 g, 83.6 mmol). The reaction mixture was stirred at 0 °C for 10 min. A solution of the above aldehyde in DCM (10 mL) was added dropwise. The mixture was warmed up to 23 °C and stirred for 30 min. The reaction was poured into saturated NaHCO3 (aq). The aqueous layer was extracted by DCM. The combined organic extracts were washed with brine and dried over anhydrous Na2SO4, concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) gave the dibromide (5.12 g, 78% for two steps) as a colorless oil. Rf value (EtOAc/hexane 1:4) 0.8; 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 6.35 (d, J = 9.4 Hz, 1H), 5.64 (ddd, J = 17.3, 10.4, 7.9 Hz, 1H), 5.38–5.11 (m, 2H), 4.67 (d, J = 6.8 Hz, 1H), 4.50 (d, J = 6.8 Hz, 1H), 3.89 (dd, J = 7.7, 5.9 Hz, 1H), 3.37 (s, 3H), 2.72–2.55 (m, 1H), 1.32–0.82 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 140.4, 135.7, 119.0, 93.6, 88.8, 79.6, 55.5, 42.9, 15.2.
To a stirred solution of the above dibromide (3.88 g, 10.1 mmol) in THF (30 mL), we added n-BuLi (1.6 M in hexanes, 19.0 mL, 30.3 mmol) at −78 °C. The mixture was stirred at −78 °C for 15 min before methyl chloroformate (2.34 mL, 30.3 mmol) was added. The reaction mixture was stirred at −78 °C for 1 h before pouring it into saturate NH4Cl (aq). The organic layer was separated and the aqueous layer was extracted by Et2O (3 × 50 mL). The organic extracts were combined, washed by water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) produced alkynyl ester 21 (2.14 g, 99%) as a pale yellow oil. Rf value (EtOAc/hexane 1:10) 0.45; [α]D20 = +82 (c = 1.22, CHCl3); IR (film, cm−1): 2950, 2888, 1716, 1644, 1435, 1225, 1156, 1031, 918; 1H NMR (400 MHz, CDCl3) δ 5.64 (ddd, J = 17.0, 10.6, 7.7 Hz, 1H), 5.37–5.13 (m, 2H), 4.63 (d, J = 6.9 Hz, 1H), 4.49 (d, J = 6.9 Hz, 1H), 4.03–3.87 (m, 1H), 3.67 (s, 3H), 3.33 (s, 3H), 2.79–2.63 (m, 1H), 1.16 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 153.9, 134.7, 119.8, 93.5, 90.3, 78.5, 73.8, 55.5, 52.3, 31.3, 15.6.
(4R,5S,2Z)-methyl-5-(methoxymethoxy)-3,4-dimethylhepta-2,6-dienoate (22): To a suspension of CuI (5.42 g, 28.4 mmol) in THF (60 mL), we added MeLi (28.5 mL, 45.6 mmol) at −60 °C. The mixture was slowly warmed up to 0 °C to obtain a clear solution. A soln of alkynyl ester 21 (2.41 g, 11.4 mmol) in THF (5 mL) was added slowly at −60 °C and stirred at −40 °C for 2 h. AcOH (2.74 mL, 47.9 mmol) was added to quench the reaction, followed by sat NH4Cl (50 mL). The organic layer was separated and the aqueous layer was extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) produced Z-enoate 22 (2.50 g, 96%) as a colorless oil. Rf value (EtOAc/hexane 1:10) 0.45; [α]D20 = +56 (c = 0.3, CHCl3); IR (film, cm−1): 2951, 2889, 1718, 1646, 1227, 1157, 1031, 1093, 919, 859; 1H NMR (300 MHz, CDCl3) δ 5.71 (brs, 1H), 5.28–5.19 (m, 1H), 4.66 (d, J = 7 Hz, 1H), 4.47 (d, J = 7 Hz, 1H), 4.13 (dq, J = 8.8, 7 Hz, 1H), 3.95 (t, J = 8.5 Hz, 1H), 3.65 (s, 3H), 3.33 (s, 3H), 1.88 (s, 3H), 0.98 (d, J = 7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 166.5, 161.8, 136.6, 119.2, 93.5, 80.1, 55.7, 50.7, 38.3, 30, 19.9, 15.1.
(2R,3R,Z)-6-(tert-butyldimethylsilyloxy)-2-(methoxymethoxy)-3,4-dimethylhex-4-enal (6): To a stirred soln of ester 22 (2.43 g, 10.6 mmol) in DCM (50 mL), we added DIBAL-H (31.9 mL, 31.9 mmol) at −78 °C. The reaction mixture was stirred 1 h, before the addition of 20 mL saturated NH4Cl (aq) and 20 mL 25% Rochelle salt solution. The mixture was stirred at rt for 4 h and extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% Et2O/hexanes) afforded the allyl alcohol (2.02 g, 95%). Rf value (EtOAc/hexane 1:4) 0.25; [α]D20 = +106 (c = 2, CHCl3); IR (film, cm−1): 3349, 2962, 2823, 1613, 1444, 1227, 1152, 1028, 916; 1H NMR (400 MHz, CDCl3): 5.69 (t, J = 7.1 Hz, 1H), 5.62–5.50 (m, 1H), 5.26 (d, J = 10.1 Hz, 1H), 5.20 (d, J = 17.1 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.35 (d, J = 6.9 Hz, 1H), 4.18 (dd, J = 8.8, 7.0 Hz, 1H), 3.82–3.75 (m, 2H), 3.26 (s, 3H), 2.85–2.75 (m, 2H). 1.67 (s, 3H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 141.9, 136.3, 126.4, 119.7, 92.8, 78.4, 57, 55.5, 38.1, 18.1, 15.1.
To a stirred solution of the above alcohol (1.45 g, 7.24 mmol) in DCM (50 mL), we added imidazole (739 mg, 10.9 mmol) and TBSCl (1.2 g, 7.96 mmol) at 0 °C. The reaction was warmed up to rt and stirred for 1 h, before pouring it into a mixture of sat NaHCO3 (50 mL) and crushed ice. The mixture was extracted with ethers (3 × 60 mL) and the organic layer was washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo to give the crude TBS ether as a clear oil, which was used for the next step without further purification. Flash chromatography on silica gel (25% EtOAc/hexanes) afforded the silyl ether (2.28 g, 99%). Rf value (EtOAc/hexane 1:4) 0.85; [α]D20 = +34 (c = 1.05, CHCl3); IR (film, cm−1): 2960 2821, 1607, 1227, 1152, 1091, 916; 1H NMR (300 MHz, CDCl3): 5.62–5.53 (m, 1H), 5.38 (t, J = 7.1 Hz, 1H), 5.25–5.15 (m, 2H), 4.62 (d, J = 6.9 Hz, 1H), 4.39 (d, J = 6.9 Hz, 1H), 4.32–4.23 (m, 1H), 4.2–4.1 (m, 1H), 3.82 (t, J = 8.5 Hz, 1H), 3.30 (s, 3H), 2.72–2.62 (m, 1H). 1.69 (d, J = 1 Hz, 3H), 0.91 (d, J = 7.0 Hz, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 137.7, 137, 127.3, 118.7, 93.2, 79.8, 60, 55.3, 38.8, 25.9, 18.7, 18.3, 15.1, −5.2.
To a stirred solution of above olefin (1.09 g, 3.47 mmol) in dioxane (24 mL) and water (8 mL), we added 2,6-lutidine (2.02 mL, 17.4 mmol), OsO4 (2.5% in t-BuOH, 1.74 mL, 0.14 mmol) and NaIO4 (2.97 g, 12.9 mmol) at 0 °C. The mixture was stirred at 0 °C for 18 h before saturated NaHCO3 (10 mL) and NaS2O3 (10 mL) were added. The mixture was stirred for another 30 min, extracted by EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% EtOAc/hexanes) produced the aldehyde 6 (736 mg, 67%). Rf value (EtOAc/hexane 1:10) 0.65; [α]D20 = +28 (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.53 (dd, J = 3.1, 0.5 Hz, 1H), 5.41 (t, J = 6.2 Hz, 1H), 4.61 (dd, J = 22.5, 6.9 Hz, 3H), 4.23 (dd, J = 13.0, 7.0 Hz, 1H), 4.17–4.04 (m, 1H), 3.70 (dd, J = 8.2, 3.3 Hz, 1H), 3.35 (d, J = 0.6 Hz, 4H), 3.00 (p, J = 7.2 Hz, 1H), 1.69 (s, 4H), 1.01 (d, J = 7.0 Hz, 4H), 0.90 (s, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 202.2, 136.0, 128.2, 96.8, 84.7, 59.3, 55.9, 34.9, 25.9, 18.9, 18.3, 14.3, −5.3.
Coupling product (23): To a stirred solution of sulfone 5 (628 mg, 0.94 mmol) in DME (30 mL), we added KHMDS (1.85 mL, 0.5 M soln in toluene, 0.93 mmol) at −78 °C. The reaction mixture was stirred for 30 min, before a soln of aldehyde 6 (357 mg, 1.13 mmol) in DME (5 mL) was transferred in. The reaction mixture was stirred for another 30 min, before it was warmed up to rt. The reaction was quenched by sat NH4Cl (10 mL) at −78 °C. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhydrous MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give coupling product 23 as a colorless oil (544 mg, 76% yield). Rf value (EtOAc/hexane 1:10): 0.5; [α]D20 = +14 (c = 0.6, CHCl3); IR (film, cm−1) 2928, 1455, 1248, 1108; 1H NMR (400 MHz, CDCl3) δ 5.73–5.54 (m, 1H), 5.40 (t, J = 6.1 Hz, 1H), 5.23 (dd, J = 15.4, 8.5 Hz, 1H), 4.88 (d, J = 32.2 Hz, 2H), 4.68 (d, J = 6.8 Hz, 1H), 4.56 (d, J = 5.1 Hz, 2H), 4.41 (d, J = 6.8 Hz, 1H), 4.21 (d, J = 6.1 Hz, 3H), 4.14 (dd, J = 8.7, 3.7 Hz, 1H), 3.86 (t, J = 8.3 Hz, 2H), 3.41 (q, J = 9.8 Hz, 2H), 3.30 (s, 3H), 2.35–2.09 (m, 8H), 1.63 (s, 3H), 1.44 (s, 3H), 1.35 (s, 3H), 1.13 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 0.9 (s, 9H), 0.87 (s, 9H), 0.12–−0.07 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.50, 138.19, 131.28, 130.98, 128.21, 127.41, 127.29, 125.83, 114.25, 107.15, 93.11, 81.53, 79.29, 78.34, 74.98, 73.45, 70.52, 60.11, 55.31, 46.97, 43.68, 39.56, 36.49, 28.56, 26.44, 25.90, 25.80, 19.16, 18.27, 17.97, 15.76, 13.52, −4.59, −4.65, −5.15. MS (ESI, m/z) [M + Na]+ 783.5.
Aldehyde (3): To a stirred solution of benzyl ether 23 (550 mg, 0.72 mmol) in THF (10 mL) and allyl ethyl ether (1 mL), we transferred a soln of lithium metal (50 mg, 7.2 mmol) in liquid ammonia (12 mL) in portions at −78 °C. The reaction was carefully monitored by TLC and stopped immediately after the solution became slightly blue. Ammonium chloride (2 g) was added to quench the reaction. The mixture was allowed to warm up to rt to evaporate the ammonia, before water (10 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the alcohol as a colorless oil (402 mg, 83% yield), along with the recovered starting material. Rf value (EtOAc/hexane 1:4): 0.43; [α]D20 = +36.0 (c = 1.2, CHCl3); IR (film, cm−1) 3410, 2954, 1253, 1096; 1H NMR (400 MHz, CDCl3) δ5.69–5.62 (m, 1H), 5.37 (t, J = 4.2 Hz, 1H), 5.25 (dd, J = 15.5, 8.6 Hz, 1H), 4.95 (s, 1H), 4.87 (s, 1H), 4.62 (d, J = 6.8 Hz, 1H), 4.36 (d, J = 6.8 Hz, 1H), 4.28 (dd, J = 8.0, 5.2 Hz, 1H), 4.22–4.18 (m 1H), 4.2–4.12 (m, 1H), 3.9–3.82 (m, 1H), 3.80 (t, J = 8.7 Hz, 1H), 3.53 (dd, J = 12.0, 9.8 Hz, 1H), 3.38–3.33 (m, 1H), 3.30 (s, 3H), 2.58–2.50 (m, 1H), 2.32–2.04 (m, 6H), 1.68 (s, 3H), 1.45 (s, 3H), 1.36 (s, 3H), 1.06 (s, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.90 (s, 9H), 0.87 (s, 9H), 0.06–0.03 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.0, 138.0, 131.6, 131.4, 127.2, 114.6, 107.0, 93.0, 82.6, 79.1, 75.9, 70.8, 65.4, 59.8, 55.3, 43.5, 39.7, 39.1, 36.1, 28.6, 26.6, 25.9, 25.8, 18.6, 18.3, 18.0, 15.4, −4.6, −5.2.
To a suspension of the above alcohol (352 mg, 0.53 mmol) and sodium bicarbonate (265 mg, 3.2 mmol) in DCM (20 mL), we added Dess–Martin periodinane (445 mg, 1.05 mmol) at rt. The reaction mixture was stirred for 1 h before it was poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo to give the crude aldehyde 3 (352 mg, quantitative), which was used directly in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1H), 5.70–5.65 (m, 1H), 5.41 (t, J = 5.3 Hz, 1H), 5.28 (dd, J = 15.5, 8.6 Hz, 1H), 4.97 (s, 1H), 4.91 (s, 1H), 4.66 (d, J = 6.8 Hz, 1H), 4.37 (d, J = 6.8 Hz, 1H), 4.30 (dd, J = 8.0, 5.2 Hz, 1H), 4.21–4.17 (m 2H), 3.90–3.82 (m, 2H), 3.30 (s, 3H), 2.7–2.65 (m, 1H), 2.25–2.12 (m, 6H), 1.69 (s, 3H), 1.50 (s, 3H), 1.46 (s, 3H), 1.20 (s, 3H), 0.92-0.87 (m, 21H), 0.1–0.05 (m, 12H).
(3S,4S)-4-(tert-butyldimethylsilyloxy)-3-methylpentanal (16): To a stirred solution of alcohol 12 (7.31 g, 73 mmol) in DMF (70 mL), we added imidazole (5.96 g, 87.6 mmol) and TBSCl (11 g, 73 mmol) at 0 °C. The reaction was warmed up to rt and stirred for 6 h. The reaction mixture was then poured into a mixture of sat NaHCO3 (50 mL) and crushed ice. The mixture was extracted with ethers (3 × 60 mL) and the organic layer was washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo to give the crude TBS ether as a clear oil, which was used for the next step without further purification. To a stirred solution of thus obtained olefin in THF (60 mL), we added the BH3·THF complex (73 mL, 1 M soln in THF, 73 mmol) at 0 °C. The reaction mixture was allowed to warm up to rt and stirred for 6 h. NaOH (10 mL) and H2O2 (15 mL, 70% soln) were added and the mixture was refluxed for 1 h. The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:20) provided corresponding alcohol 16 as a colorless oil (11.4 g, 67% for two steps). Rf value (EtOAc/hexane 1:10): 0.53; [α]D20 = +2.3 (c = 1.0, CHCl3); IR (film, cm−1) 3340, 2932, 2858, 1463, 1254, 1053; 1H NMR (400 MHz, CDCl3) δ3.77–3.71 (m, 1H), 3.69–3.62 (m, 1H), 3.59–3.53 (m, 1H), 3.19 (m, 1H), 1.71–1.66 (m, 2H), 1.39–1.34 (m, 1H), 1.1 (d, J = 6.4 Hz, 3H), 0.87 (s, 9H), 0.81 (d, J = 6.7 Hz, 3H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 61.8, 38.5, 27.3, 35.2, 25.7, 18.2, 17.9, 17.2, −4.7, −5.1.
(3S,4R,6S,7S)-7-(tert-butyldimethylsilyloxy)-3,6-dimethyloct-1-en-4-ol (17): To a stirred solution of DMSO (2.7 mL, 37.8 mmol) in DCM (70 mL), we added oxalyl chloride (2 mL, 22.7 mmol) at −78 °C. After 10 min of stirring, a solution of alcohol 16 (3.5 g, 15.1 mmol) in DCM (10 mL) was transferred in at the same temperature. The reaction mixture was stirred for 30 min before Et3N (10.5 mL, 75.5 mmol) was added. After stirring at −78 °C for 1 h, the reaction mixture was allowed to warm up to 0 °C for 30 min, before it was poured into sat NaHCO3 soln (30 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the aldehyde as a colorless oil (3.03 g, 87% yield), which was used in the next step immediately. Rf value (EtOAc/hexane 1:10): 0.85.
To a stirred mixture of potassium tert-butoxide (8.7 mL, 1.0 M soln in THF, 8.7 mmol) and trans-2-butene (1.4 mL, 14.5 mmol) in THF (30 mL), we added n-butyllithium (5.5 mL, 1.6 M soln in THF, 8.7 mmol) at −78 °C. After complete addition of n-butyllithium, the mixture was stirred at −45 °C for 10 min. The resulting orange solution was cooled to −78 °C again and a solution of (−)-Ipc2BOMe (3.3 g, 10.4 mmol) in THF (10 mL) was added dropwise. After 30 min of stirring, boron trifluoride etherate (1.5 mL, 11.6 mmol) was added dropwise. Then, the above aldehyde (1.34 g, 5.8 mmol) in THF (5 mL) was transferred in. The mixture was stirred at −78 °C for 3 h before NaOH (6.8 mL, 3 M soln) and H2O2 (4.7 mL, 70% soln) were added. The contents were refluxed for 1 h. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the alcohol 17 as a colorless oil (1.28 g, 77% yield). Rf value (EtOAc/hexane 1:10): 0.63; [α]D20 = +6.3 (c = 0.67, CHCl3); IR (film, cm−1) 3411, 2959, 1462, 1045; 1H NMR (400 MHz, CDCl3) δ5.86–5.77 (m, 1H), 5.1–5.03 (m, 2H), 3.84–3.77 (m, 1H), 3.6–3.54 (m, 1H), 2.21–2.18 (m, 1H), 2.13 (d, J = 4.9 Hz, 1H), 1.82–1.74 (m, 1H), 1.67–1.6 (m, 1H), 1.28–1.2 (m, 1H), 1.06 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 6.4 Hz, 3H), 0.88 (s, 9H), 0.92 (d, J = 9.1 Hz, 3H); 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 140.4, 115.5, 72.8, 71.2, 43.7, 37.0, 36.4, 25.7, 19.1, 17.9, 16.5, 16.2, −4.4, −4.9; MS (ESI, m/z) [M + Na]+ 309.
(3S,4R,6S,7S)-7-(tert-butyldimethylsilyloxy)-4-(4-methoxybenzyloxy)-3,6-dimethyloctan-1-ol (18): To a stirred solution of alcohol 17 (710 mg, 2.5 mmol) in DMF (15 mL), we added NaH (60%, 150 mg, 3.7 mmol) at 0 °C. The mixture was stirred for 30 min before PMBCl (0.5 mL, 3.7 mmol) was added at 0 °C. After stirring at rt overnight, water (4 mL) and Et2NH (2 mL) were added and the mixture was stirred for 1h, before it was poured into sat NaHCO3 (aq). The mixture was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the PMB ether as a colorless oil (854 mg, 84% yield). Rf value (EtOAc/hexane 1:10): 0.75; [α]D20 = +5.0 (c = 1, CHCl3); IR (film, cm−1) 2959, 1645, 1059; 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 5.89–5.82 (m, 1H), 5.08 (d, J = 18.0 Hz, 1H), 5.06 (d, J = 9.4 Hz, 1H), 4.41 (AB, JAB = 11.2 Hz, ΔυAB = 33.1 Hz, 2H), 3.84 (s, 3H), 3.69–3.64 (m, 1H), 3.43–3.4 (m, 1H), 2.55–2.5 (m, 1H), 1.62–1.56 (m, 2H), 1.32–1.29 (m, 1H), 1.08 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 6.4 Hz, 3H), 0.96 (s, 9H), 0.92 (d, J = 9.1 Hz, 3H); 0.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.0, 140.7, 131.1, 129.4, 129.3, 114.7, 113.7, 80.8, 70.9, 55.4, 37.2, 34.6, 33.5, 25.9, 20.7, 18.1, 15.2, 15.0, −4.1, −4.7.
To a stirred solution of the above olefin (1.45 g, 3.6 mmol) in THF (30 mL), we added 9-BBN (14.2 mL, 0.5 M soln in THF, 7.2 mmol) at 0 °C. The reaction mixture was allowed to warm up to rt and stirred for 3 h. NaOH (0.6 mL) and H2O2 (4 mL) were added and the mixture was refluxed for 1 h. The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. Column chromatography provided alcohol 18 as a colorless oil (1.21 g, 79%). Rf value (EtOAc/hexane 1:1): 0.68; [α]D20 = +3.7 (c = 1, CHCl3); IR (film, cm−1) 3403, 2931, 1613, 1040; 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.6 Hz, 1H), 6.86 (d, J = 8.6 Hz, 1H), 4.45 (s, 2H), 3.81 (s, 3H), 3.75–3.65 (m, 2H), 3.61–3.55 (m, 1H), 3.38–3.33 (m, 1H), 1.88–1.80 (m, 1H), 1.7–1.6 (m, 2H), 1.55–1.45 (m, 2H), 1.4–1.32 (m, 1H), 1.02 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.4 Hz, 3H), 0.92 (s, 9H), 0.85 (d, J = 9.1 Hz, 3H), 0.03 (s, 3H), 0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 129.3, 113.6, 81.2, 71.2, 70.9, 60.1, 55.1, 37.3, 34.6, 34.3, 32.8, 32.1, 27.3, 25.8, 22.6, 18, 15.5, 15.1, −4.8; MS (ESI, m/z) [M+Na]+ 447.4.
Sulfone (4): To a stirred solution of alcohol 18 (880 mg, 2.1 mmol), 1-(4-Hydroxyphenyl)-1H-tetrazole-5-thiol (739 mg, 4.1 mmol) and triphenylphosphine (814 mg, 3.11 mmol) in THF (20 mL), we added DIAD (0.72 mL, 3.7 mmol) at 0 °C. The reaction mixture was warmed up to rt and stirred overnight, before it was poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography provided the sulfide as a colorless oil (1.03 g, 85%). Rf value (EtOAc/hexane 1:4): 0.56; [α]D20 = +1.2 (c = 0.5, CHCl3); IR (film, cm−1) 2956, 2886, 1614, 1074; 1H NMR (400 MHz, CDCl3) δ 7.53–7.57 (m, 5H), 7.23 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 4.45 (AB, JAB = 11.1 Hz, ΔυAB = 18 Hz, 2H), 3.78 (s, 3H), 3.66–3.63 (m, 1H), 3.55–3.49 (m, 1H), 3.37–3.31 (m, 2H), 1.93–1.89 (m, 2H), 1.7–1.62 (m, 1H), 1.62–1.55 (m, 1H), 1.52–1.48 (m, 1H), 1.31–1.25 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 1 (d, J = 7 Hz, 3H), 0.86 (d, J = 6.8 Hz, 1H), 0.84 (s, 9H), 0.01 (s, 3H), 0 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 129.3, 113.6, 81.2, 71.2, 70.9, 60.1, 55.1, 37.3, 34.6, 34.3, 32.8, 32.1, 27.3, 25.8, 22.6, 18.0, 15.5, 15.1, −4.8. To a stirred solution of thus obtained sulfide (353 mg, 0.57 mmol) in ethanol (9 mL), we added a solution of ammonium molybdate (320 mg, 0.26 mmol) in hydrogen peroxide (1.6 mL) and water (0.8 mL) at rt. The reaction mixture was stirred for 12 h and poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:12) to give the sulfone 4 as a colorless oil (352 mg, 95% yield). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +1.8 (c = 1, CHCl3); IR (film, cm−1) 2931, 2857, 1612, 1513, 1249, 1074; 1H NMR (500 MHz, CDCl3) δ7.72–7.7 (m, 2H), 7.64–7.6 (m, 3H), 7.27 (d, J = 8.5 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 4.47 (AB, JAB = 11.2 Hz, ΔυAB = 21.8 Hz, 2H), 3.93–3.87 (m, 1H), 3.83 (s, 3H), 3.79–3.71 (m, 2H), 3.4–3.37 (m, 1H), 2.1–2.04 (m, 1H), 2.02–1.86 (m, 2H), 1.66–1.61 (m, 1H), 1.58–1.54 (m, 1H), 1.42–1.38 (m, 1H), 1.10 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 6.4 Hz, 3H), 0.92 (d, J = 3.5 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.3, 153.4, 133.1, 131.4, 130.6, 129.7, 129.4, 125.1, 113.8, 80.8, 71.2, 71.0, 55.3, 54.6, 37.2, 36.6, 34.6, 33.4, 25.8, 24.2, 20.4, 18.1, 15.5, 15.2, −4.1, −4.7; MS (ESI, m/z) [M + Na]+ 639.3. HRMS (ESI) [M+Na]+ calcd for C31H48N4O5SSiNa 639.3012, found 639.3008.
Coupling product (24): To a stirred solution of aldehyde 3 (422 mg, 0.64 mmol) and sulfone 4 (324 mg, 0.53 mmol) in DME (15 mL), we added KHMDS (1.6 mL, 0.5 M soln in toluene, 0.8 mmol) at −65 °C. The reaction mixture was stirred for 1 h at −65 °C, before it was warmed up to rt. The reaction was quenched by sat NH4Cl (5 mL) at −65 °C and the organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:20) to give the olefin 24 as a colorless oil (376 mg, 67% yield). Rf value (EtOAc/hexane 1:10): 0.48; [α]D20 = +22.5 (c = 0.55, CHCl3); IR (film, cm−1) 2857, 1729, 1612, 1513, 1257, 1081; 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 5.75–5.67 (m, 2H), 5.48 (d, J = 15.5 Hz, 1H), 5.41 (t, J = 3.9 Hz, 1H), 5.3–5.26 (dd, J = 15.5, 8.5 Hz, 1H), 4.96 (s, 1H), 4.89 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.44 (AB, JAB = 11.1 Hz, ΔυAB = 20.6 Hz, 2H), 4.4 (d, J = 6.8 Hz, 1H), 4.34 (dd, J = 12.8, 7.1 Hz, 1H), 4.2 (dd, J = 12.8, 3.9 Hz, 1H), 3.94–3.87 (m, 2H), 3.86–3.83 (m, 1H), 3.83 (s, 3H), 3.7–3.66 (m, 1H), 3.4–3.32 (m, 1H), 3.34 (s, 3H), 2.69–2.65 (m, 1H), 2.34–2.28 (m, 2H), 2.25–2.18 (m, 3H), 2.14–2.1 (m, 1H), 1.9–1.83 (m, 2H), 1.74–1.7 (m, 1H), 1.72 (s, 3H), 1.65–1.6 (m, 1H), 1.58–1.53 (m, 1H), 1.49 (s, 3H), 1.4 (s, 3H), 1.21 (s, 3H), 1.08 (d, J = 9.7 Hz, 3H), 0.95–0.85 (m, 36H), 0.1–0.04 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 159, 143.6, 138.0, 134, 131.6, 131.4, 131.1, 130, 129.3, 127.3, 114.3, 113.7, 107.2, 93.1, 82.1, 81.5, 81.2, 79.2, 71.1, 70.8, 70.7, 59.8, 55.4, 55.2, 43.8, 39.7, 39.2, 37.5, 35.7, 35.6, 32.8, 29.7, 28.5, 26.6, 26, 25.9, 20.6, 20.4, 18.7, 18.4, 18, 15.5, 15.4, 15, −4.1, −4.4, −4.5, −4.6, −4.7, −5.1; MS (ESI, m/z) [M + Na]+ 1081.7. HRMS (ESI) [M + Na]+ calcd for C60H110O9Si3Na 1081.7355, found 1081.7367.
Diol product (25): To a stirred solution of PMB ether 24 (204 mg, 0.2 mmol) in DCM (20 mL) and pH 7.0 buffer (1.6 mL), we added DDQ (88 mg, 0.4 mmol) at 0 °C and the reaction mixture was stirred at that temperature for 1 h. The reaction was quenched by sat NaHCO3 (5 mL) and the organic layer was separated. The aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with sat NaHCO3, water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:15) to give the corresponding alcohol as a colorless oil (138 mg, 76% yield). Rf value (EtOAc/hexane 1:4): 0.67; [α]D20 = +30.1 (c = 0.55, CHCl3); IR (film, cm−1) 3429, 2931, 1619, 1252, 1079; 1H NMR (500 MHz, CDCl3) δ 5.77–5.67 (m, 2H), 5.48 (d, J = 15.5 Hz, 1H), 5.41 (t, J = 5.2 Hz, 1H), 5.28 (dd, J = 15.5, 8.3 Hz, 1H), 4.99 (s, 1H), 4.88 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.4 (d, J = 6.8 Hz, 1H), 4.34 (dd, J = 12.9, 7.3 Hz, 1H), 4.2 (dd, J = 12.9, 4.1 Hz, 1H), 3.92–3.86 (m, 2H), 3.86–3.72 (m, 2H), 3.56–3.52 (m, 1H), 3.34 (s, 3H), 2.7–2.65 (m, 1H), 2.45–2.4 (m, 1H), 2.32–2.28 (m, 2H), 2.28–2.2 (m, 3H), 2.14–2.1 (m, 1H), 1.92–1.88 (m, 1H), 1.71 (s, 3H), 1.68–1.55 (m, 2H), 1.48 (s, 3H), 1.4 (s, 3H), 1.2 (s, 3H), 1.12 (d, J = 6.3 Hz, 3H), 1.04–0.87 (m, 36H), 0.11–0.06 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 143.6, 138.1, 134.1, 131.7, 131.4, 129.9, 127.3, 114.3, 107.2, 93.1, 82.1, 81.5, 79.2, 73.2, 72.3, 70.8, 59.9, 55.4, 43.8, 39.7, 39.2, 38.8, 36.3, 36.0, 35.5, 35.4, 29.7, 28.5, 26.6, 26, 25.9, 20.5, 18.7, 18.4, 18, 15.5, 15.4, −4.4, −4.5, −4.8, −5.1.
To a stirred solution of the above TBS ether (115 mg, 0.12 mmol) in methanol (4 mL), we added ammonium fluoride (125 mg, 3.37 mmol) at rt and the reaction mixture was stirred at that temperature for 8 h. Et2O (30 mL) was added to precipitate the ammonium fluoride, which was removed by suction filtration. The crude product was purified by flash chromatography (EtOAc/hexane 1:4) to give the alcohol 25 as a colorless oil (94 mg, 95% yield). Rf value (EtOAc/hexane 2:1): 0.36; [α]D20 = +37.4 (c = 0.5, CHCl3); IR (film, cm−1) 3434, 2956, 2931, 1644, 1374, 1253, 1077; 1H NMR (500 MHz, CDCl3) δ5.77–5.68 (m, 3H), 5.45 (d, J = 15.5 Hz, 1H), 5.24 (dd, J = 15.5, 8.4 Hz, 1H), 4.94 (s, 1H), 4.85 (s, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.37 (d, J = 6.8 Hz, 1H), 4.23 (dd, J = 11.6, 8.4 Hz, 1H), 3.9–3.82 (m, 2H), 3.82–3.75 (m, 2H), 3.51–3.49 (m, 1H), 3.29 (s, 3H), 2.85 (brs, 1H), 2.84–2.80 (m, 1H), 2.4–2.35 (m, 1H), 2.3–2.12 (m, 5H), 2.1–2.05 (m, 1H), 1.9–1.84 (m, 2H), 1.71 (s, 3H), 1.65–1.55 (m, 2H), 1.44 (s, 3H), 1.38 (s, 3H), 1.18 (s, 3H), 1.08 (d, J = 6.1 Hz, 3H), 0.94–0.82 (m, 27H), 0.1–0.02 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 143.4, 142.6, 134.0, 132.8, 130.6, 129.8, 126.1, 114.3, 107.1, 92.5, 82.0, 81.3, 77.8, 73.1, 72.2, 70.6, 65.8, 57.0, 55.5, 43.6, 39.5, 38.7, 38.4, 36.2, 36.0, 35.4, 35.3, 28.4, 26.5, 25.8, 20.4, 18.1, 18.0, 15.5, 15.2, −4.5, −4.7, −4.9, −5.1; MS (ESI, m/z) [M + Na]+ 845.5. HRMS (ESI) [M + Na]+ calcd for C46H88O8Si2Na 847.5916, found 847.5922.
Macrolactone(26): To a stirred solution of allyl alcohol 25 (107 mg, 0.13 mmol) in DCM (10 mL), we added activated MnO2 (126 mg, 90%, 1.3 mmol) at rt, and the reaction was stirred at rt for 5 h. Suction filtration gave a crude aldehyde (105 mg) that was used for the next step without further purification. Rf value (EtOAc/hexane 1:2): 0.73.
To a stirred solution of the above aldehyde (105 mg, 0.12 mmol) and 2-methyl-2-butene (2 mL) in tert-butanol (8 mL), we added a solution of NaH2PO4.H2O (200 mg) and NaClO2 (200 mg), dropwise, in H2O (2 mL) at 0 °C. In addition, the reaction mixture was allowed to warm up to rt and stirred for 30 min. The reaction was poured into water (5 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd Na2SO4 and evaporated in vacuo. The crude product was purified by flash chromatography (MeOH/chloroform 3:100) to give the seco-acid as a colorless oil (97 mg, 89% yield for two steps). Rf value (EtOAc/hexane 1:2): 0.68.
To the solution of thus obtained seco-acid in THF (4 mL), we added DIPEA (0.33 mL, 1.91 mmol) and 2,4,6-trichlorobenzoyl chloride (0.2 mL, 1.27 mmol) at rt. The reaction was stirred for 3 h at that temperature, before the THF solvent was removed by vacuo. The the residue toluene (10 mL) was added and the solution was transferred to a stirred solution of DMAP (388 mg, 3.18 mmol) in toluene (150 mL) at rt over 16 h, through a syringe pump. The resulting mixture was stirred at rt for 36 h and poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:40) provided macrolactone 26 as a colorless oil (65 mg, 69%). Rf value (EtOAc/hexane 1:10): 0.48; [α]D20 = −18 (c = 0.2, CHCl3); IR (film, cm−1) 2927, 1711, 1155, 1033; 1H NMR (500 MHz, CDCl3) δ 5.75 (d, J = 0.6 Hz, 1H), 5.62–5.55 (m, 2H), 5.48 (d, J = 15.6 Hz, 1H), 5.32 (dd, J = 15.6, 8.3 Hz, 1H), 4.99 (s, 1H), 4.89 (s, 1H), 4.86–4.88 (m, 1H), 4.67 (d, J = 6.8 Hz, 1H), 4.53 (d, J = 6.8 Hz, 1H), 4.15–4.10 (m, 1H), 3.96 (dd, J = 7.9, 5.8 Hz, 1H), 3.88 (dd, J = 7.9, 4.6 Hz, 1H), 3.84–3.79 (m, 1H), 3.79–3.76 (m, 1H), 3.38 (s, 3H), 2.22–2.05 (m, 8H), 1.91–1.85 (m, 1H), 1.9 (d, J = 0.6 Hz, 3H), 1.84–1.72 (m, 2H), 1.62–1.56 (m, 1H), 1.48 (s, 3H), 1.40 (s, 3H), 1.22 (s, 3H), 1.12 (d, J = 7.2 Hz, 3H), 1.05 (d, J = 6.9 Hz, 3H), 0.93–0.78 (m, 24H), 0.07–0.03 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 166.2, 160, 142.1, 134.9, 130.8, 130.1, 129.6, 118.3, 114.5, 107.2, 93.7, 81.9, 79.9, 79.8, 75.8, 70.9, 69.8, 55.5, 42.8, 40.6, 38.8, 38.3, 37.3, 36.6, 35.8, 35.6, 34.7, 29.7, 28.5, 26.6, 25.8, 24.7, 23.3, 22.7, 21.8, 20.7, 19.5, 18.1, 15.8, 15.1, 14.0, 7.9, −4, −4.4, −4.5, −4.7. MS (ESI, m/z) [M + Na]+ 843.5. HRMS (ESI) [M + Na]+ calcd for C46H84O8Si2Na 843.5603, found 843.5611.
Macrolactone alcohol (27): To a stirred solution of macrolactone 26 (52 mg, 0.063 mmol) in THF (4 mL), we added pyridine (1 mL) followed by HF.pyridine complex (70%, 0.5 mL) at 0 °C, and the reaction was warmed up to rt and stirred for 10 h. The reaction mixture was cooled to 0 °C again and sat NaHCO3 (20 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was washed with sat NaHCO3, water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:2) provided diol as a colorless oil (33 mg, 89%). Rf value (EtOAc/hexane 2:1): 0.55; [α]D20 = −12 (c = 0.18, CHCl3); IR (film, cm−1) 3456, 2926, 1707, 1155, 1032; 1H NMR (500 MHz, CDCl3) δ 5.75 (s, 1H), 5.66–5.55 (m, 2H), 5.53 (d, J = 15.6 Hz, 1H), 5.44 (dd, J = 15.6, 8.3 Hz, 1H), 5.11 (s, 1H), 4.99–4.95 (m, 1H), 4.96 (s, 1H), 4.69 (d, J = 6.8 Hz, 1H), 4.55 (d, J = 6.8 Hz, 1H), 4.12–4.08 (m, 1H), 4.07–4.03 (m, 2H), 3.87–3.83 (m, 1H), 3.77–3.72 (m, 1H), 3.4 (s, 3H), 2.4 (brs, 1H), 2.36 (dd, J = 14.4, 2.8 Hz, 1H), 2.29–2.25 (m, 2H), 2.23–2.2 (m, 2H), 2.14–2.10 (m, 1H), 2.08–2.04 (m, 1H), 1.92 (d, J = 1.1 Hz, 3H), 1.9–1.87 (m, 1H), 1.8–1.72 (m, 2H), 1.56–1.52 (m, 1H), 1.49 (s, 3H), 1.40 (s, 3H), 1.22 (s, 3H), 1.17 (d, J = 7.0 Hz, 3H), 1.12 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.5, 160.7, 143.5, 134.5, 131.7, 129.9, 129.2, 118.2, 114.8, 107.3, 93.9, 81.9, 80.3, 79.6, 74.9, 69.5, 69.2, 55.6, 43.1, 40.1, 39.1, 37.4, 36.0, 35.9, 35.4, 32.9, 28.5, 26.7, 21.9, 20.2, 20.0, 15.2, 15.1, 14.9.
A solution of the above acetonide (12 mg, 0.020 mmol) in HOAc (0.8 mL) and water (0.2 mL) was heated at 55 °C for 3 h, before the solvents were removed in vacuo. Column chromatography (MeOH/chloroform 1:20) provided alcohol 27 as a colorless oil (11 mg, 81%). Rf value (EtOAc/hexane 4:1): 0.22; [α]D20 = −8 (c = 0.2, CHCl3); IR (film, cm−1) 3500, 2956, 1712, 1034; 1H NMR (500 MHz, CDCl3) δ 5.75 (s, 1H), 5.78–5.62 (m, 2H), 5.58–5.5 (m, 2H), 5.07 (s, 1H), 5.01 (s, 1H), 4.97–4.93 (m, 1H), 4.69 (d, J = 6.8 Hz, 1H), 4.52 (d, J = 6.8 Hz, 1H), 4.08 (t, J = 6.4 Hz, 1H), 4.01–3.97 (m, 1H), 3.86–3.83 (m, 1H), 3.82–3.76 (m, 1H), 3.72–3.7 (m, 2H), 3.4 (s, 3H), 2.58 (brs, 1H), 2.55 (d, J = 14.1 Hz, 1H), 2.42–2.3 (m, 4H), 2.23–1.98 (m, 1H), 1.95–1.9 (m, 1H), 2.05–2.02 (m, 1H), 2–1.95 (m, 1H), 1.98 (d, J = 1.1 Hz, 3H), 1.89–1.85 (m, 1H), 1.81–1.75 (m, 1H), 1.5–1.45 (m, 1H), 1.21 (s, 3H), 1.15 (d, J = 6.5 Hz, 3H), 1.1 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.5, 163.7, 144, 138.5, 132.4, 128.9, 125.8, 117.5, 116.3, 94.2, 79.1, 74.8, 73.8, 73.2, 69.2, 68.9, 55.9, 41.8, 40.4, 40.5 37.2, 37, 35.5, 35.2, 32.9, 22.5, 21, 19.9, 16.1, 15, 14.1. MS (ESI, m/z) [M + Na]+ 575.3. HRMS (ESI) [M + Na]+ calcd for C31H52O8Na 575.3560, found 575.3555.
TES ether derivative (28): To a stirred solution of the above diol 27 (7 mg, 0.013 mmol) in DCM (2 mL), we added bromocatechol borane (0.65 mL, 0.1 M soln in DCM, 0.065 mmol) at −78 °C, and the mixture was stirred at that temperature for 1h, before it was quenched with sat NaHCO3 (5 mL). The organic layer was separated and the aq layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over anhyd Na2SO4 and concentrated in vacuo. Column chromatography (MeOH/chloroform 1:20) provided the corresponding alcohol as a colorless oil (4.8 mg, 72%). Rf value (EtOAc/hexane/MeOH 8:2:1): 0.33; [α]D20 = −21 (c = 0.074, CHCl3); IR (film, cm−1) 3430, 2956, 1706; 1H NMR (500 MHz, CDCl3) δ 5.9–5.85 (m, 1H), 5.84–5.78 (m, 1H), 5.82 (s, 1H), 5.75–5.69 (m, 1H), 5.58 (d, J = 15.8 Hz, 1H), 5.05 (s, 1H), 5.05–5.02 (m, 1H), 5.02 (s, 1H), 3.96–3.92 (m, 1H), 3.81–3.73 (m, 2H), 3.62–3.58 (m, 2H), 2.65–2.55 (m, 2H), 2.5–2.44 (m, 1H), 2.42–2.38 (m, 2H), 2.18–2.1 (m, 2H), 2.02–1.98 (m, 1H), 1.92 (s, 3H), 1.7–1.64 (m, 2H), 1.43–1.40 (m, 1H), 1.19 (s, 3H), 1.07 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 7.1 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H).
To a stirred solution of the above alcohol (3.2 mg, 0.006 mmol) and DMAP (18 mg, 0.14 mmol) in DCM (2 mL), we added TESCl (16 µL, 0.1 mmol) at 0 °C, and the reaction was stirred at 0 °C for 30 min, before sat NaHCO3 (5 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:20) provided tris-TES ether 28 as a colorless oil (4.9 mg, 92%). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +11 (c = 0.22, CHCl3); IR (film, cm−1) 3430, 2896, 1079; 1H NMR (500 MHz, CDCl3) δ 5.84-5.78 (m, 1H), 5.64 -5.60 (m, 3H), 5.42 (d, J = 15.5 Hz, 1H), 4.91 (brs, 2H), 4.76 (dt, J = 5.0, 2.5 Hz, 1H), 4.1–4.06 (m, 1H), 4.02–3.98 (m, 1H), 3.88–3.84 (m, 1H), 3.75–3.70 (m, 1H), 3.6–3.56 (m, 1H), 3.52 (brs, 1H), 2.53–2.49 (m, 1H), 2.4–2.36 (m, 2H), 2.3–2.25 (m, 1H), 2.18–2.08 (m, 2H), 2.07–2.01 (m, 4H), 1.85 (brs, 3H), 1.85–1.8 (m, 1H), 1.4–1.35 (m, 1H), 1.18 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 0.96–0.9 (m, 33H), 0.8 (d, J = 6.9 Hz, 3H), 0.6–0.53 (m, 18H); MS (ESI, m/z) [M+Na]+ 873.33. HRMS (ESI) [M + Na]+ calcd for C47H90O7Si3Na 873.5892, found 873.5888.
Iriomoteolide-1a (1): To a stirred solution of diol 28 (3 mg, 3.5 μmol) in DCM (1 mL), we added Dess–Martin periodinane (0.12 mL, 0.3 M soln in DCM, 0.035 mmol) at rt, and the reaction was stirred at rt for 1 h. Direct column chromatography (EtOAc/hexane 1:15) provided the corresponding ketone as a colorless oil (1.9 mg, 65%, 90% BRSM), along with the recovered starting material (1 mg). Rf value (EtOAc/hexane 1:4): 0.75; 1H NMR (500 MHz, CDCl3) δ 5.85–5.80 (m, 1H), 5.63 (s, 1H), 5.61–5.55 (m, 1H), 5.46 (d, J = 15.6 Hz, 1H), 5.37 (dd, J = 15.6, 8.0 Hz, 1H), 5.01 (s, 1H), 4.91–4.87 (m, 1H), 4.85 (s, 1H), 4.18–4.13 (m, 2H), 4.08 (s, 1H), 3.79–3.75 (m, 1H), 3.69–3.66 (m, 1H), 3.42 (d, J = 15.0 Hz, 1H), 3.12 (d, J = 15.0 Hz, 1H), 2.2–2.16 (m, 2H), 2.1–2 (m, 4H), 1.97–1.92 (m, 2H), 1.87 (brs, 3H), 1.85–1.81 (m, 1H), 1.46 (s, 3H), 1.1 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.6 Hz, 3H), 0.98–0.92 (m, 30H), 0.85 (d, J = 7.0 Hz, 3H), 0.61–0.54 (m, 18H). To a stirred solution of the above ketone (3.0 mg, 0.0035 mmol) in THF (0.6 mL), we added a HF·Py solution (0.1 mL containing 1 mL 70% HF·Py: 1.1 mL pyridine: 2.4 mL THF) at rt. After 1 h, the reaction was quenched with sat NaHCO3 and extracted with diethyl ether (3 × 10 mL). The combined organic layer was washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (MeOH/chloroform 1:25) to give iriomoteolide-1a (1) (1.0 mg, 56%) as a colorless oil, along with the isomerized product, iriomoteolide-1b (2) (0.3 mg, 17%), as a colorless oil.
Iriomoteolide-1a (1) Rf value (EtOAc/hexane 2:1): 0.4. [α]D20 = −12.0 (c = 0.10, CHCl3); IR (film, cm−1) 3456, 2926, 1707, 1032; 1H NMR (500 MHz, CDCl3) δ5.9–5.88 (m, 1H), 5.88–5.83 (m, 1H), 5.83–5.8 (m, 1H), 5.78 (s, 1H), 5.68 (dd, J = 15.4, 6.8 Hz, 1H), 5.05–4.99 (m, 1H), 4.88 (d, J = 1.6 Hz, 1H), 4.86 (d, J = 1.4 Hz, 1H), 4.07 (dd, J1 = J2 = 6.8 Hz, 1H), 4.03–3.97 (m, 1H), 4–3.95 (m, 1H), 3.89–3.83 (m, 1H), 3.29 (brs, 1H), 2.67 (brs, 1H), 2.37–2.3 (m, 1H), 2.32–2.28 (m, 1H), 2.25–2.2 (m, 3H), 2.18–2.13 (m, 2H), 2.05–2 (m, 1H), 1.96 (s, 3H), 2–1.92 (m, 1H), 1.84–1.78 (m, 1H), 1.56–1.5 (m, 1H), 1.4–1.35 (m, 1H), 1.33 (s, 3H), 1.12 (d, J = 6.4 Hz, 3H), 1.1 (d, J = 7 Hz, 3H), 1.01 (d, J = 7 Hz, 3H), 0.9 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.3, 160, 141.7, 134.8, 132.7, 129.6, 127.2, 118.8, 110.8, 99.1, 77.2, 74.9, 74.4, 70.3, 69.3, 40.6, 39.7, 37.8, 37.3, 36.7, 35.5, 34.2, 20.8, 20.6, 20, 15.8, 15.7, 14.9; MS (ESI, m/z) [M + Na]+ 529.24. HRMS (ESI) [M + Na]+ calcd for C29H46O7Na 529.3141, found 529.3139.
Iriomoteolide-1b (2) Rf value (EtOAc/hexane 2:1): 0.32; [α]D20 = −78 (c = 0.03, CHCl3); IR (film, cm−1) 3703, 2965, 1810, 1694; 1H NMR (500 MHz, CDCl3) δ 6.32 (s, 1H), 5.82 (s, 1H), 5.83–5.78 (m, 1H), 5.73–5.68 (m, 2H), 5.54 (d, J = 15.7 Hz, 1H), 4.98 (dt, J = 7.5, 3.0 Hz, 1H), 4.55 (s, 1H), 4.17–4.1 (m, 1H), 3.90 (m, 1H), 3.82–3.75 (m, 1H), 3.75–3.7 (m, 1H), 2.35–2.3 (m, 2H), 2.3–2.22 (m, 2H), 2.27 (s, 3H), 2.12–2.08 (m, 2H), 1.99 (s, 3H), 1.98–1.9 (m, 2H), 1.66–1.6 (m, 1H), 1.48 (s, 3H), 1.42–1.35 (m, 1H), 1.18 (d, J = 7.1 Hz, 3H), 1.12 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 6.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 200.7, 167.2, 160.1, 159.3, 136, 132.9, 129.8, 126.5, 120.5, 118.6, 77.6, 76.9, 75.5, 69.4, 68.2, 48.3, 40.9, 40.4, 36.2, 35.6, 31.5, 30.9, 22.6, 21.6, 20.3, 20.1, 15.7, 15, 13.7; MS (ESI, m/z) [M + Na]+ 529.33. HRMS (ESI) [M + Na]+ calcd for C29H46O7Na 529.3141, found 529.3135.
Alcohol (32): To a suspension of CuI (2.18 g, 11.4 mmol) in THF (30 mL), we added MeLi (14.3 mL, 22.8 mmol) at −60 °C. The mixture was slowly warmed up to 0 °C to obtain a clear solution and cooled back to −60 °C. TMSCl (1.5 mL, 11.4 mmol) was added dropwise and stirred at this temperature for 5 min. A soln of alkynyl ester 21 (1.2 g, 5.7 mmol) in THF (5 mL) was added dropwise at −60 °C. After addtion, the reaction mixture was warmed up to 0 °C slowly and stirred for 30 min. Then, the reaction mixture was poured into sat NH4Cl (30 mL) and crushed ice. The organic layer was separated and the aqueous layer was extracted by Et2O (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) produced an inseparable mixture of E-enoate and Z-isomer (1.2 g, 92%) as a colorless oil. Rf value (EtOAc/hexane 1:10) 0.45.
To the above stirred enoate (1.15 g, 5.04 mmol) in DCM (50 mL), we added DIBAL-H (20 mL, 20 mmol) at −78 °C. The reaction mixture was stirred 1h before addition of 10 mL saturated NH4Cl (aq) and 20 mL 25% Rochelle salt solution. The mixture was stirred at rt, until a clear soln was obtained. The organic phase was separated and the aq phase was extracted with Et2O (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (15% Et2O/hexanes) afforded the E-allyl alcohol 32 (0.71 g, 68%) and Z-isomer (0.3 g, 29%). Rf value (EtOAc/hexane 1:4) 0.2; [α]D20 = +27 (c = 0.2, CHCl3); IR (film, cm−1): 3350, 2965, 1624, 1456, 1210, 1008, 914; 1H NMR (400 MHz, CDCl3) δ 5.55 (ddd, J = 17.2, 10.3, 8.1 Hz, 1H), 5.44 (t, J = 6.7 Hz, 1H), 5.26–5.09 (m, 2H), 4.62 (d, J = 6.9 Hz, 1H), 4.41 (d, J = 6.9 Hz, 1H), 4.11 (d, J = 6.7 Hz, 2H), 3.86 (t, J = 8.2 Hz, 1H), 3.27 (s, 3H), 2.35–2.16 (m, 1H), 2.10 (s, 1H), 1.63 (s, 3H), 0.92 (d, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 140.28, 136.49, 125.11, 118.63, 93.38, 79.84, 77.19 58.99, 55.37, 46.73, 15.5, 13.36. MS (ESI, m/z) [M+Na]+ 223.1.
Aldehyde (33): To a stirred solution of the above alcohol (0.7 g, 3.5 mmol) (1.45 g, 7.24 mmol) in DCM (30 mL), we added imidazole (357 mg, 5.26 mmol) and TBSCl (0.58 g, 3.82 mmol) at 0 °C. The reaction was warmed up to rt and stirred for 1 h, before pouring into a mixture of sat NaHCO3 (50 mL) and crushed ice. The mixture was extracted with ethers (3 × 30 mL) and the organic layer was washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo to give the crude TBS ether as a clear oil. The resulting crude product was used for the next step without further purification. Flash chromatography on silica gel (25% EtOAc/hexanes) afforded the silyl ether (1.1 g, 100%). Rf value (EtOAc/hexane 1:4) 0.8; [α]D20 = +12 (c = 0.55, CHCl3); IR (film, cm−1): 2970, 2811, 1610, 1230, 1155, 925; 1H NMR (400 MHz, CDCl3) δ 5.59 (ddd, J = 17.2, 10.4, 8.0 Hz, 1H), 5.39 (t, J = 6.0 Hz, 1H), 5.26–5.10 (m, 2H), 4.65 (d, J = 6.8 Hz, 1H), 4.44 (d, J = 6.8 Hz, 1H), 4.20 (d, J = 6.1 Hz, 2H), 3.89 (t, J = 8.1 Hz, 1H), 3.30 (s, 3H), 2.36–2.16 (m, 1H), 1.62 (d, J = 0.7 Hz, 3H), 0.95 (d, J = 7.1 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 137.81, 136.65, 126.04, 118.29, 93.45, 79.81, 60.09, 55.33, 46.64, 25.86, 18.24, 15.29, 13.66, −5.20.
To a stirred solution of the above olefin (1.05 g, 3.34 mmol) (1.09 g, 3.47 mmol) in dioxane (20 mL) and water (7 mL), we added 2,6-lutidine (1.9 mL, 16.7 mmol), OsO4 (2.5% in t-BuOH, 1.6 mL, 0.13 mmol) and NaIO4 (2.85 g, 12.4 mmol) at 0 °C. The mixture was stirred at 0 °C for 16 h before saturated NaHCO3 (10 mL) and Na2S2O3 (10 mL) were added. The mixture was stirred for another 30 min, extracted by EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% EtOAc/hexanes) produced the aldehyde 33 (655 mg, 62%), which was used for the next step quickly.
Aldehyde (34): To a stirred solution of sulfone 5 (628 mg, 0.94 mmol) in DME (30 mL), we added KHMDS (1.85 mL, 0.5 M soln in toluene, 0.93 mmol) at −78 °C. The reaction mixture was stirred for 30 min, before a soln of aldehyde 33 (357 mg, 1.13 mmol) in DME (5 mL) was transferred in. The reaction mixture was stirred for another 30 min, before it was warmed up to rt. The reaction was quenched by sat NH4Cl (10 mL) at −78 °C. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give olefin as a colorless oil (544 mg, 76% yield). Rf value (EtOAc/hexane 1:10): 0.5; [α]D20 = +14 (c = 0.6, CHCl3); IR (film, cm−1) 2928, 1455, 1248, 1108; 1H NMR (400 MHz, CDCl3) δ 5.73–5.54 (m, 1H), 5.40 (t, J = 6.1 Hz, 1H), 5.23 (dd, J = 15.4, 8.5 Hz, 1H), 4.88 (d, J = 32.2 Hz, 2H), 4.68 (d, J = 6.8 Hz, 1H), 4.56 (d, J = 5.1 Hz, 2H), 4.41 (d, J = 6.8 Hz, 1H), 4.21 (d, J = 6.1 Hz, 3H), 4.14 (dd, J = 8.7, 3.7 Hz, 1H), 3.86 (t, J = 8.3 Hz, 2H), 3.41 (q, J = 9.8 Hz, 2H), 3.30 (s, 3H), 2.35–2.09 (m, 8H), 1.63 (s, 3H), 1.44 (s, 3H), 1.35 (s, 3H), 1.13 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 0.9 (s, 9H), 0.87 (s, 9H), 0.12–−0.07 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.50, 138.19, 131.28, 130.98, 128.21, 127.41, 127.29, 125.83, 114.25, 107.15, 93.11, 81.53, 79.29, 78.34, 74.98, 73.45, 70.52, 60.11, 55.31, 46.97, 43.68, 39.56, 36.49, 28.56, 26.44, 25.90, 25.80, 19.16, 18.27, 17.97, 15.76, 13.52, −4.59, −4.65, −5.15.
To a stirred solution of the above benzyl ether (520 mg, 0.68 mmol) in THF (20 mL) and allyl ethyl ether (1 mL), we transferred a soln of lithium metal (100 mg, 14.4 mmol) in liquid ammonia (30 mL) in portions at −78 °C. The reaction was carefully monitored by TLC and stopped immediately after the solution became slightly blue. Ammonium chloride (3 g) was added to quench the reaction. The mixture was allowed to warm up to rt to evaporate ammonia, before water (10 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the alcohol as a colorless oil (356 mg, 78% yield), along with the recovered starting material. Rf value (EtOAc/hexane 1:4): 0.4; [α]D20 = +12 (c = 1, CHCl3); IR (film, cm−1) 3430, 2965, 1255, 1087; 1H NMR (400 MHz, CDCl3) δ 5.70–5.55 (m, 1H), 5.38 (t, J = 6.0 Hz, 1H), 5.23 (dd, J = 15.5, 8.4 Hz, 1H), 4.93 (s, 1H), 4.85 (s, 1H), 4.67 (d, J = 6.8 Hz, 1H), 4.40 (d, J = 6.8 Hz, 1H), 4.19 (d, J = 5.6 Hz, 3H), 3.85 (dd, J = 11.1, 5.3 Hz, 2H), 3.51 (dd, J = 11.8, 3.6 Hz, 1H), 3.34 (dd, J = 11.5, 9.5 Hz, 1H), 3.28 (s, 3H), 2.37–2.03 (m, 10H), 1.60 (s, 3H), 1.43 (s, 3H), 1.34 (s, 3H), 1.04 (s, 3H), 0.97–0.91 (m, 3H), 0.87 (s, 9H), 0.86 (s, 9H), 0.04 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 143, 138.15, 131.21, 131.01, 125.82, 114.54, 106.99, 93.07, 82.56, 79.24, 75.94, 70.70, 65.40, 60.10, 55.29, 46.92, 43.44, 39.63, 36.07, 28.59, 26.60, 25.88, 25.78, 18.60, 18.25, 17.95, 15.70, 13.52, −4.62, −4.66, −5.17.
To a suspension of the above alcohol (139 mg, 0.21 mmol) and sodium bicarbonate (106 mg, 1.26 mmol) in DCM (15 mL), we added Dess–Martin periodinane (176 mg, 0.42 mmol) at rt. The reaction mixture was stirred for 1 h, before it was poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo to give the crude aldehyde 34 (138 mg, quantitative), which was used directly in the next step without further purification.
Alcohol (36): To a stirred mixture of potassium tert-butoxide (13.9 mL, 1.0 M soln in THF, 13.9 mmol) and trans-2-butene (2.2 mL, 23.2 mmol) in THF (40 mL), we added n-butyllithium (8.8 mL, 1.6 M soln in THF, 13.9 mmol) at −78 °C. After complete addition of n-butyllithium, the mixture was stirred at −45 °C for 10 min. The resulting orange solution was cooled to −78 °C again and a solution of (−)-Ipc2BOMe (5.3 g, 16.6 mmol) in THF (10 mL) was added dropwise. After 30 min of stirring, boron trifluoride etherate (2.4 mL, 18.6 mmol) was added dropwise. Then, the aldehyde 35 (2.14 g, 9.3 mmol) in THF (5 mL) was transferred in. The mixture was stirred at −78 °C for 3 h, before NaOH (11 mL, 3 M soln) and H2O2 (7.5 mL, 70% soln) were added. The contents were refluxed for 1 h. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:20) to give the alcohol 36-favored desired syn product (8:1 dr) as a colorless oil (1.92 g, 72% yield). Rf value (EtOAc/hexane 1:10): 0.6; [α]D20 = +1.5 (c = 0.33, CHCl3); IR (film, cm−1) 3410, 2965, 1456, 1044; 1H NMR (500 MHz, CDCl3) δ 5.81 (ddd, J = 17.8, 10.4, 7.6 Hz, 1H), 5.07–5.05 (m, 1H), 5.04–5.01 (m, 1H), 5.02–5.00 (m, 1H), 3.76 (qd, J = 6.5, 3.5 Hz, 1H), 3.46 (dd, J = 8.5, 3.6 Hz, 1H), 3.24 (d, J = 3.6 Hz, 1H), 2.30–2.21 (m, 1H), 1.80–1.69 (m, 1H), 1.48–1.30 (m, 3H), 1.08 (d, J = 6.3 Hz, 3H), 1.03 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 0.84 (d, J = 7.0 Hz, 3H), 0.06 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 141.36, 114.43, 73.75, 72.83, 44.16, 38.41, 36.93, 18.30, 17.99, 16.98, 15.22, −4.72, −5.09; MS (ESI, m/z) [M + Na]+ 309.
Alcohol (37): To a stirred solution of alcohol 36 (1.78 g, 6.25 mmol) in DMF (25 mL), we added NaH (60%, 375 mg, 9.25 mmol) at 0 °C. The mixture was stirred for 30 min before PMBCl (1.25 mL, 9.25 mmol) was added at 0 °C. After stirring at rt over night, water (10 mL) and Et2NH (5 mL) were added and the mixture was stirred for 1h, before it was poured into sat NaHCO3 (aq). The mixture was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the PMB ether as a colorless oil (2.11 g, 83% yield). Rf value (EtOAc/hexane 1:10): 0.7; [α]D20 = +3.2 (c = 0.35, CHCl3); IR (film, cm−1) 2952, 1654, 1062; 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.6 Hz, 1H), 6.87 (d, J = 8.6 Hz, 1H), 5.94–5.83 (m, 1H), 5.09–4.99 (m, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.39 (d, J = 11.0 Hz, 1H), 3.81 (s, 1H), 3.67 (tt, J = 9.7, 4.9 Hz, 1H), 3.37 (ddd, J = 10.2, 5.0, 2.4 Hz, 1H), 2.60–2.52 (m, 1H), 1.65 (tt, J = 13.5, 5.0 Hz, 1H), 1.51 (ddd, J = 13.4, 10.3, 2.8 Hz, 1H), 1.27 (ddd, J = 13.7, 10.9, 2.6 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 1.03 (d, J = 6.9 Hz, 3H), 0.88 (brs, 9H), 0.79 (d, J = 6.7 Hz, 3H), 0.02 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 158.93, 140.66, 131.08, 129.18, 114.21, 113.57, 80.37, 72.47, 71.02, 55.16, 40.42, 36.03, 35.42, 25.79, 20.94, 18.01, 15.45, 12.96, −4.35, −4.91.
To a stirred solution of the above olefin (1.89 g, 4.68 mmol) in THF (30 mL), we added 9-BBN (18.5 mL, 0.5 M soln in THF, 9.36 mmol) at 0 °C. The reaction mixture was allowed to warm up to rt and stirred for 3 h. NaOH (1 mL) and H2O2 (5.5 mL) were added and the mixture was refluxed for 1 h. The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. Column chromatography provided alcohol 37 as a colorless oil (1.55 g, 77%). Rf value (EtOAc/hexane 1:1): 0.65; [α]D20 = +1.5 (c = 0.2, CHCl3); IR (film, cm−1) 3411, 2926, 1609, 1038; 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.5 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.38 (d, J = 11.0 Hz, 1H), 3.78 (d, J = 2.1 Hz, 2H), 3.73–3.63 (m, 1H), 3.59 (d, J = 12.3 Hz, 1H), 3.36 (d, J = 10.1 Hz, 1H), 2.93 (s, 1H), 2.05 (dd, J = 7.9, 5.6 Hz, 1H), 1.76 (dt, J = 21.2, 7.1 Hz, 1H), 1.69–1.51 (m, 1H), 1.45–1.31 (m, 1H), 1.18 (dd, J = 13.4, 10.9 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 0.88 (d, J = 8.1 Hz, 3H), 0.87 (s, 9H), 0.76 (d, J = 6.7 Hz, 3H), 0.02 (s, 3H), 0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.05, 130.55, 129.32, 113.63, 80.86, 72.38, 71.19, 61.80, 55.12, 36.18, 34.81, 33.45, 33.11, 25.80, 20.75, 18.01, 17.32, 13.21, −4.34, −4.86. MS (ESI, m/z) [M + Na]+ 447.4.
Sulfone (38): To a stirred solution of alcohol 37 (1.5 g, 3.57 mmol), 1-(4-Hydroxyphenyl)-1H-tetrazole-5-thiol (1.26 g, 6.97 mmol) and triphenylphosphine (1.38 g, 5.29 mmol) in THF (30 mL), we added DIAD (1.2 mL, 6.29 mmol) at 0 °C. The reaction mixture was warmed up to rt and stirred overnight, before it was poured into sat NaHCO3 (30 mL). The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography provided the sulfide as a colorless oil (1.7 g, 83%). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +2.2 (c = 0.25, CHCl3); IR (film, cm−1) 2962, 2878, 1616, 1069; 1H NMR (500 MHz, CDCl3) δ 7.58–7.51 (m, 1H), 7.22 (d, J = 8.7 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H), 4.50 (d, J = 11.1 Hz, 1H), 4.36 (d, J = 11.1 Hz, 1H), 3.77 (s, 1H), 3.68 (qd, J = 6.1, 3.8 Hz, 1H), 3.53 (ddd, J = 12.9, 8.9, 5.6 Hz, 1H), 3.41–3.32 (m, 1H), 2.09–2.00 (m, 1H), 1.63 (ddt, J = 10.5, 8.9, 4.4 Hz, 1H), 1.54 (ddd, J = 13.3, 10.0, 3.0 Hz, 1H), 1.19 (ddd, J = 13.6, 10.5, 2.8 Hz, 1H), 1.06 (d, J = 6.2 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.87 (s, 9H), 0.78 (d, J = 6.7 Hz, 3H), 0.01 (s, J = 8.2 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 158.90, 154.35, 133.63, 130.92, 129.92, 129.64, 129.09, 123.71, 113.57, 80.20, 72.31, 71.08, 55.15, 36.20, 34.40, 33.69, 31.92, 30.96, 25.80, 20.68, 18.02, 15.52, 13.39, −4.32, −4.84.
To a stirred solution of thus obtained sulfide (1.41 g, 2.28 mmol) in ethanol (35 mL), we added a solution of ammonium molybdate (1.28 g, 1.04 mmol) in hydrogen peroxide (6.5 mL) and water (3 mL) at rt. The reaction mixture was stirred for 12 h and poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give sulfone 38 as a colorless oil (1.4 g, 94% yield). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +0.6 (c = 0.3, CHCl3); IR (film, cm−1) 2928, 2860, 1609, 1511, 1252, 1068; 1H NMR (500 MHz, CDCl3) δ 7.70–7.65 (m, 1H), 7.63–7.56 (m, 2H), 7.24 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 4.49 (d, J = 11.1 Hz, 1H), 4.39 (d, J = 11.1 Hz, 1H), 3.80 (s, 3H), 3.68 (tt, J = 6.3, 3.1 Hz, 1H), 3.39 (dt, J = 9.8, 3.0 Hz, 1H), 2.21–2.11 (m, 1H), 2.07 (ddd, J = 9.7, 7.4, 3.2 Hz, 1H), 1.74 (ddd, J = 12.0, 9.2, 7.3 Hz, 1H), 1.63 (dtd, J = 9.7, 6.5, 3.1 Hz, 1H), 1.56 (ddd, J = 13.2, 9.9, 3.0 Hz, 1H), 1.18 (ddd, J = 13.5, 10.6, 3.0 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.87 (s, 9H), 0.78 (d, J = 6.7 Hz, 3H), 0.03 (s, 3H), 0.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.01, 153.29, 132.95, 131.29, 130.62, 129.57, 129.26, 124.99, 113.67, 79.96, 72.23, 71.19, 55.18, 54.71, 36.20, 34.18, 33.41, 25.78, 24.00, 20.63, 18.01, 15.65, 13.43, −4.33, −4.86.
Coupling product (39): To a stirred solution of the above solfone (194 mg, 0.32 mmol) and aldehyde 34 (253 mg, 0.38 mmol) in DME (15 mL), we added KHMDS (1 mL, 0.5 M soln in toluene, 0.5 mmol) at −65 °C. The reaction mixture was stirred for 1 h at −65 °C before it was warmed up to rt. The reaction was quenched by sat NH4Cl (5 mL) at −65 °C and the organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:20) to give the pure E-olefin 39 as a colorless oil (180 mg, 54% yield) and corresponding Z-isomer (21 mg, 6%). Rf value (EtOAc/hexane 1:10): 0.45; [α]D20 = +11 (c = 0.35, CHCl3); IR (film, cm−1) 2861, 1735, 1622, 1515, 1263, 1080; 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 6.24–6.13 (m, 1H), 5.97 (d, J = 15.6 Hz, 1H), 5.93 (t, J = 6.1 Hz, 1H), 5.78 (dd, J = 15.4, 8.5 Hz, 1H), 5.42 (d, J = 39.9 Hz, 1H), 5.22 (d, J = 6.8 Hz, 1H), 5.02 (d, J = 11.1 Hz, 1H), 4.93 (dd, J = 20.6, 9.0 Hz, 1H), 4.74 (d, J = 6.0 Hz, 1H), 4.40 (t, J = 8.5 Hz, 2H), 4.33 (s, 3H), 4.25–4.17 (m, 1H), 3.88 (d, J = 5.5 Hz, 1H), 3.84 (s, 3H), 2.91–2.75 (m, 2H), 2.72 (dd, J = 12.4, 6.9 Hz, 2H), 2.63 (d, J = 12.9 Hz, 1H), 2.42–2.31 (m, 1H), 2.16 (s, 3H), 1.99 (s, 3H), 1.90 (s, 3H), 1.60 (d, J = 6.2 Hz, 3H), 1.49 (d, J = 7.0 Hz, 3H), 1.42 (m, 27H), 1.32 (d, J = 6.6 Hz, 3H), 0.58–0.53 (m, 18H).
To a stirred solution of the above PMB ether (163 mg, 0.16 mmol) in DCM (10 mL) and pH 7.0 buffer (0.8 mL), we added DDQ (70 mg, 0.32 mmol) at 0 °C and the reaction mixture was stirred at that temperature for 1 h. The reaction was quenched by sat NaHCO3 (5 mL) and the organic layer was separated. The aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with sat NaHCO3, water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:15) to give the corresponding alcohol as a colorless oil (119 mg, 79% yield). Rf value (EtOAc/hexane 1:4): 0.6; [α]D20 = +21 (c = 0.45, CHCl3); IR (film, cm−1) 3440, 2926, 1621, 1245, 1088; 1H NMR (400 MHz, CDCl3) δ 5.72–5.60 (m, 1H), 5.51–5.36 (m, 3H), 5.25 (dd, J = 15.4, 8.4 Hz, 1H), 4.97 (s, 1H), 4.88 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.42 (d, J = 6.8 Hz, 1H), 4.21 (d, J = 6.1 Hz, 2H), 3.94 (dd, J = 9.2, 2.9 Hz, 1H), 3.91–3.84 (m, 2H), 3.74 (dd, J = 6.2, 3.4 Hz, 1H), 3.62 (d, J = 7.4 Hz, 1H), 3.44 (d, J = 3.3 Hz, 1H), 3.30 (s, 3H), 2.62–2.49 (m, 1H), 2.38–2.09 (m, 8H), 2.06 (dd, J = 5.3, 3.1 Hz, 1H), 2.04–2.00 (m, 1H), 1.62 (s, 3H), 1.45 (s, 3H), 1.35 (s, 3H), 1.25 (s, 3H), 1.09 (d, J = 6.2 Hz, 3H), 0.89 (s, 12H), 0.88 (s, 15H), 0.82 (d, J = 6.8 Hz, 3H), 0.13–−0.03 (m, 18H). To a stirred solution of the above TBS ether (114 mg, 0.12 mmol) in methanol (4 mL), we added ammonium fluoride (124 mg, 3.37 mmol) at rt and the reaction mixture was stirred at that temperature for 8 h. Et2O (30 mL) was added to precipitate the ammonium fluoride, which was removed by suction filtration. The crude product was purified by flash chromatography (EtOAc/hexane 1:4) to give the alcohol 39 as a colorless oil (90 mg, 90% yield). Rf value (EtOAc/hexane 2:1): 0.35; [α]D20 = +24 (c = 0.3, CHCl3); IR (film, cm−1) 3445, 2966, 2928, 1652, 1376, 1251, 1071; 1H NMR (400 MHz, CDCl3) δ 5.80–5.57 (m, 1H), 5.49 (t, J = 7.0 Hz, 1H), 5.45 (d, J = 15.6 Hz, 1H), 5.23 (dd, J = 15.5, 8.4 Hz, 1H), 4.92 (s, 1H), 4.84 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.42 (d, J = 6.8 Hz, 1H), 4.16 (d, J = 6.7 Hz, 2H), 3.91–3.86 (m, 1H), 3.85 (dd, J = 8.9, 3.2 Hz, 1H), 3.77 (dd, J = 6.3, 3.2 Hz, 1H), 3.52 (d, J = 9.4 Hz, 1H), 3.30 (s, 3H), 2.35–2.11 (m, 6H), 2.08 (dd, J = 15.0, 3.1 Hz, 1H), 1.96–1.82 (m, 1H), 1.74 (d, J = 8.0 Hz, 1H), 1.68 (s, 3H), 1.59–1.48 (m, 1H), 1.44 (s, 3H), 1.35 (s, 3H), 1.17 (s, 3H), 1.09 (d, J = 6.3 Hz, 3H), 0.96 (d, J = 7.1 Hz, 3H), 0.89 (s, 9H), 0.87 (s, 9H), 0.84 (d, J = 7.0 Hz, 3H), 0.09–0.00 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 143.35, 140.99, 133.85, 131.41, 130.87, 129.89, 124.82, 114.29, 107.06, 93.20, 81.99, 81.24, 79.35, 77.12, 73.18, 72.90, 70.61, 59.19, 55.36, 47.04, 43.54, 39.49, 39.41, 38.88, 37.16, 36.16, 35.43, 28.40, 26.51, 25.79, 25.71, 20.44, 17.97, 17.65, 15.82, 13.92, 13.49, −4.57, −4.66, −4.77, −5.07. MS (ESI, m/z) [M + Na]+ 845.5.
Macrolactone (40): To a stirred solution of allyl alcohol 39 (85 mg, 0.104 mmol) in DCM (10 mL), we added activated MnO2 (36 mg, 90%, 0.416 mmol) at rt, and the reaction was stirred at rt for 3 h. Another portion of MnO2 (36 mg, 90%, 0.416 mmol) was added and was stirred for two more hours. Suction filtration gave a crude aldehyde (83 mg) that was used for the next step without further purification. Rf value (EtOAc/hexane 1:2): 0.7. To a stirred solution of thus obtained aldehyde (83 mg, 0.1 mmol) and 2-methyl-2-butene (2 mL) in tert-butanol (8 mL), we added a solution of NaH2PO4.H2O (200 mg) and NaClO2 (200 mg), dropwise, in H2O (2 mL) at 0 °C. In addition, the reaction mixture was allowed to warm up to rt and stirred for 20 min. The reaction was poured into water (5 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd Na2SO4 and evaporated in vacuo. The crude product was purified by flash chromatography (MeOH/chloroform 3:100) to give the seco-acid as a colorless oil (77 mg, 88% yield for two steps). Rf value (EtOAc/hexane 1:2): 0.6.
To the solution of thus obtained seco-acid in THF (5 mL), we added DIPEA (0.24 mL, 1.38 mmol) and 2,4,6-trichlorobenzoyl chloride (0.14 mL, 0.92 mmol) at rt. The reaction was stirred for 3 h at that temperature, before the THF solvent was removed by vacuo. The the residue toluene (10 mL) was added and the solution was transferred to a stirred solution of DMAP (280 mg, 2.3 mmol) in toluene (180 mL) at rt over 16 h, through a syringe pump. The resulting mixture was stirred at rt for 48 h and poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:40) provided macrolactone 40 as a colorless oil (56 mg, 75%). Rf value (EtOAc/hexane 1:10): 0.5; [α]D20 = +16 (c = 0.3, CHCl3); IR (film, cm−1) 2930, 1715, 1166, 1029; 1H NMR (400 MHz, CDCl3) δ 5.70–5.56 (m, 3H), 5.37 (d, J = 15.7 Hz, 1H), 5.28 (dd, J = 15.6, 7.5 Hz, 1H), 4.99 (t, J = 6.1 Hz, 1H), 4.96 (s, 1H), 4.84 (s, 1H), 4.66 (d, J = 6.7 Hz, 1H), 4.53 (d, J = 6.7 Hz, 1H), 4.11 (dd, J = 7.5, 3.4 Hz, 1H), 3.96–3.88 (m, 1H), 3.79–3.71 (m, 1H), 3.63 (dt, J = 9.1, 5.9 Hz, 1H), 3.47 (dd, J = 14.8, 7.7 Hz, 1H), 3.38 (s, 3H), 2.52 (dd, J = 7.0, 3.6 Hz, 1H), 2.36–2.23 (m, 1H), 2.19 (s, 3H), 2.18–2.11 (m, 2H), 2.12–2.05 (m, 2H), 1.93–1.77 (m, 1H), 1.68–1.58 (m, 1H), 1.45 (s, 3H), 1.37 (s, 3H), 1.22 (s, 3H), 1.18 (d, J = 7.0 Hz, 3H), 1.03 (d, J = 6.1 Hz, 3H), 0.88 (s, 9H), 0.87 (s, 9H), 0.84 (d, J = 6.6 Hz, 3H), 0.06–0 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 166.21, 160.37, 141.98, 133.78, 130.36, 129.80, 129.66, 128.10, 117.71, 114.67, 107.00, 93.88, 81.80, 79.80, 78.93, 77.12, 73.77, 72.17, 71.36, 55.45, 48.73, 43.06, 39.77, 37.12, 36.09, 35.47, 34.96, 33.04, 28.34, 26.59, 25.77, 20.44, 19.81, 18.00, 17.43, 15.70, 13.68, 13.06, −4.34, −4.58, −4.70, −4.90. MS (ESI, m/z) [M + Na]+ 843.5.
Diastereomer(2E, 19S) (29): To a stirred solution of TBS ether 40 (56 mg, 0.068 mmol) in THF (4 mL), we added pyridine (1 mL) followed by HF.pyridine complex (70%, 0.5 mL) at 0 °C, and the reaction was warmed up to rt and stirred for 16 h. The reaction mixture was cooled to 0 °C again and sat NaHCO3 was added until the bubbles disappeared. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with sat NaHCO3, water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:2) provided diol as a colorless oil (31 mg, 76%). Rf value (EtOAc/hexane 2:1): 0.5. A solution of the above acetonide (31 mg, 0.052 mmol) in HOAc (2.4 mL) and water (0.6 mL) was heated at 55 °C for 3 h before the solvents were removed in vacuo. Column chromatography (MeOH/chloroform 1:20) provided the vicinal alcohol as a colorless oil (18 mg, 64%). Rf value (EtOAc/hexane 4:1): 0.2. [α]D20 = −9 (c = 0.14, CHCl3); IR (film, cm−1) 3442, 2962, 1715, 1639, 1431, 1016; 1H NMR (400 MHz, CDCl3) δ 5.76–5.65 (m, 1H), 5.60 (s, 1H), 5.54 (t, J = 7.8 Hz, 1H), 5.51 (d, J = 7.6 Hz, 1H), 5.38 (dd, J = 15.6, 7.3 Hz, 1H), 5.03 (s, 1H), 5.05-5 (m, 1H), 4.97 (s, 1H), 4.68 (d, J = 6.7 Hz, 1H), 4.57 (d, J = 6.8 Hz, 1H), 4.12 (dd, J = 7.3, 3.5 Hz, 1H), 3.74 (dd, J = 6.3, 3.7 Hz, 1H), 3.63 (d, J = 6.2 Hz, 1H), 3.58 (d, J = 10.3 Hz, 1H), 3.39 (s, 3H), 2.56 (dd, J = 6.9, 3.5 Hz, 2H), 2.36 (d, J = 14.5 Hz, 2H), 2.28 (d, J = 13.0 Hz, 1H), 2.17 (s, 3H), 2.13–1.94 (m, 4H), 1.8–1.65 (m, 1H), 1.6–1.40 (m, 2H), 1.47 (brs, 1H), 1.16 (d, J = 7.0 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 160.79, 143.10, 135.04, 130.43, 129.83, 129.61, 117.55, 115.48, 94.16, 78.74, 77.12, 74.58, 73.59, 70.74, 68.04, 55.46, 48.29, 42.94, 39.72, 37.99, 36.48, 35.89, 35.82, 32.90, 19.87, 19.29, 18.46, 15.34, 14.09, 13.2.
To a stirred solution of MOM ether (11 mg, 0.02 mmol) in DCM (2 mL), we added B-bromocatechol borane (0.5 mL, 0.2 M soln in DCM, 0.1 mmol) at −78 °C, and the mixture was stirred at that temperature for 1h, before it was quenched with sat NaHCO3 (5 mL). The organic layer was separated and the aq layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over anhyd Na2SO4 and concentrated in vacuo. Column chromatography (MeOH/chloroform 1:20) provided the corresponding alcohol as a colorless oil (6.7 mg, 66%). Rf value (EtOAc/hexane/MeOH 8:2:1): 0.3.
To a stirred solution of thus obtained alcohol (3 mg, 0.0059 mmol) and DMAP (9 mg, 0.074 mmol) in DCM (2 mL), we added TESCl (0.01 mL, 0.059 mmol) at 0 °C, and the reaction was stirred at 0 °C for 30 min before sat NaHCO3 (5 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:20) provided tris-TES ether as a colorless oil (4.4 mg, 88%). Rf value (EtOAc/hexane 1:4): 0.5.
To a stirred solution of thus obtained diol (2.4 mg, 2.8 μmol) in DCM (1 mL), we added DMSO (40 μL, 0.141 mmol), DIPEA (49 μL, 0.282 mmol) and SO3.Py (22.4 mg, 0.141 mmol) at 0 °C. The mixture was stirred at rt for 30 min. The solvent was removed by high vacuum and THF (1 mL) was added, followed by a HF·Py solution (0.3 mL containing 1 mL 70% HF·Py: 1.1 mL pyridine: 2.4 mL THF) at rt. The reaction mixture was stirred for 1h, before it was quenched with sat NaHCO3 and extracted with diethyl ether (3 × 10 mL). The combined organic layer was washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (MeOH/chloroform 1:25) to give diastereomer 29 (1 mg, 71%) as a colorless oil. Rf value (EtOAc/hexane 2:1): 0.3. [α]D20 = +35.7 (c = 0.042, CH2Cl2); IR (film, cm−1) 3465, 2980, 2360, 1695, 1452, 1369, 1222, 1004; 1H NMR (500 MHz, CDCl3) δ 5.77–5.70 (m, 1H), 5.67 (d, J = 18.6 Hz, 2H), 5.58–5.51 (m, 1H), 5.47 (dd, J = 16.0, 7.4 Hz, 1H), 4.86 (s, 1H), 4.84 (s, 1H), 4.83–4.79 (m, 1H), 4.21 (dd, J = 8.2, 3.8 Hz, 1H), 4.07–3.95 (m, 1H), 3.80 (dd, J = 6.3, 3.4 Hz, 1H), 3.35 (s, 1H), 3.06 (brs, 1H), 2.67 (dd, J = 6.8, 3.9 Hz, 1H), 2.50 (d, J = 14.4 Hz, 1H), 2.33 (m, 1H), 2.20 (m, 2H), 2.18 (d, J = 0.8 Hz, 3H), 2.15–2.08 (m, 2H), 2.07–1.96 (m, 2H), 1.94–1.81 (m, 1H), 1.78–1.69 (m, 1H), 1.51–1.45 (m, 1H), 1.45–1.37 (m, 1H), 1.30 (s, 3H), 1.15 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 7.1 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 159.2, 141.4, 134.1, 130.5, 130.3, 129.6, 118.2, 110.9, 99.2, 73.1, 69.7, 48.6, 38.3, 37.85, 37.1, 36.5, 35.7, 35.3, 33.6, 24.6, 23.2, 19.5, 18.7, 14.8, 14.3, 10.5. MS (ESI, m/z) [M + Na]+ 529.31.
Diastereomer (2E, 9R) (30): [α]D20 = +18.8 (c = 0.07, CH2Cl2); IR (film, cm−1) 3380, 2924, 1690, 1684,1541, 1508, 1458, 1212; 1H NMR (500 MHz, CDCl3) δ 9.42 (s, 1H), 6.22 (d, J = 11.0 Hz, 1H), 5.73–5.59 (m, 3H), 5.52 (dd, J = 15.6, 8.1 Hz, 1H), 5.02–4.91 (m, 2H), 4.89 (s, 1H), 4.42 (dd, J = 8.0, 3.6 Hz, 1H), 3.85 (dd, J = 6.3, 3.4 Hz, 1H), 3.56–3.50 (m, 1H), 3.46–3.38 (m, 1H), 2.63 (dd, J = 6.8, 4.0 Hz, 1H), 2.31–2.10 (m, 6H), 2.19 (d, J = 0.9 Hz, 3H), 2.04 (dd, J = 13.8, 9.3 Hz, 1H), 1.94 (dd, J = 14.2, 9.1 Hz, 1H), 1.80 (s, 3H), 1.54–1.45 (m, 1H), 1.38–1.28 (m, 1H), 1.16 (d, J = 6.9 Hz, 3H), 1.11 (d, J = 6.4 Hz, 3H), 0.90–0.80 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 191.2, 167.0, 161.0, 153.3, 143.0, 135.5, 130.7, 130.5, 115.6, 115.3, 75.8, 73.7, 69.5, 48.5, 45.6, 42.7, 40.2, 35.9, 35.9, 35.7, 35.1, 32.1, 20.5, 19.9, 16.7, 16.5, 14.9. MS (ESI, m/z) [M + Na]+ 529.33.
Diastereomer(4S, 5R) (31): [α]D20 = −15 (c = 0.03, CH2Cl2); IR (film, cm−1) 3470, 2987, 2975, 2360, 1684, 1255, 1005; 1H NMR (500 MHz, CDCl3) δ 5.87 (s, 1H), 5.86–5.74 (m, 3H), 5.57 (dd, J = 15.7, 7.2 Hz, 1H), 5.12 (dd, J = 11.5, 5.8 Hz, 1H), 4.88 (s, 1H), 4.85 (s, 1H), 4.16–4.07 (m, 1H), 3.94–3.85 (m, 1H), 3.80 (dt, J = 10, 6.2 Hz, 1H), 3.42 (dq, J = 13.9, 6.9 Hz, 1H), 2.76 (brs, 1H), 2.45–2.32 (m, 3H), 2.33–2.22 (m, 3H), 2.18 (d, J = 11.2 Hz, 1H), 2.08–1.93 (m, 4H), 1.89 (s, 3H), 1.79–1.7 (m, 1H), 1.5–1.41 (m, 1H), 1.13 (d, J = 6.3 Hz, 3H), 1.06 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.3, 155.5, 141.5, 134.66, 132.0, 130.0, 128.4, 120.4, 111.1, 99.4, 75.1, 70.3, 69.7, 53.3, 42.6, 39.6, 36.8, 36.8, 35.8, 34.8, 34.1, 32.8, 21.9, 19.7, 19.0, 16.7, 15.8, 14.8. MS (ESI, m/z) [M + Na]+ 529.33.


{kind=link}
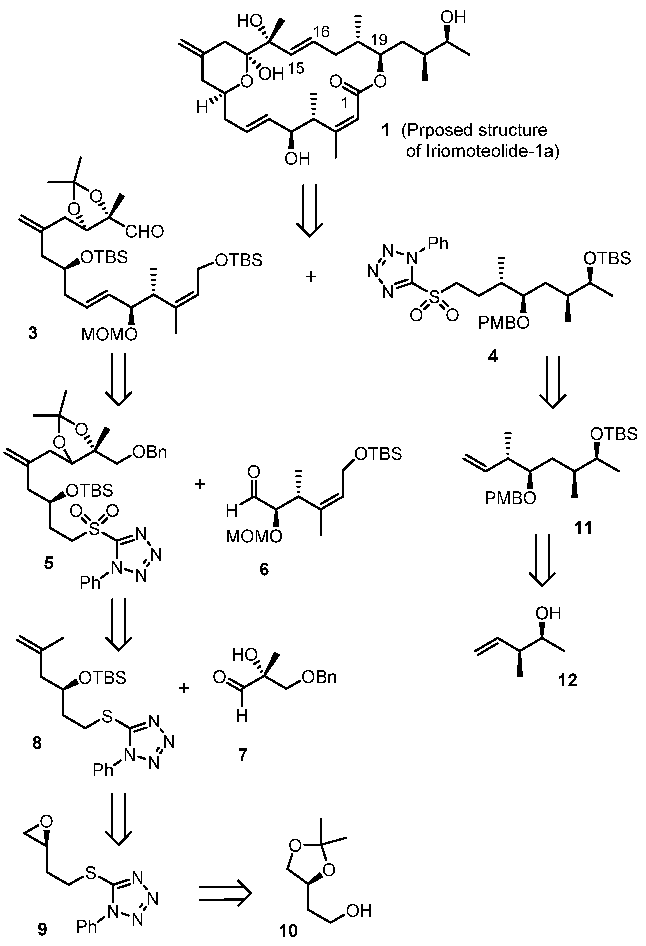{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
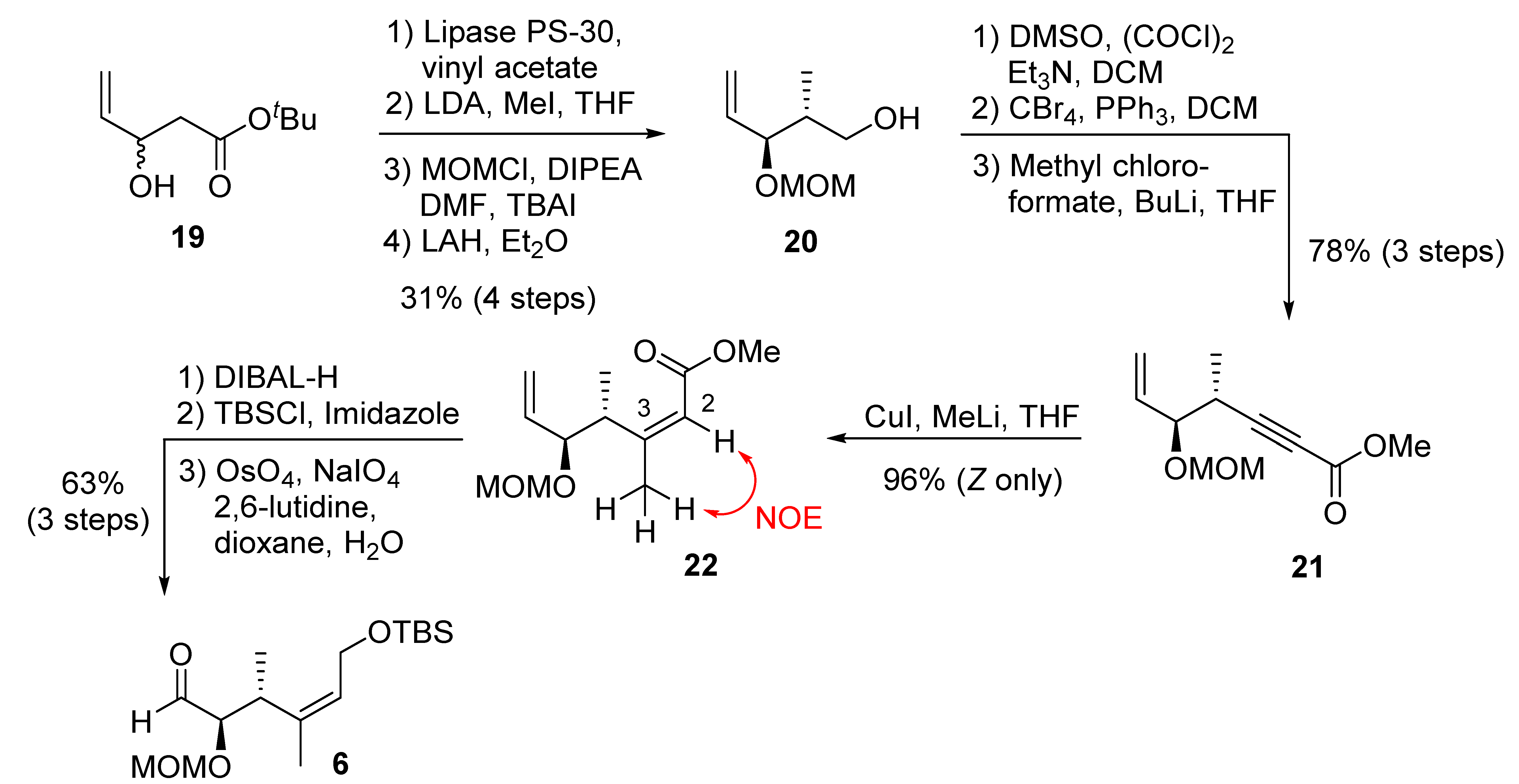{kind=link}
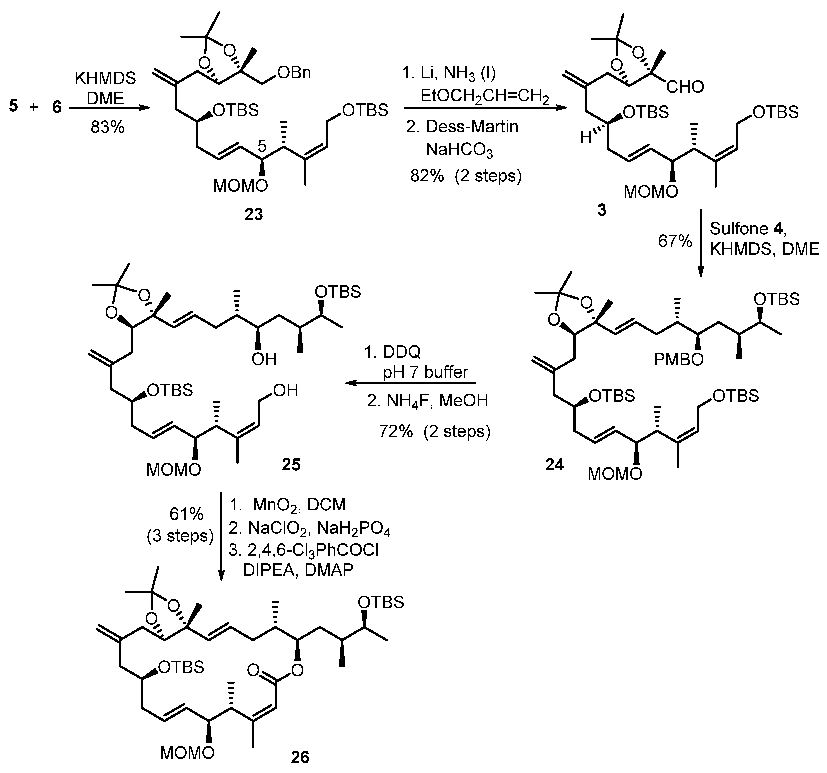{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












