
Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry


Abstract
:1. Introduction
2. Pharmacological Characteristics
2.1. Antitumor Effects
2.1.1. Anti-Lymphoma Effect
2.1.2. Anti-Small Cell Lung Cancer Effect
2.1.3. Anti-Ovarian Carcinoma Effect
2.1.4. Anti-Prostate Cancer Effect
2.1.5. Anti-Soft Tissue Sarcoma Effect
2.1.6. Anti-Breast Cancer Effect
2.1.7. Anti-Hepatobiliary Pancreatic Carcinoma Effect
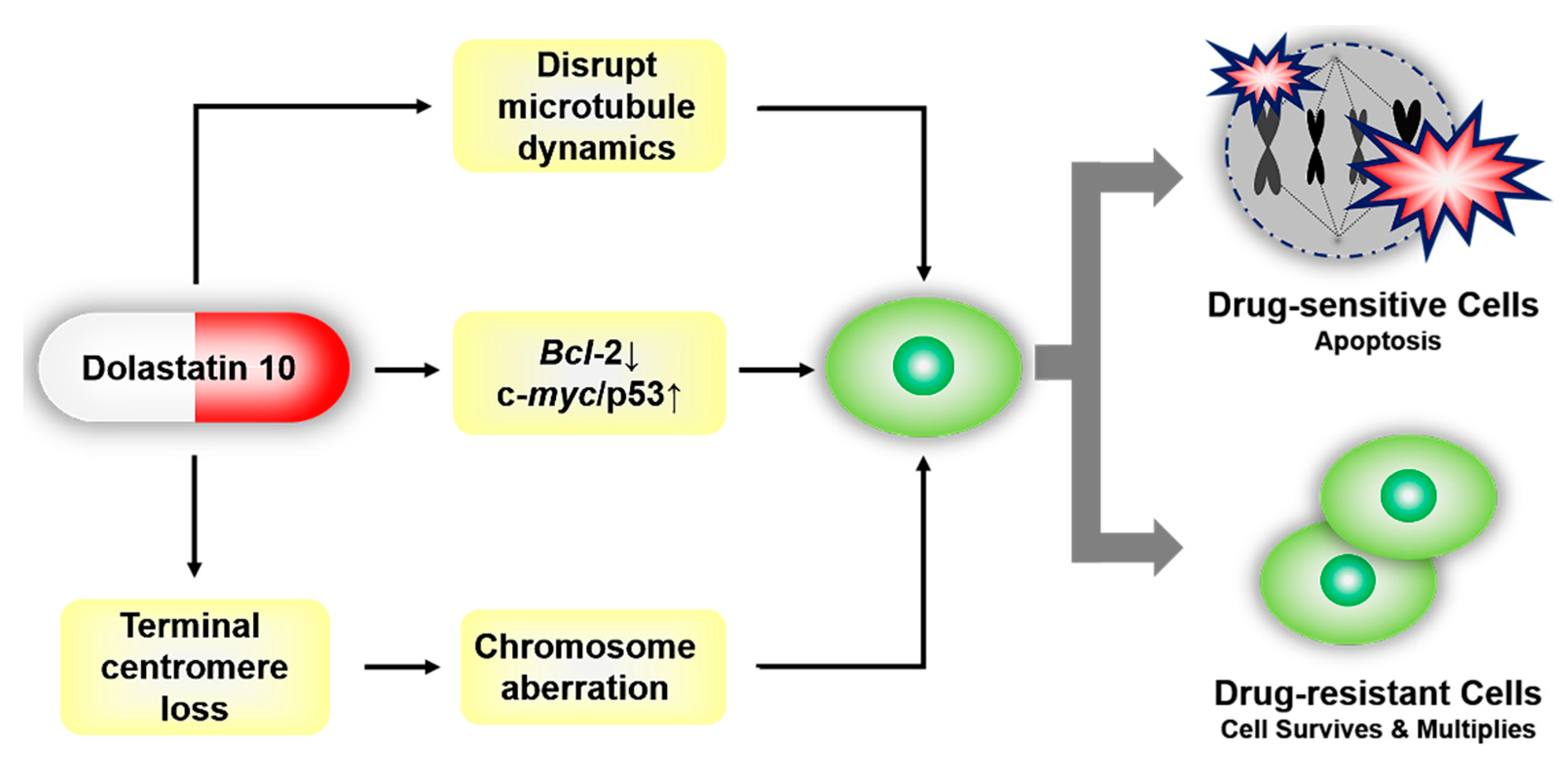
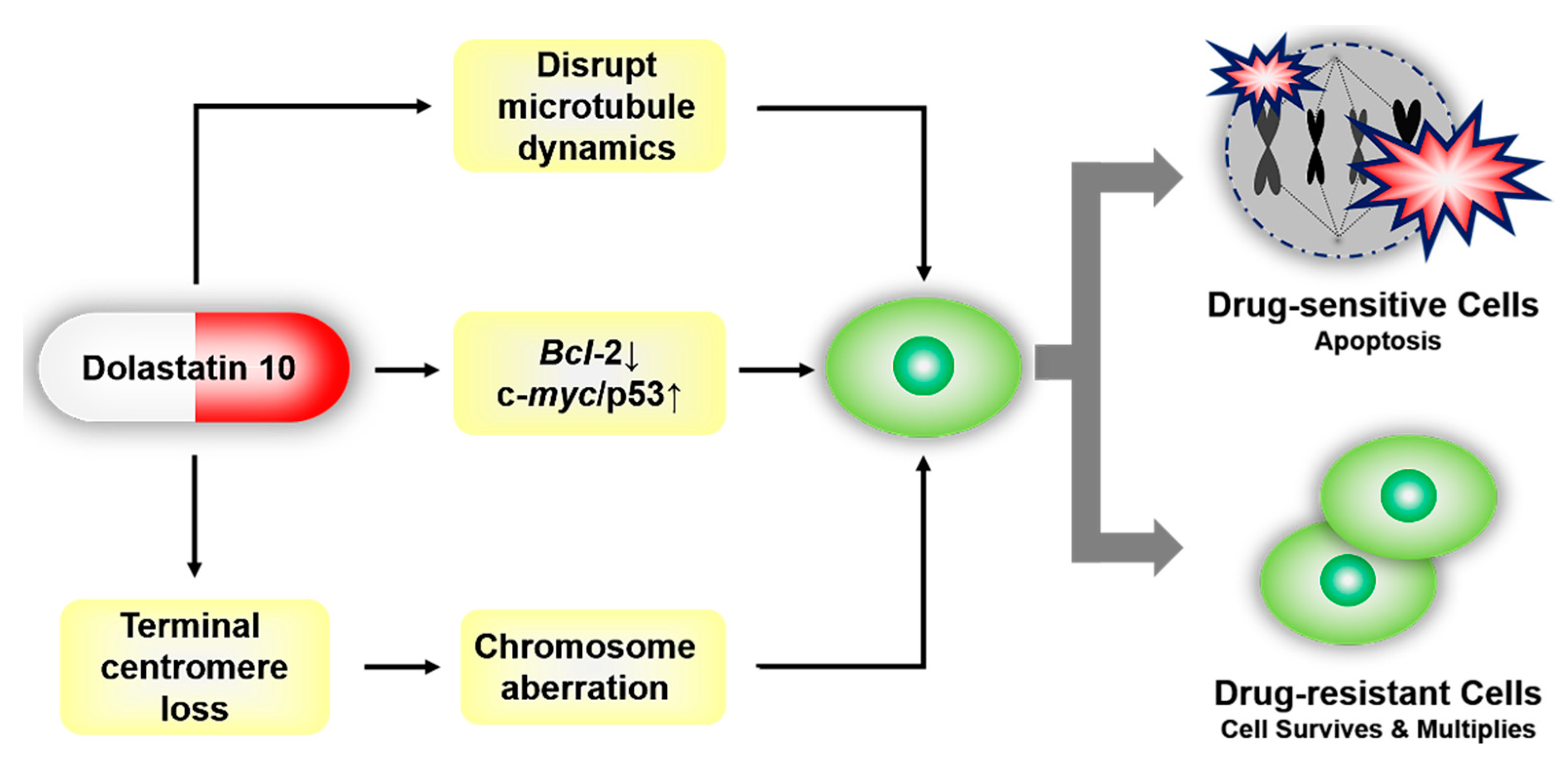
2.2. Mechanisms of Inducing Apoptosis
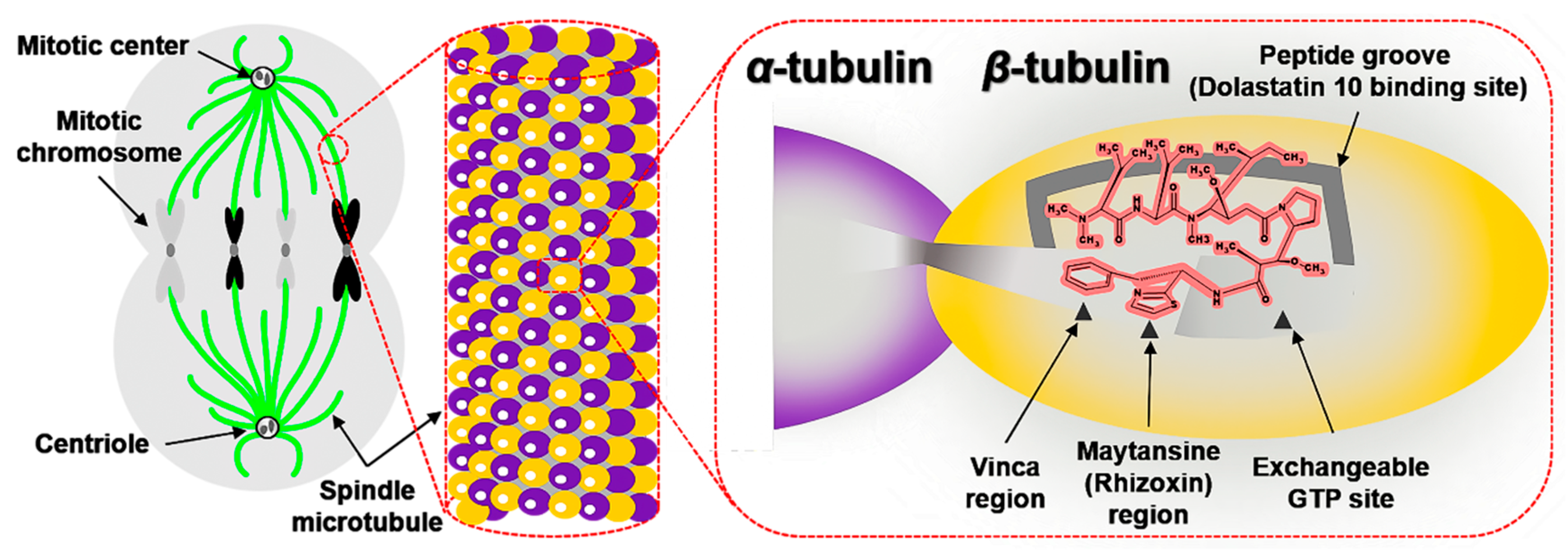
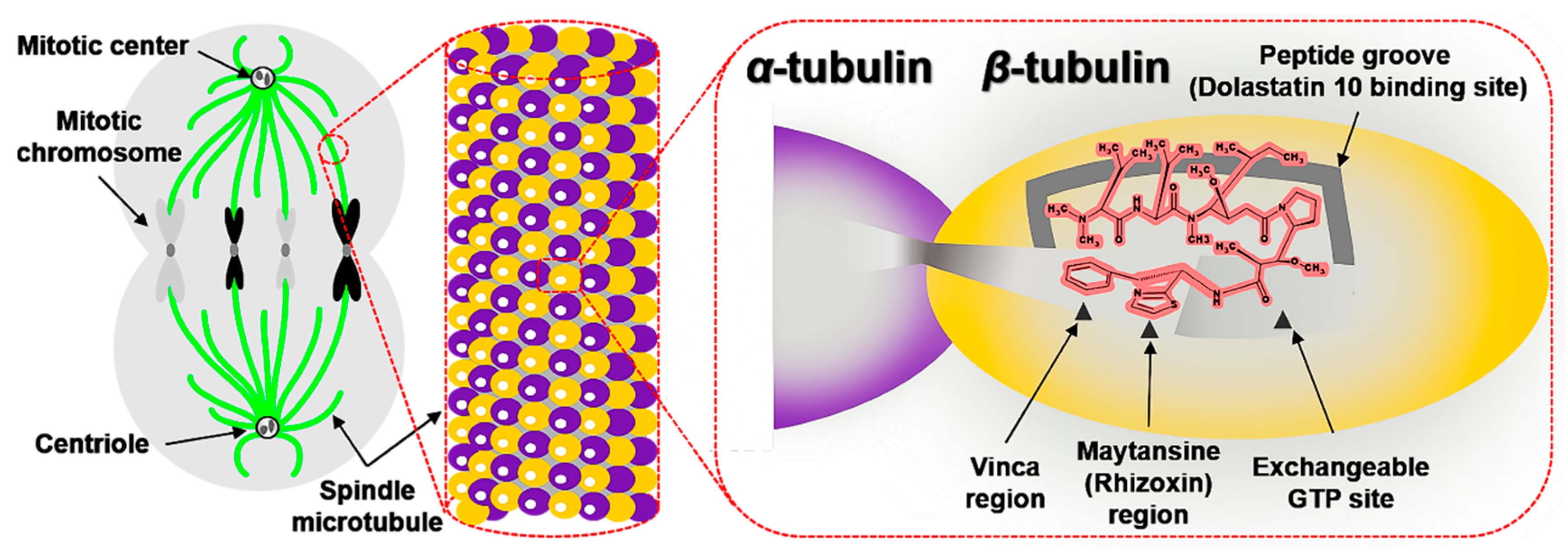
2.2.1. Interactions with β-tubulin
2.2.2. Regulating the Expression of Bcl-2, c-myc, and p53
2.2.3. The Loss of Telomeric Repeats and Induction of Chromosome Aberrations
2.3. Pharmacokinetics and Pharmacodynamics
2.4. Toxicity
2.5. Drug Resistance
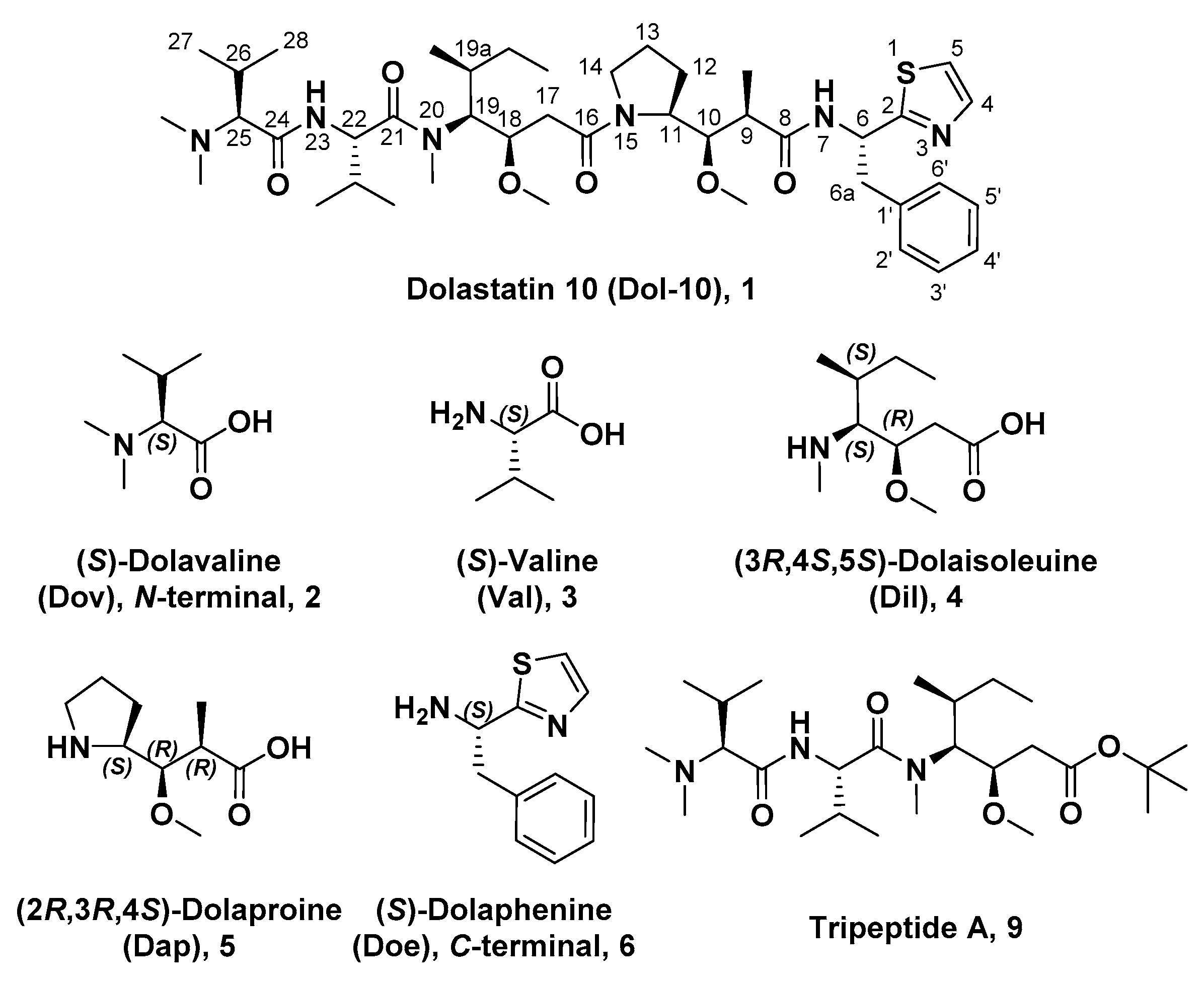
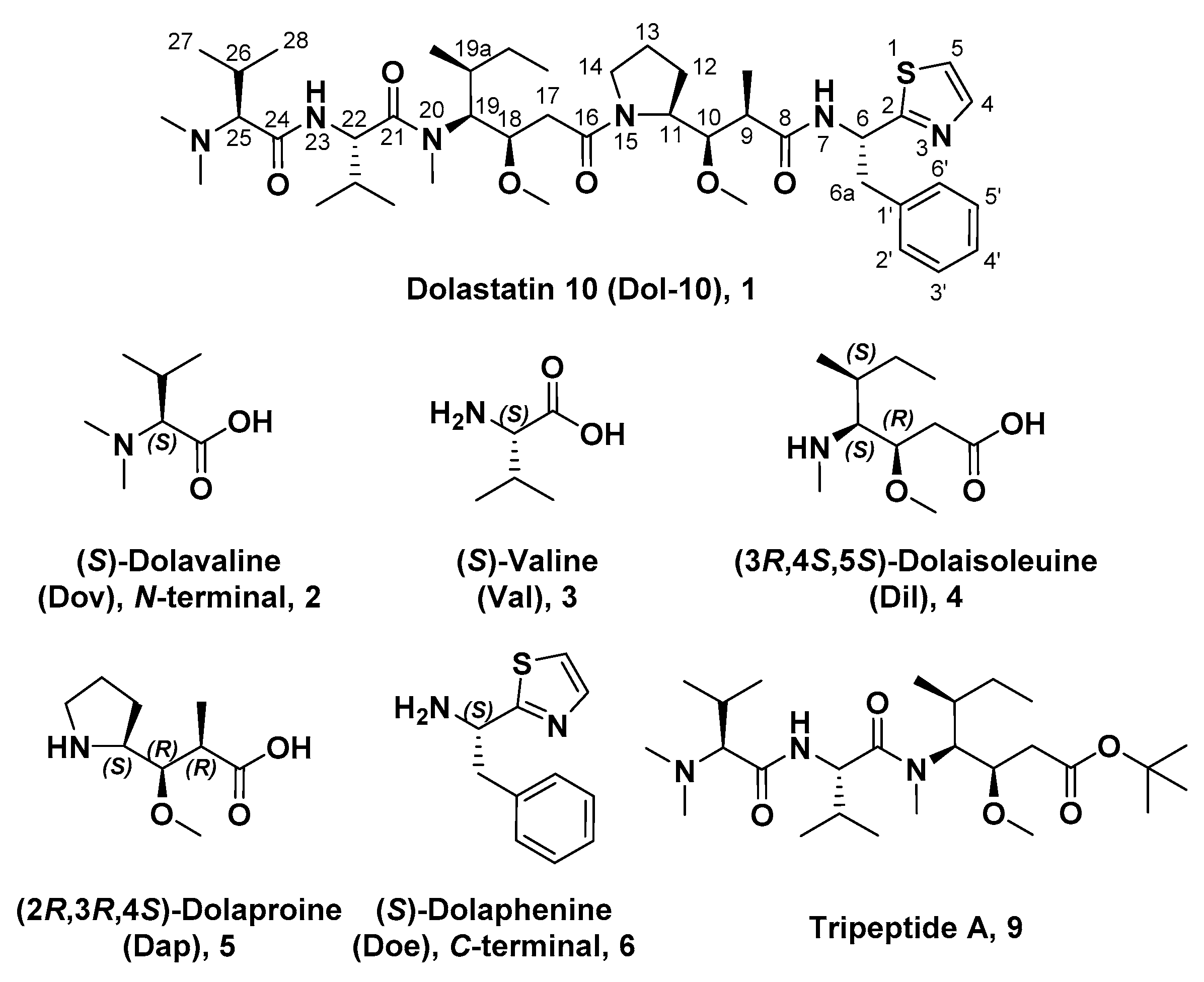
3. Medicinal Chemistry
3.1. Antitumor Derivatives
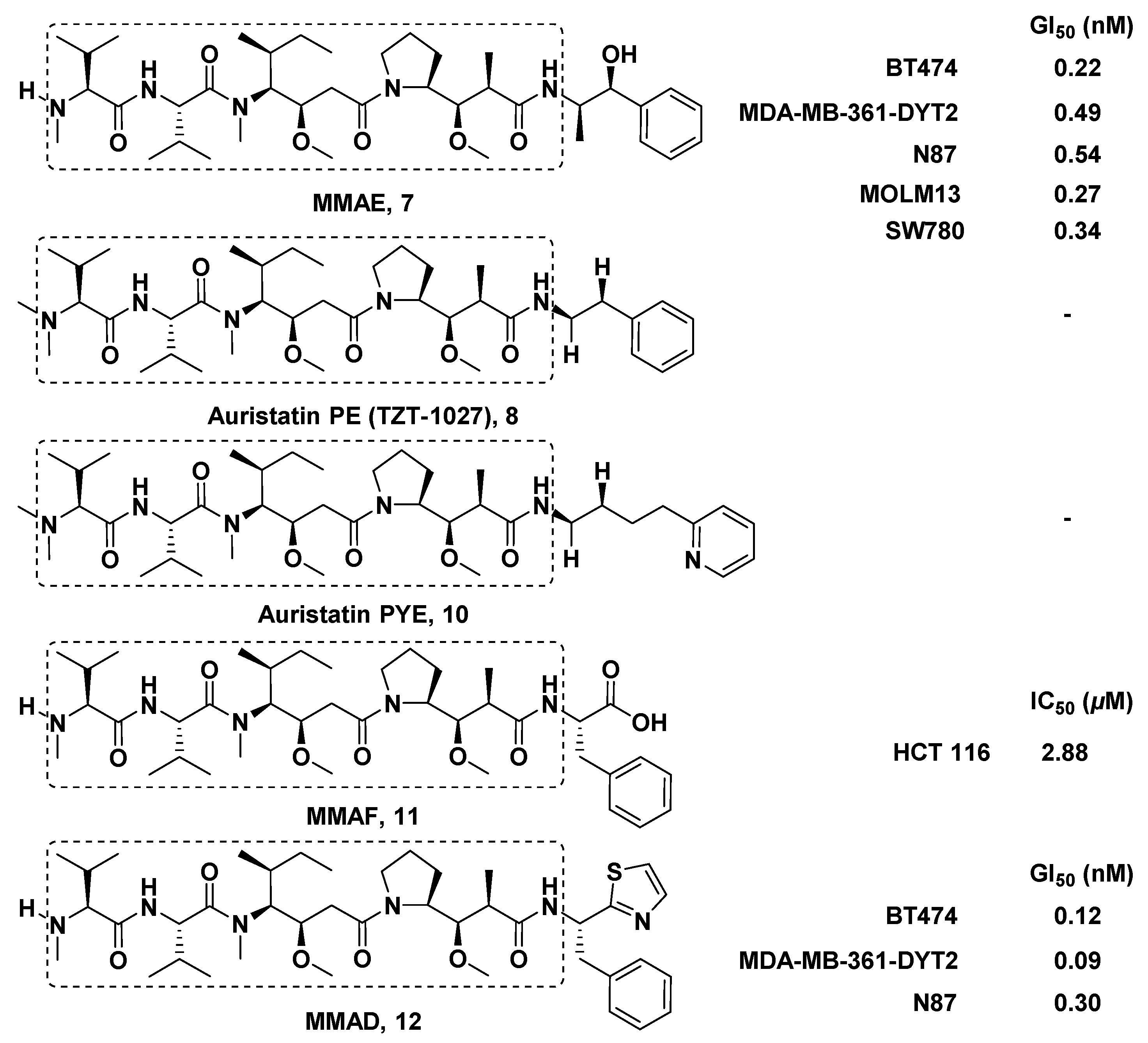
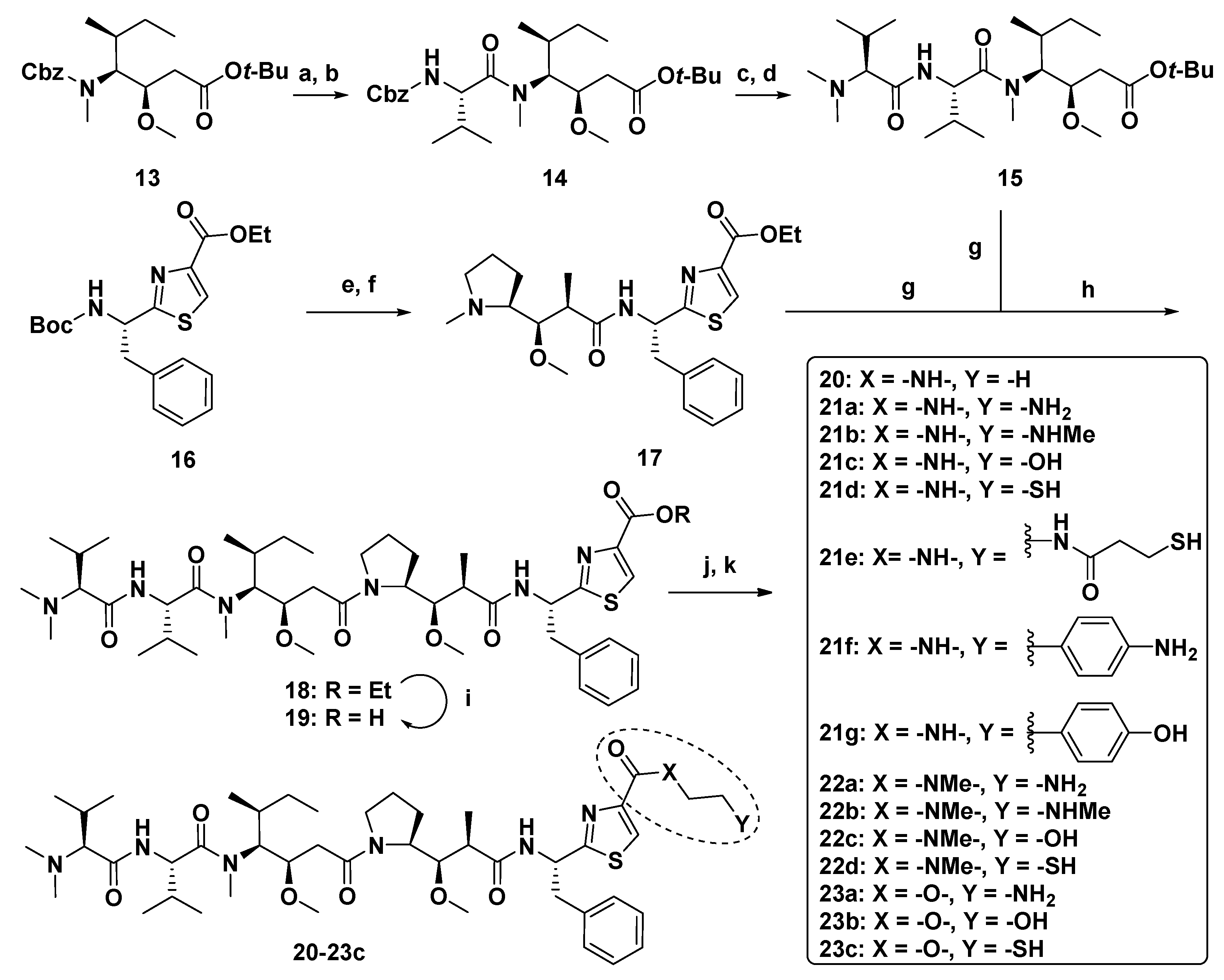
3.1.1. Doe Unit Modified Derivatives
3.1.2. Dap Unit Modified Derivatives
3.1.3. Dil Unit Modified Derivatives
3.1.4. Val Unit Modified Derivatives
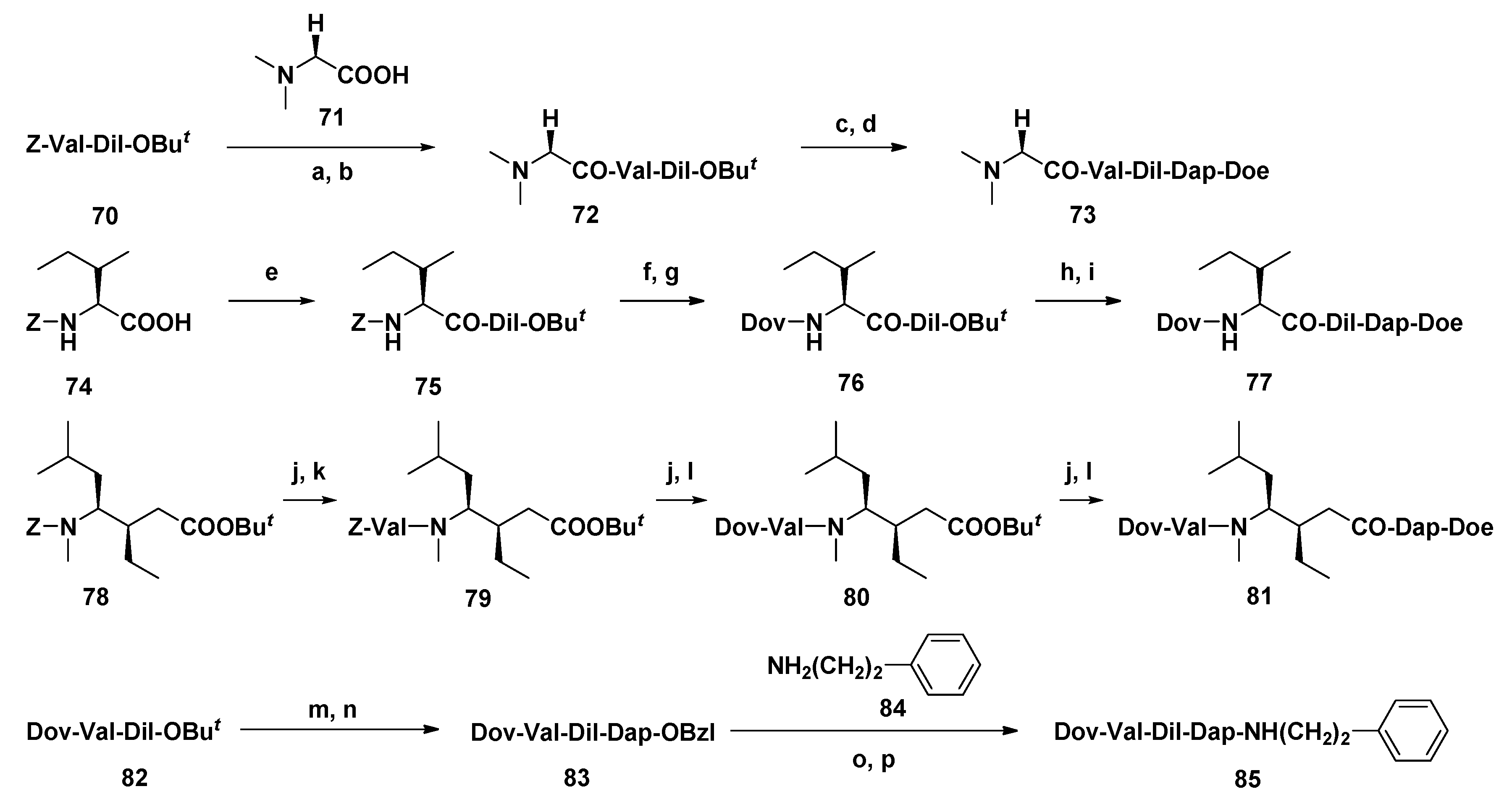
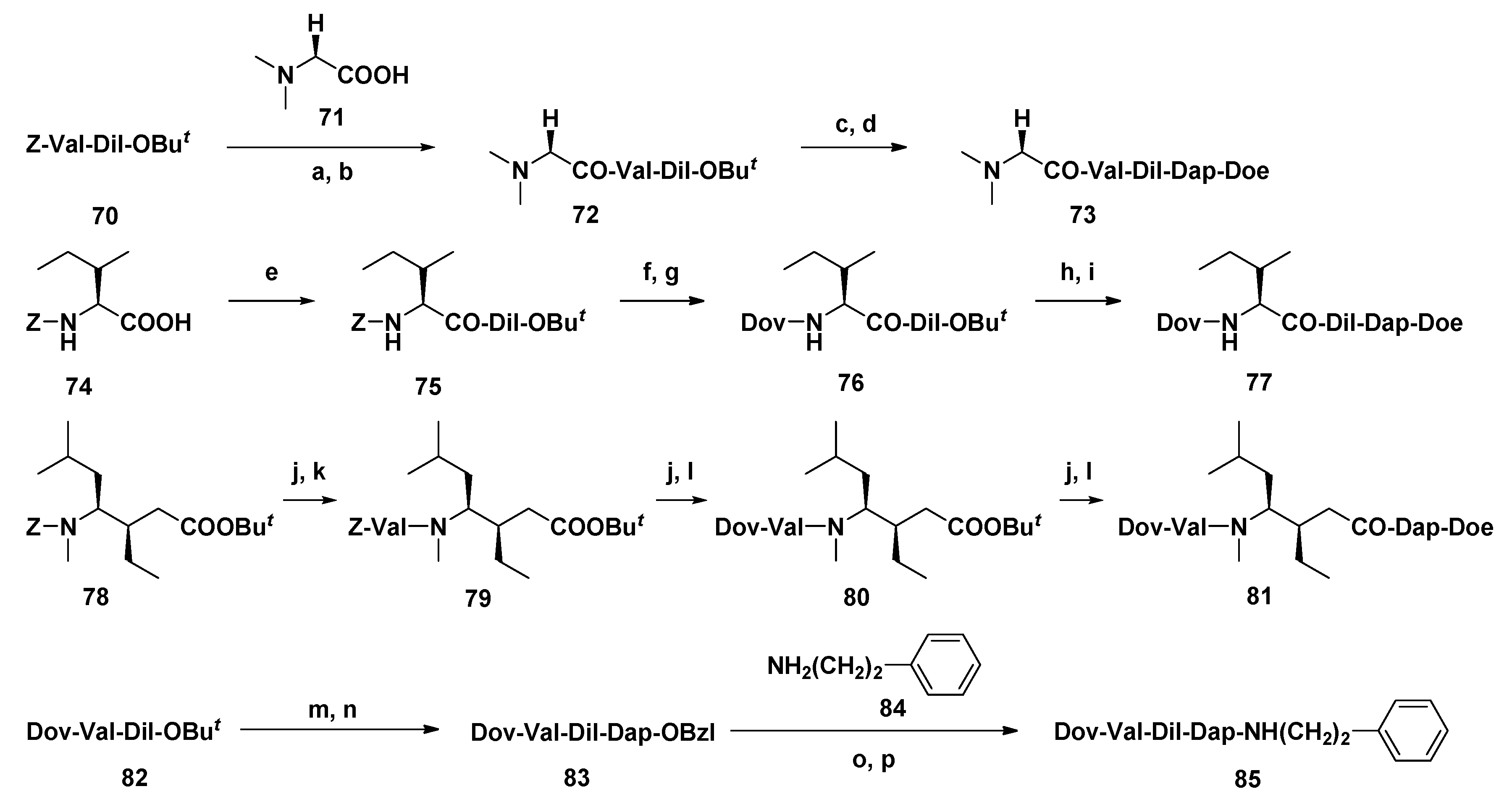
3.1.5. Dov Unit Modified Derivatives
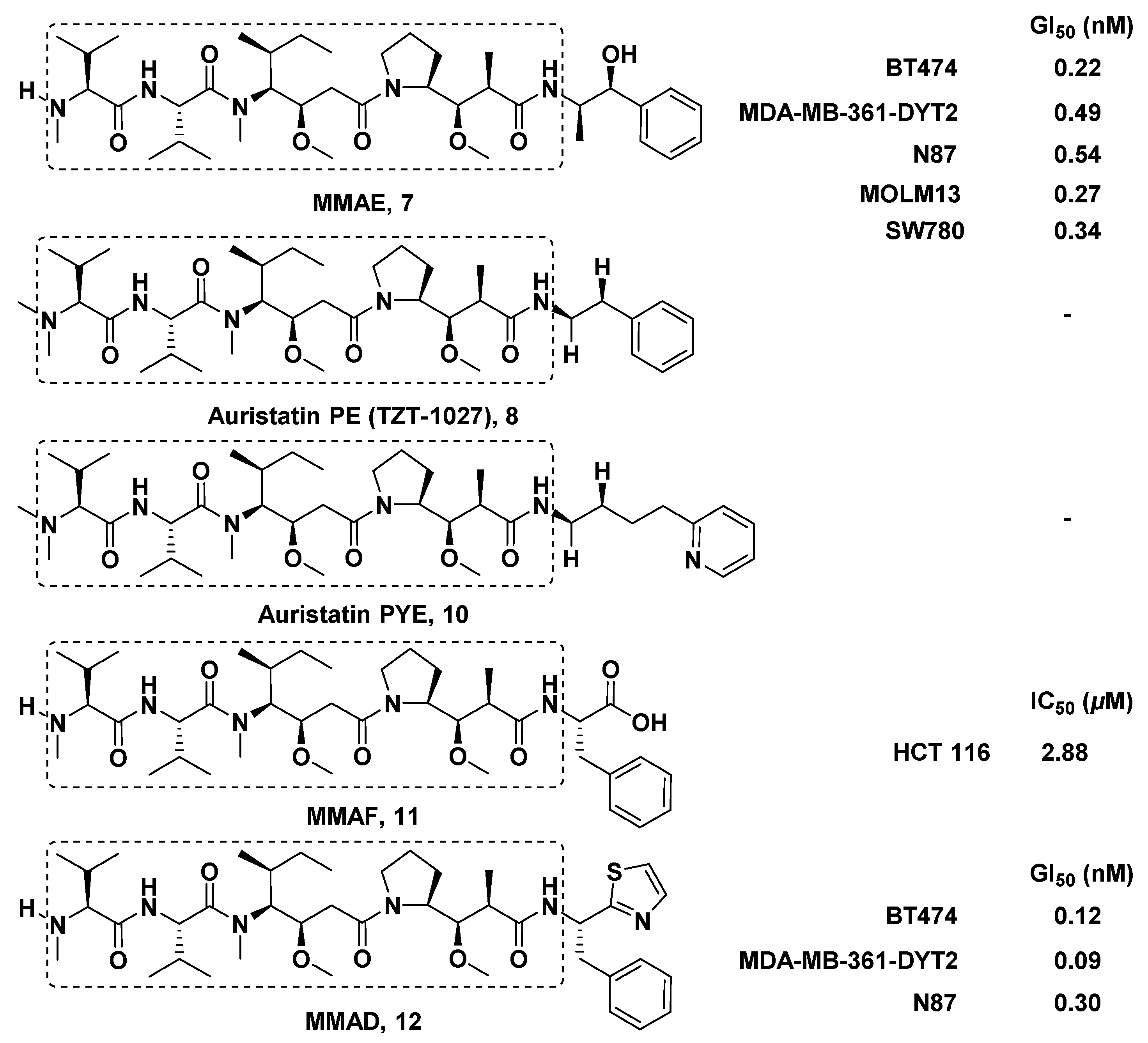
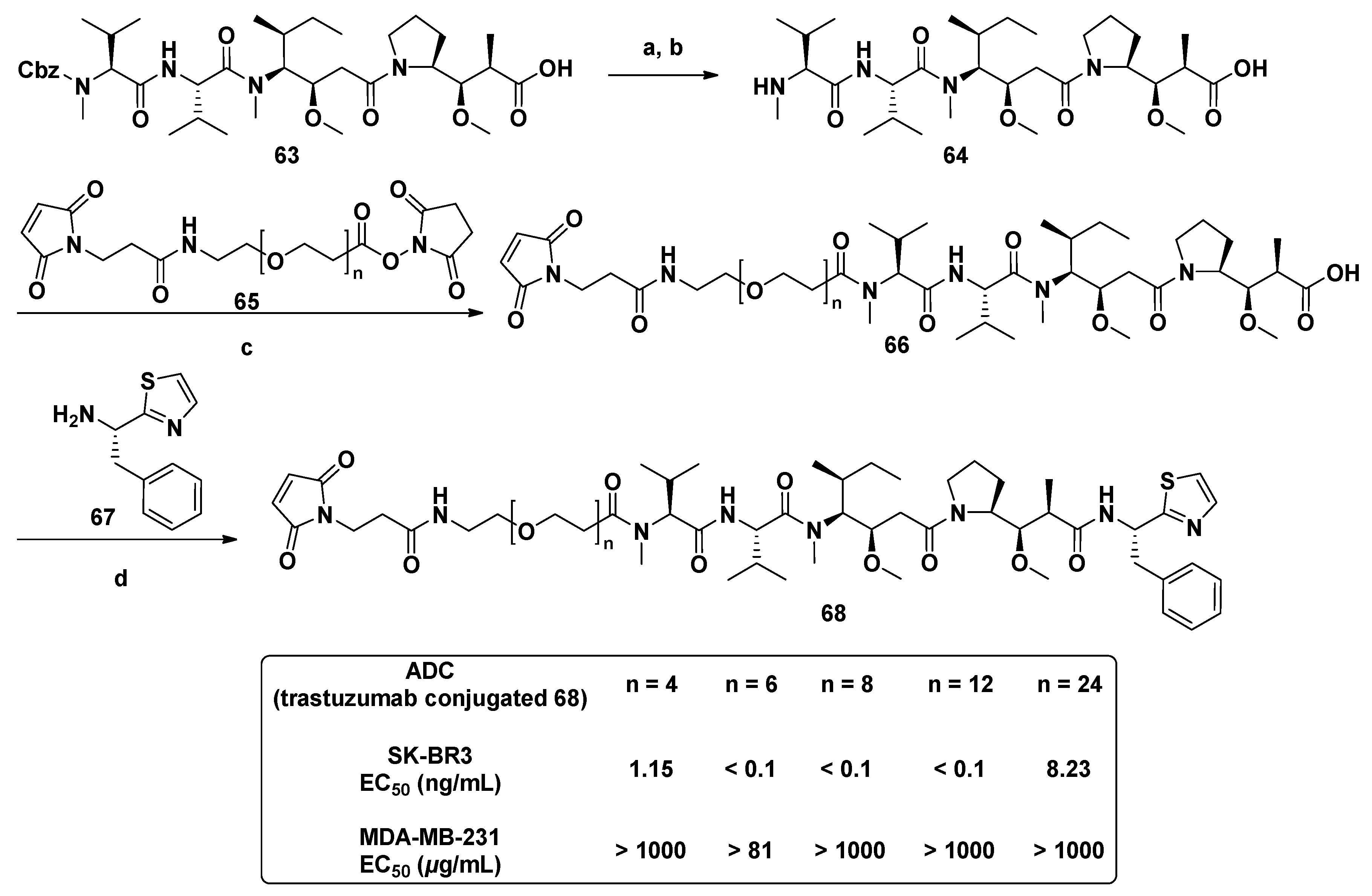
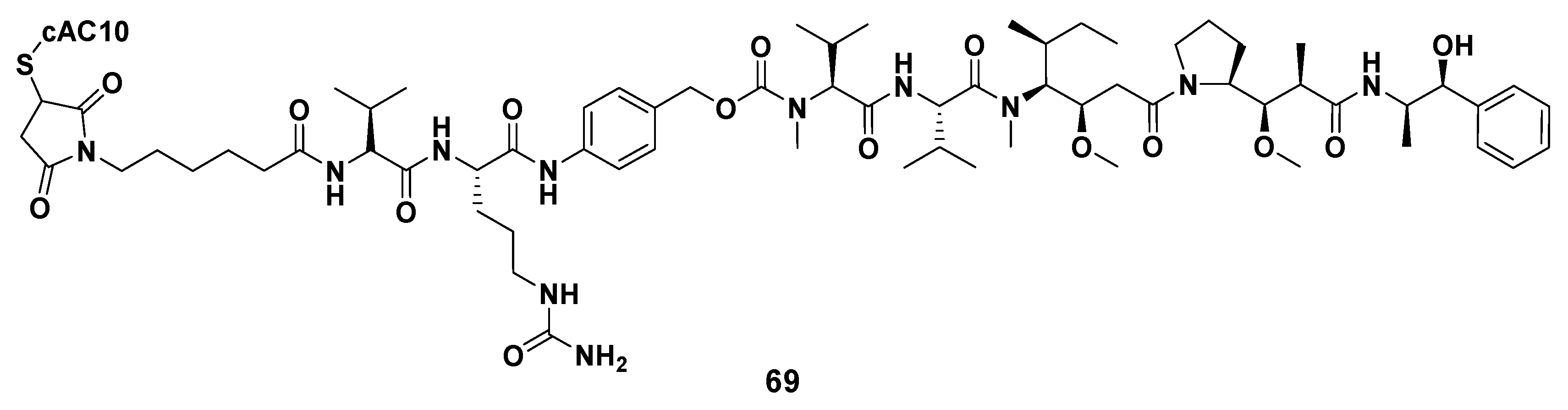
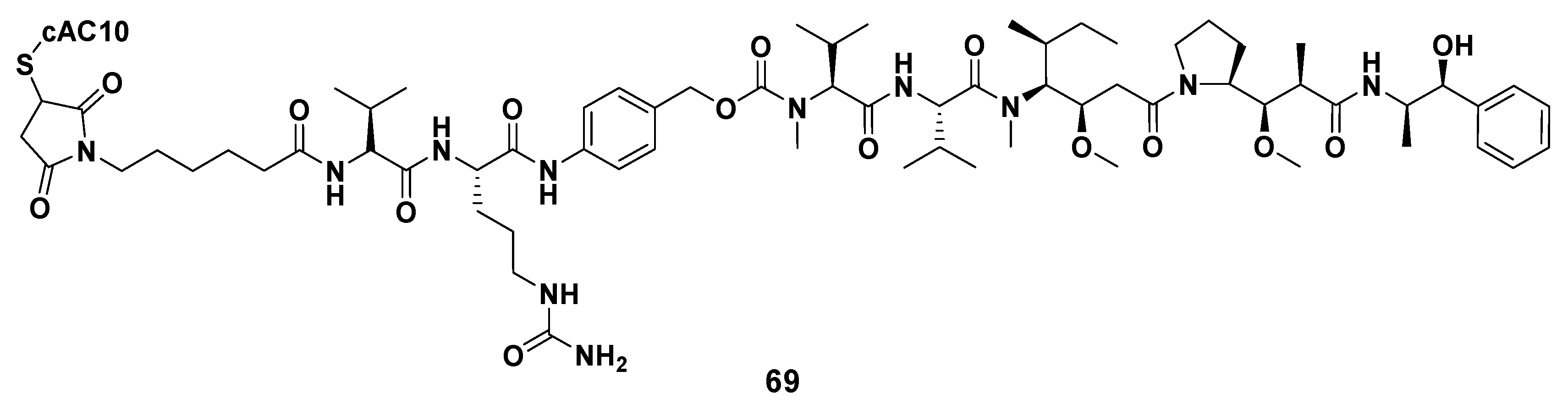
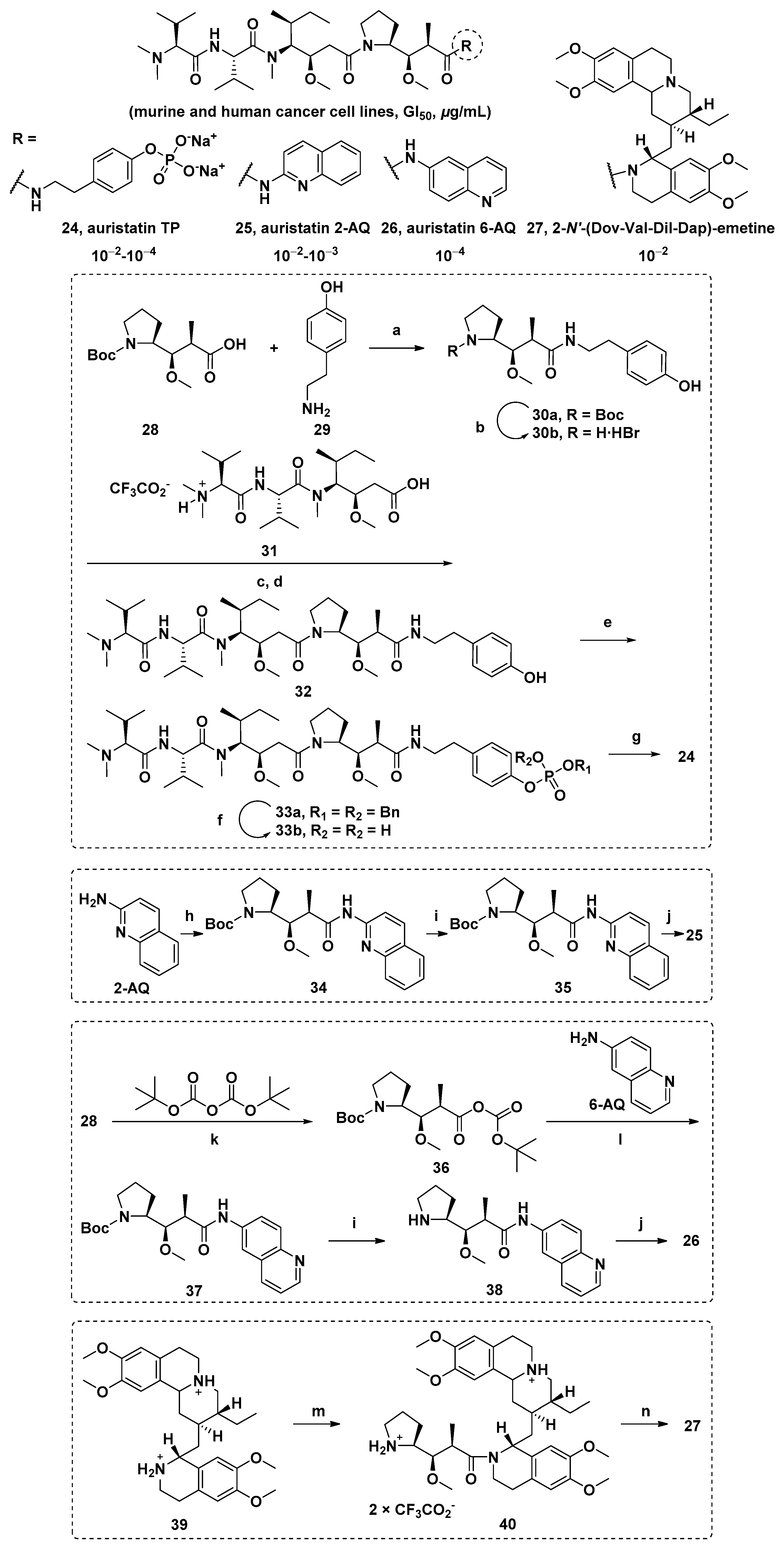
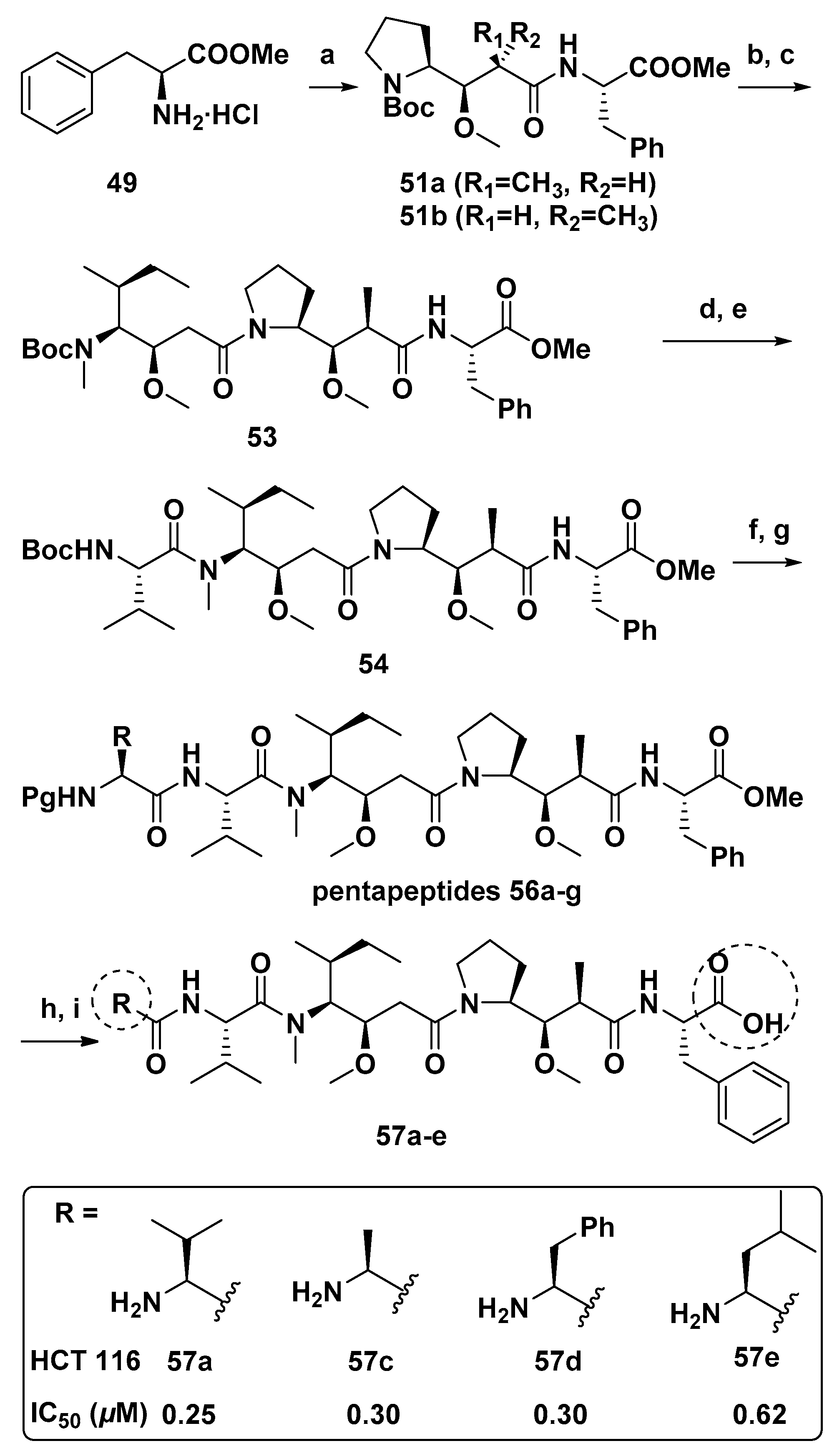
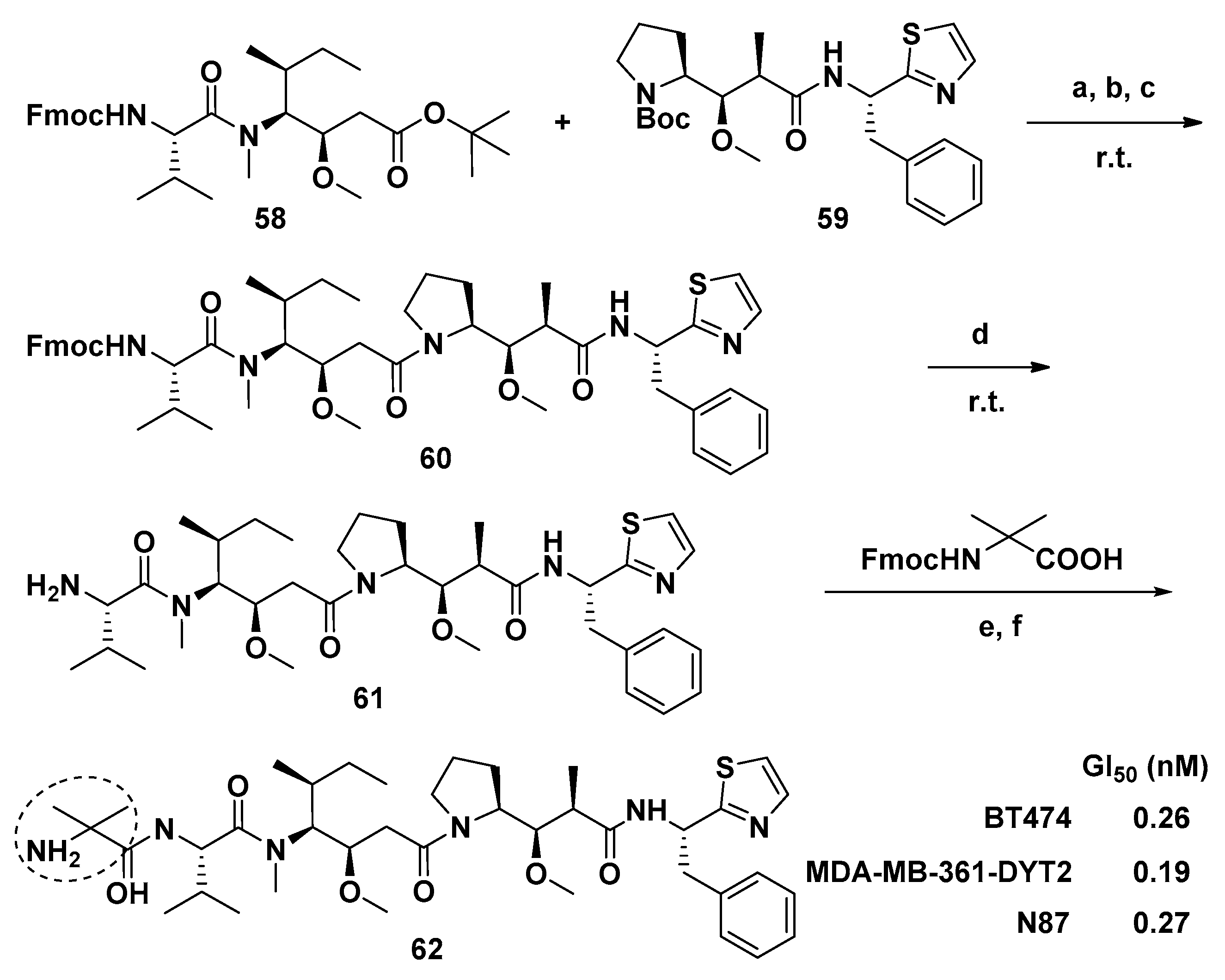
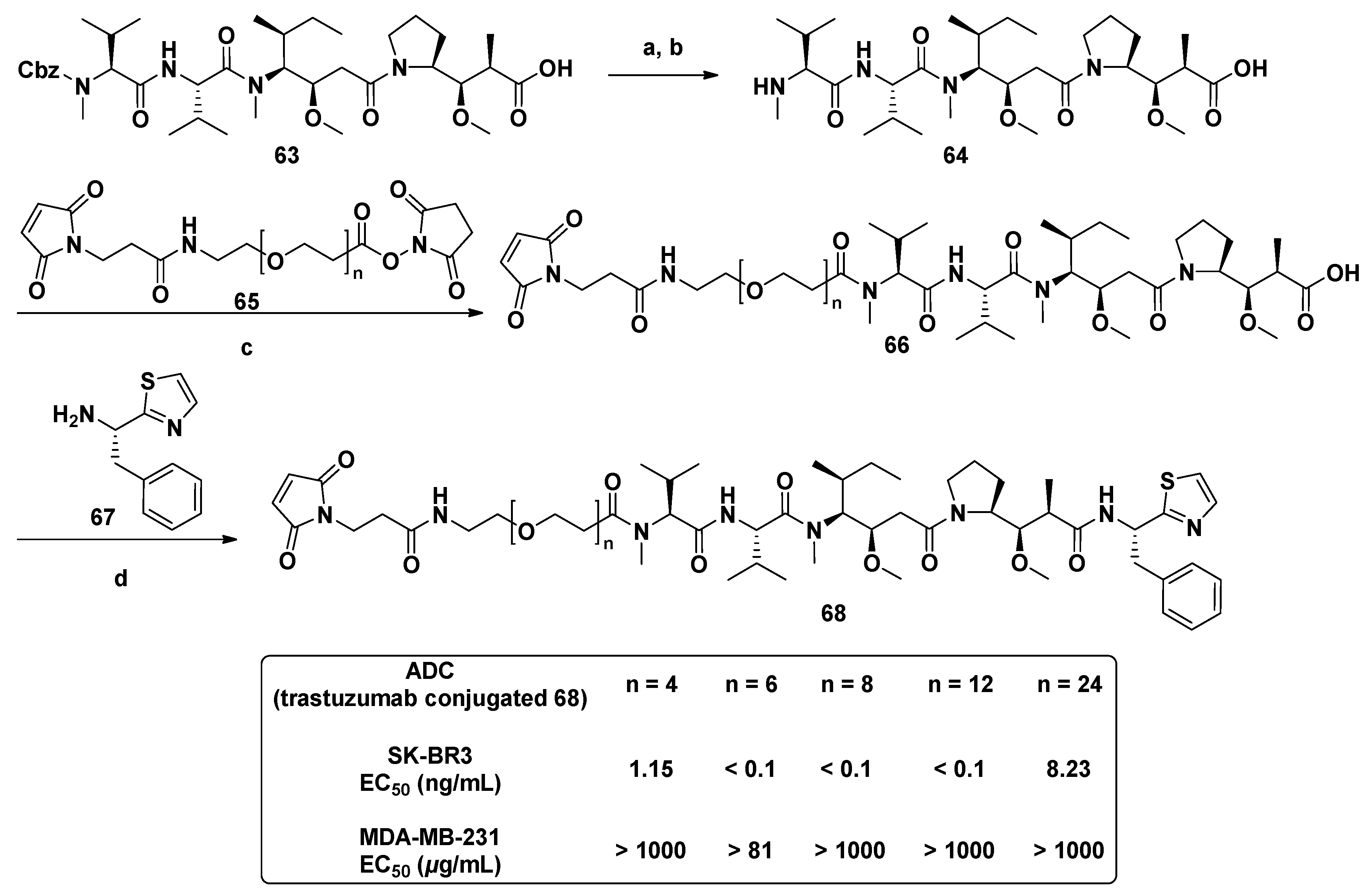
3.1.6. Multiunit Modified Derivatives
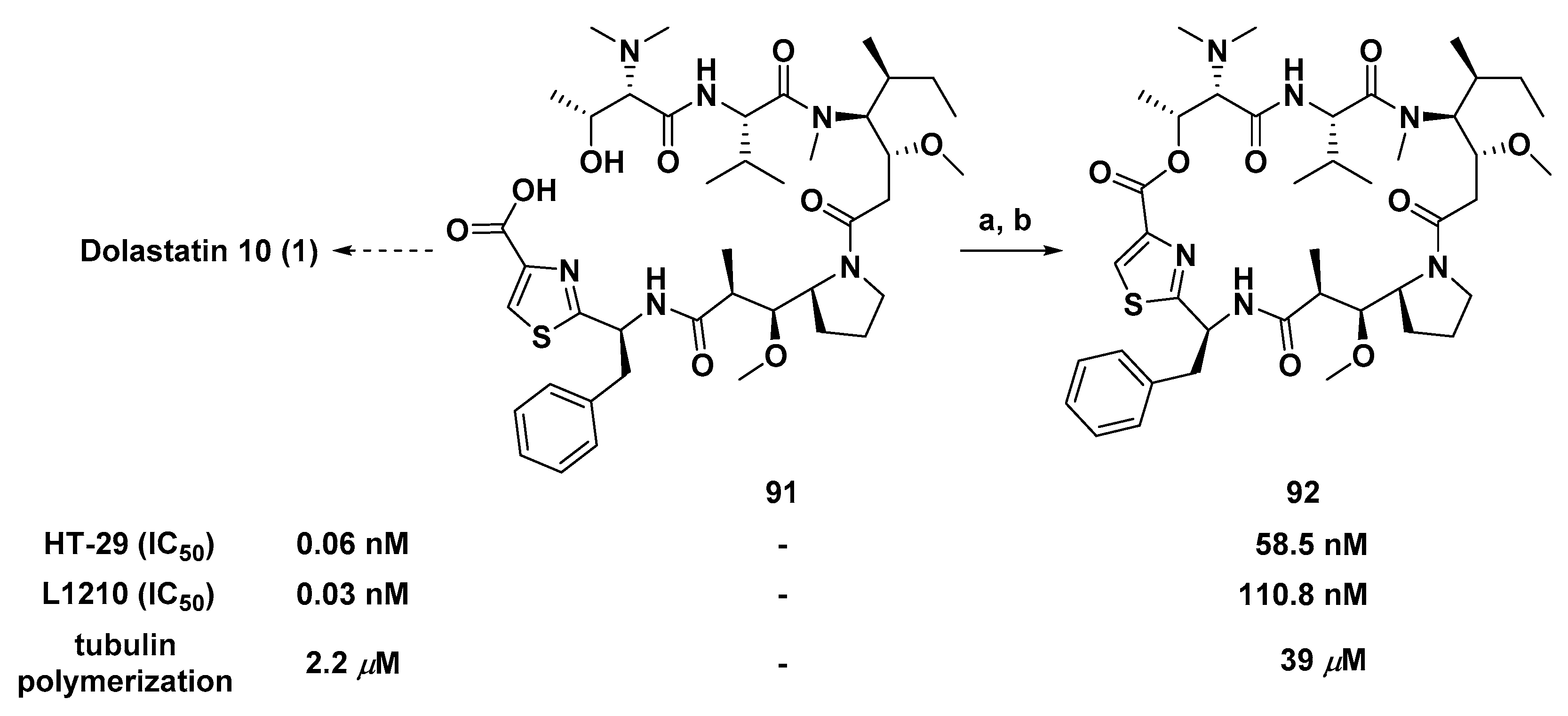
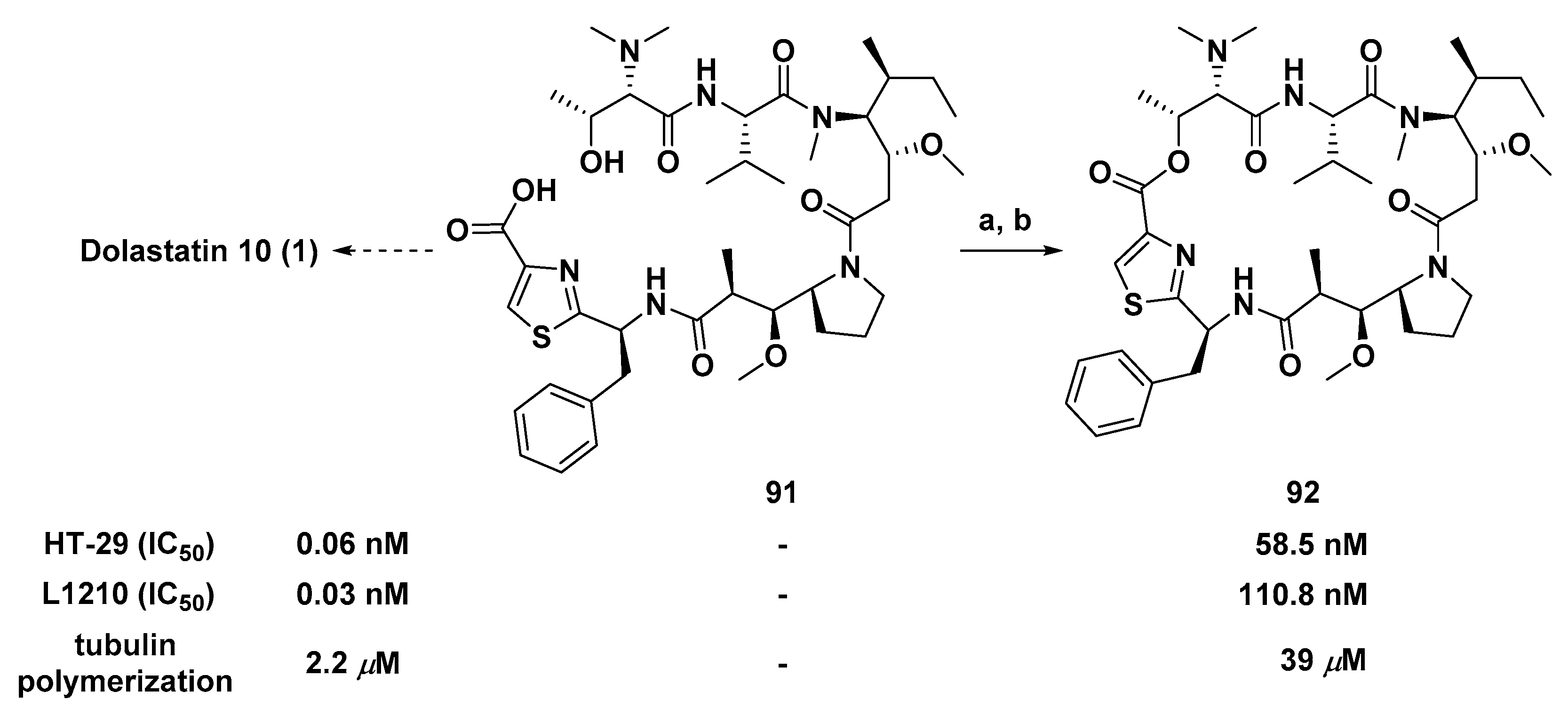
3.1.7. Cyclic Analogs
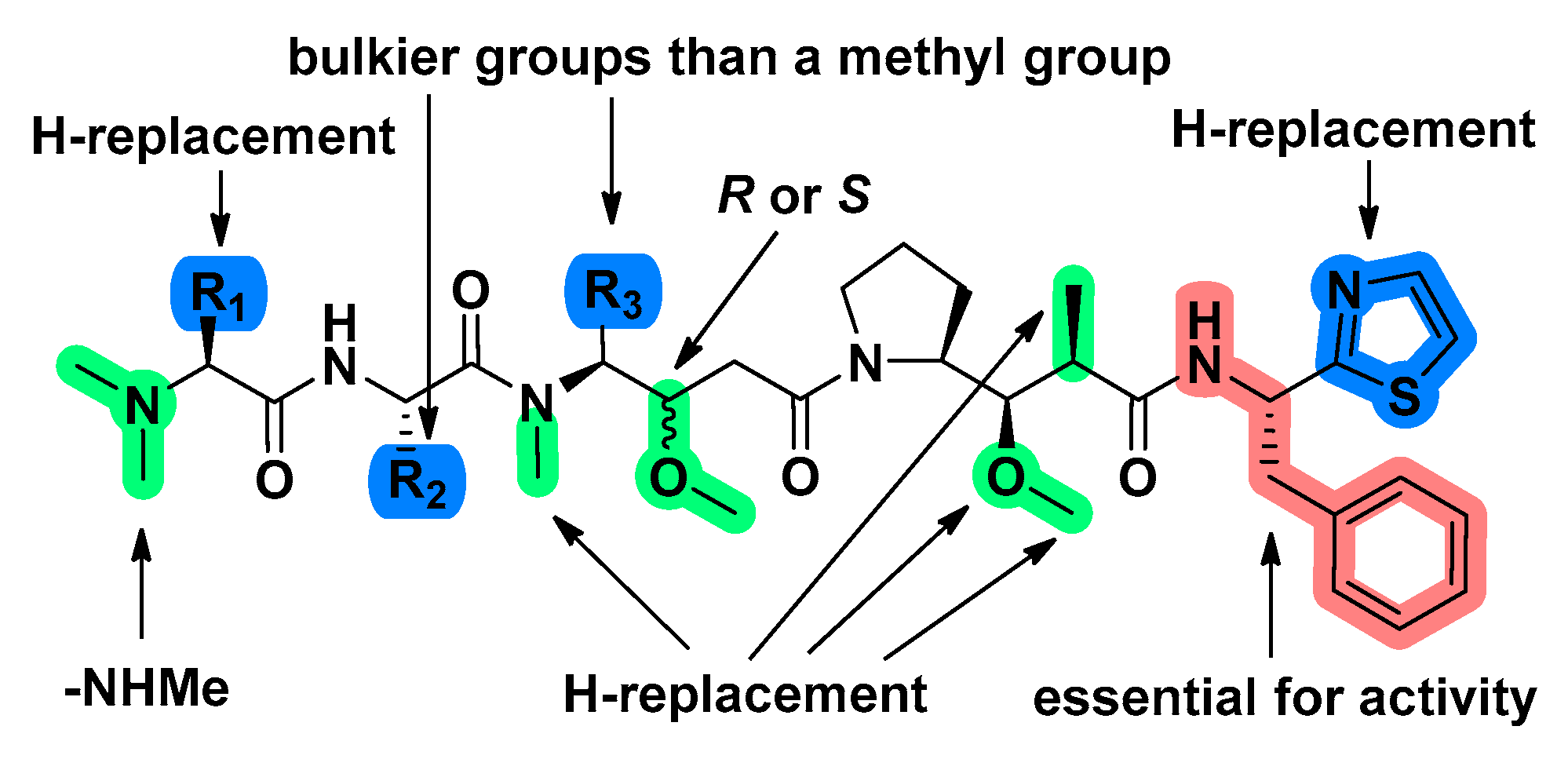
3.2. Structure-Activity Relationship
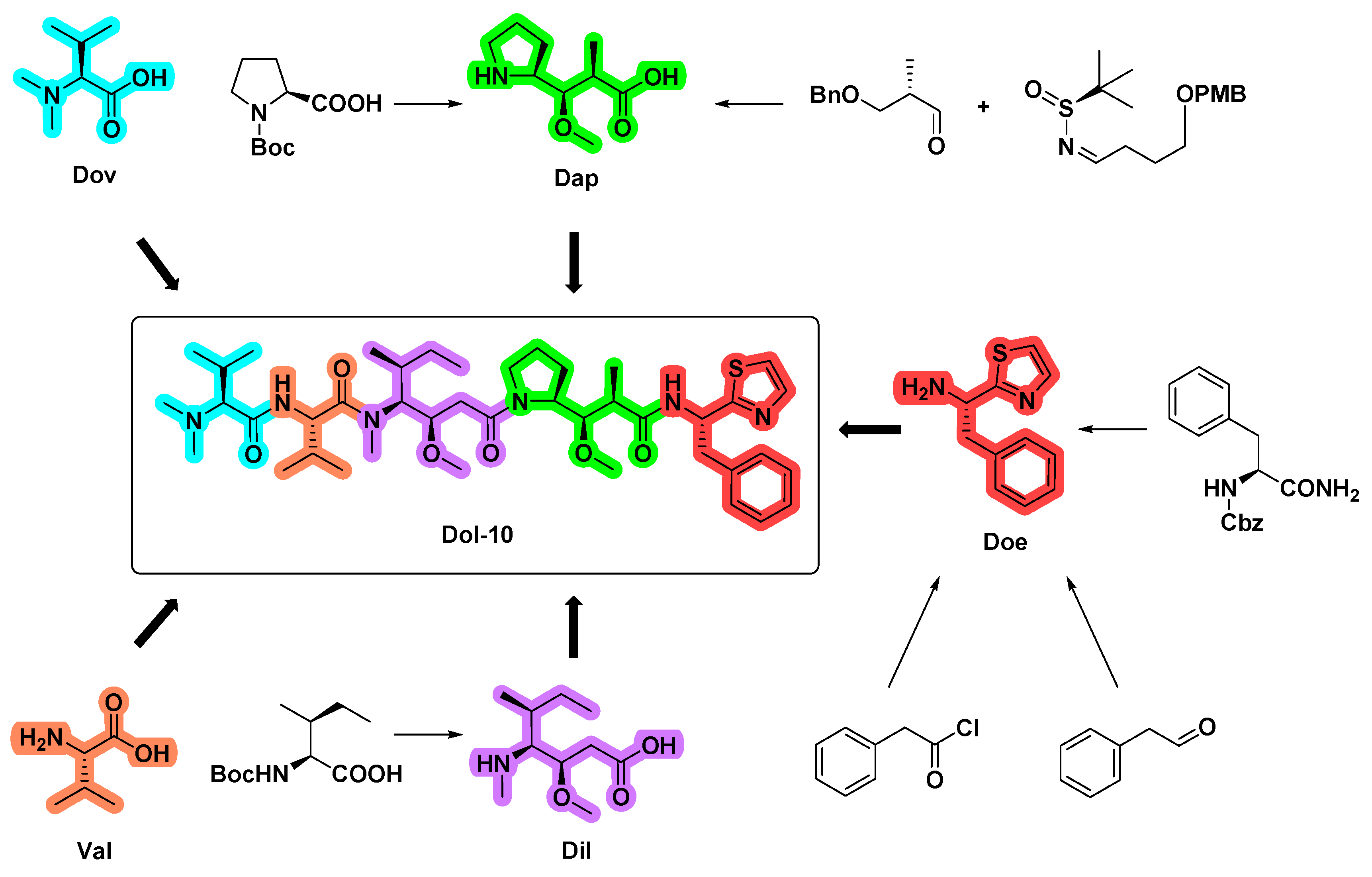
4. Synthetic Chemistry
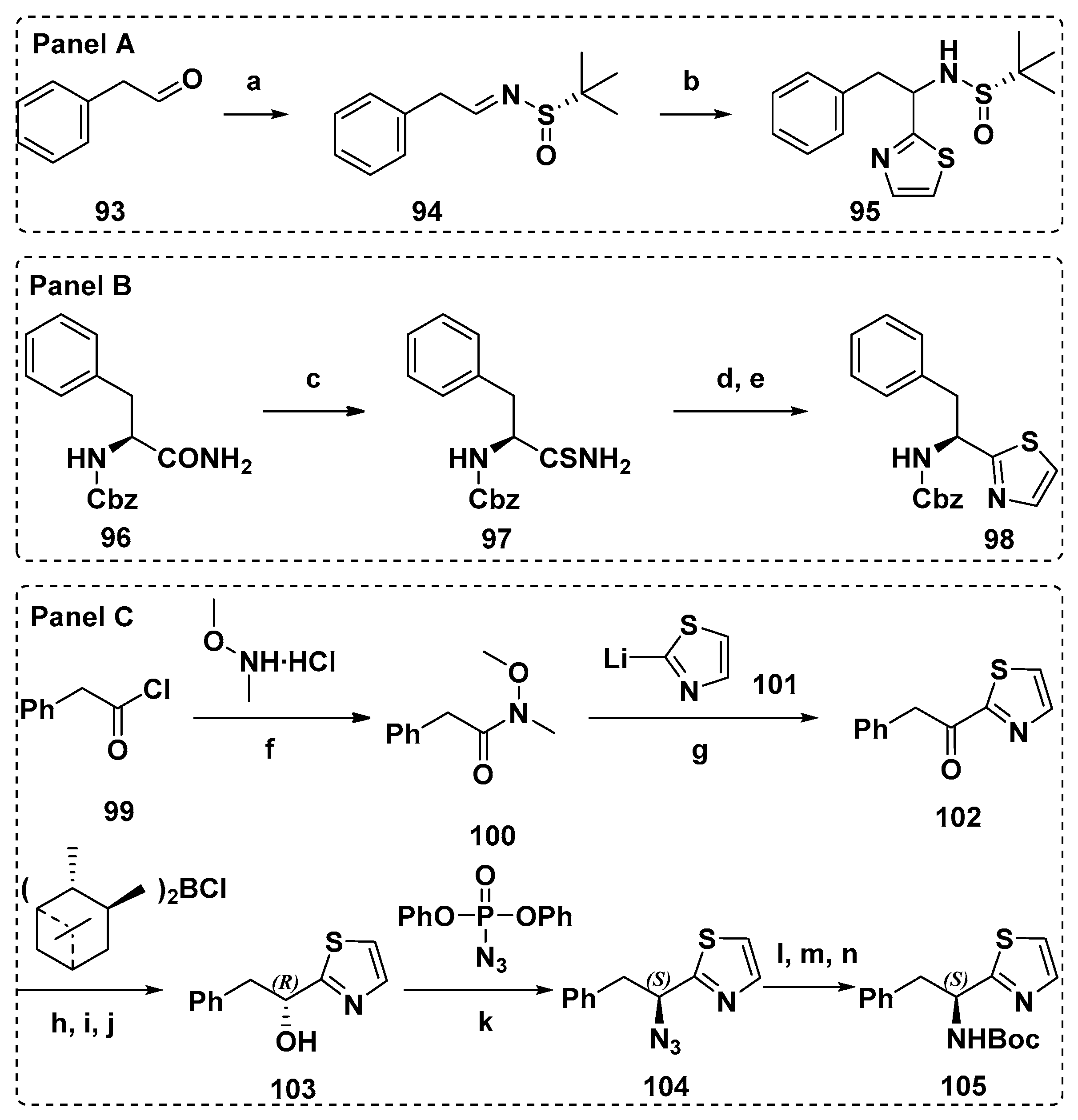
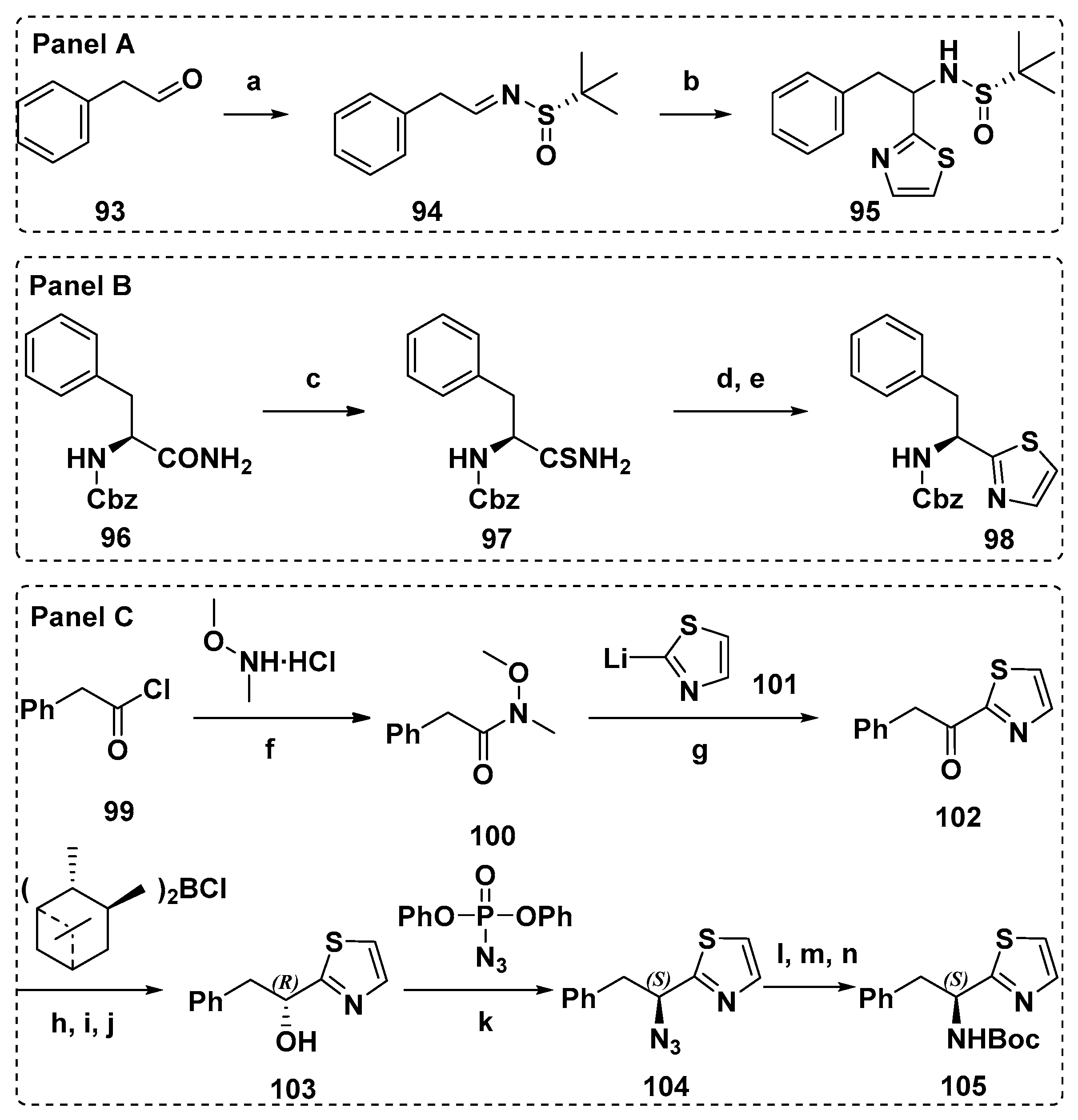
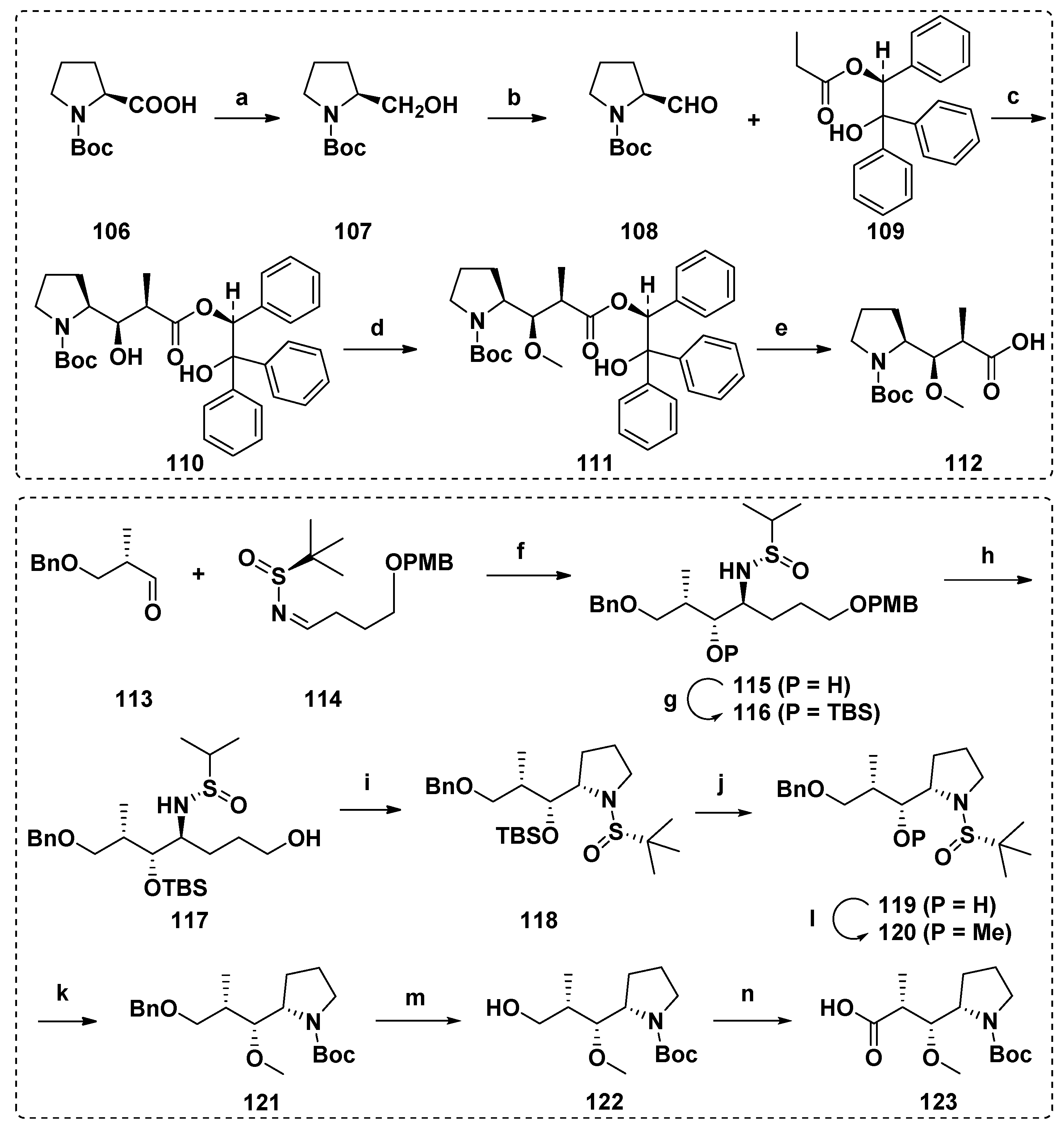
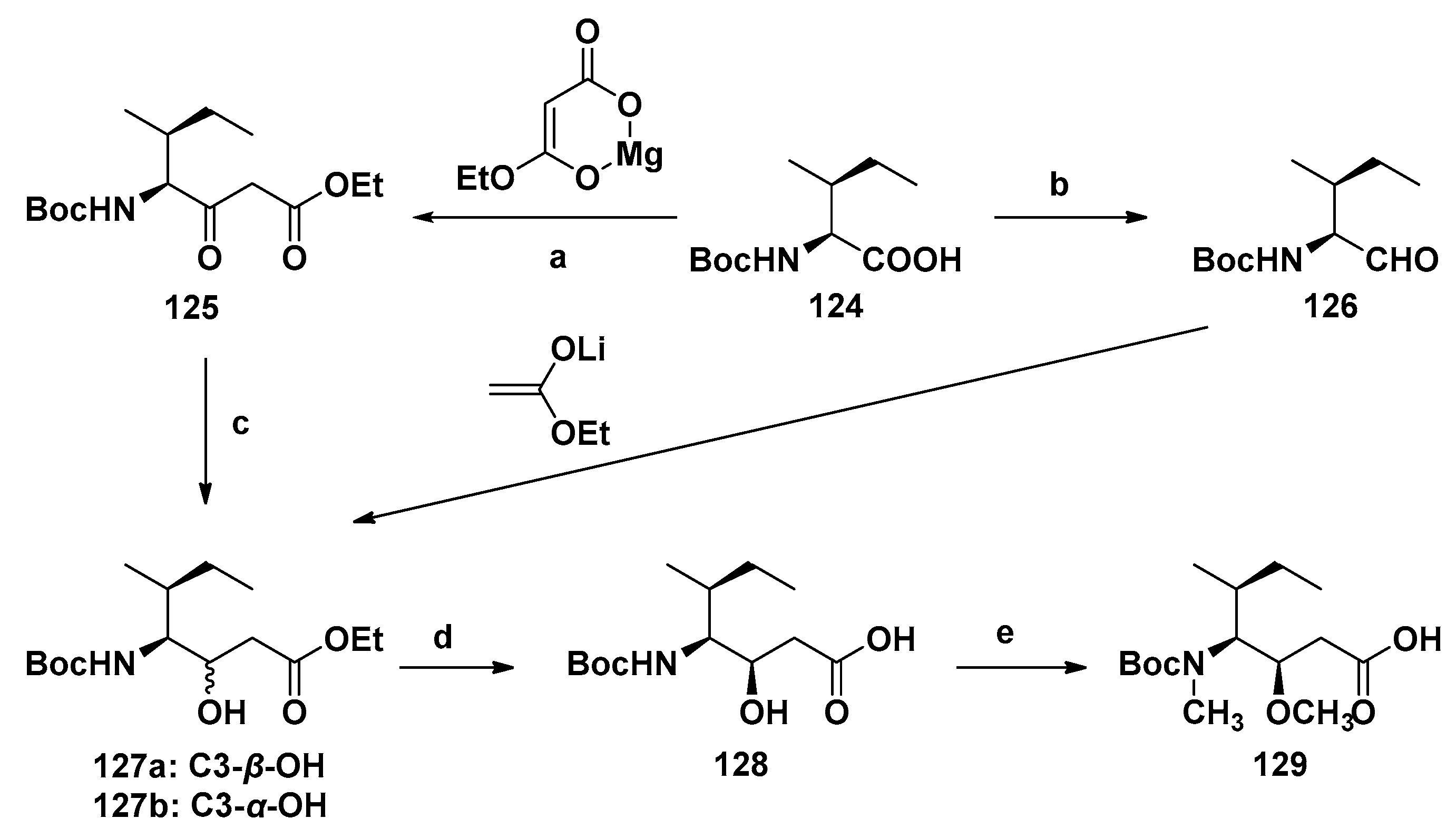
4.1. Synthesis of the Doe Unit
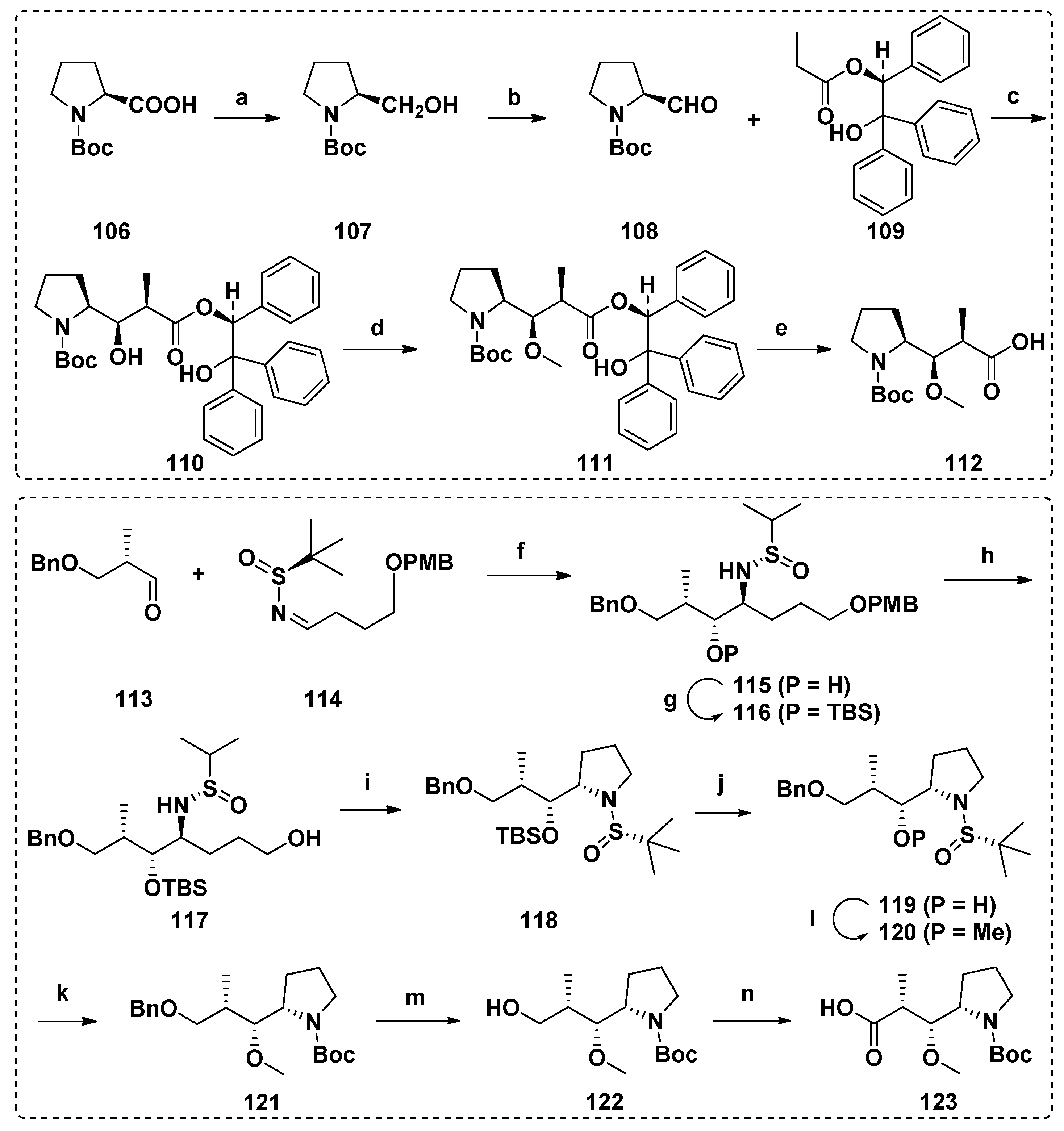
4.2. Synthesis of the Dap Unit
4.3. Synthesis of the Dil Unit
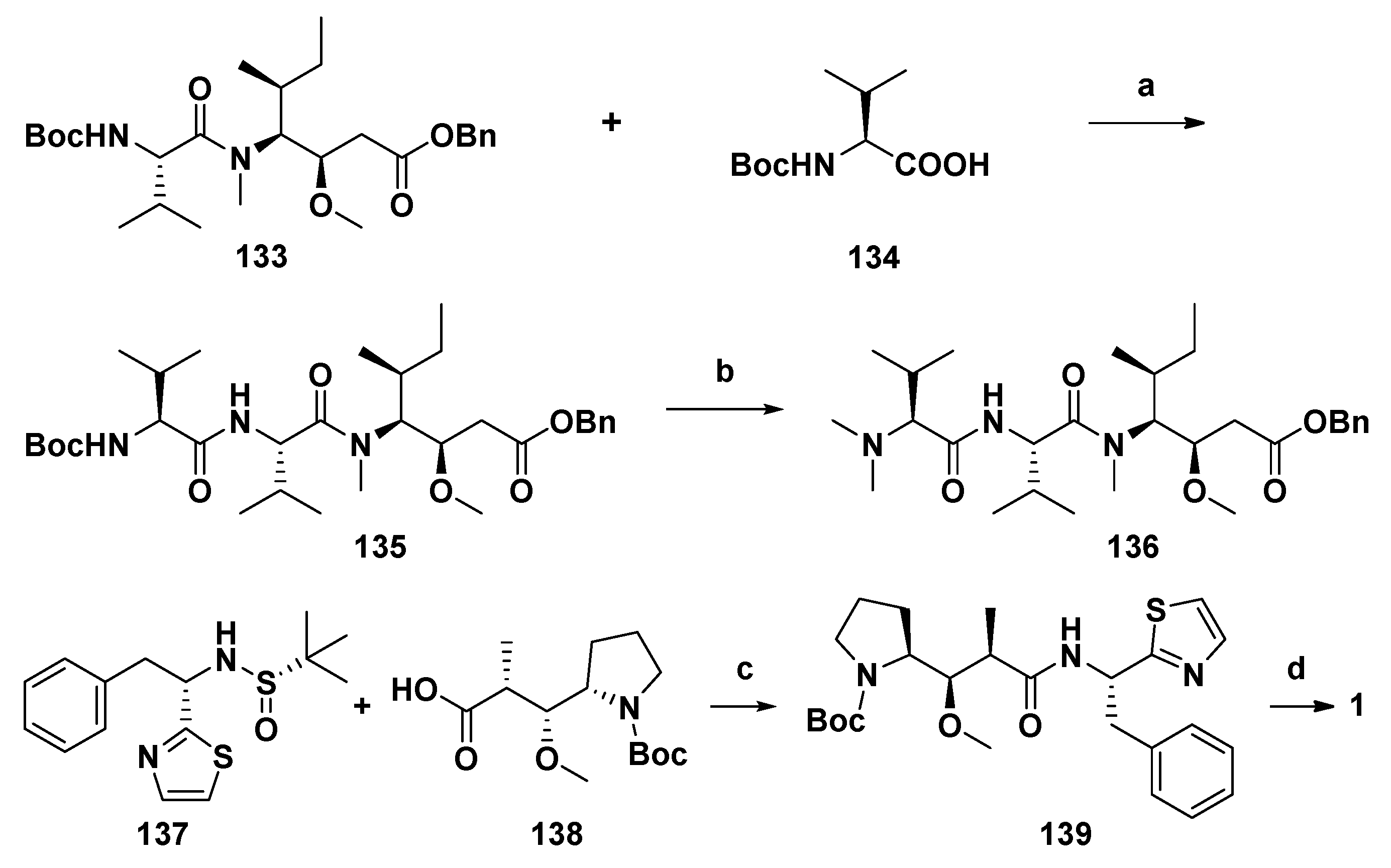
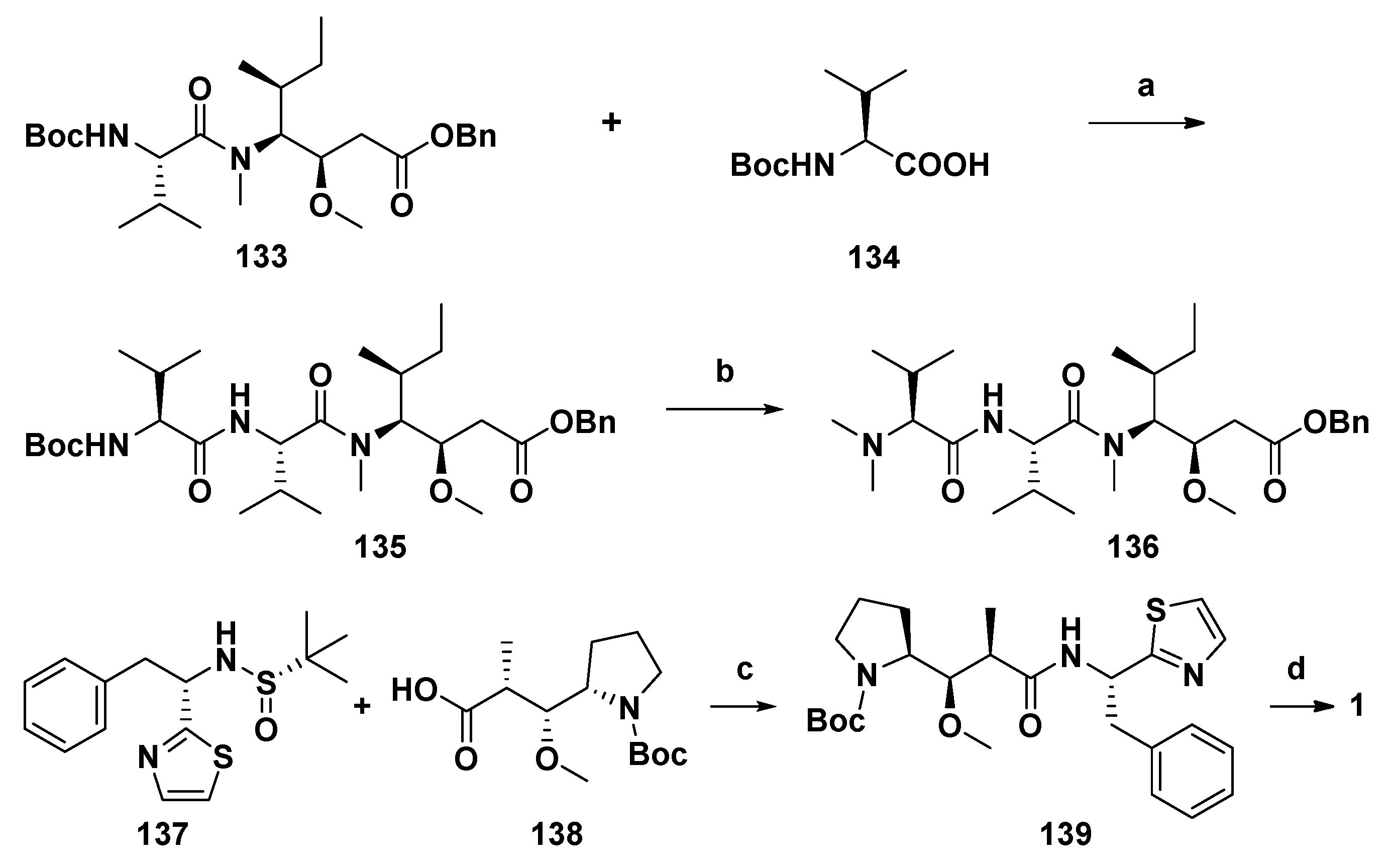
4.4. Synthesis of Dolastatin 10
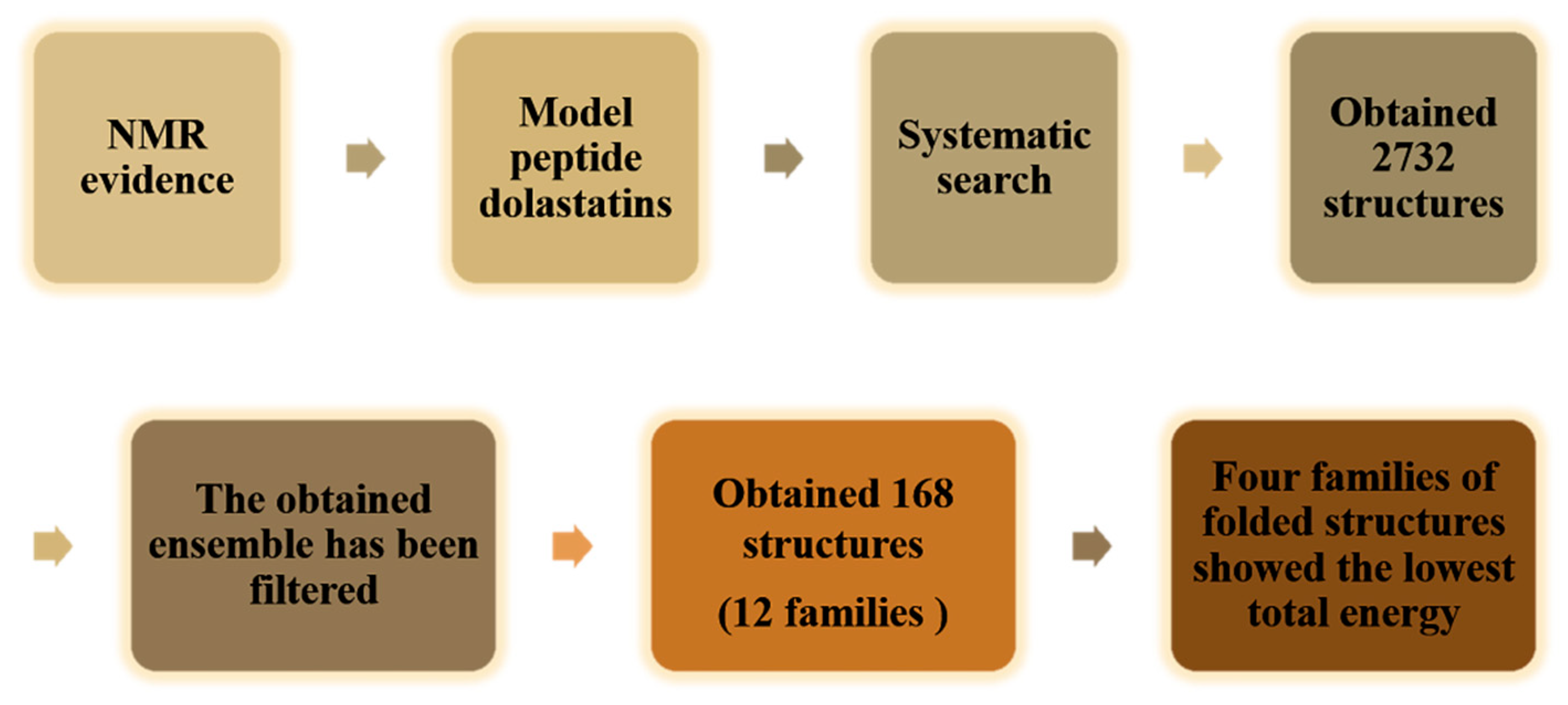
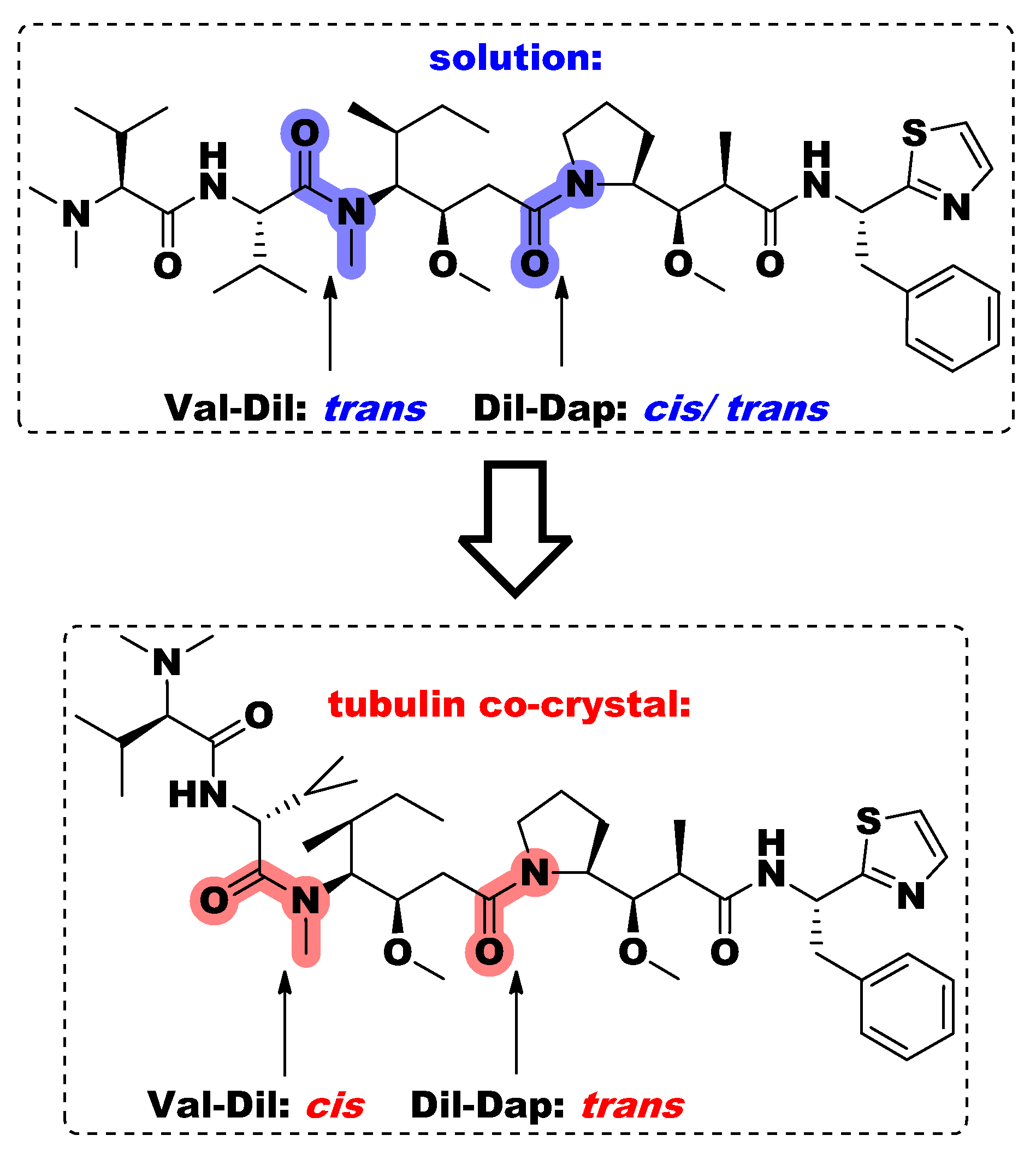
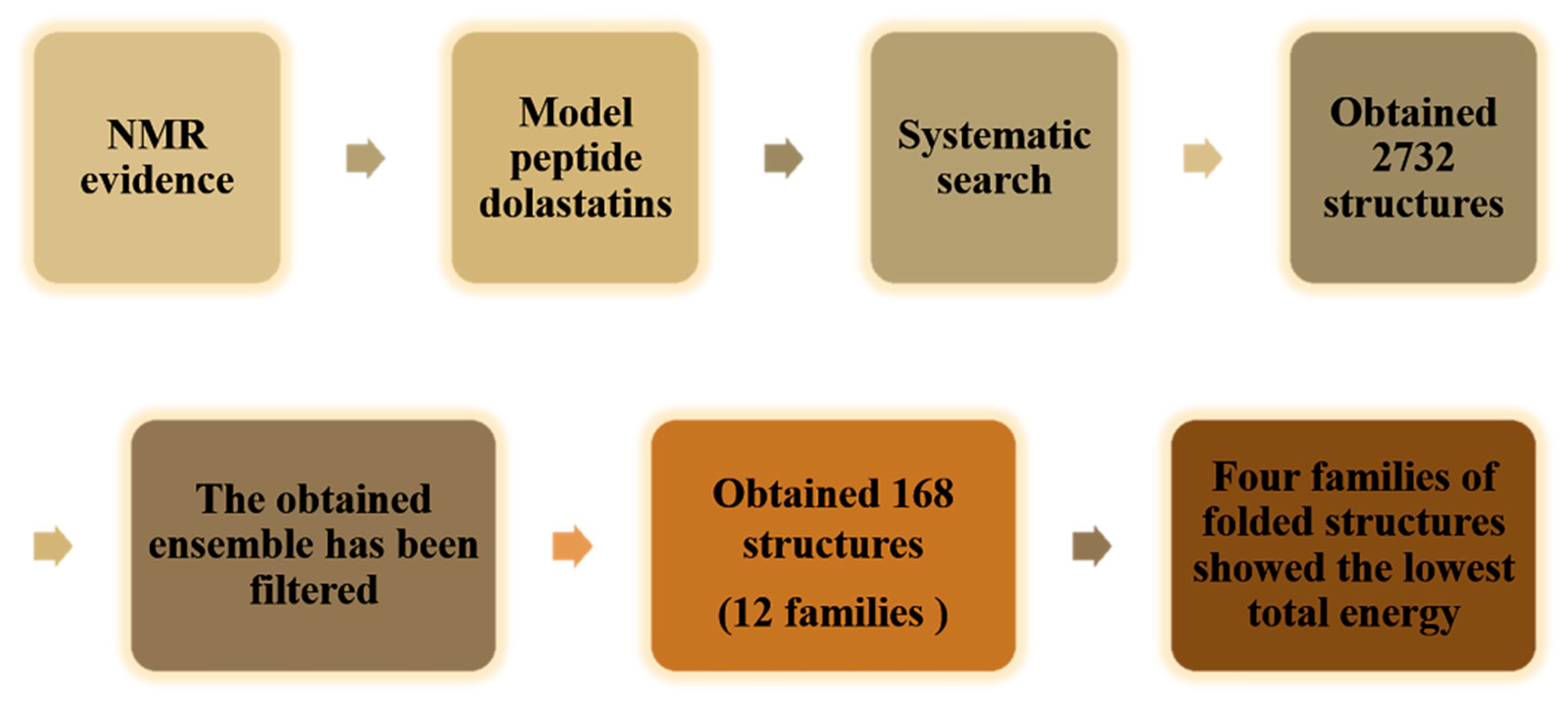
4.5. Conformational Study
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 19, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: The First Two Decades of XXI Century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Stonik, V.A. Marine Natural Products: A Way to New Drugs. Acta Nat. 2009, 1, 15–25. [Google Scholar] [CrossRef]
- Paterson, I.; Anderson, E.A. The Renaissance of Natural Products as Drug Candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2008, 8, 69–85. [Google Scholar] [CrossRef]
- Jing, Q.; Hu, X.; Ma, Y.; Mu, J.; Liu, W.; Xu, F.; Li, Z.; Bai, J.; Hua, H.; Li, D. Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification. Mar. Drugs 2019, 17, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, B.; Lu, Y.; Guo, Y.; Sun, J.; Wei, B.; Zhang, H.; Wang, H. Quorum Sensing Inhibitors from Marine Microorganisms and Their Synthetic Derivatives. Mar. Drugs 2019, 17, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, A.; Naughton, L.M.; Montánchez, I.; Dobson, A.D.W.; Rai, D.K. Current Status and Future Prospects of Marine Natural Products (MNPs) as Antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Zhang, Z.; Cui, W. Marine-Derived Natural Compounds for the Treatment of Parkinson’s Disease. Mar. Drugs 2019, 17, 221. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Nay, B.; Yang, M.; Ni, Y.; Wang, H.; Yao, L.; Li, X. Marine sponges of the genus Stelletta as promising drug sources: Chemical and biological aspects. Acta Pharm. Sin. B 2019, 9, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Livingston, D.M.; Kung, A.L. Composition and Method for Imaging Cells. US20090185977A1, 23 July 2009. [Google Scholar]
- Nalli, Y.; Gupta, S.; Khajuria, V.; Singh, V.P.; Sajgotra, M.; Ahmed, Z.; Thakur, N.L.; Ali, A. TNF-α and IL-6 inhibitory effects of cyclic dipeptides isolated from marine bacteria Streptomyces sp. Med. Chem. Res. 2017, 26, 93–100. [Google Scholar] [CrossRef]
- Zheng, L.; Xu, Y.; Lin, X.; Yuan, Z.; Liu, M.; Cao, S.; Zhang, F.; Linhardt, R.J. Recent Progress of Marine Polypeptides as Anticancer Agents. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Sable, R.; Parajuli, P.; Jois, S. Peptides, Peptidomimetics, and Polypeptides from Marine Sources: A Wealth of Natural Sources for Pharmaceutical Applications. Mar. Drugs 2017, 15, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, E.; Andersen, J.H. Screening for Marine Natural Products with Potential as Chemotherapeutics for Acute Myeloid Leukemia. Curr. Pharm. Biotechnol. 2015, 17, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.-H.; Wang, Y.-J.; Sheng, J.; Wang, F.; Zheng, Y.; Lin, X.-K.; Sun, M. Antitumor Peptides from Marine Organisms. Mar. Drugs 2011, 9, 1840–1859. [Google Scholar] [CrossRef] [Green Version]
- Daqiao, Y.; Jinxu, W.; Laihao, L.; Xianqing, Y.; Haixia, M. Development of Co-production of Polysaccharides and Polypeptides from Marine Organisms—A Review. Chin. Fish. Qual. Stand. 2019, 9, 01–08. [Google Scholar]
- Cao, W.L.; Song, J.X.R.; Li, F.Q.C. Research advances on marine antitumor peptides dolastatin 10. J. Med. Postgrad. 2011, 24, 1208–1211. [Google Scholar]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef]
- Cui, Q.; Chen, J.R.; Jiang, X.Y.; Guan, L.L.; Liu, S.M.; Kong, L.C.; Hong-Xia, M.A. Advances in the application of marine bioactive peptide drugs. Chin. J. Mar. Drugs 2019, 38, 54–60. [Google Scholar]
- Ma, W.; Qin, T.; Sun, Y. The classification and advances of bioactive peptides. Chin. J. Inj. Rep. Wound Heal. 2019, 14, 149–152. [Google Scholar]
- Festa, C.; Marino, S.D.; D’Auria, M.; Monti, M.C.; Bucci, M.; Vellecco, V.; Debitus, C.; Zampella, A. Anti-inflammatory cyclopeptides from the marine sponge Theonella swinhoei. Tetrahedron 2012, 68, 2851–2857. [Google Scholar] [CrossRef]
- Donia, M.; Hamann, M.T. Marine natural products and their potential applications as anti-infective agents. Lancet Infect. Dis. 2003, 3, 338–348. [Google Scholar] [CrossRef]
- Ram Singh, M.S.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med. Chem. 2008, 8, 603–617. [Google Scholar] [CrossRef]
- de Castro, R.J.S.; Sato, H.H. Biologically active peptides: Processes for their generation, purification and identification and applications as natural additives in the food and pharmaceutical industries. Food Res. Int. 2015, 74, 185–198. [Google Scholar] [CrossRef]
- Cavé, A.; Cortes, D.; Figadère, B.; Laurens, A.; Pettit, G.R.; Herz, W.; Kirby, G.W.; Moore, R.E.; Steglich, W.; Tamm, C. [Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products] Fortschritte der Chemie organischer Naturstoffe Progress in the Chemistry of Organic Natural Products. Dolastatins 1997, 70, 1–79. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Fujii, Y.; Kizu, H.; Boyd, M.R.; Boettner, F.E.; Doubek, D.L.; Schmidt, J.M.; Chapuis, J.-C.; et al. Antineoplastic Agents. Part 247. The Dolastatins. Part 18. Isolation of Dolastatins 10–15 from the Marine Mollusc Dolabella auricularia. Tetrahedron 1993, 49, 9151–9170. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Tubulin-Interactive Natural Products as Anticancer Agents. J. Nat. Prod. 2009, 72, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, R.; Roach, M.C.; Jayaram, S.K.; Barkoczy, J.; Pettit, G.R.; Luduena, R.F.; Hamel, E. Differential effects of active isomers, segments, and analogs of dolastatin 10 on ligand interactions with tubulin. Biochem. Pharmacol. 1993, 45, 1503–1515. [Google Scholar] [CrossRef]
- Bai, R.; Pettit, G.R.; Hamel, E. Structure-activity studies with chiral isomers and with segments of the antimitotic marine peptide dolastatin 10. Biochem. Pharmacol. 1990, 40, 1859–1864. [Google Scholar] [CrossRef]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Maki, A.; Mohammad, R.; Raza, S.; Saleh, M.; Govindaraju, K.D.; Pettit, G.R.; al-Katib, A. Effect of dolastatin 10 on human non-Hodgkin’s lymphoma cell lines. Anticancer Drugs 1996, 7, 344–350. [Google Scholar] [CrossRef]
- Kalemkerian, G.P.; Ou, X.; Adil, M.R.; Rosati, R.; Khoulani, M.M.; Madan, S.K.; Pettit, G.R. Activity of dolastatin 10 against small-cell lung cancer in vitro and in vivo: Induction of apoptosis and bcl-2 modification. Cancer Chemother. Pharmacol. 1999, 43, 507–515. [Google Scholar] [CrossRef]
- Turner, T.; Jackson, W.H.; Pettit, G.R.; Wells, A.; Kraft, A.S. Treatment of human prostate cancer cells with dolastatin 10, a peptide isolated from a marine shell-less mollusc. Prostate 1998, 34, 175–181. [Google Scholar] [CrossRef]
- Pettit, G.R.; Srirangam, J.K.; Williams, M.D.; Durkin, K.P.M.; Barlozzari, T.; Kling, A.; Janssen, B.; Haupt, A. Dolastatin Peptides. US6323315B1, 27 December 2001. [Google Scholar]
- Pettit, G.R.; Kamano, Y.; Fujii, Y.; Herald, C.L.; Inoue, M.; Brown, P.; Gust, D.; Kitahara, K.; Schmidt, J.M.; Doubek, D.L.; et al. Marine Animal Biosynthetic Constituents For Cancer Chemotherapy. J. Nat. Prod. 1981, 44, 482–485. [Google Scholar] [CrossRef]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Lloydwilliams, P.; Herald, D.L.; Burkett, D.D.; Clewlow, P.J. Antineoplastic Agents.189. The Absolute-Configuration and Synthesis of Natural (-)-Dolastatin-10. J. Am. Chem. Soc. 1989, 111, 5463–5465. [Google Scholar] [CrossRef]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar]
- Pitot, H.C.; McElroy, E.A.; Reid, J.M.; Windebank, A.J.; Sloan, J.A.; Erlichman, C.; Bagniewski, P.G.; Walker, D.L.; Rubin, J.; Goldberg, R.M.; et al. Phase I trial of dolastatin-10 (NSC 376128) in patients with advanced solid tumors. Clin. Cancer Res. 1999, 5, 525–531. [Google Scholar] [PubMed]
- Yamamoto, N.; Andoh, M.; Kawahara, M.; Fukuoka, M.; Niitani, H. Phase I study of TZT-1027, a novel synthetic dolastatin 10 derivative and inhibitor of tubulin polymerization, given weekly to advanced solid tumor patients for 3 weeks. Cancer Sci. 2009, 100, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Horti, J.; Juhász, E.; Monostori, Z.; Maeda, K.; Eckhardt, S.; Bodrogi, I. Phase I study of TZT-1027, a novel synthetic dolastatin 10 derivative, for the treatment of patients with non-small cell lung cancer. Cancer Chemother. Pharmacol. 2008, 62, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Hoffman, M.A.; Blessing, J.A.; Lentz, S.S. A phase II trial of dolastatin-10 in recurrent platinum-sensitive ovarian carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2003, 89, 95–98. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Kepler, J.A.; Pettit, G.R.; Hamel, E. Sustained intracellular retention of dolastatin 10 causes its potent antimitotic activity. Mol. Pharmacol. 2000, 57, 180–187. [Google Scholar]
- Beckwith, M.; Urba, W.J.; Longo, D.L. Growth Inhibition of Human Lymphoma Cell Lines by the Marine Products, Dolastatins 10 and 15. J. Natl. Cancer Inst. 1993, 85, 483–488. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Pettit, G.R.; Almatchy, V.P.; Wall, N.; Varterasian, M.; Ai-Katib, A. Synergistic interaction of selected marine animal anticancer drugs against human diffuse large cell lymphoma. Anti-Cancer Drugs 1998, 9, 149–156. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Glode, M.; Du, W.; Kraft, A.; Hudes, G.; Wright, J.; Hussain, M. Phase II study of dolastatin-10 in patients with hormone-refractory metastatic prostate adenocarcinoma. Clin. Cancer Res. 2000, 6, 4205. [Google Scholar]
- Von, M.M.; Balcerzak, S.P.; Kraft, A.S.; Edmonson, J.H.; Okuno, S.H.; Davey, M.; Mclaughlin, S.; Beard, M.T.; Rogatko, A. Phase II Trial of Dolastatin-10, a Novel Anti-Tubulin Agent, in Metastatic Soft Tissue Sarcomas. Sarcoma 2004, 8, 107–111. [Google Scholar]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investig. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef]
- Kindler, H.L.; Tothy, P.K.; Wolff, R.; McCormack, R.A.; Abbruzzese, J.L.; Mani, S.; Wade-Oliver, K.T.; Vokes, E.E. Phase II trials of dolastatin-10 in advanced pancreaticobiliary cancers. Investig. New Drugs 2005, 23, 489–493. [Google Scholar] [CrossRef]
- Hadfield, J.A.; Ducki, S.; Hirst, N.; McGown, A.T. Tubulin and microtubules as targets for anticancer drugs. Prog. Cell Cycle Res. 2003, 5, 309–325. [Google Scholar]
- Perez, E.A.; Shang, X.; Burlingame, S.M.; Okcu, M.F.; Ge, N.; Russell, H.V.; Egler, R.A.; David, R.D.; Vasudevan, S.A.; Yang, J.; et al. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Morris, P.G.; Fornier, M.N. Microtubule Active Agents: Beyond the Taxane Frontier. Clin. Cancer Res. 2008, 14, 7167–7172. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, E.; Kavallaris, M. Microtubules: A dynamic target in cancer therapy. Iubmb Life 2008, 60, 165–170. [Google Scholar] [CrossRef]
- Kavallaris, M.; Verrills, N.M.; Hill, B.T. Anticancer therapy with novel tubulin-interacting drugs. Drug Resist. Updat. 2001, 4, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.S.; Wolff, J. Localization of the Vinblastine-binding Site on β-Tubulin. J. Biol. Chem. 1996, 271, 14707–14711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kobayashi, H.; Hashimoto, Y.; Shirai, R.; Hirata, A.; Hayashi, K.; Hamada, Y.; Shioiri, T.; Iwasaki, S. Interaction of marine toxin dolastatin 10 with porcine brain tubulin: Competitive inhibition of rhizoxin and phomopsin A binding. Chem. Interact. 1994, 93, 175–183. [Google Scholar] [CrossRef]
- Luduena, R.F.; Roach, M.C.; Prasad, V.; Pettit, G.R. Interaction of dolastatin 10 with bovine brain tubulin. Biochem. Pharmacol. 1992, 43, 539–543. [Google Scholar] [CrossRef]
- Bai, R.L.; Pettit, G.R.; Hamel, E. Binding of dolastatin 10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990, 265, 17141–17149. [Google Scholar] [CrossRef]
- Roach, M.C.; Ludueña, R.F. Different effects of tubulin ligands on the intrachain cross-linking of beta 1-tubulin. J. Biol. Chem. 1984, 259, 12063–12071. [Google Scholar] [CrossRef]
- Ludueña, R.F.; Roach, M.C. Contrasting effects of maytansine and vinblastine on the alkylation of tubulin sulfhydryls. Arch. Biochem. Biophys. 1981, 210, 498–504. [Google Scholar] [CrossRef]
- Little, M.; Ludueña, R.F. Location of two cysteines in brain beta 1-tubulin that can be cross-linked after removal of exchangeable GTP. Biochim. Biophys. Acta 1987, 912, 28–33. [Google Scholar] [CrossRef]
- Ludueña, R.F.; Roach, M.C.; Prasad, V.; Lacey, E. Effect of phomopsin a on the alkylation of tubulin. Biochem. Pharmacol. 1990, 39, 1603–1608. [Google Scholar] [CrossRef]
- Mitra, A.; Sept, D. Localization of the antimitotic peptide and depsipeptide binding site on beta-tubulin. Biochemistry 2004, 43, 13955–13962. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Covell, D.G.; Taylor, G.F.; Kepler, J.A.; Copeland, T.D.; Nguyen, N.Y.; Pettit, G.R.; Hamel, E. Direct photoaffinity labeling by dolastatin 10 of the amino-terminal peptide of beta-tubulin containing cysteine 12. J. Biol. Chem. 2004, 279, 30731–30740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, A.; Diwakaran, H.; Redman, B.; Al-Asfar, S.; Pettit, G.R.; Mohammad, R.M.; Al-Katib, A. The bcl-2 and p53 oncoproteins can be modulated by bryostatin 1 and dolastatins in human diffuse large cell lymphoma. Anti-Cancer Drugs 1995, 6, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Basu, A.; Croce, C.M. Serine-70 is one of the critical sites for drug-induced Bcl2 phosphorylation in cancer cells. Cancer Res. 1998, 58, 1609. [Google Scholar]
- Pathak, S.; Multani, A.S.; Ozen, M.; Richardson, M.A.; Newman, R.A. Dolastatin-10 induces polyploidy, telomeric associations and apoptosis in a murine melanoma cell line. Oncol. Rep. 1998, 5, 373–379. [Google Scholar] [CrossRef]
- Pathak, S.; Risin, S.; Brown, N.; Berry, K. Telomeric association of chromosomes is an early manifestation of programmed cell death. Int. J. Oncol. 1994, 4, 323–328. [Google Scholar] [CrossRef]
- Pathak, S.; Dave, B.J.; Gagos, S. Chromosome alterations in cancer development and apoptosis. In Vivo 1994, 8, 843–850. [Google Scholar]
- Aherne, G.W.; Hardcastle, A.; Valenti, M.; Bryant, A.; Rogers, P.; Pettit, G.R.; Srirangam, J.K.; Kelland, L.R. Antitumour evaluation of dolastatins 10 and 15 and their measurement in plasma by radioimmunoassay. Cancer Chemother. Pharmacol. 1996, 38, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Garteiz, D.A.; Madden, T.; Beck, D.E.; Huie, W.R.; McManus, K.T.; Abbruzzese, J.L.; Chen, W.; Newman, R.A. Quantitation of dolastatin-10 using HPLC/electrospray ionization mass spectrometry: Application in a phase I clinical trial. Cancer Chemother. Pharmacol. 1998, 41, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Madden, T.; Tran, H.T.; Beck, D.; Huie, R.; Newman, R.A.; Pusztai, L.; Wright, J.J.; Abbruzzese, J.L. Novel marine-derived anticancer agents: A phase I clinical, pharmacological, and pharmacodynamic study of dolastatin 10 (NSC 376128) in patients with advanced solid tumors. Clin. Cancer Res. 2000, 6, 1293–1301. [Google Scholar]
- Mirsalis, J.C.; Schindler-Horvat, J.; Hill, J.R.; Tomaszewski, J.E.; Donohue, S.J.; Tyson, C.A. Toxicity of dolastatin 10 in mice, rats and dogs and its clinical relevance. Cancer Chemother. Pharmacol. 1999, 44, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D.H.; MacEwen, G.E.; Phillips, B.S.; Hershey, E.A.; Burgess, K.M.; Pettit, G.R.; Vail, D.M. Preclinical study of dolastatin-10 in dogs with spontaneous neoplasia. Cancer Chemother. Pharmacol. 2002, 49, 251–255. [Google Scholar] [CrossRef]
- Fayette, J.; Coquard, I.R.; Alberti, L.; Boyle, H.; Méeus, P.; Decouvelaere, A.-V.; Thiesse, P.; Sunyach, M.-P.; Ranchère, D.; Blay, J.-Y. ET-743: A novel agent with activity in soft-tissue sarcomas. Curr. Opin. Oncol. 2006, 18, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Toppmeyer, D.L.; Slapak, C.A.; Croop, J.; Kufe, D.W. Role of P-glycoprotein in dolastatin 10 resistance. Biochem. Pharmacol. 1994, 48, 609–612. [Google Scholar] [CrossRef]
- Akaiwa, M.; Martin, T.; Mendelsohn, B.A. Synthesis and Evaluation of Linear and Macrocyclic Dolastatin 10 Analogues Containing Pyrrolidine Ring Modifications. ACS Omega 2018, 3, 5212–5221. [Google Scholar] [CrossRef] [PubMed]
- Dugal-Tessier, J.; Barnscher, S.D.; Kanai, A.; Mendelsohn, B.A. Synthesis and Evaluation of Dolastatin 10 Analogues Containing Heteroatoms on the Amino Acid Side Chains. J. Nat. Prod. 2017, 80, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
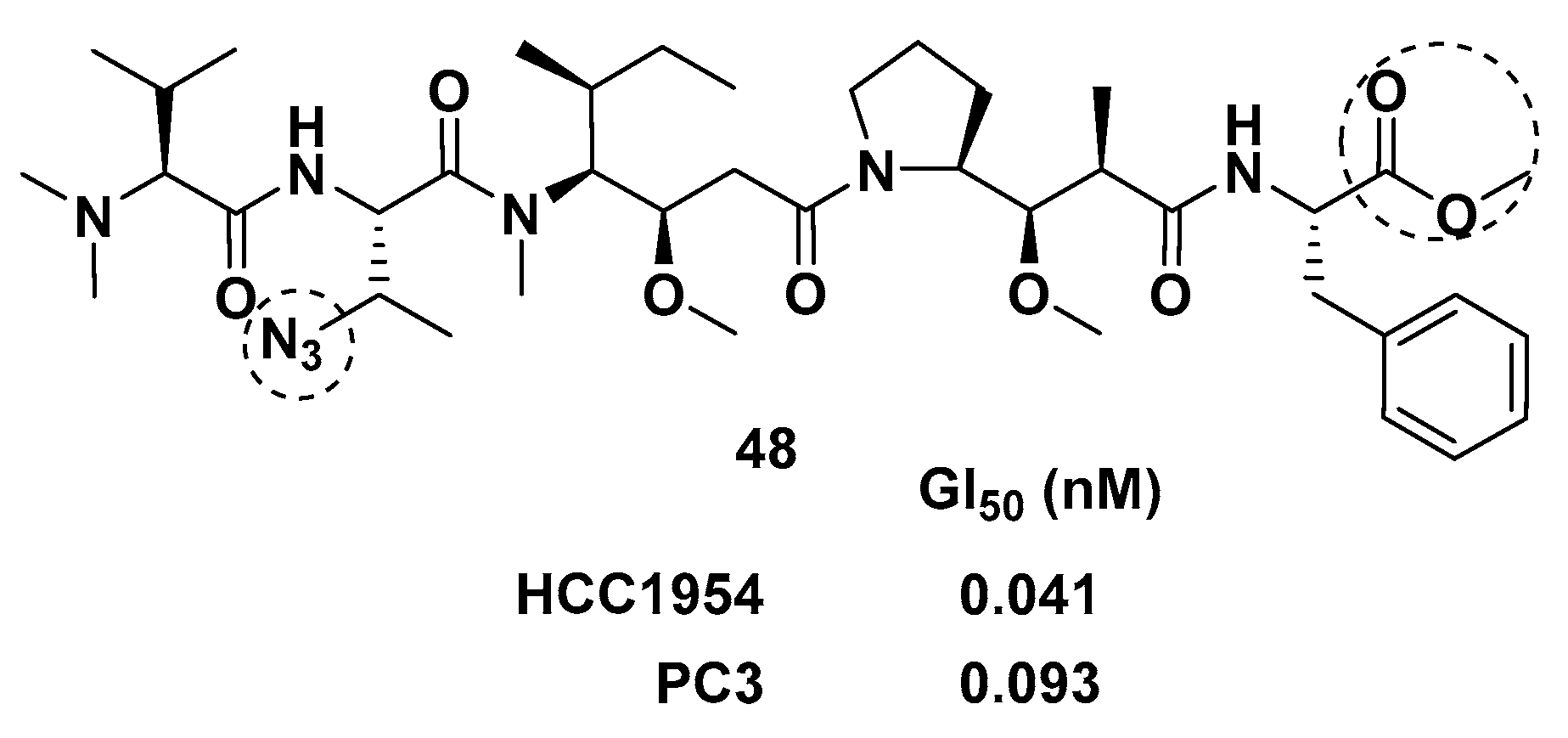
- Maderna, A.; Doroski, M.; Subramanyam, C.; Porte, A.; Leverett, C.A.; Vetelino, B.C.; Chen, Z.; Risley, H.; Parris, K.; Pandit, J.; et al. Discovery of Cytotoxic Dolastatin 10 Analogues with N-Terminal Modifications. J. Med. Chem. 2014, 57, 10527–10543. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Hogan, F.; Bai, R.; Chapuis, J.C.; et al. Antineoplastic agents 365. Dolastatin 10 SAR probes. Anticancer Drug Des. 1998, 13, 243–277. [Google Scholar] [PubMed]
- Shnyder, S.D.; Cooper, P.A.; Millington, N.J.; Pettit, G.R.; Bibby, M.C. Auristatin PYE, a novel synthetic derivative of dolastatin 10: Activity and mechanistic studies in a colon adenocarcinoma model. Cancer Res. 2005, 65, 806–807. [Google Scholar]
- Akashi, Y.; Okamoto, I.; Suzuki, M.; Tamura, K.; Iwasa, T.; Hisada, S.; Satoh, T.; Nakagawa, K.; Ono, K.; Fukuoka, M. The novel microtubule-interfering agent TZT-1027 enhances the anticancer effect of radiation in vitro and in vivo. Br. J. Cancer 2007, 96, 1532–1539. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, J.; Minami, M.; Kobayashi, M. Antitumor activity of TZT-1027 (Soblidotin). Anticancer Res. 2006, 26, 1973–1981. [Google Scholar]
- Natsume, T.; Watanabe, J.-I.; Koh, Y.; Fujio, N.; Ohe, Y.; Horiuchi, T.; Saijo, N.; Nishio, K.; Kobayashi, M. Antitumor activity of TZT-1027 (Soblidotin) against vascular endothelial growth factor-secreting human lung cancer in vivo. Cancer Sci. 2003, 94, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Yokosaka, S.; Izawa, A.; Sakai, C.; Sakurada, E.; Morita, Y.; Nishio, Y. Synthesis and evaluation of novel dolastatin 10 derivatives for versatile conjugations. Bioorganic Med. Chem. 2018, 26, 1643–1652. [Google Scholar] [CrossRef]
- Pettit, G.R.; Hogan, F.; Toms, S. Antineoplastic Agents. 592. Highly Effective Cancer Cell Growth Inhibitory Structural Modifications of Dolastatin 10. J. Nat. Prod. 2011, 74, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Melody, N.; Chapuis, J.-C. Antineoplastic Agents. 607. Emetine Auristatins. J. Nat. Prod. 2020, 83, 1571–1576. [Google Scholar] [CrossRef]
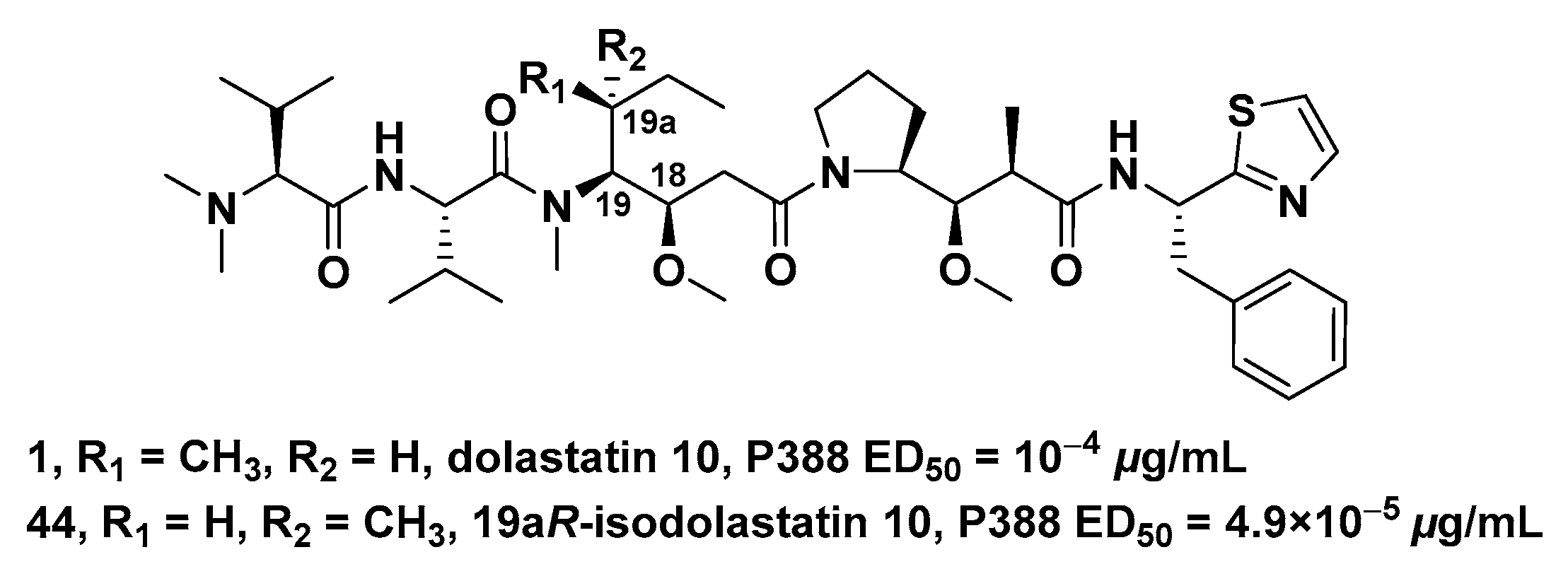
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Burkett, D.D. Chiral modifications of dolastatin 10: The potent cytostatic peptide (19aR)-isodolastatin 10. J. Med. Chem. 1990, 33, 3132–3133. [Google Scholar] [CrossRef]
- Nie, X.-D.; Mao, Z.-Y.; Zhou, W.; Si, C.-M.; Wei, B.-G.; Lin, G.-Q. A diastereoselective approach to amino alcohols and application for divergent synthesis of dolastatin 10. Org. Chem. Front. 2020, 7, 76–103. [Google Scholar] [CrossRef]
- Poncet, J.; Busquet, M.; Roux, F.; Pierre, A.; Atassi, G.; Jouin, P. Synthesis and Biological Activity of Chimeric Structures Derived from the Cytotoxic Natural Compounds Dolastatin 10 and Dolastatin 15. J. Med. Chem. 1998, 41, 1524–1530. [Google Scholar] [CrossRef]
- Wang, X.; Dong, S.; Feng, D.; Chen, Y.; Ma, M.; Hu, W. Synthesis and biological activity evaluation of dolastatin 10 analogues with N-terminal modifications. Tetrahedron 2017, 73, 2255–2266. [Google Scholar] [CrossRef]
- Zhou, W.; Nie, X.-D.; Zhang, Y.; Si, C.-M.; Zhou, Z.; Sun, X.; Wei, B.-G. A practical approach to asymmetric synthesis of dolastatin 10. Org. Biomol. Chem. 2017, 15, 6119–6131. [Google Scholar] [CrossRef] [Green Version]
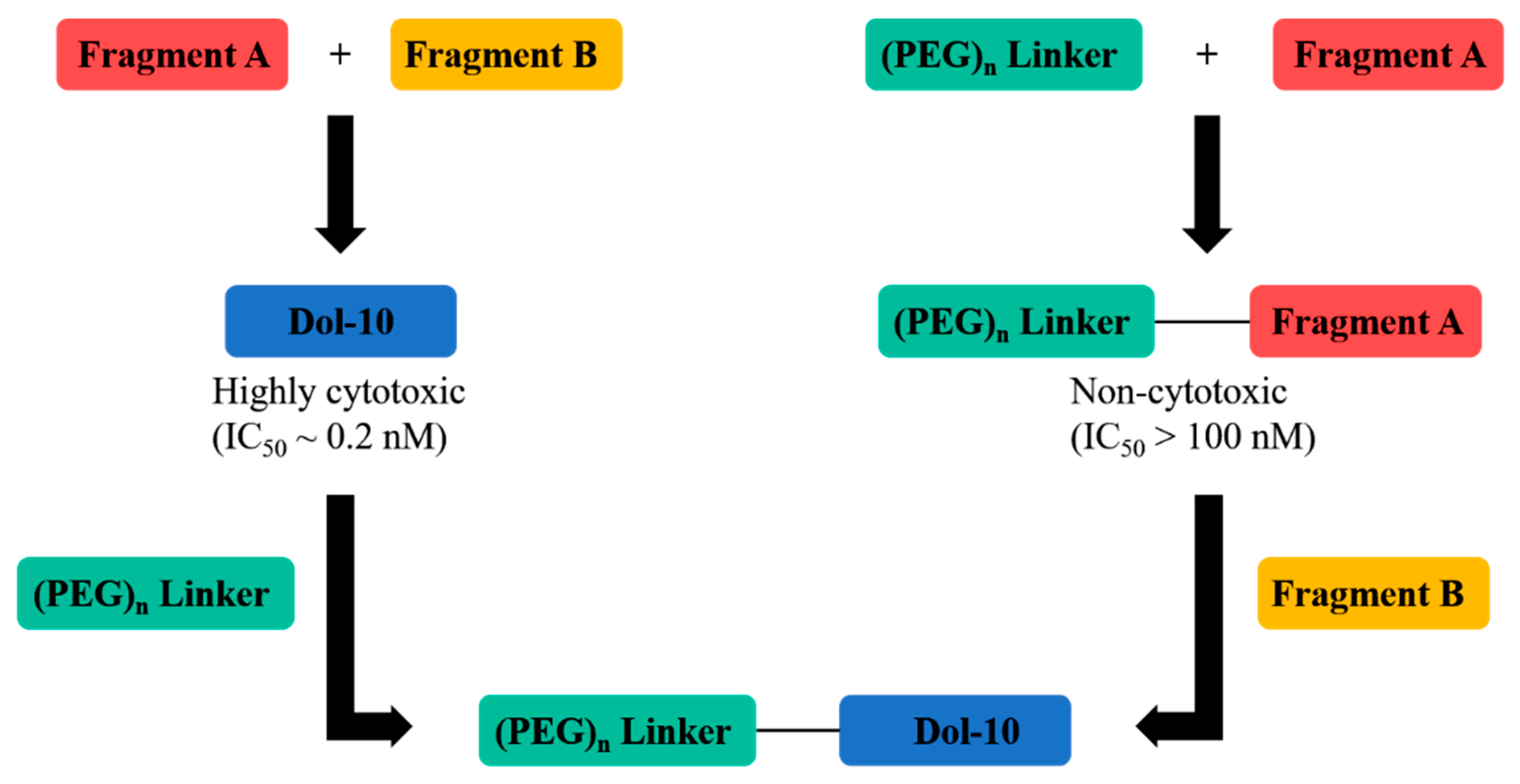
- Yang, K.; Chen, B.; Gianolio, D.A.; Stefano, J.E.; Busch, M.; Manning, C.; Alving, K.; Gregory, R.C.; Brondyk, W.H.; Miller, R.J.; et al. Convergent synthesis of hydrophilic monomethyl dolastatin 10 based drug linkers for antibody–drug conjugation. Org. Biomol. Chem. 2019, 17, 8115–8124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Chen, M.X.R.; Fang-qiu, L.I. Application of fluorescence imaging in the research of tumor. J. Med. Postgrad. 2009, 22, 195–197. [Google Scholar]
- Lee, J.-W.; Stone, R.L.; Lee, S.J.; Nam, E.J.; Roh, J.-W.; Nick, A.M.; Han, H.-D.; Shahzad, M.M.; Kim, H.-S.; Mangala, L.S.; et al. EphA2 Targeted Chemotherapy Using an Antibody Drug Conjugate in Endometrial Carcinoma. Clin. Cancer Res. 2010, 16, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Han, H.D.; Shahzad, M.M.K.; Kim, S.W.; Mangala, L.S.; Nick, A.M.; Lu, C.; Langley, R.R.; Schmandt, R.; Kim, H.-S.; et al. EphA2 Immunoconjugate as Molecularly Targeted Chemotherapy for Ovarian Carcinoma. J. Natl. Cancer Inst. 2009, 101, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, L.; Castel, V.; Bisogno, G.; Casanova, M.; Marquez-Vega, C.; Chisholm, J.C.; Doz, F.; Moreno, L.; Ruggiero, A.; Gerber, N.U.; et al. Phase II results from a phase I/II study to assess the safety and efficacy of weekly nab-paclitaxel in paediatric patients with recurrent or refractory solid tumours: A collaboration with the European Innovative Therapies for Children with Cancer Network. Eur. J. Cancer 2020, 135, 89–97. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Kohn, E.C.; Lorusso, P.; Houston, N.D.; Coleman, R.L.; Buzoianu, M.; Robbie, G.; Lechleider, R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Investig. New Drugs 2012, 31, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.A.; Czuczman, M.S. Novel antibodies in the treatment of non-Hodgkin’s lymphoma. Neth. J. Med. 2009, 67, 311. [Google Scholar]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Melody, N.; Chapuis, J.-C. Antineoplastic Agents. 603. Quinstatins: Exceptional Cancer Cell Growth Inhibitors. J. Nat. Prod. 2017, 80, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Melody, N.; Chapuis, J.-C. Antineoplastic Agents. 604. The Path of Quinstatin Derivatives to Antibody Drug Conjugates. J. Nat. Prod. 2017, 80, 2447–2452. [Google Scholar] [CrossRef]
- Pettit, G.R.; Melody, N.; Chapuis, J.C. Antineoplastic Agents. 605. Isoquinstatins. J. Nat. Prod. 2018, 81, 451–457. [Google Scholar] [CrossRef]
- Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Synthesis and Antitumor Activity of Novel Dolastatin 10 Analogs. Chem. Pharm. Bull. 1995, 43, 1706–1718. [Google Scholar] [CrossRef] [Green Version]
- Shioiri, T.; Hayashi, K.; Hamada, Y. Stereoselective synthesis of dolastatin 10 and its congeners. Tetrahedron 1993, 49, 1913–1924. [Google Scholar] [CrossRef]
- Poncet, J.; Hortala, L.; Busquet, M.; Guéritte-Voegelein, F.; Thoret, S.; Pierré, A.; Atassi, G.; Jouin, P. Synthesis and antiproliferative activity of a cyclic analog of dolastatin 10. Bioorg. Med. Chem. Lett. 1998, 8, 2855–2858. [Google Scholar] [CrossRef]
- Alattia, T.; Roux, F.; Poncet, J.; Cavé, A.; Jouin, P. Conformational study of dolastatin 10. Tetrahedron 1995, 51, 2593–2604. [Google Scholar] [CrossRef]
- Sone, H.; Kigoshi, H.; Yamada, K. Isolation and stereostructure of dolastatin I, a cytotoxic cyclic hexapeptide from the Japanese sea hare Dolabella auricularia. Tetrahedron 1997, 53, 8149–8154. [Google Scholar] [CrossRef]
- Tomioka, K.; Satoh, M.; Taniyama, D.; Kanai, M.; Iida, A. ChemInform Abstract: Enantioselective Addition of Thiazolyllithium to Aldimines with the Aid of Chiral Ligand. Asymmetric Synthesis of (S)-Doe, a Component of Marine Natural Product, Dolastatin 10. Cheminform 1998, 29. [Google Scholar] [CrossRef]
- Hamada, Y.; Hayashi, K.; Shioiri, T. Efficient stereoselective synthesis of dolastatin 10, an antineoplastic peptide from a sea hare. Tetrahedron Lett. 1991, 32, 931–934. [Google Scholar] [CrossRef]
- Kazmaier, U.; Burkhart, J.L. A Straightforward Approach to Protected (S)-Dolaphenine (Doe), the Unusual Amino Acid Component of Dolastatin 10. Synthesis 2011, 2011, 4033–4036. [Google Scholar] [CrossRef]
- Mordant, C.; Reymond, S.; Tone, H.; Lavergne, D.; Touati, R.; Ben Hassine, B.; Ratovelomanana-Vidal, V.; Genet, J.-P. Total Synthesis of Dolastatin 10 Through Ruthenium-Catalyzed Asymmetric Hydrogenations. ChemInform 2007, 38, 6115–6123. [Google Scholar] [CrossRef]
- Kiyoshi, T.; Motomu, K.; Kenji, K. An expeditious synthesis of dolastatin 10. Tetrahedron Lett. 1991, 32, 2395–2398. [Google Scholar]
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Lloyd-Williams, P.; Herald, D.L.; Burkett, D.D.; Clewlow, P.J. ChemInform Abstract: Antineoplastic Agents. Part 189. The Absolute Configuration and Synthesis of Natural (-)-Dolastatin 10. ChemInform 1989, 20. [Google Scholar] [CrossRef]
- Benedetti, E.; Carlomagno, T.; Fraternali, F.; Hamada, Y.; Hayashi, K.; Paolillo, L.; Shioiri, T. Conformational analysis of dolastatin 10: An nmr and theoretical approach. Biopolymers 1995, 36, 525–538. [Google Scholar] [CrossRef]
- Fantucci, P.; Mattioli, E.; Marino, T.; Russo, N. The Conformational Properties of (-)-Dolastatin 10, a Powerful Antineoplastic Agent. Mol. Electron. 1994, 11, 205–209. [Google Scholar] [CrossRef]
- Fantucci, P.; Marino, T.; Russo, N.; Villa, A.M. Conformational behaviour of the antineoplastic peptide dolastatin-10 and of two mutated derivatives. J. Comput. Mol. Des. 1995, 9, 425–438. [Google Scholar] [CrossRef]
- Pettit, R.K.; Pettit, G.R.; Hazen, K.C. Specific Activities of Dolastatin 10 and Peptide Derivatives against Cryptococcus neoformans. Antimicrob. Agents Chemother. 1998, 42, 2961–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fennell, B.J.; Carolan, S.; Pettit, G.R.; Bell, A. Effects of the antimitotic natural product dolastatin 10, and related peptides, on the human malarial parasite Plasmodium falciparum. J. Antimicrob. Chemother. 2003, 51, 833–841. [Google Scholar] [CrossRef] [Green Version]
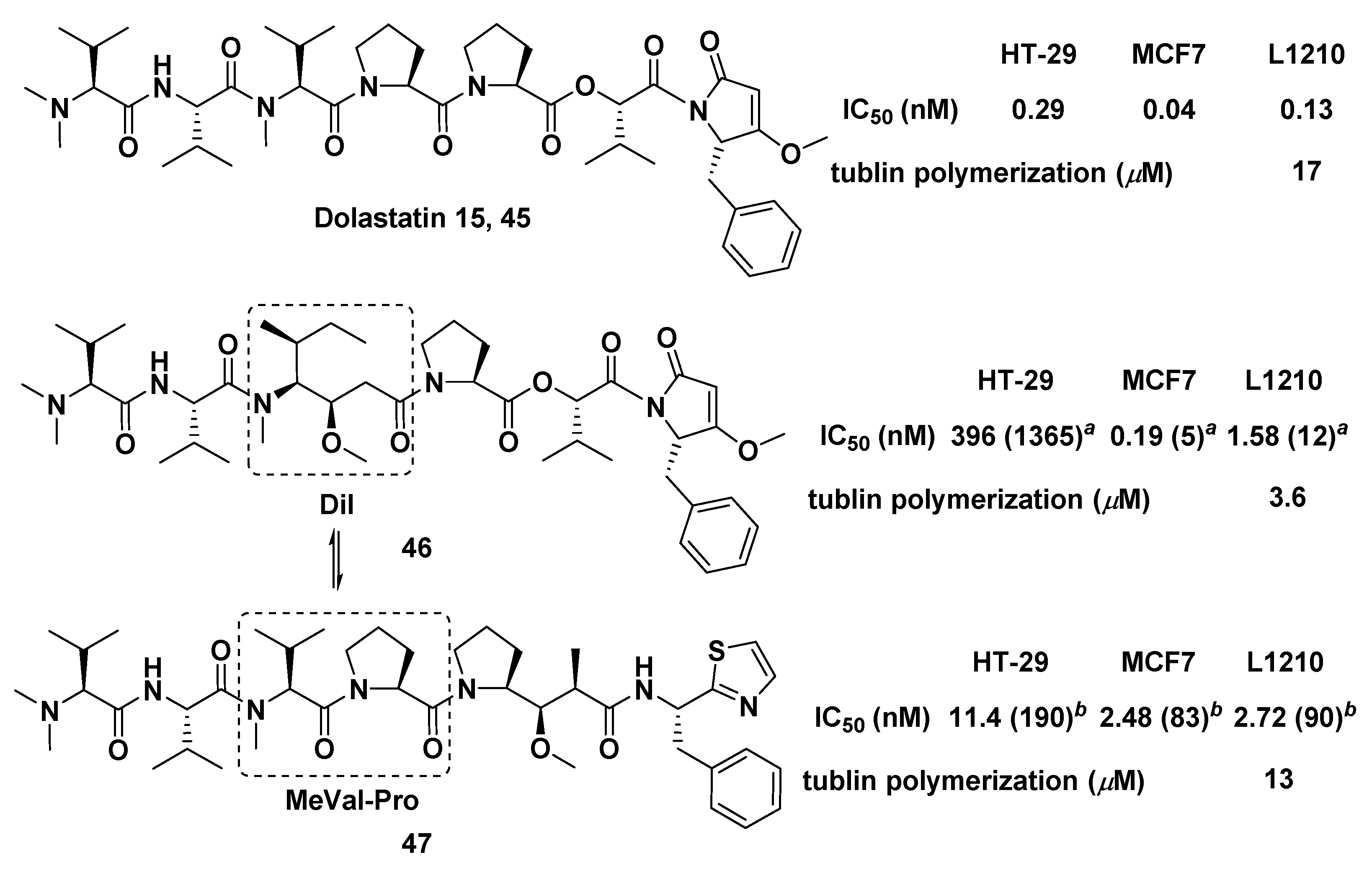

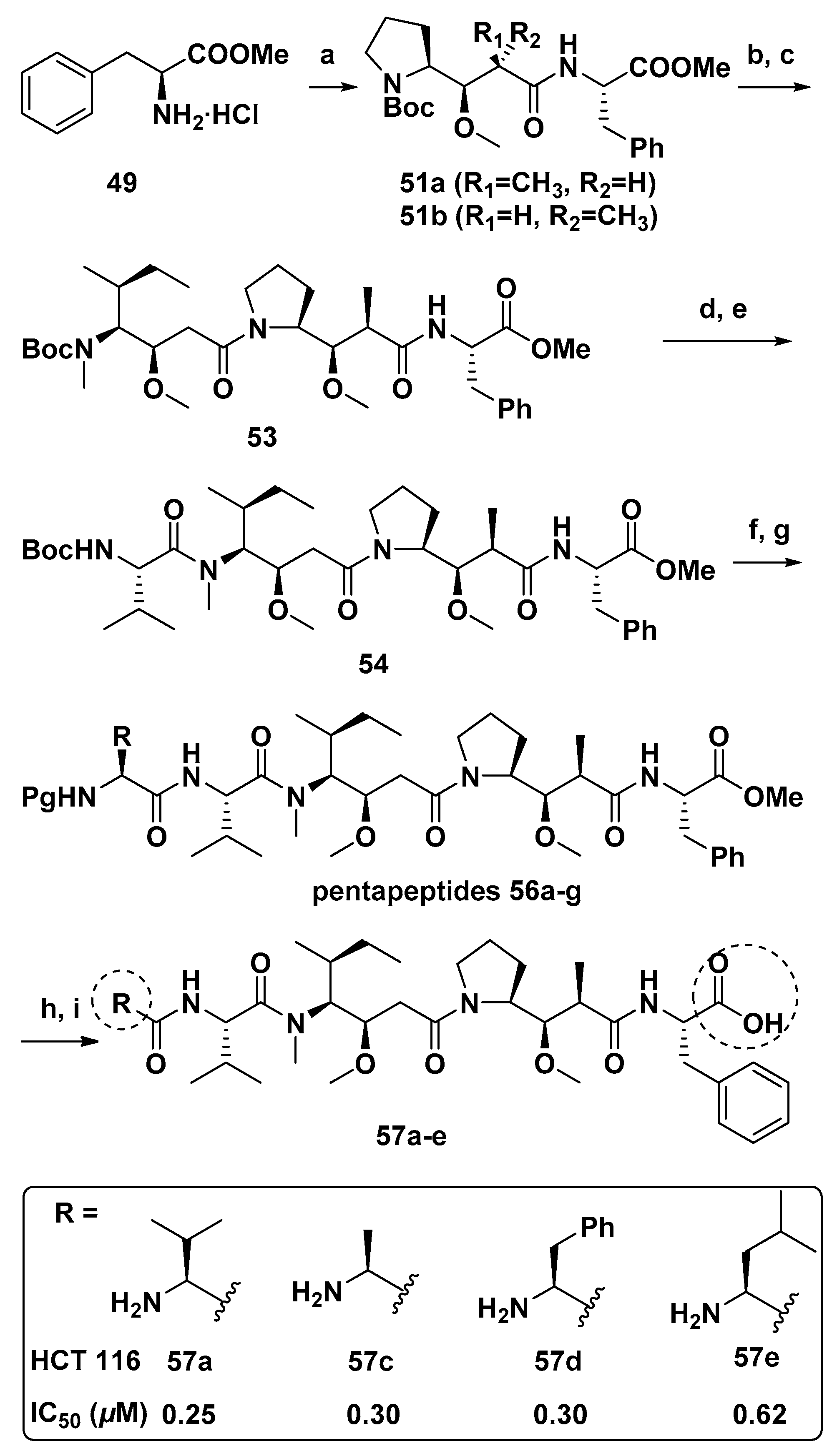
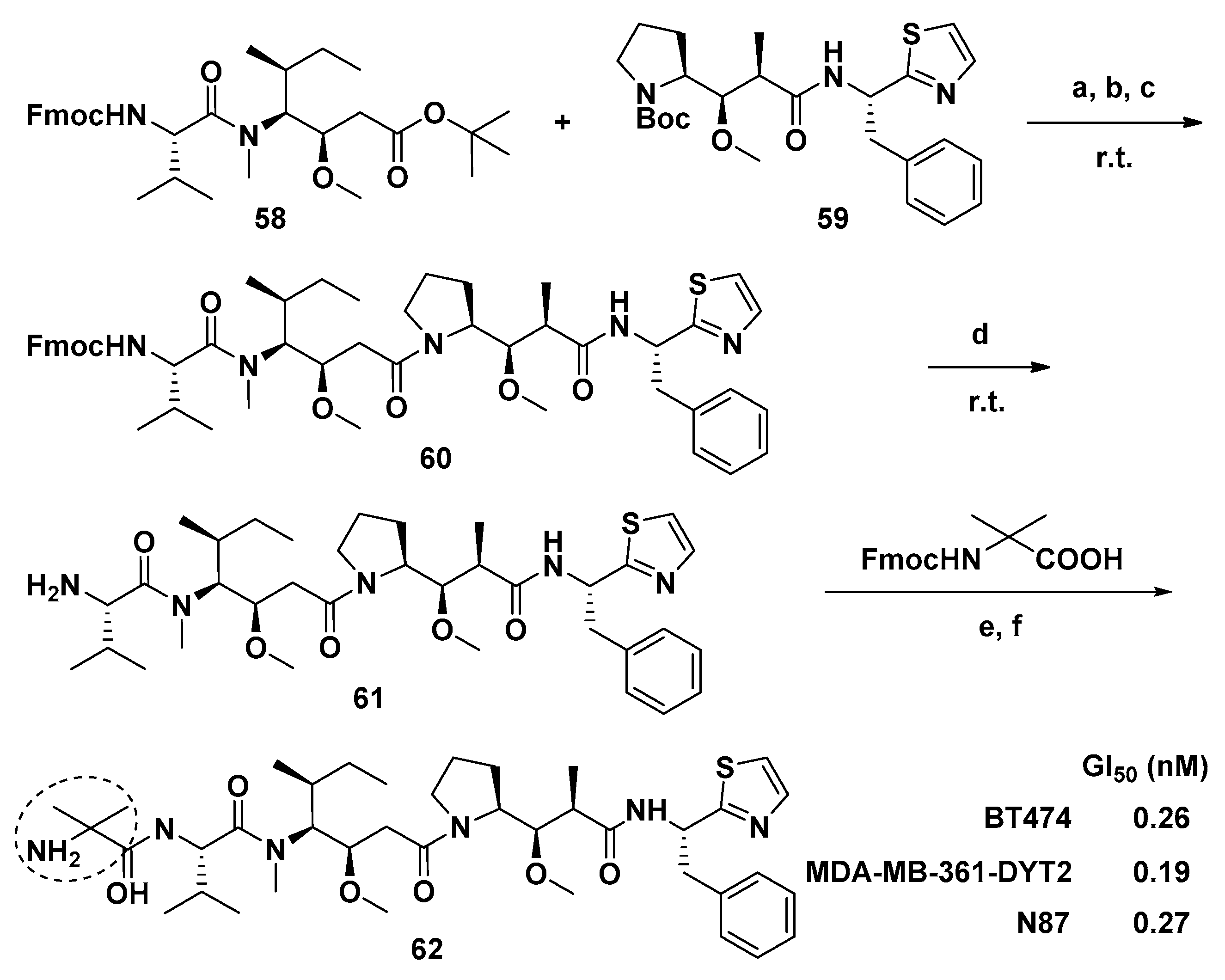
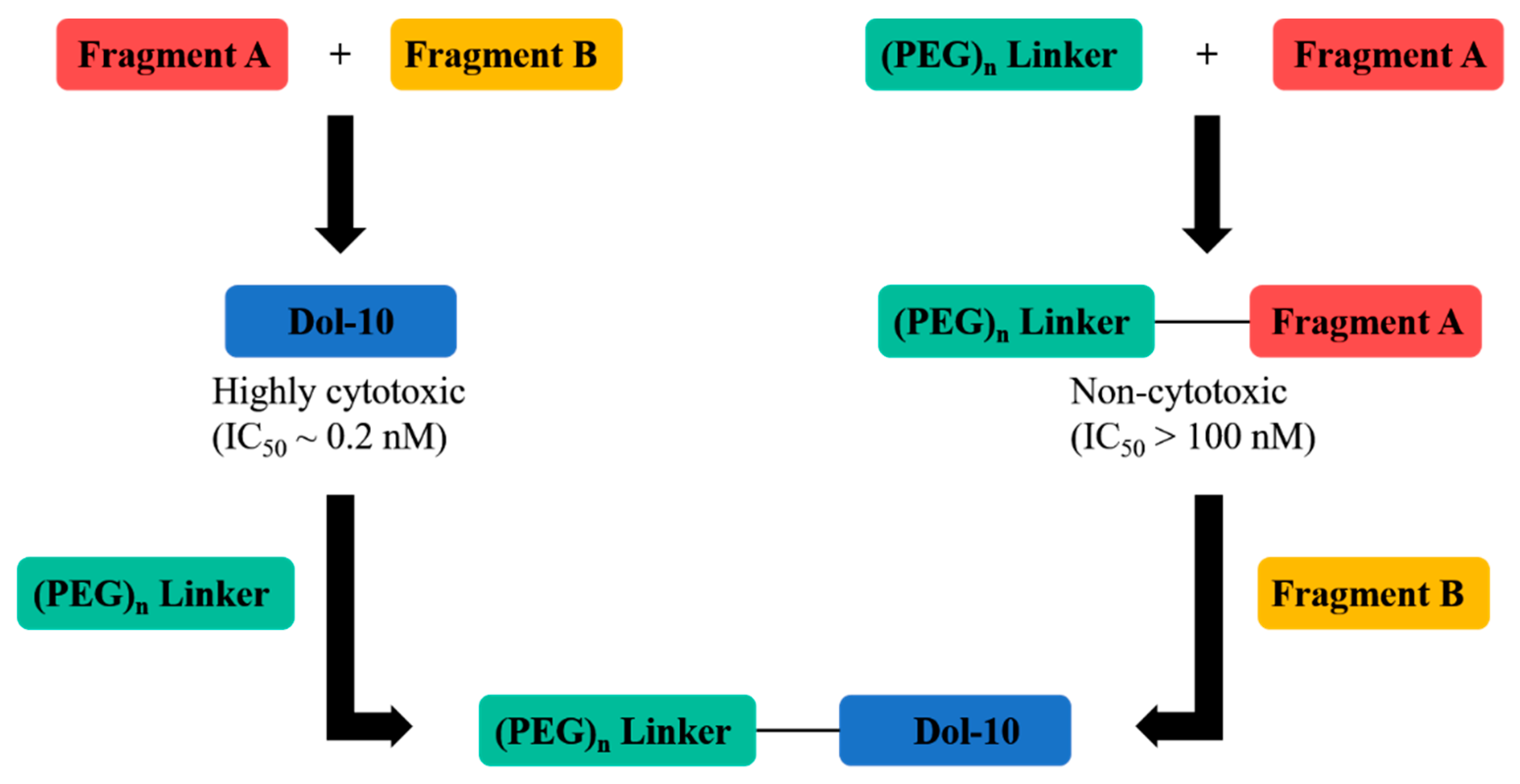





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Use | Development State | Evaluation Outcome * | Reference |
|---|---|---|---|
| Anti-lymphoma | Preclinical | ++ | [36,49,50,51] |
| Anti-lung cancer | Preclinical | ++ | [37] |
| Anti-ovarian carcinoma | Phase II | + | [48] |
| Anti-prostate cancer | Preclinical, phase II | ++, + (respectively) | [36,52] |
| Anti-soft tissue sarcoma | Phase II | - | [53] |
| Anti-breast cancer | Phase II | - | [54] |
| Anti-hepatobiliary pancreatic carcinoma | Phase II | - | [55] |
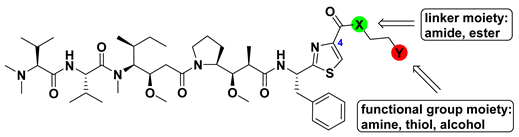
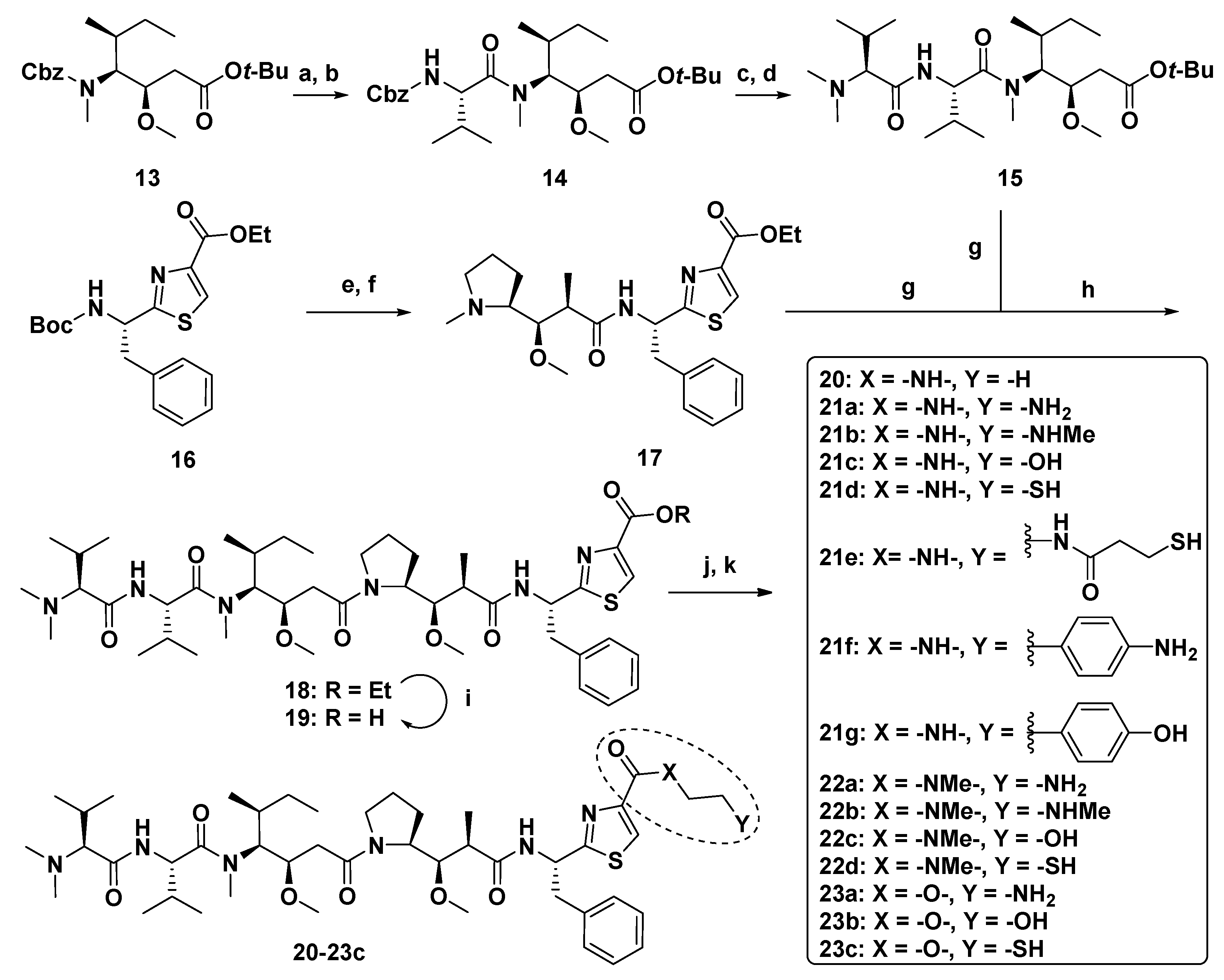
| Compound | -X- | -Y | IC50 (nM) | ||
|---|---|---|---|---|---|
| SKOV-3 | A549 | L1210 | |||
| MMAE (7) | - | - | 0.66 | 1.3 | 2.1 |
| 18 | -O- | -H | 0.024 | - | - |
| 20 | -NH- | -H | 0.55 | 0.74 | 6.5 |
| 21a | -NH- | -NH2 | >10 | >10 | >50 |
| 21b | -NH- | -NHMe | >10 | 9.9 | 27 |
| 21c | -NH- | -OH | 4.7 | 1.7 | 28 |
| 21d | -NH- | -SH | 0.44 | 1.9 | 31 |
| 21e | -NH- | 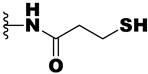 | >10 | >10 | >50 |
| 21f | -NH- |  | 0.27 | 1.1 | 4.1 |
| 21g | -NH- | 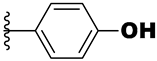 | 0.37 | 1.3 | 5.6 |
| 22a | -NMe- | -NH2 | 0.56 | 0.81 | 12 |
| 22b | -NMe- | -NHMe | 0.19 | 1.9 | 14 |
| 22c | -NMe- | -OH | 1.5 | 1.6 | 8.5 |
| 22d | -NMe- | -SH | 1.1 | 1.2 | 7.0 |
| 23a | -O- | -NH2 | 0.85 | 0.83 | 29 |
| 23b | -O- | -OH | 0.086 | 1.8 | 4.7 |
| 23c | -O- | -SH | 0.13 | 6.9 | 14 |
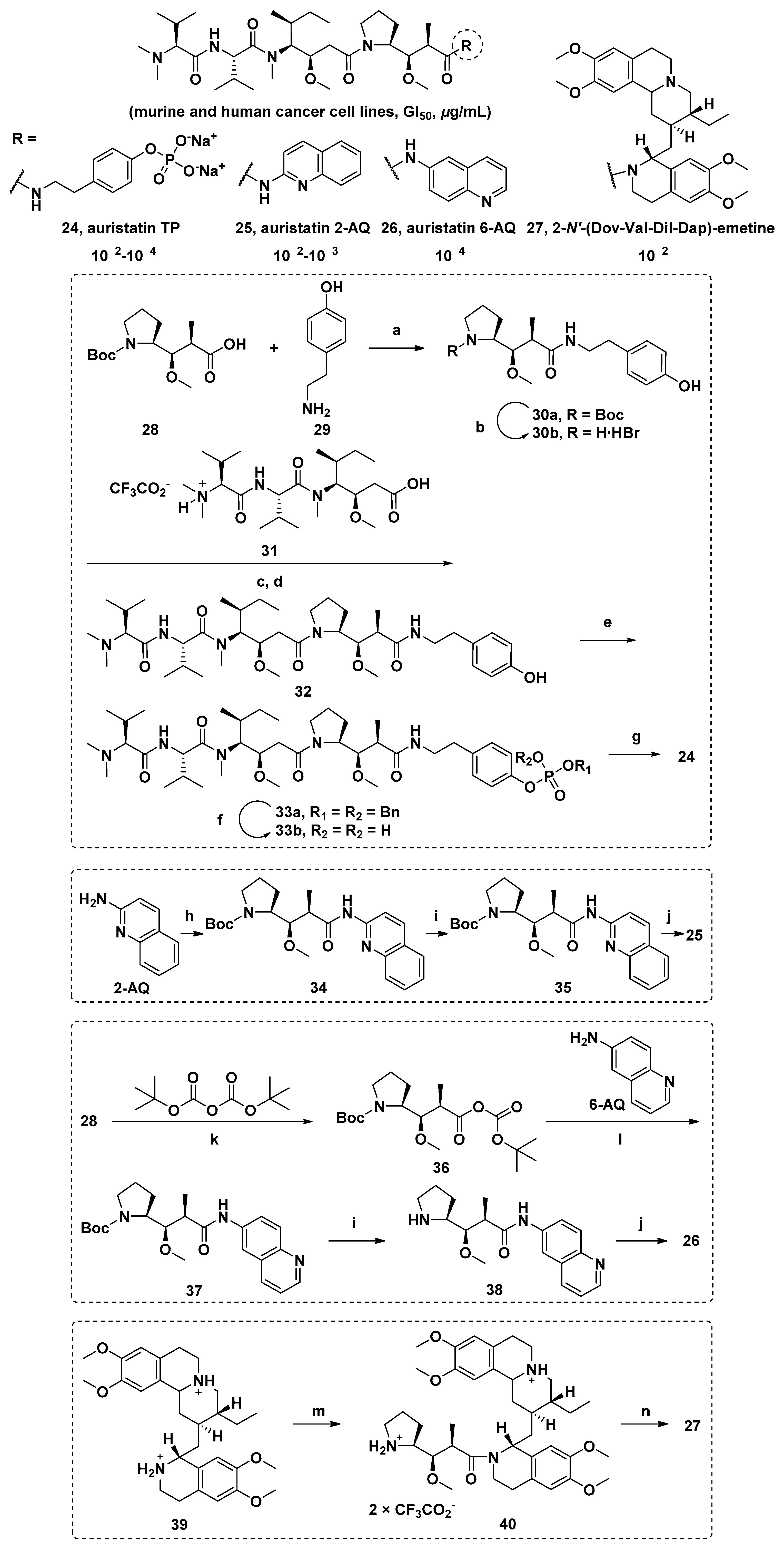
| Compound | Cell Line b | ||||||
|---|---|---|---|---|---|---|---|
| P388 | NCI-H460 | KM20L2 | DU-145 | BXPC-3 | MCF-7 | SF-268 | |
| 24 | <0.001 (<1.2) | 0.00088 (1.05) | 0.00061 (0.72) | 0.00054 (0.64) | 0.046 (54.6) | 0.00068 (0.81) | 0.00125 (1.48) |
| 25 | 0.031 (42.8) | 0.0160 (22.1) | 0.0077 (10.6) | 0.023 (31.8) | 0.029 (40.1) | 0.0046 (6.35) | 0.029 (40.1) |
| 26 | 0.0026 (3.59) | 0.00036 (0.50) | 0.00025 (0.35) | 0.00030 (0.41) | 0.00031 (0.43) | 0.00014 (0.19) | 0.00016 (0.22) |
| 27 | - | - | - | - | - | - | - |
| (-) | (198.0) | (35.82) | (12.26) | (386.5) | (35.82) | (30.17) | |
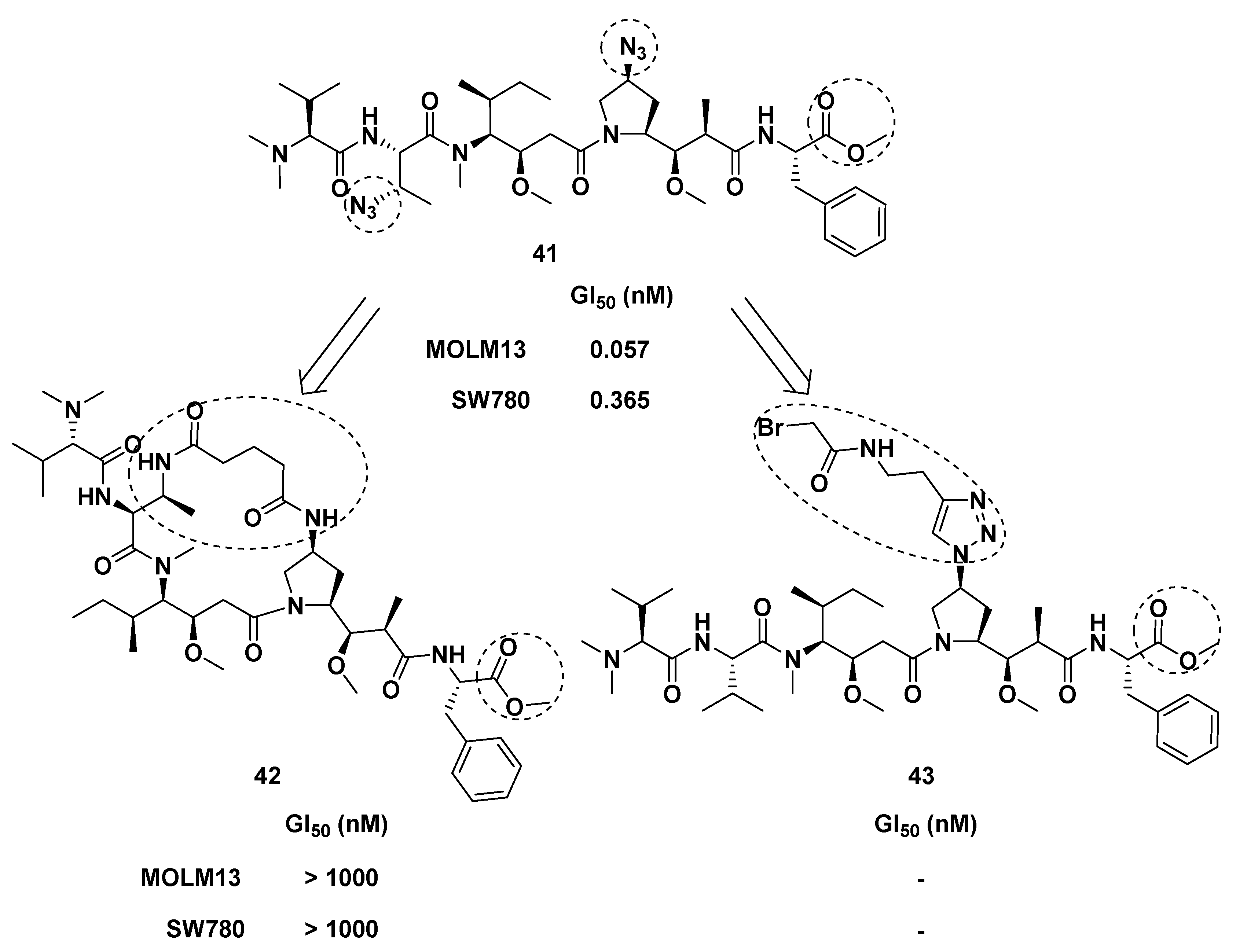
| Compound | Modified Position | ILSmax * | Optimal Dose (mg/kg per inj.) |
|---|---|---|---|
| 1 | 50 | 0.05 | |
| 73 | 1 | 94 | 0.5 |
| 77 | 2 | 80 | 0.5 |
| 81 | 3 | 74 | 2.0 |
| 85 | 5 | 83 | 0.5 |
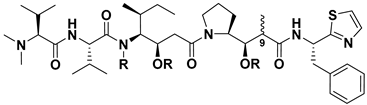
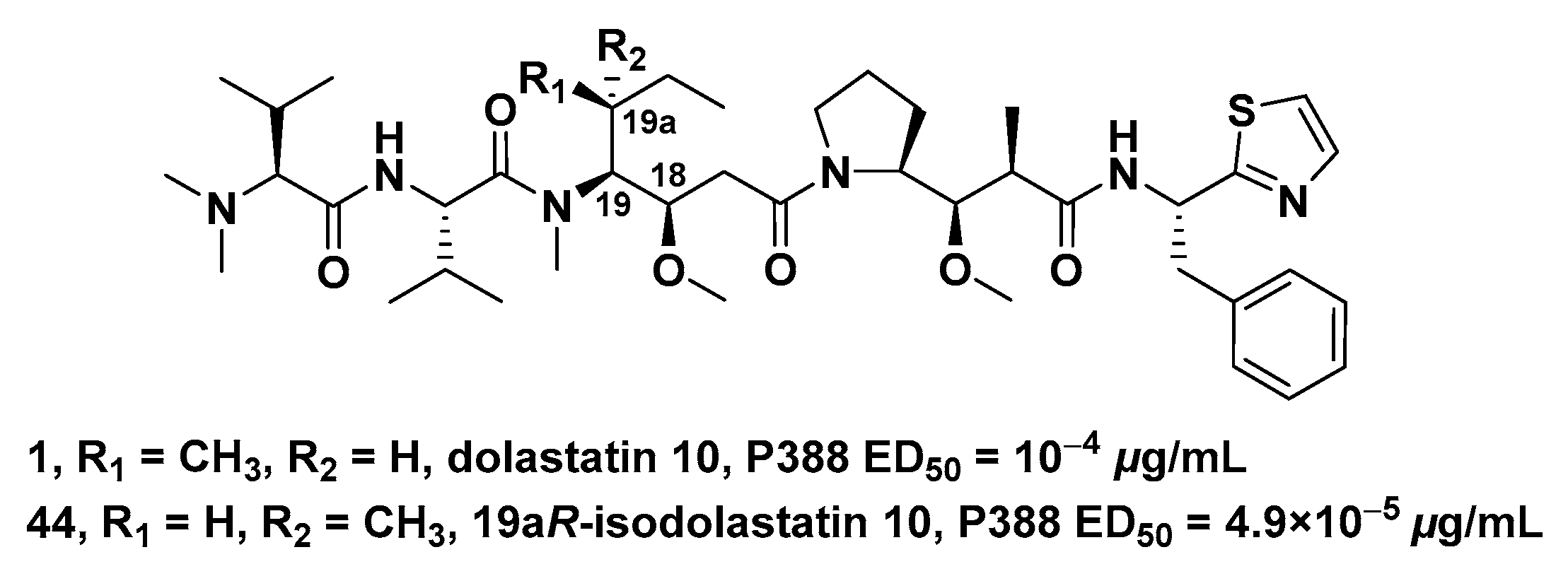
| Compound | R | C9-CH3 | IC50 (μg/mL) | |
|---|---|---|---|---|
| 1 | Dol-10 | CH3 | β | 2.95 × 10−4 |
| 86 | trisnordolastatin 10 | H | β | 1.0 × 10−1 |
| 87 | 9-epi-trisnordolastatin 10 | H | α | 50 |
| 88 | Boc-Val-Dil-Dap-Doe | 4.0 × 10−3 | ||
| 89 | Boc-Dil-Dap-Doe | 11 | ||
| 90 | Boc-Dap-Doe | CH3 | β | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, G.; Wang, Y.; Hua, H.; Li, D.; Tang, C. Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry. Mar. Drugs 2021, 19, 363. https://doi.org/10.3390/md19070363
Gao G, Wang Y, Hua H, Li D, Tang C. Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry. Marine Drugs. 2021; 19(7):363. https://doi.org/10.3390/md19070363
Chicago/Turabian StyleGao, Gang, Yanbing Wang, Huiming Hua, Dahong Li, and Chunlan Tang. 2021. "Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry" Marine Drugs 19, no. 7: 363. https://doi.org/10.3390/md19070363
APA StyleGao, G., Wang, Y., Hua, H., Li, D., & Tang, C. (2021). Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry. Marine Drugs, 19(7), 363. https://doi.org/10.3390/md19070363