3.2. General Experimental Procedures
3.2.1. Synthesis of (3R,5R)-2,2,5-trimethyloct-7-en-3-ol (S11)
To a solution of cyclic sulfate 6 (620 mg, 2.98 mmol, 1.0 eq.) and CuI (57 mg, 0.3 mmol, 0.1 eq.) in dry THF (1 mL) at −20 °C, allylmagnesium bromide (1.0 M in THF, 14.9 mL, 14.9 mmol, 5.0 eq.) was added under an argon atmosphere. The purple-colored reaction mixture was allowed to stir at −20 °C for 7 h before it was allowed to warm to room temperature and then become concentrated in vacuo. The solid residue was redissolved in Et2O (30 mL) and treated with 20% aqueous H2SO4 (10 mL) solution. The contents of the flask were then stirred vigorously for another 12 h before the phases were separated. The aqueous layer was extracted with Et2O (3 × 30 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 20:1) to afford alcohol S11 (380 mg, 75%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 10:1), iodine and PMA stain. = +36.0 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.86–5.67 (m, 1H), 5.14–4.91 (m, 2H), 3.29 (dd, J = 10.3, 1.6 Hz, 1H), 2.30–2.13 (m, 1H), 1.92–1.82 (m, 1H), 1.81–1.72 (m, 1H), 1.53 (s, 1H), 1.46–1.34 (m, 1H), 1.18 (ddd, J = 14.3, 10.3, 4.1 Hz, 1H), 0.93 (d, J = 6.7 Hz, 3H), 0.87 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 137.2, 116.1, 77.7, 39.9, 38.6, 35.1, 29.9, 25.7, 21.0; HRMS (ESI) calculated for C11H22ONa+ [M + Na]+ 193.1563, found 193.1565.
3.2.2. Synthesis of tert-butyldimethyl(((3R,5R)-2,2,5-trimethyloct-7-en-3-yl)oxy)silane (10)
To a solution of alcohol S11 (2.8 g, 16.5 mmol, 1.0 eq.) in dry DCM (30 mL, 0.55 M), Et3N (33 mmol, 4.6 mL, 2.0 eq.) and TBSOTf (21.5 mmol, 4.9 mL, 1.3 eq.) were added at 0 °C. The reaction mixture was allowed to stir at 0 °C for 2 h before it was diluted with DCM (20 mL) and quenched with a saturated aqueous solution of NH4Cl (30 mL). The aqueous layer was extracted with DCM (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes) to furnish silyl ether 10 (4.5 g, 96%) as a colorless oil. TLC: Rf = 0.95 (hexanes), iodine and PMA stain. = +7.4 (c 0.02, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.83–5.73 (m, 1H), 5.04–4.99 (m, 2H), 3.34 (dd, J = 7.5, 2.8 Hz, 1H), 2.22–2.16 (m, 1H), 1.80–1.72 (m, 1H), 1.72–1.64 (m, 1H), 1.45 (ddd, J = 14.3, 9.3, 2.8 Hz, 1H), 1.21 (ddd, J = 14.3, 7.5, 4.2 Hz, 1H), 0.90 (d, J = 6.5 Hz, 3H), 0.90 (s, 9H), 0.86 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 137.3, 116.1, 78.6, 41.0, 40.5, 36.0, 29.8, 26.6, 26.4, 20.9, 18.7, −3.2, −3.6; HRMS (ESI) calculated for C17H36OSi Na+ [M + Na]+ 307.2428, found 307.2425.
3.2.3. Synthesis of (4R,6R)-6-((tert-butyldimethylsilyl)oxy)-4,7,7-trimethyloctan-1-ol (11)
To a solution of alkene 10 (0.5 g, 1.7 mmol, 1.0 eq.) in dry THF (3 mL, 0.17 M), 9-BBN (0.5 M in THF, 3.52 mmol, 7.0 mL, 2.0 eq.) was added at 0 °C under an argon atmosphere. The reaction mixture was stirred at room temperature for 8 h before a saturated aqueous solution of NaHCO3 (10 mL) and 30% H2O2 (2 mL) were added sequentially at 0 °C and stirred for another 12 h at room temperature. The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1) to afford alcohol 11 (473 mg, 89%) as a colorless oil. TLC: Rf = 0.5 (hexanes/EtOAc = 4:1), PMA stain. = +10.8 (c 0.01, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.63 (td, J = 6.6, 1.5 Hz, 2H), 3.30 (dd, J = 7.3, 2.9 Hz, 1H), 1.73–1.58 (m, 1H), 1.59–1.53 (m, 1H), 1.52–1.36 (m, 4H), 1.19 (ddd, J = 14.2, 7.3, 4.4 Hz, 1H), 1.06–0.94 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.88 (s, 9H), 0.84 (s, 9H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 78.5, 63.7, 41.7, 35.9, 32.2, 30.3, 29.9, 26.5, 26.3, 20.9, 18.6, −3.3, −3.7; HRMS (ESI) calculated for C17H38O2SiNa+ [M + Na]+ 325.2533, found 325.2528.
3.2.4. Synthesis of (4R,6R)-6-((tert-butyldimethylsilyl)oxy)-4,7,7-trimethyloctanal (12)
To a solution of alcohol 11 (1.0 g, 3.3 mmol, 1.0 eq.) and TEMPO (51 mg, 0.33 mmol, 0.1 eq.) in DCM (30 mL), a solution of NaBr (2.0 g, 19.8 mmol, 6.0 eq.) and NaHCO3 (1.7 g, 19.8 mmol, 6.0 eq.) were added in water (50 mL), followed by NaClO (1 M, 3.3 mL, 1.0 eq.) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and then quenched with a saturated aqueous solution of Na2S2O3 (3 mL) and extracted with DCM (3 × 30 mL). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 20:1) to afford aldehyde 12 (842 mg, 85%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 10:1), PMA stain. = +11.5 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3) δ 9.78 (t, J = 1.8 Hz, 1H), 3.30 (dd, J = 7.3, 2.9 Hz, 1H), 2.58–2.44 (m, 1H), 2.40–2.29 (m, 1H), 1.90–1.71 (m, 1H), 1.65–1.53 (m, 1H), 1.44 (ddd, J = 14.3, 9.0, 2.8 Hz, 1H), 1.34–1.25 (m, 1H), 1.22 (ddd, J = 14.3, 7.1, 4.3 Hz, 1H), 0.90 (d, J = 6.7 Hz, 3H), 0.88 (s, 9H), 0.84 (s, 9H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 202.7, 78.4, 41.6, 41.5, 36.0, 29.7, 28.0, 26.5, 26.3, 20.7, 18.6, −3.3, −3.7; HRMS (ESI) calculated for C17H38O2SiNa+ [M + Na]+ 323.2377, found 323.2367.
3.2.5. Synthesis of (4R,6S)-4-(tert-butyl)-6-methyl-1,3,2-dioxathiane 2,2-dioxide (7)
To a solution of Me
4NHB(OAc)
3 (13.7 g, 52.1 mmol, 5.0 eq.) in anhydrous CH
3CN (25 mL) and anhydrous AcOH (15 mL) at −40 °C, a solution of
9 [23] (1.5 g, 10.4 mmol, 1.0 eq.) was added in anhydrous CH
3CN (15 mL). The reaction mixture was stirred at −40 °C for 12 h, allowed to warm to ambient temperature, poured into a saturated aqueous solution of Na
2CO
3 (80 mL) and then extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1–5:1) to afford diol
S13 (1.1 g, 71%) as a white solid. [
43]
1H NMR analysis revealed the presence of a 5:1 ratio of
anti/
syn.
To a solution of
anti-diol
S13 (3.15 g, 21.5 mmol, 1.0 eq.) in dry DCM (200 mL, 0.22 M), pyridine (17.3 mL, 215.0 mmol, 10.0 eq.) and SOCl
2 (7.9 mL, 108.0 mmol, 5.0 eq.) were added sequentially at 0 °C. The reaction mixture was allowed to stir at 0 °C for 45 min before it was quenched by the addition of water (50 mL) and then extracted with DCM (3 × 100 mL). The combined organic layers were washed with saturated aqueous KHSO
4 solution (50 mL), followed by saturated aqueous NaHCO
3 solution (60 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude sulfite, which was used in the next step without further purification. To a solution of the crude sulfite in a mixture of H
2O/MeCN/CCl
4 (200 mL:200 mL:100 mL), RuCl
3·nH
2O (562 mg, 2.15 mmol, 0.1 eq.) and NaIO
4 (6.9 g, 32.3 mmol, 1.5 eq.) were added. The biphasic reaction mixture was vigorously stirred at room temperature for 2 h before it was diluted with Et
2O (60 mL) and quenched with a saturated aqueous solution of NaHCO
3 (100 mL). The aqueous layer was extracted with Et
2O (3 × 200 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1–5:1) to afford
anti-cyclic sulfate
7 (3.76 g, 84% for two steps) as an amorphous white solid. The spectral data are in accordance with those reported in literature for its enantiomer [
23]. TLC: R
f = 0.5 (hexanes/ EtOAc = 4:1), PMA stain.
= −0.06 (
c 0.03, CHCl
3).
1H NMR (400 MHz, CDCl
3) δ 4.94 (ddq,
J = 6.7, 6.1, 4.4 Hz, 1H), 4.59 (dd,
J = 11.4, 3.6 Hz, 1H), 2.30 (ddd,
J = 14.2, 11.4, 6.1 Hz, 1H), 1.75 (ddd,
J = 14.2, 4.4, 3.6 Hz, 1H), 1.63 (d,
J = 6.8 Hz, 3H), 1.00 (s, 9H).
13C NMR (100 MHz, CDCl
3) δ 88.6, 81.1, 34.3, 30.0, 25.2, 19.7. HRMS (ESI) calculated for C
8H
16SO
4Na
+ [M + Na]
+ 231.0662, found 231.0661.
3.2.6. Synthesis of (3R,5S)-2,2,5-trimethyloct-7-en-3-ol (S12)
The product S12 was synthesised according to the procedures for the synthesis of S11 from 7 (620 mg, 2.98 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 20:1) to afford S12 (370 mg, 73%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 10:1), iodine and PMA stain. = +24.9 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.78 (ddt, J = 17.4, 10.4, 7.1 Hz, 1H), 5.13–4.81 (m, 2H), 3.28 (dd, J = 10.6, 1.8 Hz, 1H), 2.21–1.85 (m, 2H), 1.75 (dtt, J = 13.6, 6.7, 3.3 Hz, 1H), 1.35 (ddd, J = 13.9, 10.6, 3.2 Hz, 1H), 1.18 (ddd, J = 14.0, 10.6, 1.8 Hz, 1H), 0.89 (d, J = 6.5 Hz, 3H), 0.87 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 137.6, 115.9, 77.4, 42.8, 38.5, 35.0, 29.7, 25.8, 18.9. HRMS (ESI) calculated for C11H22ONa+ [M + Na]+ 193.1563, found 193.1562.
3.2.7. Synthesis of tert-butyldimethyl(((3R,5S)-2,2,5-trimethyloct-7-en-3-yl)oxy)silane (13)
The product 13 was synthesised according to the procedures for the synthesis of 10 from S12 (2.9 g, 17.14 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes) to afford 13 (4.67 g, 96%) as a colorless oil. TLC: Rf = 0.95 (hexanes), iodine and PMA stain. = +11.1 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.98–5.61 (m, 1H), 5.19–4.90 (m, 2H), 3.30 (dd, J = 8.5, 1.8 Hz, 1H), 2.12–1.97 (m, 1H), 1.96–1.84 (m, 1H), 1.75–1.62 (m, 1H), 1.38 (ddd, J = 14.1, 8.5, 2.9 Hz, 1H), 1.19 (ddd, J = 14.0, 10.9, 1.8 Hz, 1H), 0.89 (s, 8H), 0.84 (d, J = 6.5 Hz, 3H), 0.84 (s, 9H), 0.04 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 137.7, 115.7, 78.6, 43.1, 40.6, 35.8, 29.6, 26.6, 26.4, 19.2, 18.7, −3.2, −3.5. HRMS (ESI) calculated for C17H36OSi Na+ [M + Na]+ 307.2428, found 307.2423.
3.2.8. Synthesis of (4S,6R)-6-((tert-butyldimethylsilyl)oxy)-4,7,7-trimethyloctan-1-ol (14)
The product 14 was synthesised according to the procedures for the synthesis of 11 from 13 (2.58 g, 9.08 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1) to afford alcohol 14 (2.33 g, 85%) as a colorless oil. TLC: Rf = 0.5 (hexanes/EtOAc = 4:1), PMA stain. = +11.1 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 3.62 (td, J = 6.8, 1.1 Hz, 2H), 3.29 (dd, J = 8.6, 1.7 Hz, 1H), 1.65–1.48 (m, 4H), 1.36 (ddd, J = 14.0, 8.6, 2.8 Hz, 1H), 1.25 (ddd, J = 7.3, 6.2, 2.5 Hz, 1H), 1.25–1.12 (m, 2H), 0.89 (s, 9H), 0.84 (d, J = 7.3 Hz, 3H), 0.83 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 78.6, 63.5, 41.0, 35.7, 34.7, 30.5, 29.3, 26.6, 26.4, 19.4, 18.7, −3.1, −3.5. HRMS (ESI) calculated for C17H38O2SiNa+ [M + Na]+ 325.2533, found 325.2534.
3.2.9. Synthesis of (4S,6R)-6-((tert-butyldimethylsilyl)oxy)-4,7,7-trimethyloctanal (15)
The product 15 was synthesised according to the procedures for the synthesis of 12 from 14 (2.0 g, 6.6 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 20:1) to afford aldehyde 15 (1.68 g, 85%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 10:1), PMA stain. = +12.7 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.76 (t, J = 1.8 Hz, 1H), 3.29 (dd, J = 8.6, 1.7 Hz, 1H), 2.51–2.30 (m, 2H), 1.68–1.43 (m, 3H), 1.35 (ddd, J = 13.9, 8.6, 2.5 Hz, 1H), 1.20 (ddd, J = 14.0, 10.4, 1.8 Hz, 1H), 0.88 (s, 9H), 0.85 (d, J = 6.1 Hz, 3H), 0.83 (s, 9H), 0.03 (s, 3H), 0.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 202.9, 78.4, 41.9, 40.7, 35.7, 30.5, 29.2, 26.5, 26.4, 19.2, 18.7, −3.2, −3.5. HRMS (ESI) calculated for C17H38O2SiNa+ [M + Na]+ 323.2377, found 323.2376.
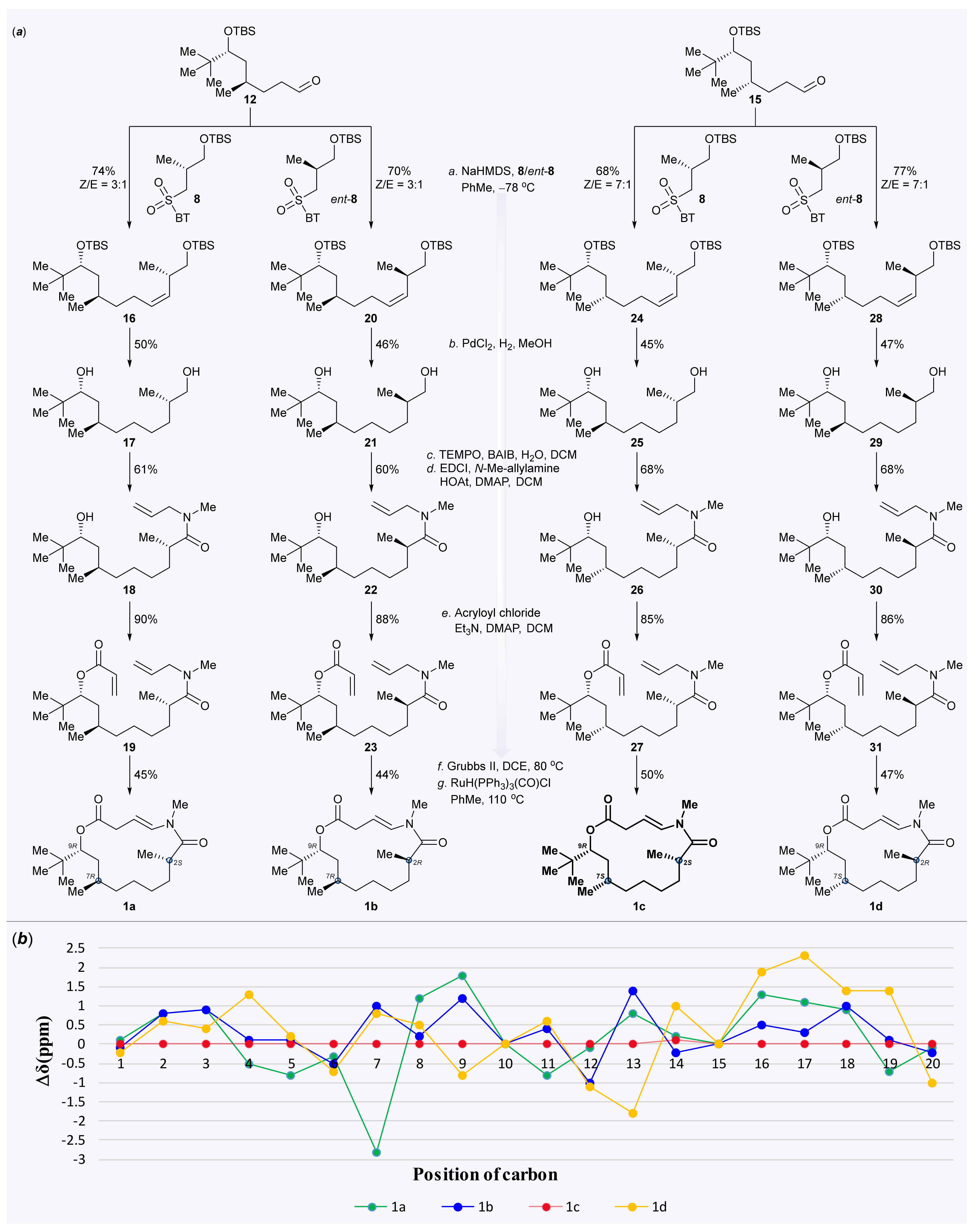
3.2.10. Synthesis of (6S,11R,13R,Z)-13-(tert-butyl)-2,2,3,3,6,11,15,15,16,16-decamethyl-4,14-dioxa-3,15-disilaheptadec-7-ene (16)
To a cooled (−78 °C) stirring solution of sulfone
8 [44] (200 mg, 0.52 mmol, 1.2 eq.) in dry toluene (4 mL, 0.1 M), NaHMDS (2 M in THF, 0.26 mL, 0.52 mmol, 1.2 eq.) was added dropwise for 1 h, followed by a solution of aldehyde
12 (130 mg, 0.43 mmol, 1.0 eq.) in dry toluene (2 mL, 0.22 M). The reaction mixture was stirred at −78 °C for 3 h and then quenched with a saturated aqueous solution of NH
4Cl (10 mL). The layers were separated and the aqueous layer was extracted with MTBE (3 × 15 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 100:1) to afford olefin
16 (149 mg, 74%) as a colorless oil. TLC: R
f = 0.6 (hexanes/ EtOAc = 80:1), iodine and PMA stain.
= +15.8 (
c 0.01, CHCl
3).
1H NMR (400 MHz, CDCl
3 as a mixture of
Z/E = 3:1) δ (5.53–5.27 (m) and 5.14 (ddt,
J = 11.0, 9.5, 1.6 Hz), 2H), 3.47 (ddd,
J = 9.7, 6.0, 4.9 Hz, 1H), 3.35 (ddd,
J = 9.8, 7.3, 5.3 Hz, 1H), 3.30 (dd,
J = 7.1, 2.9 Hz, 1H), (2.69–2.54 (m) and 2.33–2.21 (m), 1H), 2.21–2.08 (m, 1H), 2.12–1.98 (m, 1H), 2.02–1.86 (m, 1H), 1.64–1.51 (m, 1H), 1.52–1.37 (m, 2H), 1.24–1.12 (m, 1H), 1.09–0.97 (m, 1H), (0.97 (d,
J = 6.8 Hz) and 0.96 (d,
J = 6.7 Hz), 3H), 0.92 (d,
J = 6.6 Hz, 3H), 0.90 (m, 18H), 0.85 (s, 9H), 0.09 (m, 12H).
13C NMR (100 MHz, CDCl
3) δ 132.8, 132.6, 130.5, 130.4, 78.7, 78.6, 68.5, 68.2, 41.9, 41.8, 39.5, 36.7, 36.3, 36.0, 35.0, 30.2, 30.0, 29.7, 26.5, 26.4, 26.2, 25.1, 20.9, 20.8, 18.7, 18.6, 17.7, 16.9, −3.2, −3.7, −5.1, −5.1. HRMS (ESI) calculated for C
27H
58O
2Si
2Na
+ [M + Na]
+ 493.3869, found 493.3869.
3.2.11. Synthesis of (2S,7R,9R)-2,7,10,10-tetramethylundecane-1,9-diol (17)
To a solution of alkene 16 (149 mg, 0.317 mmol, 1.0 eq.) in anaerobic MeOH (10 mL, 0.03 M), PdCl2 (17 mg, 0.095 mmol, 0.3 eq.) was added under an argon atmosphere. The reaction flask was evacuated and purged with H2 three times and then the reaction was stirred at ambient temperature under a hydrogen atmosphere for 10 h. The reaction flask was then evacuated and purged with nitrogen three times. The catalyst was removed via filtration through Celite. The filter cake was rinsed thoroughly with MeOH and the filtrate was concentrated in vacuo to provide the crude product. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford diol 17 (39 mg, 50%) as a colorless oil. TLC: Rf = 0.3 (hexanes/ EtOAc = 3:1), PMA stain. = +22.6 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.48 (dd, J = 10.5, 5.8 Hz, 1H), 3.42 (dd, J = 10.5, 6.3 Hz, 1H), 3.28 (dd, J = 10.2, 1.8 Hz, 1H), 1.65–1.57 (m, 3H), 1.45–1.39 (m, 2H), 1.38–1.33 (m, 3H), 1.25–1.19 (m, 3H), 1.18–1.14 (m, 1H), 1.11–1.05 (m, 1H), 1.03–0.99 (m, 1H), 0.92 (d, J = 6.7 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H) 0.87 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 77.7, 68.4, 39.4, 35.9, 35.4, 35.1, 33.2, 29.9, 27.4, 27.1, 25.8, 21.2, 16.8. HRMS (ESI) calculated for C15H32O2Na+ [M + Na]+ 267.2295, found 267.2293.
3.2.12. Synthesis of (2S,7R,9R)-9-hydroxy-2,7,10,10-tetramethylundecanoic acid (S14)
To a solution of diol 17 (53 mg, 0.217 mmol, 1.0 eq.) in DCM (3 mL, 0.07 M), TEMPO (7 mg, 0.043 mmol, 0.2 eq.), H2O (0.2 mL, 11.0 mmol, 50 eq.) and PhI(OAc)2 (175 mg, 0.54 mmol, 2.5 eq.) were sequentially added and stirred at room temperature for 20 h before it was quenched with saturated aqueous solution of Na2S2O3 (3 mL) and extracted with DCM (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1–5:1) to afford the corresponding acid S14 (51 mg, 90%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), PMA stain. = +20.6 (c 0.01, CHCl3). 1H NMR δ 3.29 (dd, J = 10.3, 1.8 Hz, 1H), 2.52–2.36 (m, 1H), 1.77–1.64 (m, 1H), 1.67–1.57 (m, 1H), 1.49–1.37 (m, 2H), 1.40–1.27 (m, 4H), 1.25–1.13 (m, 2H), 1.16 (d, J = 7.0 Hz, 3H), 1.08–0.95 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.87 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 182.7, 77.8, 39.5, 39.3, 35.2, 35.0, 33.6, 29.8, 27.5, 26.7, 25.7, 21.1, 17.1. HRMS (ESI) calculated for C15H30O3Na+ [M + Na]+ 281.2087, found 281.2091.
3.2.13. Synthesis of (2S,7R,9R)-N-allyl-9-hydroxy-N,2,7,10,10-pentamethylundecanamide (18)
To a solution of acid S14 (39 mg, 0.15 mmol, 1.0 eq.) and N-allylmethylamine (29 μL, 0.30 mmol, 2.0 eq.) in dry DCM (2 mL, 0.08 M), HOAt (41 mg, 0.30 mmol, 2.0 eq.), DMAP (56 mg, 0.46 mmol, 3.0 eq.) and EDCI (58 mg, 0.30 mmol, 2.0 eq.) were sequentially added at 0 °C. The reaction mixture was allowed to stir at room temperature for 12 h, quenched with saturated aqueous solution of NaHCO3 (5 mL) and extracted with DCM (3 × 5 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford the amide 18 (32 mg, 68%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 2:1), iodine and PMA stain. = +26.0 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 5.82–5.66 (m, 1H), 5.24–5.04 (m, 2H), 4.07–3.94 (m, 1H), 3.94–3.90 (m, 1H), 3.34–3.20 (m, 1H), (2.97 and 2.91 (s), 3H), 2.74–2.52 (m, 1H), 1.87 (s, 1H), 1.78–1.66 (m, 1H), 1.65–1.55 (m, 1H), 1.51–1.40 (m, 1H), 1.36–1.32 (m, 1H), 1.32–1.29 (m, 1H), 1.29–1.26 (m, 1H), 1.26–1.21 (m, 2H), 1.21–1.19 (m, 1H), 1.18–1.12 (m, 1H), (1.09 and 1.08 (d, J = 6.7 Hz), 3H), 1.04–0.92 (m, 1H), (0.89 and 0.89 (d, J = 6.7 Hz), 3H), 0.87 (s, 9H). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.3 and 176.6, 133.4 and 133.1, 117.0 and 116.6, 77.4, 52.2 and 50.3, 39.5 and 39.5, 36.0 and 35.8, 35.1 and 35.0, 34.9 and 34.8, 34.4, 34.1 and 34.0, 29.5 and 29.5, 27.7 and 27.7, 26.8 and 26.8, 25.8 and 25.8, 21.2 and 21.2, 18.4 and 17.8. HRMS (ESI) calculated for C19H37NO2Na+ [M + Na]+ 334.2717, found 334.2717.
3.2.14. Synthesis of (3R,5R,10S)-11-(allyl(methyl)amino)-2,2,5,10-tetramethyl-11-oxoundecan-3-yl acrylate (19)
To a solution of alcohol 18 (32 mg, 0.1 mmol, 1.0 eq.) in dry DCM (2 mL, 0.05 M), Et3N (69 μL, 0.5 mmol, 5.0 eq.), DMAP (2.4 mg, 0.02 mmol, 0.2 eq.) and acryloyl chloride (24 μL, 0.3 mmol, 3.0 eq.) were sequentially added at 0 °C. The reaction mixture was allowed to stir at room temperature for 12 h, quenched with water (1 mL) and extracted with DCM (3 × 3 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford ester 19 (33 mg, 90%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), iodine and PMA stain. = +23.6 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 6.37 (dd, J = 17.3, 1.6 Hz, 1H), 6.11 (dd, J = 17.3, 10.4 Hz, 1H), 5.80 (dd, J = 10.4, 1.6 Hz, 1H), 5.80–5.66 (m, 1H), 5.26–5.04 (m, 2H), 4.88 (ddd, J = 7.9, 4.3, 1.1 Hz, 1H), 4.06–3.95 (m, 1H), 3.98–3.89 (m, 1H), (2.97 and 2.92 (s, 3H)), 2.75–2.52 (m, 1H), 1.71–1.58 (m, 1H), 1.51–1.36 (m, 3H), 1.37–1.23 (m, 3H), 1.27–1.10 (m, 3H), (1.09 and 1.08 (d, J = 6.8 Hz, 3H)), 1.06–0.94 (m, 1H), 0.88 (s, 9H), (0.85 and 0.84 (d, J = 6.5 Hz, 3H)). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.4 and 176.7, 166.2, 133.5 and 133.2, 130.3, 129.1, 117.0 and 116.6, 79.2, 52.2 and 50.2, 37.3, 35.9 and 35.7, 35.7, 35.0, 34.8 and 34.7, 34.4 and 34.0, 29.8, 28.1 and 28.1, 27.0 and 27.0, 26.0, 20.9, 18.24 and 17.6. HRMS (ESI) calculated for C22H39NO3Na+ [M + Na]+ 388.2822, found 388.2822.
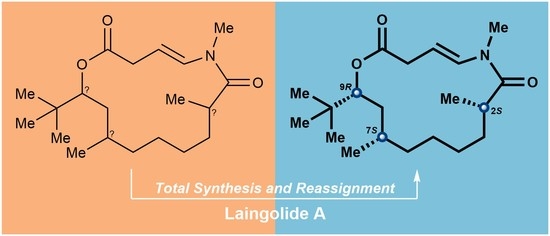
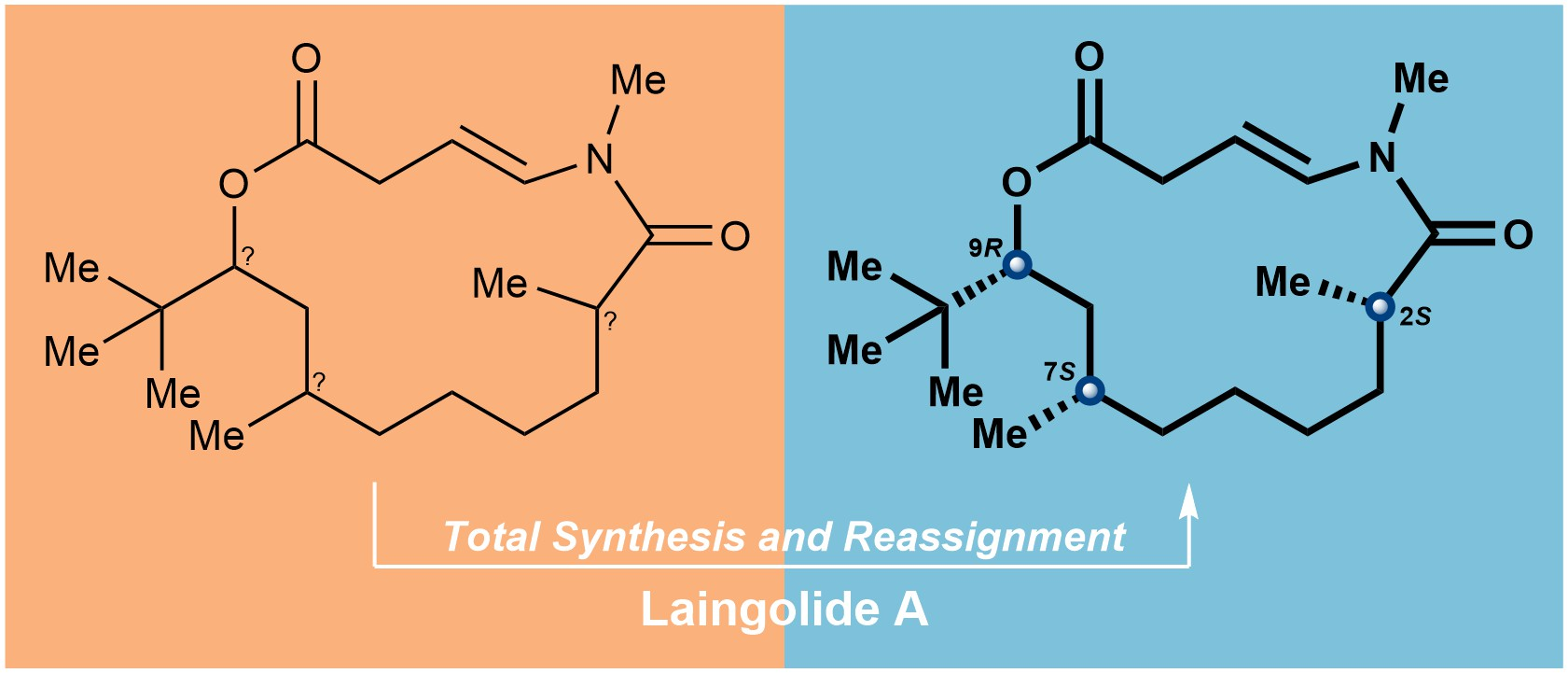
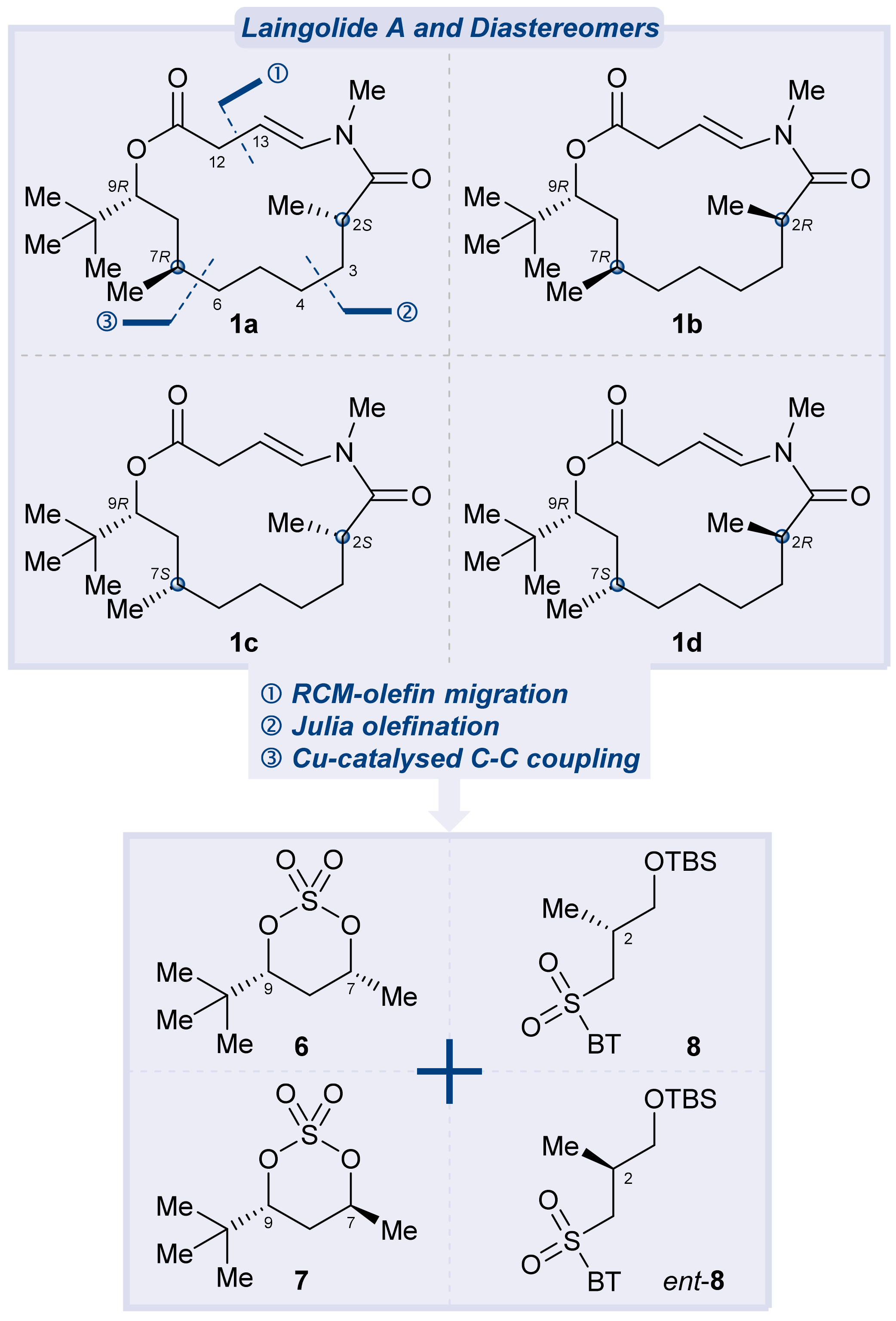
3.2.15. Synthesis of (2S,7R,9R)-laingolide A (1a)
To a solution of diene 19 (33 mg, 0.09 mmol, 1.0 eq.) in DCE (90 mL, 0.001 M) at room temperature, second-generation Grubbs catalyst (G-II) (7.6 mg, 0.009 mmol, 0.1 eq.) was added. The reaction mixture was heated at 80 °C for 24 h and then a second portion of G-II (7.6 mg, 0.009 mmol, 0.1 eq.) was added. The reaction mixture was kept at 80 °C, stirred for another 24 h and then concentrated under reduced pressure. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford an inseparable mixture of the desired product S15 and a minor unidentified byproduct as a white solid.
To a solution of the above mixture of S15 and a minor unidentified byproduct in degassed dry toluene (1 mL) under argon, a solution of RuH(PPh3)3(CO)Cl (8.6 mg, 0.009 mmol, 0.1 eq.) was added in degassed dry toluene (7 mL). The reaction mixture was heated to reflux for 24 h, cooled to room temperature, then concentrated under reduced pressure to provide the crude product. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford (2S,7R,9R)-laingolide A (1a) (13.6 mg, 45% for two steps) as a white solid. TLC: Rf = 0.6 (hexanes/ EtOAc = 2:1), UV and PMA stain. = +301 (c 0.01, MeOH). 1H NMR (400 MHz, CDCl3) δ 6.76 (d, J = 13.8 Hz, 1H), 5.21 (ddd, J = 13.8, 9.4, 6.0 Hz, 1H), 4.94 (dd, J = 11.1, 2.5 Hz, 1H), 3.17–3.08 (m, 1H), 3.11 (s, 3H), 2.98 (ddd, J = 16.0, 9.5, 0.8 Hz, 1H), 2.86–2.72 (m, 1H), 1.80–1.63 (m, 2H), 1.59–1.52 (m, 1H), 1.46–1.40 (m, 1H), 1.40–1.33 (m, 2H), 1.32–1.27 (m, 2H), 1.25–1.17 (m, 2H), 1.15 (d, J = 6.5 Hz, 3H), 0.96–0.91 (m, 1H), 0.89 (d, J = 6.8 Hz, 3H), 0.88 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 176.3, 173.3, 133.4, 104.2, 77.9, 37.7, 37.1, 35.7, 35.4, 34.3, 34.2, 31.3, 30.5, 27.6, 27.4, 26.7, 20.1, 17.2. HRMS (ESI) calculated for C20H35NO3Na+ [M + Na]+ 360.2509, found 360.2510.
3.2.16. Synthesis of (6R,11R,13R,Z)-13-(tert-butyl)-2,2,3,3,6,11,15,15,16,16-decamethyl-4,14-dioxa-3,15-disilaheptadec-7-ene (20)
Product 20 was synthesised according to the procedures for the synthesis of 16 from 12 (910 mg, 3.0 mmol, 1.0 eq.) and sulfone ent-8 (1.4 g, 3.6 mmol, 1.2 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 100:1) to afford 20 (998 mg, 70%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 80:1), iodine and PMA stain. = +1.10 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a mixture of Z/E = 3:1) δ (5.50–5.26 (m) and 5.14 (ddd, J = 10.9, 9.4, 1.5 Hz) 2H), 3.55–3.42 (m, 1H), 3.41–3.31 (m, 1H), 3.34–3.27 (m, 1H), (2.78–2.54 (m) and 2.37–2.20 (m), 1H), 2.22–2.03 (m, 1H), 2.07–1.94 (m, 1H), 1.69–1.51 (m, 1H), 1.51–1.37 (m, 2H), 1.26–1.13 (m, 1H), 1.10–0.98 (m, 1H), (0.96 (d, J = 6.7 Hz) and 0.96 (d, J = 6.7 Hz), 3H), 0.95–0.86 (m, 21H), 0.88–0.83 (m, 9H), 0.07–0.03 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 132.8, 132.6, 130.5, 130.4, 78.7, 78.6, 68.5, 68.2, 41.9, 39.5, 36.7, 36.4, 36.0, 36.0, 35.0, 30.3, 30.0, 29.8, 26.6, 26.4, 26.2, 26.2, 25.2, 20.9, 18.7, 18.7, 18.6, 18.5, 17.7, 16.9, −3.2, −3.2, −3.7, −5.1, −5.1. HRMS (ESI) calculated for C27H58O2Si2Na+ [M + Na]+ 493.3869, found 493.3866.
3.2.17. Synthesis of (2R,7R,9R)-2,7,10,10-tetramethylundecane-1,9-diol (21)
Product 21 was synthesised according to the procedures for the synthesis of 17 from 20 (250 mg, 0.53 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford 21 (59.5 mg, 46%) as a colorless oil. TLC: Rf = 0.3 (hexanes/EtOAc = 3:1), PMA stain. = +27.7 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.47 (dd, J = 10.4, 5.9 Hz, 1H), 3.39 (dd, J = 10.5, 6.5 Hz, 1H), 3.26 (dd, J = 10.3, 1.8 Hz, 1H), 1.80 (s, 2H), 1.72–1.51 (m, 2H), 1.47–1.35 (m, 2H), 1.37–1.21 (m, 5H), 1.21–1.06 (m, 2H), 1.01 (ddd, J = 18.2, 9.5, 3.4 Hz, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H), 0.86 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 77.7, 68.4, 39.5, 35.8, 35.5, 35.0, 33.2, 30.0, 27.2, 27.1, 25.8, 21.1, 16.6. HRMS (ESI) calculated for C15H32O2Na+ [M + Na]+ 267.2295, found 267.2293.
3.2.18. Synthesis of (2R,7R,9R)-9-hydroxy-2,7,10,10-tetramethylundecanoic acid (S16)
Product S16 was synthesised according to the procedures for the synthesis of S14 from 21 (258 mg, 1.06 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1–5:1) to afford S16 (246 mg, 90%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), PMA stain. = +17.7 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 3.28 (dd, J = 10.3, 1.8 Hz, 1H), 2.53–2.32 (m, 1H), 1.82–1.56 (m, 2H), 1.49–1.37 (m, 2H), 1.39–1.17 (m, 5H), 1.21–1.13 (m, 1H), 1.16 (d, J = 6.9 Hz, 3H), 1.09–0.95 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.87 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 182.8, 77.9, 39.5, 39.3, 35.4, 35.0, 33.7, 30.0, 27.6, 26.8, 25.8, 21.1, 16.9. HRMS (ESI) calculated for C15H30O3Na+ [M + Na]+ 281.2087, found 281.2088.
3.2.19. Synthesis of (2R,7R,9R)-N-allyl-9-hydroxy-N,2,7,10,10-pentamethylundecanamide (22)
Product 22 was synthesised according to the procedures for the synthesis of 18 from S16 (105 mg, 0.4 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford 22 (83.4 mg, 67%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 2:1), iodine and PMA stain. = +10.6 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 6.04–5.59 (m, 1H), 5.26–4.94 (m, 2H), 4.14–3.94 (m, 1H), 3.97–3.88 (m, 1H), 3.26 (dd, J = 10.2, 1.7 Hz, 1H), (2.97 and 2.92 (s, 3H)), 2.79–2.48 (m, 1H), 1.80–1.55 (m, 3H), 1.44–1.28 (m, 3H), 1.29–1.25 (m, 1H), 1.24 (s, 1H), 1.22–1.12 (m, 3H), (1.10 and 1.08 (d, J = 6.7 Hz, 3H)), 1.05–0.95 (m, 1H), (0.91 and 0.90 (d, J = 6.8 Hz, 3H)), 0.87 (s, 9H). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.3 and 176.7, 133.4 and 133.2, 117.0 and 116.6, 77.7, 52.1 and 50.2, 39.4, 35.9 and 35.7, 35.6, 35.0, 34.8 and 34.5, 34.2 and 34.0, 30.1 and 30.1, 28.1 and 28.1, 27.1 and 27.0, 25.8 and 25.8, 21.2, 18.2 and 17.6. HRMS (ESI) calculated for C19H37NO2Na+ [M + Na]+ 334.2717, found 334.2718.
3.2.20. Synthesis of (3R,5R,10R)-11-(allyl(methyl)amino)-2,2,5,10-tetramethylundecan-3-yl acrylate (23)
Product 23 was synthesised according to the procedures for the synthesis of 19 from 22 (63 mg, 0.2 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford 23 (64.3 mg, 88%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), iodine and PMA stain. = +8.9 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 6.37 (dd, J = 17.3, 1.6 Hz, 1H), 6.11 (dd, J = 17.3, 10.4 Hz, 1H), 5.80 (dd, J = 10.4, 1.6 Hz, 1H), 5.79–5.66 (m, 1H), 5.27–5.04 (m, 2H), 4.94–4.82 (m, 1H), 4.06–3.95 (m, 1H), 3.97–3.84 (m, 1H), (2.97 and 2.91 (s, 3H)), 2.76–2.52 (m, 1H), 1.71–1.58 (m, 1H), 1.41 (ddd, J = 7.2, 5.6, 1.9 Hz, 2H), 1.39–1.26 (m, 1H), 1.27–1.16 (m, 5H), (1.10 and 1.08 (d, J = 6.7 Hz, 1H)), 1.06–0.95 (m, 1H), 0.88 (s, 9H), (0.85 and 0.84 (d, J = 6.5 Hz, 3H)). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.4 and 176.7, 166.3, 133.5 and 133.2, 130.3, 129.1, 117.0 and 116.6, 79.2, 52.2 and 50.2, 37.2, 35.9 and 35.7, 35.5 and 35.0, 34.8 and 34.6, 34.3, 33.9, 29.7 and 29.7, 28.0 and 28.0, 26.8 and 26.8, 26.0, 20.9, 18.2 and 17.61. HRMS (ESI) calculated for C22H39NO3Na+ [M + Na]+ 388.2822, found 388.2822.
3.2.21. Synthesis of (2R,7R,9R)-laingolide A (1b)
Product S17 was synthesised according to the procedures for the synthesis of S15 from 23 (10.3 mg, 0.027 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford an inseparable mixture of the desired product S17 and a minor unidentified byproduct (9 mg) as a white solid.
(2R,7R,9R)-laingolide A (1b) was synthesised according to the procedures for the synthesis of (2S,7S,9R)-laingolide A (1a) from the above mixture of S17 and a minor unidentified byproduct. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford (2R,7R,9R)-laingolide A (1b) (4 mg, 44% for two steps) as a white solid. TLC: Rf = 0.6 (hexanes/EtOAc = 2:1), UV and PMA stain. = −76.3 (c 0.005, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.09 (d, J = 13.9 Hz, 1H), 5.16 (dt, J = 14.1, 7.2 Hz, 1H), 4.92 (dd, J = 11.9, 1.8 Hz, 1H), 3.09 (s, 3H), 3.05 (ddd, J = 13.3, 7.4, 0.9 Hz, 1H), 2.99 (ddd, J = 13.3, 6.9, 1.4 Hz, 1H), 2.91 (dt, J = 13.4, 6.5 Hz, 1H), 1.65 – 1.61 (m, 0H), 1.58 – 1.51 (m, 2H), 1.46 – 1.36 (m, 1H), 1.33 – 1.25 (m, 3H), 1.25 – 1.18 (m, 2H), 1.15 (d, J = 6.6 Hz, 3H), 1.10 – 0.99 (m, 1H), 0.88 (s, 9H), 0.85 – 0.79 (m, 1H), 0.78 (d, J = 6.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 176.5, 172.1, 133.8, 103.6, 78.5, 38.6, 37.3, 35.7, 35.4, 35.3, 34.1, 31.4, 26.7, 26.7, 26.6, 26.1, 20.9, 18.0. HRMS (ESI) calculated for C20H35NO3Na+ [M + Na]+ 360.2509, found 360.2510.
3.2.22. Synthesis of (6S,11S,13R,Z)-13-(tert-butyl)-2,2,3,3,6,11,15,15,16,16-decamethyl-4,14-dioxa-3,15-disilaheptadec-7-ene (24)
Product 24 was synthesised according to the procedures for the synthesis of 16 from 15 (780 mg, 2.6 mmol, 1.0 eq.) and sulfone 8 (1.2 g, 3.1 mmol, 1.2 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 100:1) to afford 24 (831 mg, 68%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 80:1), iodine and PMA stain. = +18.8 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a mixture of Z/E = 7:1) δ (5.45–5.28 (m) and 5.12 (ddt, J = 10.9, 9.4, 1.6 Hz), 2H), 3.46 (dd, J = 9.7, 5.9 Hz, 1H), 3.35 (dd, J = 9.8, 7.4 Hz, 1H), 3.31 (dd, J = 8.4, 1.7 Hz, 2H), (2.72–2.57 (m) and 2.33–2.21 (m), 1H), 2.17–1.96 (m, 2H), 1.68–1.54 (m, 1H), 1.41–1.25 (m, 2H), 1.29–1.15 (m, 2H), 0.96 (d, J = 6.8 Hz, 3H), 0.91 (s, 18H), 0.85 (d, J = 6.2 Hz, 12H), 0.05 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 132.5, 130.6, 78.7, 68.2, 41.2, 39.0, 35.8, 35.0, 29.4, 26.6, 26.5, 26.2, 25.4, 19.1, 17.7, −3.1, −3.5, −5.1, −5.1. HRMS (ESI) calculated for C27H58O2Si2Na+ [M + Na]+ 493.3869, found 493.3868.
3.2.23. Synthesis of (2S,7S,9R)-2,7,10,10-tetramethylundecane-1,9-diol (25)
Product 25 was synthesised according to the procedures for the synthesis of 17 from 24 (235 mg, 0.5 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford 25 (55 mg, 45%) as a colorless oil. TLC: Rf = 0.3 (hexanes/EtOAc = 3:1), PMA stain. = +14.4 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 3.48 (dd, J = 10.6, 5.8 Hz, 1H), 3.39 (ddd, J = 10.3, 6.5, 0.8 Hz, 1H), 3.27 (dd, J = 10.6, 1.8 Hz, 1H), 1.74–1.53 (m, 4H), 1.46–1.33 (m, 2H), 1.32–1.25 (m, 4H), 1.24–1.08 (m, 4H), 0.89 (d, J = 6.7 Hz, 3H), 0.86 (d, J = 7.1 Hz, 3H), 0.87 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 77.4, 68.4, 39.1, 38.5, 35.8, 34.9, 33.2, 29.7, 27.4, 27.3, 25.8, 19.0, 16.7. HRMS (ESI) calculated for C15H32O2Na+ [M + Na]+ 267.2295, found 267.2293.
3.2.24. Synthesis of (2S,7S,9R)-9-hydroxy-2,7,10,10-tetramethylundecanoic acid (S18)
Product S18 was synthesised according to the procedures for the synthesis of S14 from 25 (50 mg, 0.21 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1–5:1) to afford S18 (47.8 mg, 91%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), PMA stain. = +13.8 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.29 (dd, J = 10.6, 1.7 Hz, 1H), 2.52–2.35 (m, 1H), 1.72–1.64 (m, 1H), 1.64–1.56 (m, 1H), 1.47–1.39 (m, 1H), 1.35–1.28 (m, 5H), 1.26–1.23 (m, 1H), 1.23–1.10 (m, 2H), 1.16 (d, J = 7.0 Hz, 3H), 0.87 (s, 9H), 0.86 (d, J = 6.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 182.6, 77.6, 39.5, 38.9, 38.3, 34.9, 33.7, 29.5, 27.4, 27.0, 25.8, 19.1, 17.0. HRMS (ESI) calculated for C15H30O3Na+ [M + Na]+ 281.2087, found 281.2087.
3.2.25. Synthesis of (2S,7S,9R)-N-allyl-9-hydroxy-N,2,7,10,10-pentamethylundecanamide (26)
Product 26 was synthesised according to the procedures for the synthesis of 18 from S18 (47.8 mg, 0.182 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford 26 (42.5 mg, 75%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 2:1), iodine and PMA stain. = +16.7 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 5.82–5.65 (m, 1H), 5.30–4.98 (m, 2H), 4.07–3.92 (m, 1H), 3.91 (d, J = 4.7 Hz, 1H), 3.26 (d, J = 10.5 Hz, 1H), (2.96 and 2.90 (s, 3H)), 2.73–2.51 (m, 1H), 1.74–1.62 (m, 1H), 1.61–1.55 (m, 1H), 1.40–1.27 (m, 3H), 1.27–1.18 (m, 5H), 1.17–1.10 (m, 2H), (1.09 and1.07 (d, J = 6.9 Hz, 3H)), 0.86 (s, 9H), 0.84 (d, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.3 and 176.6, 133.4 and 133.2, 117.0 and 116.6, 77.3, 52.1 and 50.2, 39.0, 38.4, 35.9 and 35.7, 34.9 and 34.8, 34.5 and 34.2, 33.9, 29.7, 28.0 and 27.9, 27.3, 25.8, 19.0, 18.2 and 17.61. HRMS (ESI) calculated for C19H37NO2Na+ [M + Na]+ 334.2717, found 334.2721.
3.2.26. Synthesis of (3R,5S,10S)-11-(allyl(methyl)amino)-2,2,5,10-tetramethyl-11-oxoundecan-3-yl acrylate (27)
Product 27 was synthesised according to the procedures for the synthesis of 19 from 26 (116 mg, 0.372 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford 27 (115 mg, 85%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), iodine and PMA stain. = +8.8 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 6.37 (dd, J = 17.4, 1.5 Hz, 1H), 6.11 (dd, J = 17.3, 10.4 Hz, 1H), 5.80 (dd, J = 10.4, 1.7 Hz, 1H), 5.72 (ddt, J = 16.5, 11.5, 5.9 Hz, 1H), 5.24–5.02 (m, 2H), 4.89 (d, J = 10.9 Hz, 1H), 4.08–3.93 (m, 1H), 3.95–3.87 (m, 1H), (2.95 and 2.91 (s, 3H)), 2.72–2.46 (m, 1H), 1.74–1.1 (m, 1H), 1.58–1.52 (m, 1H), 1.36–1.29 (m, 1H), 1.27–1.12 (m, 8H), (1.08 and 1.06 (d, J = 6.5 Hz, 3H)), 0.86 (s, 12H). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.2 and 176.6, 166.3, 133.4 and 133.1, 130.4, 129.0, 117 and 116.6, 79.0, 52.1 and 50.2, 38.1 and 37.1, 35.9 and 35.7, 34.8 and 34.8, 34.5, 34.2, 33.9, 29.5, 27.9 and 27.9, 27.2, 26.1, 19.3, 18.2 and 17.6. HRMS (ESI) calculated for C22H39NO3Na+ [M + Na]+ 388.2822, found 388.2824.
3.2.27. Synthesis of (2S,7S,9R)-laingolide A (1c)
Product S19 was synthesised according to the procedures for the synthesis of S15 from 27 (28 mg, 0.077 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford an inseparable mixture of the desired product S19 and a minor unidentified byproduct (20 mg) as a white solid.
(2S,7S,9R)-laingolide A (1c) was synthesised according to the procedures for the synthesis of (2S,7R,9R)-laingolide A (1a) from the above mixture of S19 and a minor unidentified byproduct. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford (2S,7S,9R)-laingolide A (1c) (13 mg, 50% for two steps) as a white solid. TLC: Rf = 0.6 (hexanes/EtOAc = 2:1), UV and PMA stain. = +145.6 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.02 (dd, J = 13.7, 1.4 Hz, 1H), 5.18 (ddd, J = 13.6, 10.3, 5.7 Hz, 1H), 4.81 (dd, J = 11.3, 1.3 Hz, 1H), 3.10 (s, 3H), 3.07 (ddd, J = 12.3, 5.6, 1.5 Hz, 1H), 3.03–2.95 (m, 1H), 2.94 (dd, J = 12.3, 10.4 Hz, 1H), 1.64–1.57 (m, 1H), 1.60–1.52 (m, 1H), 1.47–1.34 (m, 1H), 1.35–1.27 (m, 1H), 1.30–1.17 (m, 4H), 1.15 (d, J = 6.5 Hz, 3H), 1.17–1.12 (m, 2H), 1.03–0.95 (m, 1H), 0.89 (s, 9H), 0.84 (d, J = 5.7 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 176.4, 172.5, 133.5, 105.0, 79.7, 37.6, 36.8, 36.6, 36.2, 35.5, 35.1, 31.2, 27.7, 26.8, 26.7, 26.2, 21.2, 18.5. HRMS (ESI) calculated for C20H35NO3Na+ [M + Na]+ 360.2509, found 360.2512.
3.2.28. Synthesis of (6R,11S,13R,Z)-13-(tert-butyl)-2,2,3,3,6,11,15,15,16,16-decamethyl-4,14-dioxa-3,15-disilaheptadec-7-ene (28)
Product 28 was synthesised according to the procedures for the synthesis of 16 from 15 (700 mg, 2.34 mmol, 1.0 eq.) and sulfone ent-8 (1.1 g, 2.8 mmol, 1.2 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 100:1) to afford 28 (847 mg, 77%) as a colorless oil. TLC: Rf = 0.6 (hexanes/EtOAc = 80:1), iodine and PMA stain. = −1.9 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a mixture of Z/E = 7:1) δ (5.50–5.26 (m), 5.11 (ddt, J = 11.0, 9.6, 1.6 Hz), 2H), 3.52–3.43 (m, 1H), 3.39–3.33 (m, 1H), 3.32–3.26 (m, 1H), (2.69–2.55 (m) and 2.32–2.20 (m), 1H), 2.14–1.90 (m, 2H), 1.69–1.49 (m, 1H), 1.38–1.32 (m, 1H), 1.31–1.24 (m, 1H), 1.26–1.12 (m, 2H), (0.97 (d, J = 6.8 Hz) and 0.95 (d, J = 6.7 Hz), 3H), 0.90 (s, 18H), 0.87–0.81 (m, 12H), 0.04 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 132.5, 130.5, 78.6, 68.2, 41.1, 39.0, 35.8, 35.0, 29.3, 26.6, 26.4, 26.1, 25.4, 19.2, 18.7, 18.5, 17.7, −3.1, −3.5, −5.1, −5.1. HRMS (ESI) calculated for C27H58O2Si2Na+ [M + Na]+ 493.3869, found 493.3866.
3.2.29. Synthesis of (2R,7S,9R)-2,7,10,10-tetramethylundecane-1,9-diol (29)
Product 29 was synthesised according to the procedures for the synthesis of 17 from 28 (250 mg, 0.53 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford 29 (60.8 mg, 47%) as a colorless oil. TLC: Rf = 0.3 (hexanes/ EtOAc = 3:1), PMA stain. = +19.1 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 3.44 (ddd, J = 10.2, 5.8, 1.7 Hz, 1H), 3.35 (ddd, J = 10.5, 6.5, 1.9 Hz, 1H), 3.24 (dt, J = 10.6, 1.5 Hz, 1H), 2.14 (d, J = 31.1 Hz, 2H), 1.68–1.50 (m, 2H), 1.42–1.30 (m, 2H), 1.30–1.19 (m, 5H), 1.17–0.98 (m, 3H), 0.87 (d, J = 6.7 Hz, 3H), 0.85–0.82 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 77.3, 68.2, 39.0, 38.4, 35.8, 34.9, 33.2, 29.6, 27.4, 27.3, 25.8, 19.0, 16.7. HRMS (ESI) calculated for C15H32O2Na+ [M + Na]+ 267.2295, found 267.2295.
3.2.30. Synthesis of (2R,7S,9R)-9-hydroxy-2,7,10,10-tetramethylundecanoic acid (S20)
Product S20 was synthesised according to the procedures for the synthesis of S14 from 29 (112 mg, 0.46 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 10:1–5:1) to afford S20 (108 mg, 91%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), PMA stain. = +8.4 (c 0.01, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.29 (dd, J = 10.6, 1.7 Hz, 1H), 2.51–2.37 (m, 1H), 1.78–1.57 (m, 2H), 1.46–1.38 (m, 1H), 1.36–1.28 (m, 5H), 1.28–1.23 (m, 1H), 1.22–1.18 (m, 1H), 1.16 (d, J = 6.9 Hz, 3H), 1.16–1.13 (m, 1H), 0.88 (s, 9H), 0.86 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 182.7, 77.6, 39.5, 39.0, 38.2, 34.9, 33.7, 29.6, 27.5, 27.0, 25.8, 19.0, 17.1. HRMS (ESI) calculated for C15H30O3Na+ [M + Na]+ 281.2087, found 281.2087.
3.2.31. Synthesis of (2R,7S,9R)-N-allyl-9-hydroxy-N,2,7,10,10-pentamethylundecanamide (30)
Product 30 was synthesised according to the procedures for the synthesis of 18 from S20 (116 mg, 0.45 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 3:1) to afford 30 (105 mg, 75%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 2:1), iodine and PMA stain. = +2.8 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 5.97–5.56 (m, 1H), 5.30–5.00 (m, 2H), 4.04–3.92 (m, 1H), 3.93 (dt, J = 4.9, 1.8 Hz, 1H), 3.27 (dt, J = 10.3, 1.4 Hz, 1H), (2.97 and 2.92 (s, 3H)), 2.79–2.43 (m, 1H), 1.79–1.53 (m, 3H), 1.38–1.23 (m, 6H), 1.21–1.13 (m, 3H), (1.10 and 1.08 (d, J = 6.8 Hz, 3H)), 0.87 (s, 9H), (0.85 and 0.85 (d, J = 6.5 Hz, 3H)). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.3 and 176.6, 133.4 and 133.2, 117.0 and 116.6, 77.4, 52.2 and 50.2, 39.1, 38.4 and 38.3, 35.9 and 35.7, 34.9 and 34.8, 34.5 and 34.3, 33.9, 29.6, 27.9 and 27.9, 27.3, 25.8, 19.0, 18.3 and 17.7. HRMS (ESI) calculated for C19H37NO2Na+ [M + Na]+ 334.2717, found 334.2716.
3.2.32. Synthesis of (3R,5S,10R)-11-(allyl(methyl)amino)-2,2,5,10-tetramethyl-11-oxoundecan-3-yl acrylate (31)
Product 31 was synthesised according to the procedures for the synthesis of 19 from 30 (88 mg, 0.28 mmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford 31 (88 mg, 86%) as a colorless oil. TLC: Rf = 0.4 (hexanes/EtOAc = 3:1), iodine and PMA stain. = +12.1 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 6.38 (dd, J = 17.3, 1.6 Hz, 1H), 6.12 (dd, J = 17.3, 10.4 Hz, 1H), 5.80 (dd, J = 10.3, 1.6 Hz, 1H), 5.78–5.66 (m, 1H), 5.33–5.04 (m, 2H), 4.90 (dd, J = 11.0, 1.2 Hz, 1H), 4.16–3.94 (m, 1H), 3.92 (dt, J = 4.9, 1.8 Hz, 1H), (2.96 and 2.91 (s, 3H)), 2.72–2.49 (m, 1H), 1.92–1.46 (m, 3H), 1.29–1.15 (m, 8H), (1.09 and 1.08 (d, J = 6.8 Hz, 3H)), 0.88 (s, 12H). 13C NMR (100 MHz, CDCl3 as a 1:1 mixture of two major conformers) δ 177.3 and 176.6, 166.4, 133.4 and 133.2, 130.4, 129.0, 117.0 and 116.6, 79.0, 52.1 and 50.2, 38.2, 37.1, 35.9 and 35.7, 34.9 and 34.8, 34.5 and 34.3, 33.9, 29.5, 28.0 and 27.9, 27.2, 26.1, 19.2, 18.2 and 17.6. HRMS (ESI) calculated for C22H39NO3Na+ [M + Na]+ 388.2822, found 388.2821.
3.2.33. Synthesis of (2R,7S,9R)-laingolide A (1d)
Product S21 was synthesised according to the procedures for the synthesis of S15 from 31 (30.9 mg, 85 μmol, 1.0 eq.). Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford an inseparable mixture of the desired product S21 and a minor unidentified byproduct (28 mg) as a white solid.
(2R,7S,9R)-laingolide A (1d) was synthesised according to the procedures for the synthesis of (2S,7R,9R)-laingolide A (1a) from the above mixture of the desired product S21 and a minor unidentified byproduct. Purification of the crude product was performed using flash chromatography on silica gel (hexanes/EtOAc = 5:1) to afford (2R,7S,9R)-laingolide A (1d) (13.4 mg, 47% for two steps) as a white solid. TLC: Rf = 0.6 (hexanes/EtOAc = 2:1), UV and PMA stain. = −55.3 (c 0.01, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.74 (d, J = 13.9 Hz, 1H), 5.21 (ddd, J = 13.8, 8.6, 6.3 Hz, 1H), 4.86 (dd, J = 10.9, 1.3 Hz, 1H), 3.20 (ddd, J = 16.5, 6.3, 1.4 Hz, 1H), 3.11 (s, 3H), 3.07 (ddd, J = 16.5, 8.7, 1.0 Hz, 1H), 2.68 (dqd, J = 12.9, 6.6, 4.4 Hz, 1H), 1.63–1.50 (m, 1H), 1.49–1.33 (m, 3H), 1.29–1.25 (m, 1H), 1.25–1.23 (m, 2H), 1.22–1.16 (m, 2H), 1.14 (d, J = 6.4 Hz, 4H), 1.14–1.05 (m, 1H), 0.88 (s, 9H), 0.85 (d, J = 5.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 176.6, 171.9, 132.6, 106.8, 80.5, 38.7, 37.5, 36.2, 35.6, 35.0, 33.7, 32.2, 26.9, 26.6, 25.3, 24.9, 18.9, 16.6. HRMS (ESI) calculated for C20H35NO3Na+ [M + Na]+ 360.2509, found 360.2513.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



