
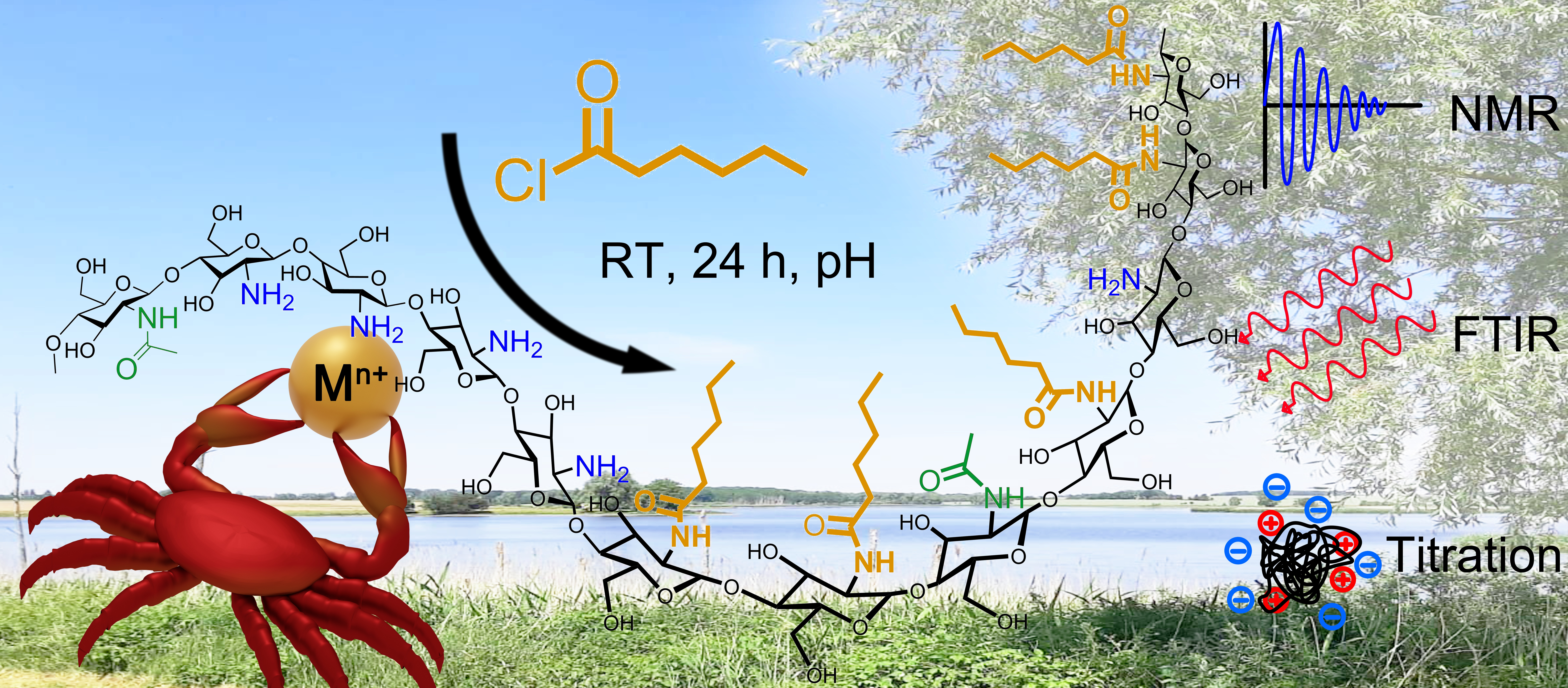
A Complementary and Revised View on the N-Acylation of Chitosan with Hexanoyl Chloride

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
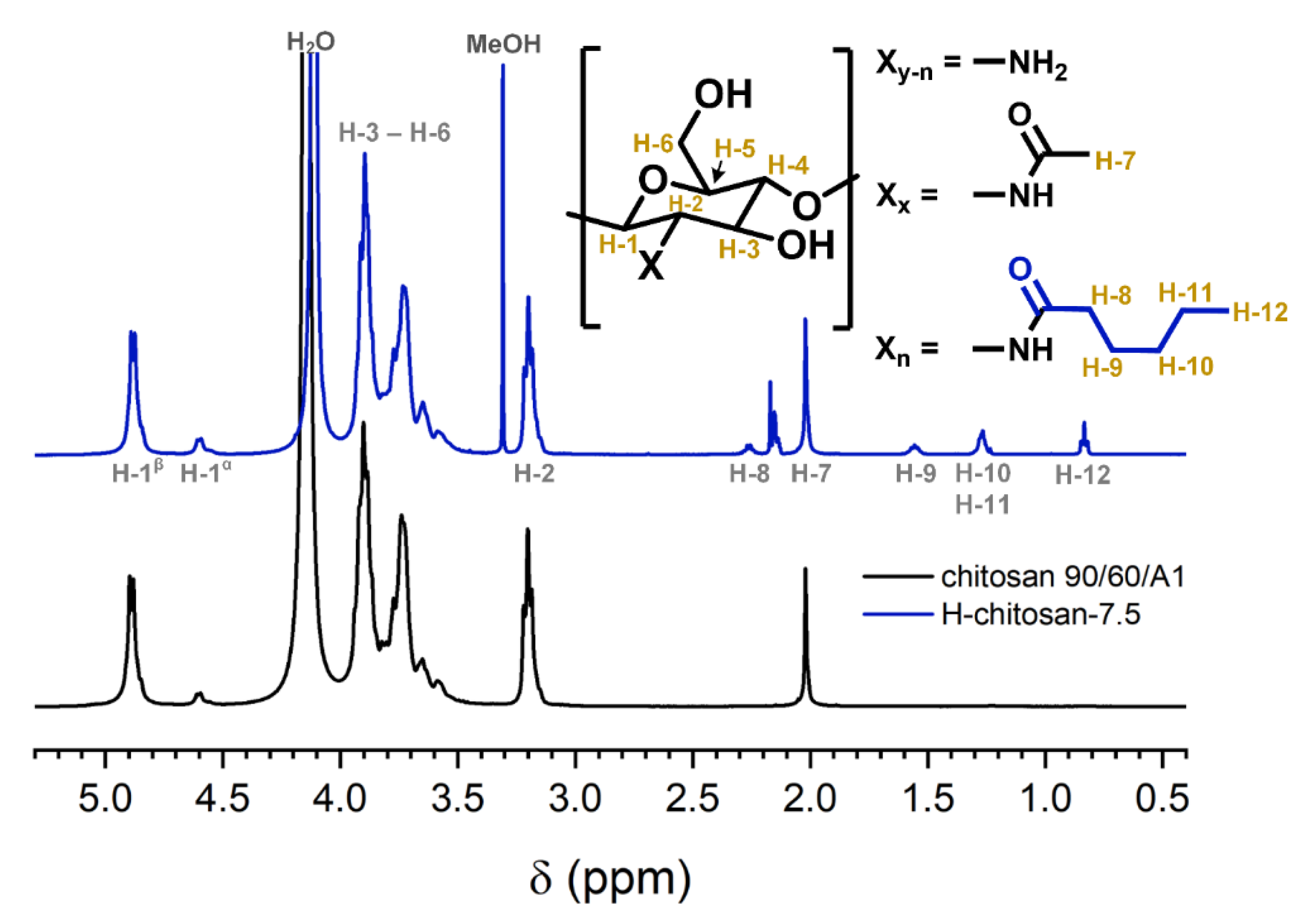
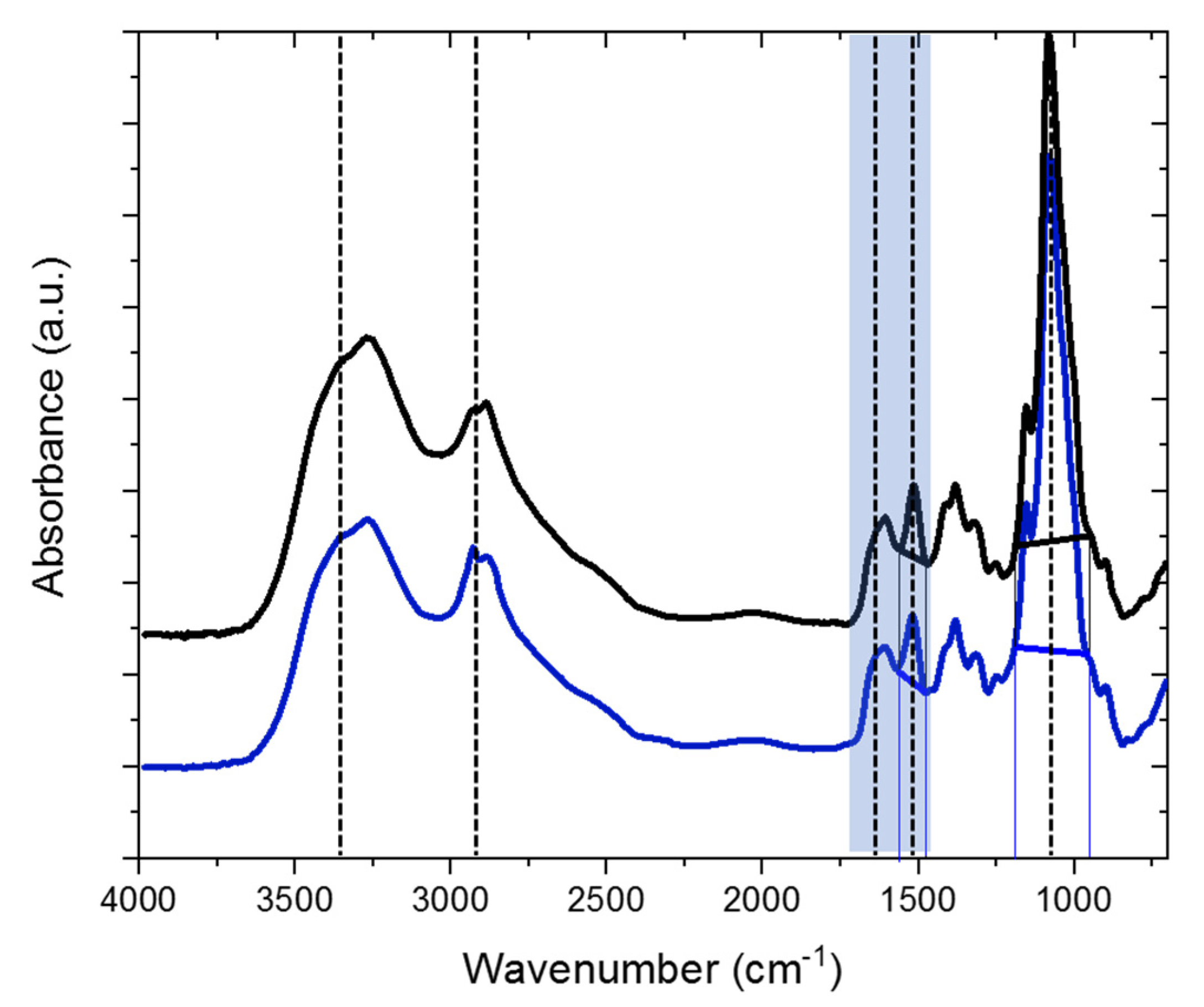
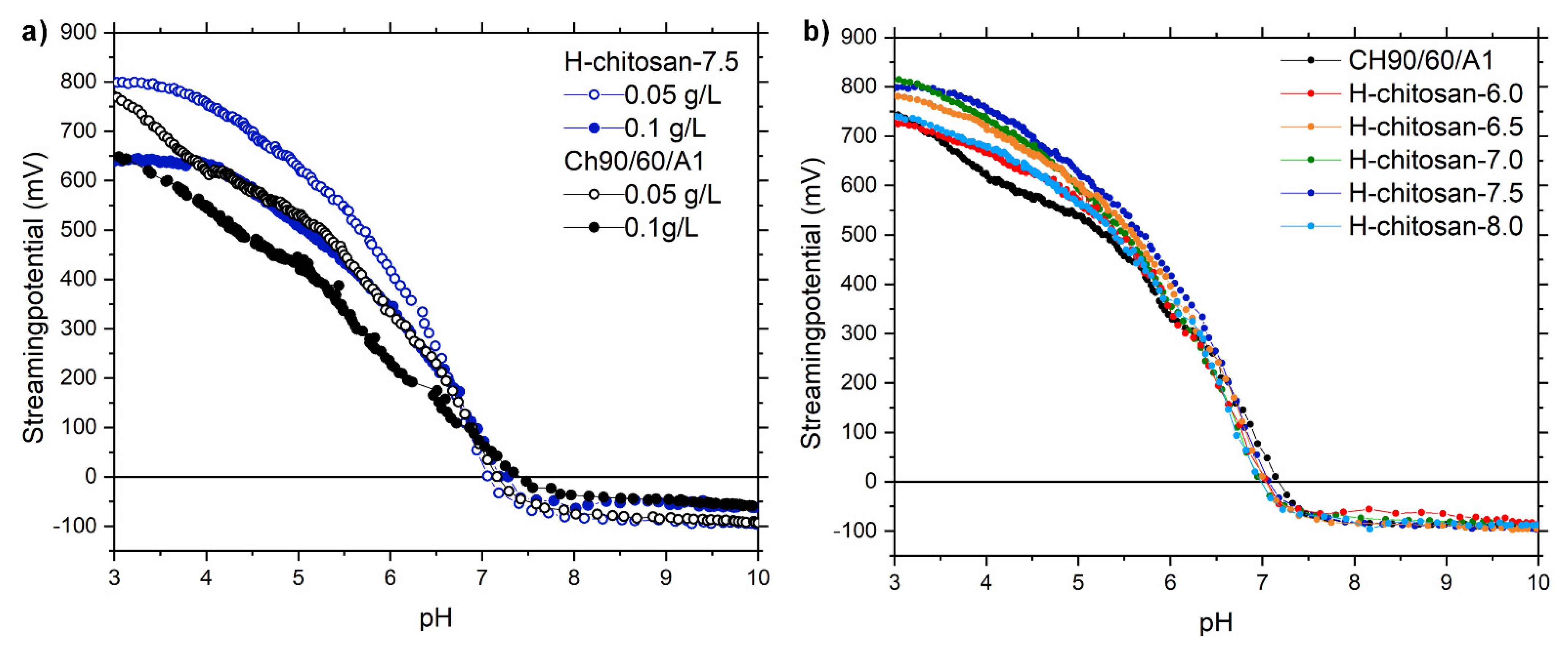
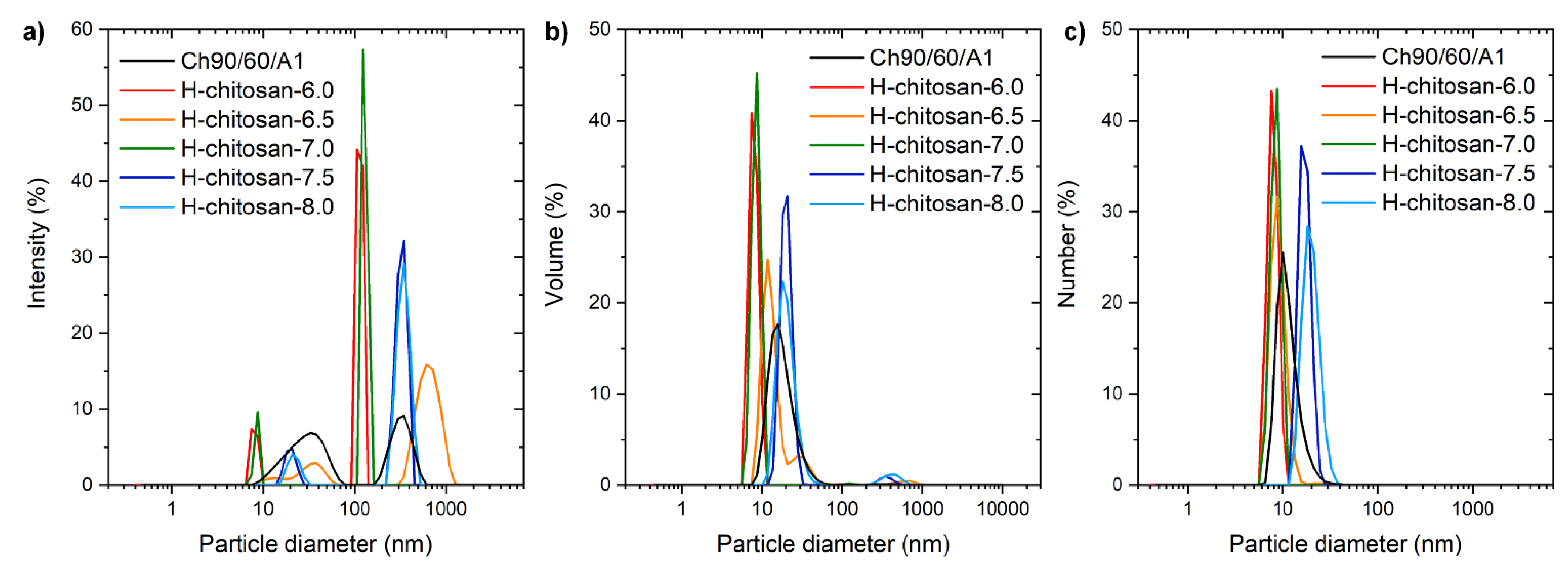
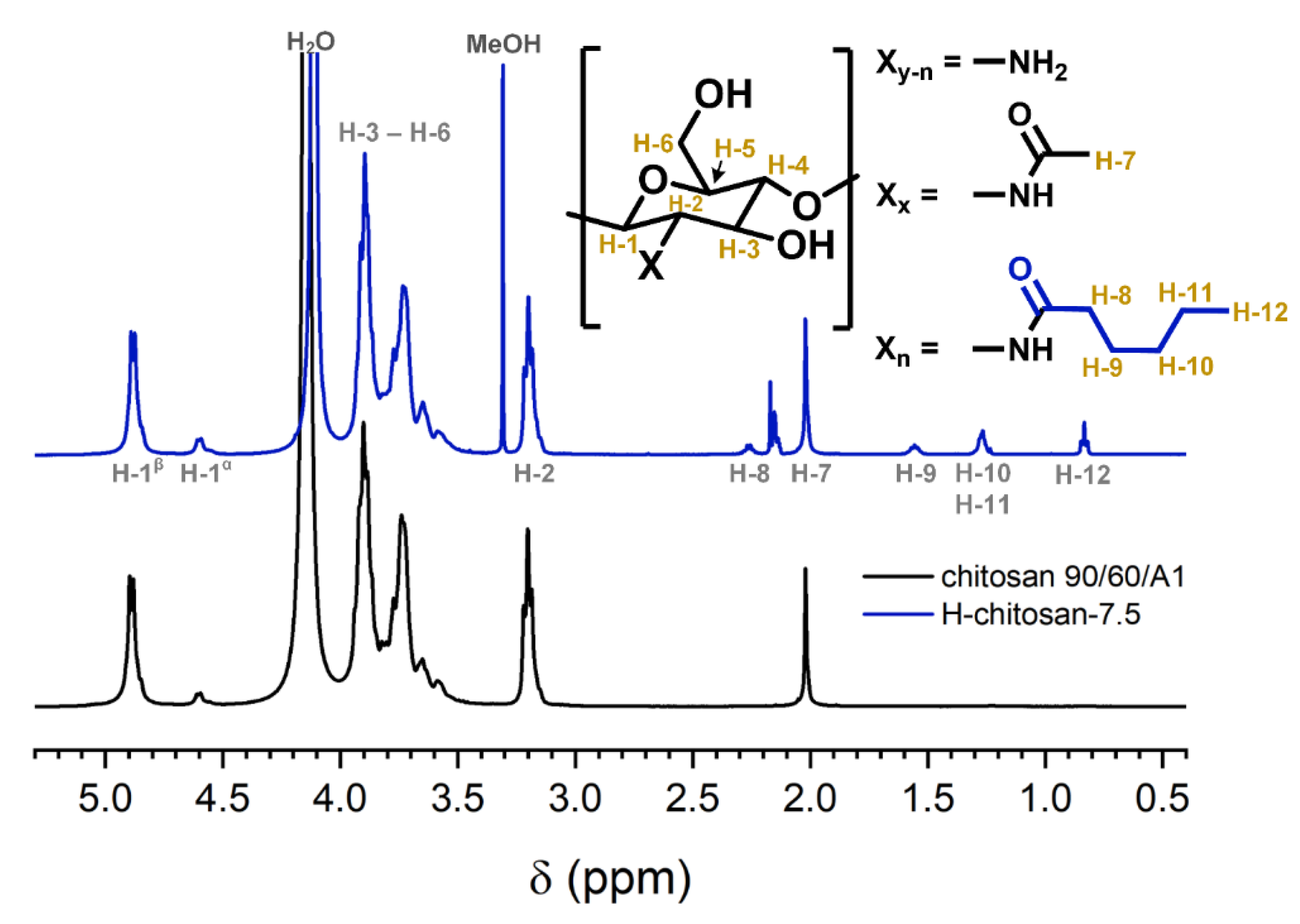
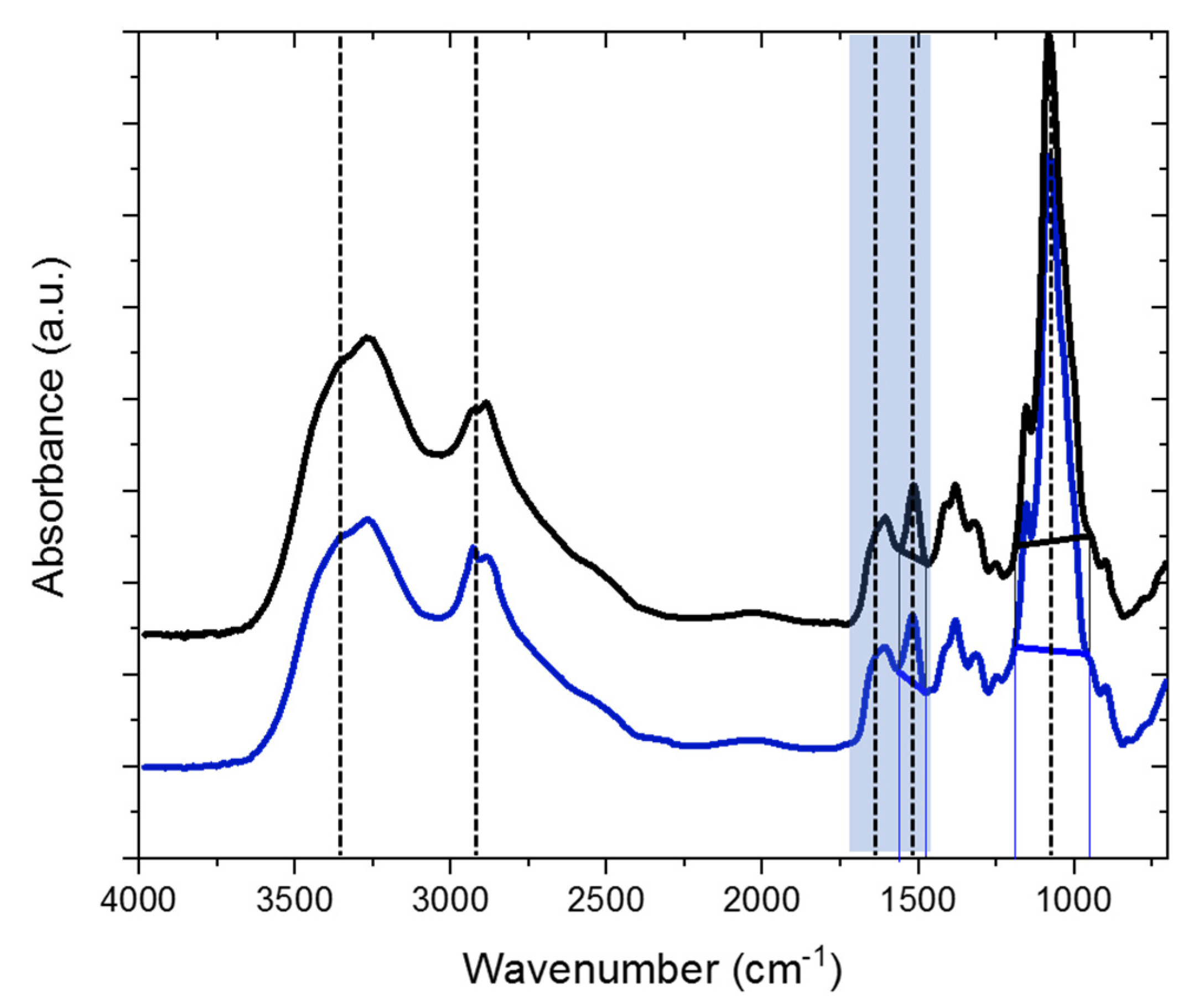
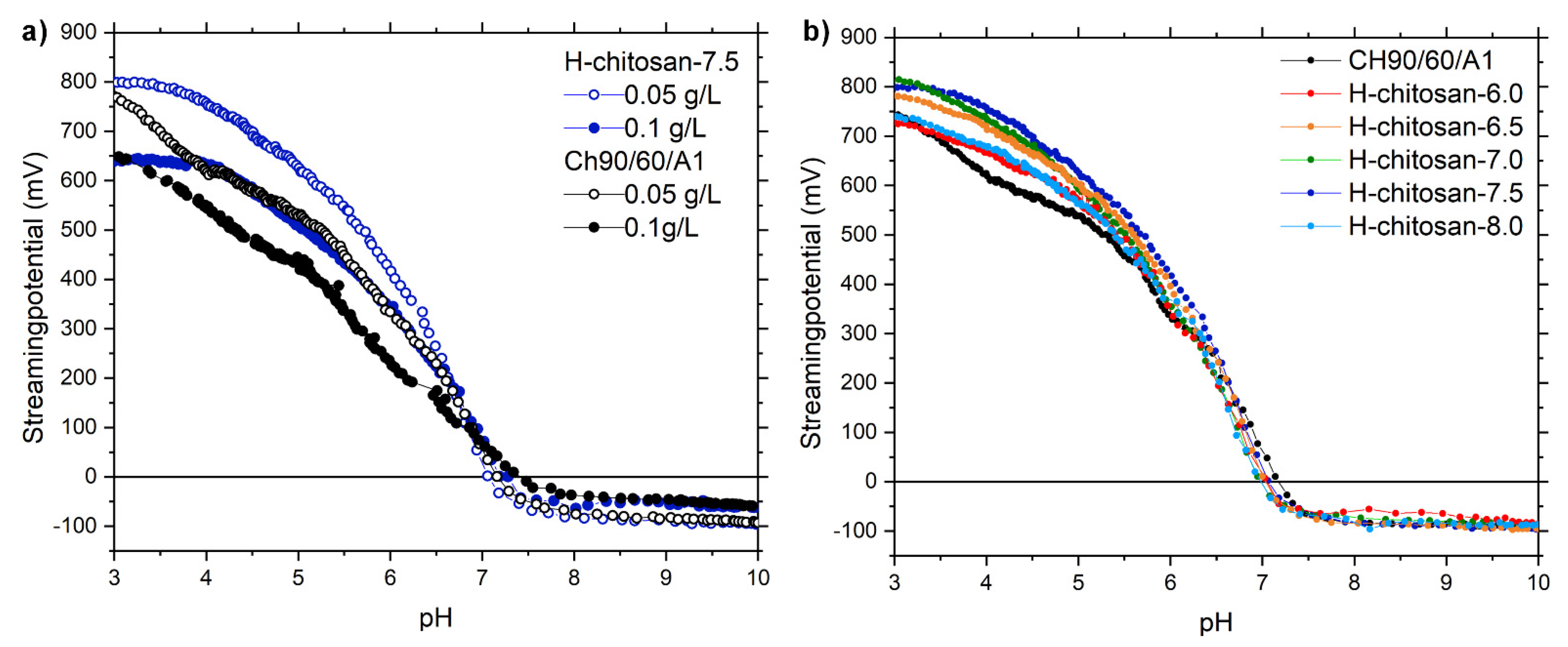
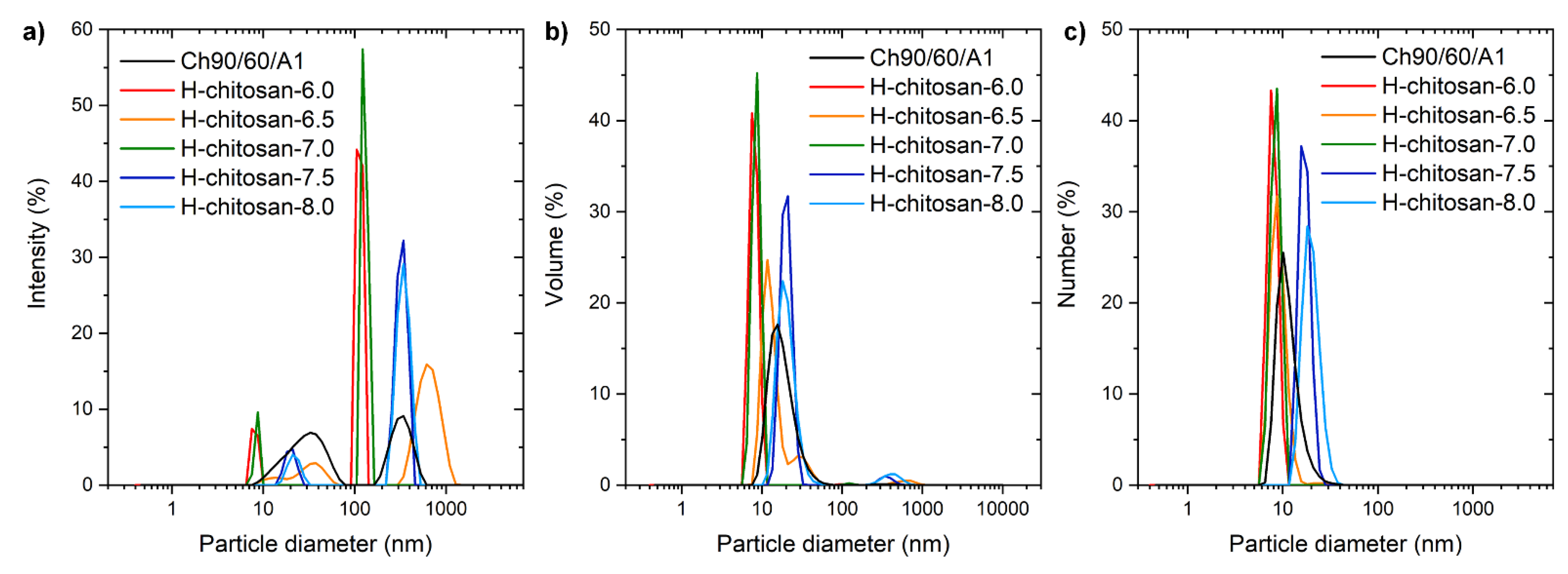
2.1. Characterization of the H-Chitosan Samples
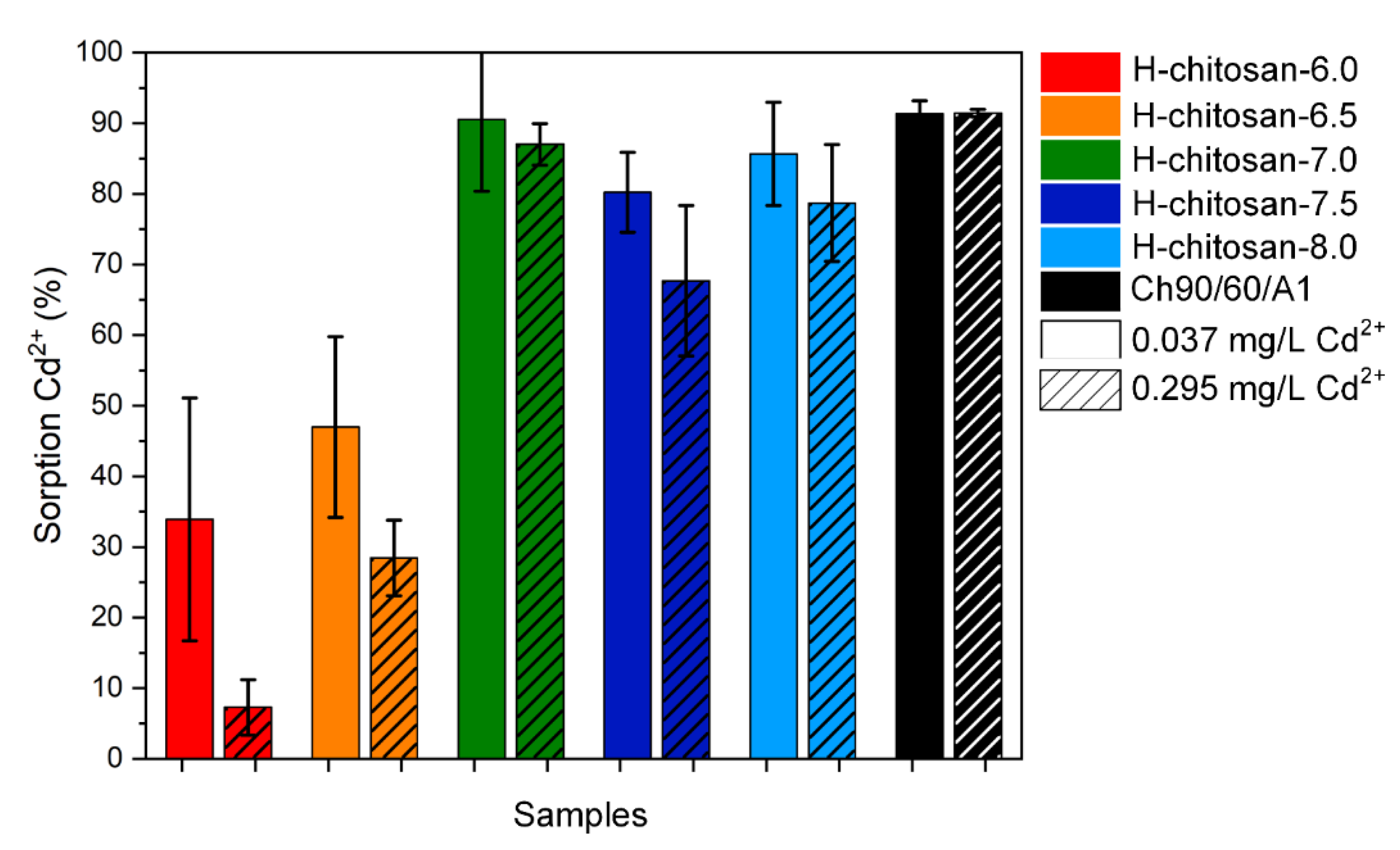
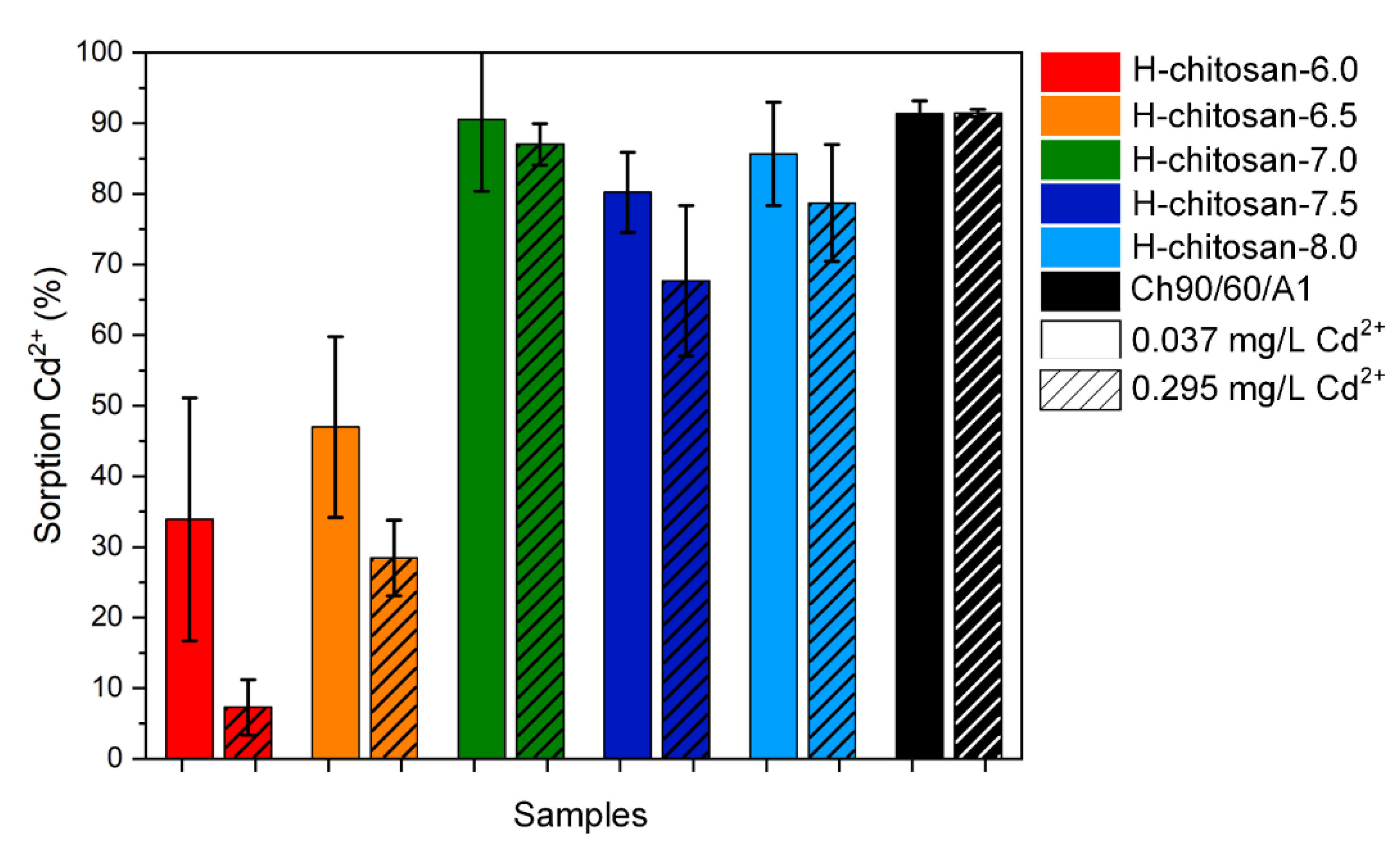
2.2. Adsorption Experiments with CdSO4(aq)
3. Materials and Methods
3.1. Materials
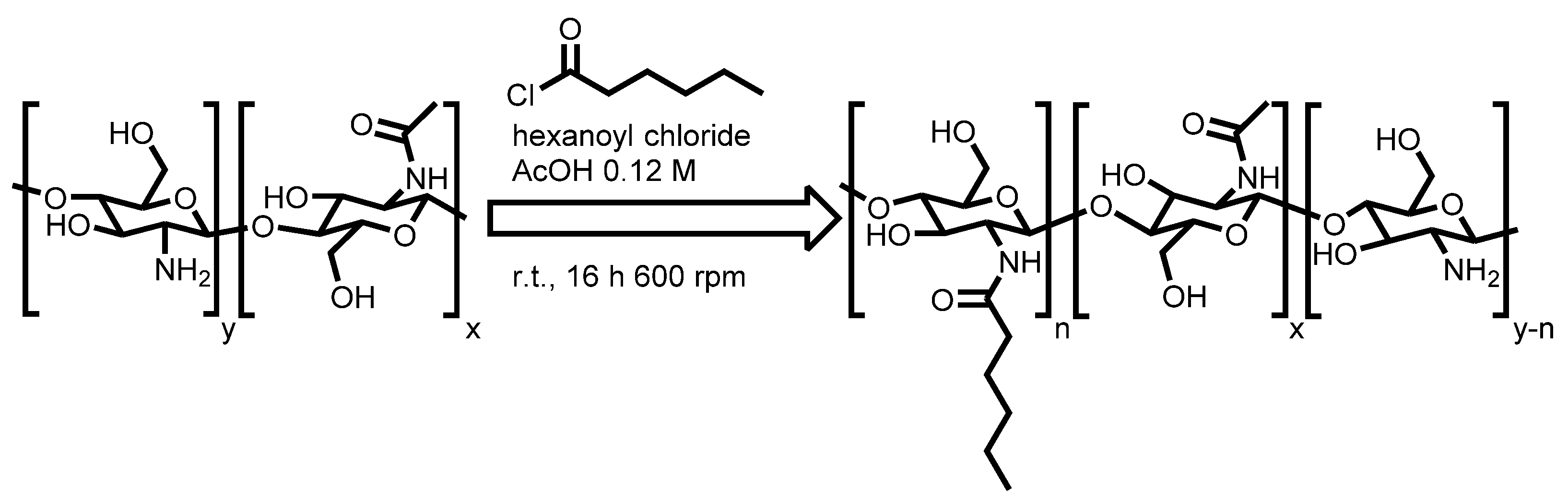
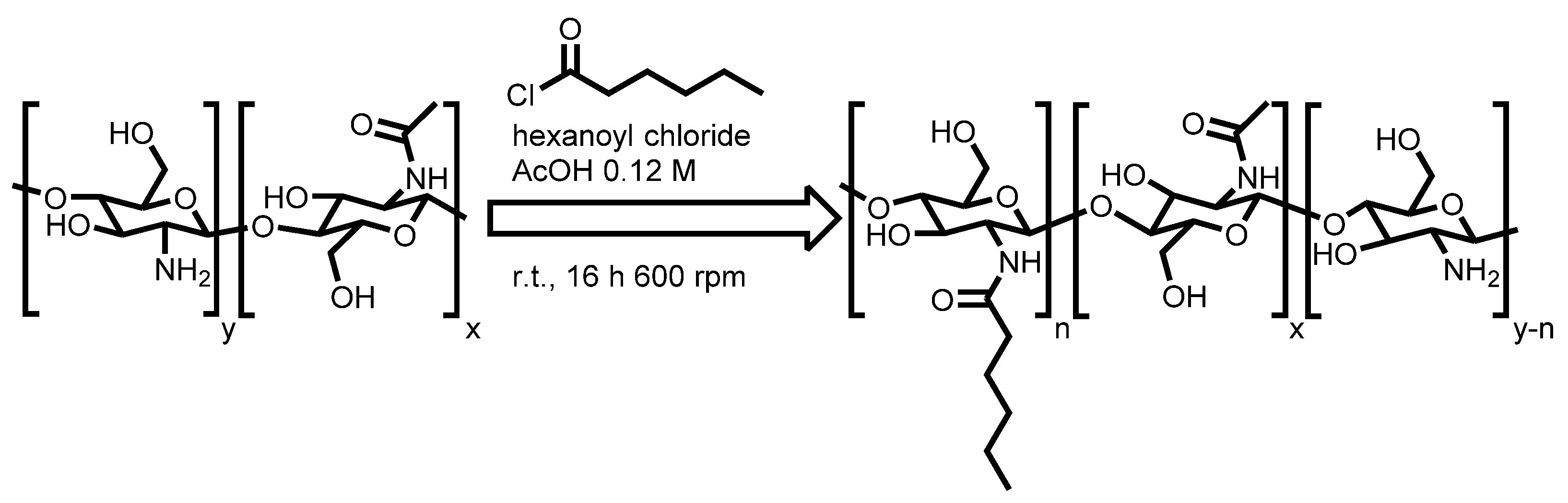
3.2. Synthesis of H-Chitosan
3.3. Adsorption Experiments with CdSO4
3.4. Characterization and Analysis
3.4.1. H-NMR Measurements
3.4.2. FTIR Measurements
Method 1
Method 2
3.4.3. Potentiometric Titration and Zetapotential vs. pH
3.4.4. Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES)
3.4.5. Nitrogen Sorption Studies
3.4.6. Dynamic Light Scattering
3.4.7. pH Measurement
3.4.8. X-ray Diffraction Measurements (XRD)
3.4.9. Oscillating Rotational Rheometer Measurements
3.4.10. Thermogravimetric Analysis (TG)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fenice, M.; Gorrasi, S. Advances in Chitin and Chitosan Science. Molecules 2021, 26, 1805. [Google Scholar] [CrossRef]
- Upadhyay, U.; Sreedhar, I.; Singh, S.A.; Patel, C.M.; Anitha, K.L. Recent Advances in Heavy Metal Removal by Chitosan Based Adsorbents. Carbohydr. Polym. 2021, 251, 117000. [Google Scholar] [CrossRef] [PubMed]
- Borchert, K.B.L.; Steinbach, C.; Schwarz, S.; Schwarz, D. A Comparative Study on the Flocculation of Silica and China Clay with Chitosan and Synthetic Polyelectrolytes. Mar. Drugs 2021, 19, 102. [Google Scholar] [CrossRef]
- Ghiorghita, C.-A.; Borchert, K.B.L.; Vasiliu, A.-L.; Zaharia, M.-M.; Schwarz, D.; Mihai, M. Porous Thiourea-Grafted-Chitosan Hydrogels: Synthesis and Sorption of Toxic Metal Ions from Contaminated Waters. Colloids Surf. A Physicochem. Eng. Asp. 2020, 607, 125504. [Google Scholar] [CrossRef]
- Haufe, S.; Bohrisch, J.; Schwarz, D.; Bratskaya, S.Y.; Steinbach, C.; Schwarz, S. Flocculation Efficiency of Reacetylated Water Soluble Chitosan versus Commercial Chitosan. Colloids Surf. A Physicochem. Eng. Asp. 2017, 532, 222–227. [Google Scholar] [CrossRef]
- Weißpflog, J.; Gündel, A.; Vehlow, D.; Steinbach, C.; Müller, M.; Boldt, R.; Schwarz, S.; Schwarz, D. Solubility and Selectivity Effects of the Anion on the Adsorption of Different Heavy Metal Ions onto Chitosan. Molecules 2020, 25, 2482. [Google Scholar] [CrossRef]
- Xu, J.; McCarthy, S.P.; Gross, R.A.; Kaplan, D.L. Chitosan Film Acylation and Effects on Biodegradability. Macromolecules 1996, 29, 3436–3440. [Google Scholar] [CrossRef]
- Bagheri-Khoulenjani, S.; Taghizadeh, S.M.; Mirzadeh, H. An Investigation on the Short-Term Biodegradability of Chitosan with Various Molecular Weights and Degrees of Deacetylation. Carbohydr. Polym. 2009, 78, 773–778. [Google Scholar] [CrossRef]
- VandeVord, P.J.; Matthew, H.W.T.; DeSilva, S.P.; Mayton, L.; Wu, B.; Wooley, P.H. Evaluation of the Biocompatibility of a Chitosan Scaffold in Mice. J. Biomed. Mater. Res. 2002, 59, 585–590. [Google Scholar] [CrossRef]
- Weißpflog, J.; Vehlow, D.; Müller, M.; Kohn, B.; Scheler, U.; Boye, S.; Schwarz, S. Characterization of Chitosan with Different Degree of Deacetylation and Equal Viscosity in Dissolved and Solid State—Insights by Various Complimentary Methods. Int. J. Biol. Macromol. 2021, 171, 242–261. [Google Scholar] [CrossRef]
- Pestov, A.; Bratskaya, S. Chitosan and Its Derivatives as Highly Efficient Polymer Ligands. Molecules 2016, 21, 330. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.K.; Roberts, G.A.F. Determination of the degree of N-acetylation of chitosan. In Proceedings of the First International Conference on Chitin/Chitosan; MIT Sea Grant Program. Massachusetts Institute of Technology: Cambridge, MA, USA; pp. 421–429.
- Layek, B.; Singh, J. N-Hexanoyl, N-Octanoyl and N-Decanoyl Chitosans: Binding Affinity, Cell Uptake, and Transfection. Carbohydr. Polym. 2012, 89, 403–410. [Google Scholar] [CrossRef]
- Le Tien, C.; Lacroix, M.; Ispas-Szabo, P.; Mateescu, M.-A. N-Acylated Chitosan: Hydrophobic Matrices for Controlled Drug Release. J. Control. Release 2003, 93, 1–13. [Google Scholar] [CrossRef]
- Zong, Z.; Kimura, Y.; Takahashi, M.; Yamane, H. Characterization of Chemical and Solid State Structures of Acylated Chitosans. Polymer 2000, 41, 899–906. [Google Scholar] [CrossRef]
- Naidoo, S.; Nomadolo, N.; Matshe, W.M.R.; Cele, Z.; Balogun, M. Exploring the Potential of N-Acylated Chitosan for the Removal of Toxic Pollutants from Wastewater. IOP Conf. Ser. Mater. Sci. Eng. 2019, 655, 012047. [Google Scholar] [CrossRef]
- Han, J.; Guenier, A.-S.; Salmieri, S.; Lacroix, M. Alginate and Chitosan Functionalization for Micronutrient Encapsulation. J. Agric. Food Chem. 2008, 56, 2528–2535. [Google Scholar] [CrossRef]
- Piegat, A.; Goszczyńska, A.; Idzik, T.; Niemczyk, A. The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives. Molecules 2019, 24, 3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugebauer, W.A.; Neugebauer, E.; Brzezinski, R. Determination of the Degree of N-Acetylation of Chitin-Chitosan with Picric Acid. Carbohydr. Res. 1989, 189, 363–367. [Google Scholar] [CrossRef]
- Curotto, E.; Aros, F. Quantitative Determination of Chitosan and the Percentage of Free Amino Groups. Anal. Biochem. 1993, 211, 240–241. [Google Scholar] [CrossRef] [PubMed]
- Vårum, K.M.; Antohonsen, M.W.; Grasdalen, H.; Smidsrød, O. Determination of the Degree of N-Acetylation and the Distribution of N-Acetyl Groups in Partially N-Deacetylated Chitins (Chitosans) by High-Field n.m.r. Spectroscopy. Carbohydr. Res. 1991, 211, 17–23. [Google Scholar] [CrossRef]
- Hexanoic Acid. Available online: https://bmrb.io/metabolomics/mol_summary/show_data.php?id=bmse000351 (accessed on 16 May 2021).
- Clayden, J.; Greeves, N.; Warren, S. Organische Chemie, 2nd ed.; Springer Spektrum: Heidelberg, Germany, 2013; ISBN 978-3-642-34715-3. [Google Scholar]
- Moore, G.K.; Roberts, G.A.F. Determination of the Degree of N-Acetylation of Chitosan. Int. J. Biol. Macromol. 1980, 2, 115–116. [Google Scholar] [CrossRef]
- Lin, A.; Chen, J.; Liu, Y.; Deng, S.; Wu, Z.; Huang, Y.; Ping, Q. Preparation and Evaluation of N-Caproyl Chitosan Nanoparticles Surface Modified with Glycyrrhizin for Hepatocyte Targeting. Drug Dev. Ind. Pharm. 2009, 35, 1348–1355. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, J.T.; Park, H.J. Size-Controlled Self-Aggregated N-Acyl Chitosan Nanoparticles as a Vitamin C Carrier. Carbohydr. Polym. 2012, 88, 1087–1092. [Google Scholar] [CrossRef]
- Pa, J.-H.; Yu, T.L. Light Scattering Study of Chitosan in Acetic Acid Aqueous Solutions. Macromol. Chem. Phys. 2001, 202, 985–991. [Google Scholar] [CrossRef]
- WHO. Guidelines for Drinking-Water Quality, 3rd ed.; Volume 1—Recommendations. Available online: http://www.who.int/water_sanitation_health/publications/gdwq3rev/en/ (accessed on 29 April 2021).
- Hasan, S.; Krishnaiah, A.; Ghosh, T.K.; Viswanath, D.S.; Boddu, V.M.; Smith, E.D. Adsorption of Divalent Cadmium (Cd(II)) from Aqueous Solutions onto Chitosan-Coated Perlite Beads. Ind. Eng. Chem. Res. 2006, 45, 5066–5077. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Chitosan | DS%NMR * | DS%FTIR-1 ** | DS%FTIR-2 *** |
|---|---|---|---|
| H-chitosan-6.0 | 1.6% | 16.6% | 2.9% |
| H-chitosan-6.5 | 2.1% | 20.8% | 1.8% |
| H-chitosan-7.0 | 2.7% | 20.1% | 5.3% |
| H-chitosan-7.5 | 3.0% | 19.9% | 3.8% |
| H-chitosan-8.0 | 2.9% | 19.5% | 3.0% |
| Concentration | pH0 | pHdis 90/60/A1 | pHdis H-Chitosan-7.5 | c (mmol/L) 90/60/A1 | c (mmol/L) H-Chitosan-7.5 | DS%Pot-1 |
|---|---|---|---|---|---|---|
| 0.01 g/L | 1.0 | 2.9 | 2.9 | 6.2 | 5.4 | 8.7% |
| 0.05 g/L | 1.0 | 2.2 | 2.2 | 5.8 | 5.0 | 8.6% |
| 0.10 g/L | 1.0 | 2.0 | 2.0 | 5.7 | 5.0 | 8.8% |
| H-Chitosan | DS%NMR | DS%Pot-1 * | DS%Pot-2 ** |
|---|---|---|---|
| H-chitosan-6.0 | 1.6% | 8.2% | 6.9% |
| H-chitosan-6.5 | 2.1% | 9.8% | 9.9% |
| H-chitosan-7.0 | 2.7% | 10.8% | 11.1% |
| H-chitosan-7.5 | 3.0% | 8.6% | 7.7% |
| H-chitosan-8.0 | 2.9% | 8.5% | 6.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis, B.; Gerlach, N.; Steinbach, C.; Haro Carrasco, K.; Oelmann, M.; Schwarz, S.; Müller, M.; Schwarz, D. A Complementary and Revised View on the N-Acylation of Chitosan with Hexanoyl Chloride. Mar. Drugs 2021, 19, 385. https://doi.org/10.3390/md19070385
Reis B, Gerlach N, Steinbach C, Haro Carrasco K, Oelmann M, Schwarz S, Müller M, Schwarz D. A Complementary and Revised View on the N-Acylation of Chitosan with Hexanoyl Chloride. Marine Drugs. 2021; 19(7):385. https://doi.org/10.3390/md19070385
Chicago/Turabian StyleReis, Berthold, Niklas Gerlach, Christine Steinbach, Karina Haro Carrasco, Marina Oelmann, Simona Schwarz, Martin Müller, and Dana Schwarz. 2021. "A Complementary and Revised View on the N-Acylation of Chitosan with Hexanoyl Chloride" Marine Drugs 19, no. 7: 385. https://doi.org/10.3390/md19070385
APA StyleReis, B., Gerlach, N., Steinbach, C., Haro Carrasco, K., Oelmann, M., Schwarz, S., Müller, M., & Schwarz, D. (2021). A Complementary and Revised View on the N-Acylation of Chitosan with Hexanoyl Chloride. Marine Drugs, 19(7), 385. https://doi.org/10.3390/md19070385