
Integrated Omics Strategy Reveals Cyclic Lipopeptides Empedopeptins from Massilia sp. YMA4 and Their Biosynthetic Pathway


 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
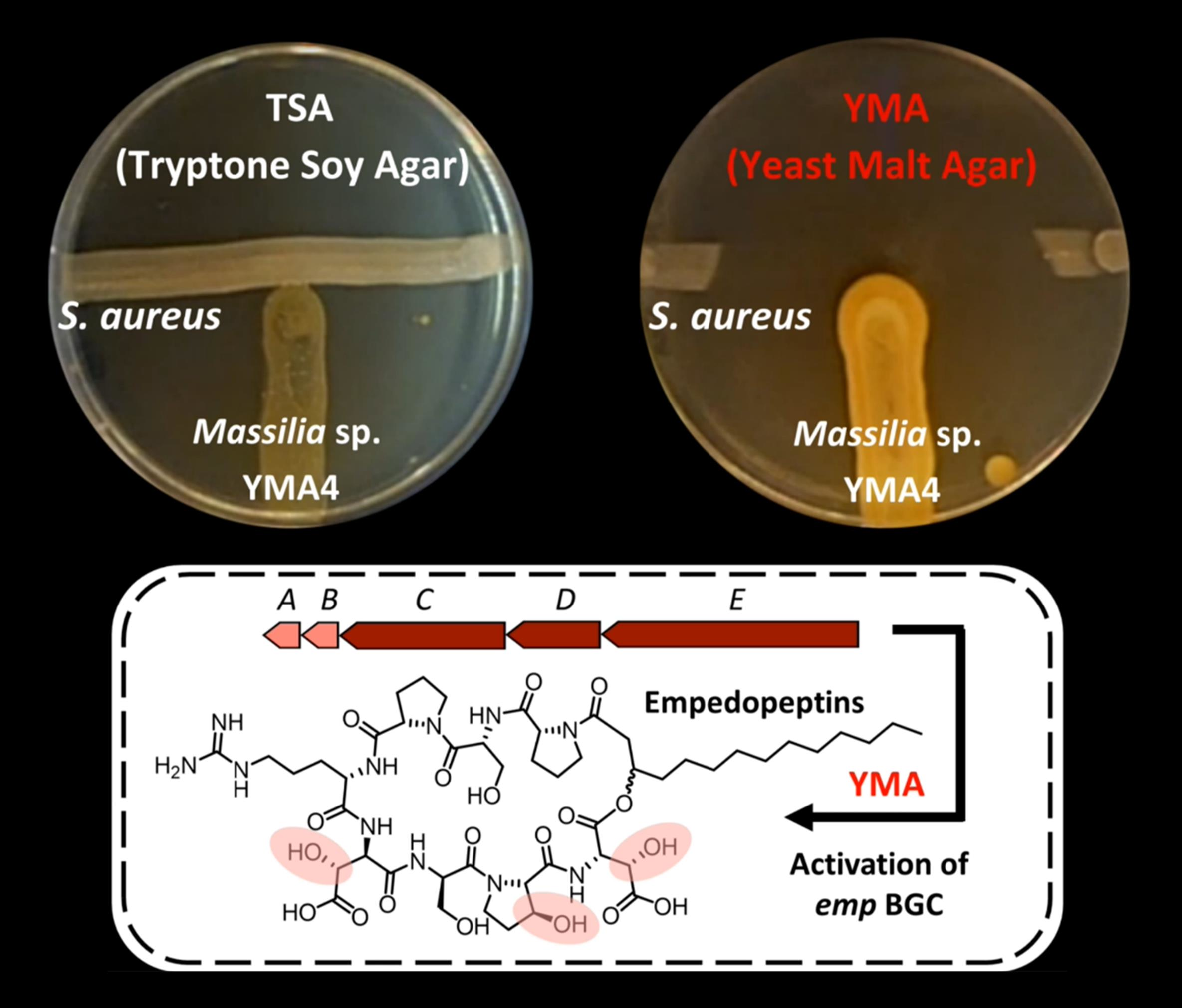
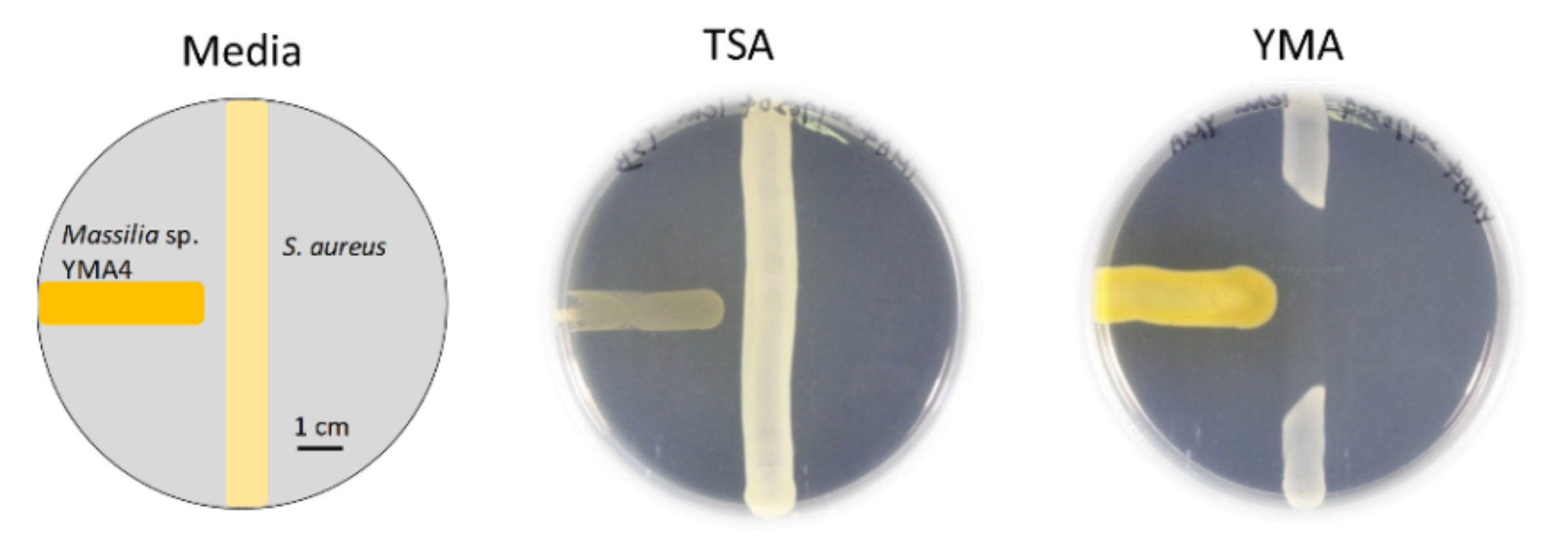
2.1. Antibiotic Activity of Massilia sp. YMA4
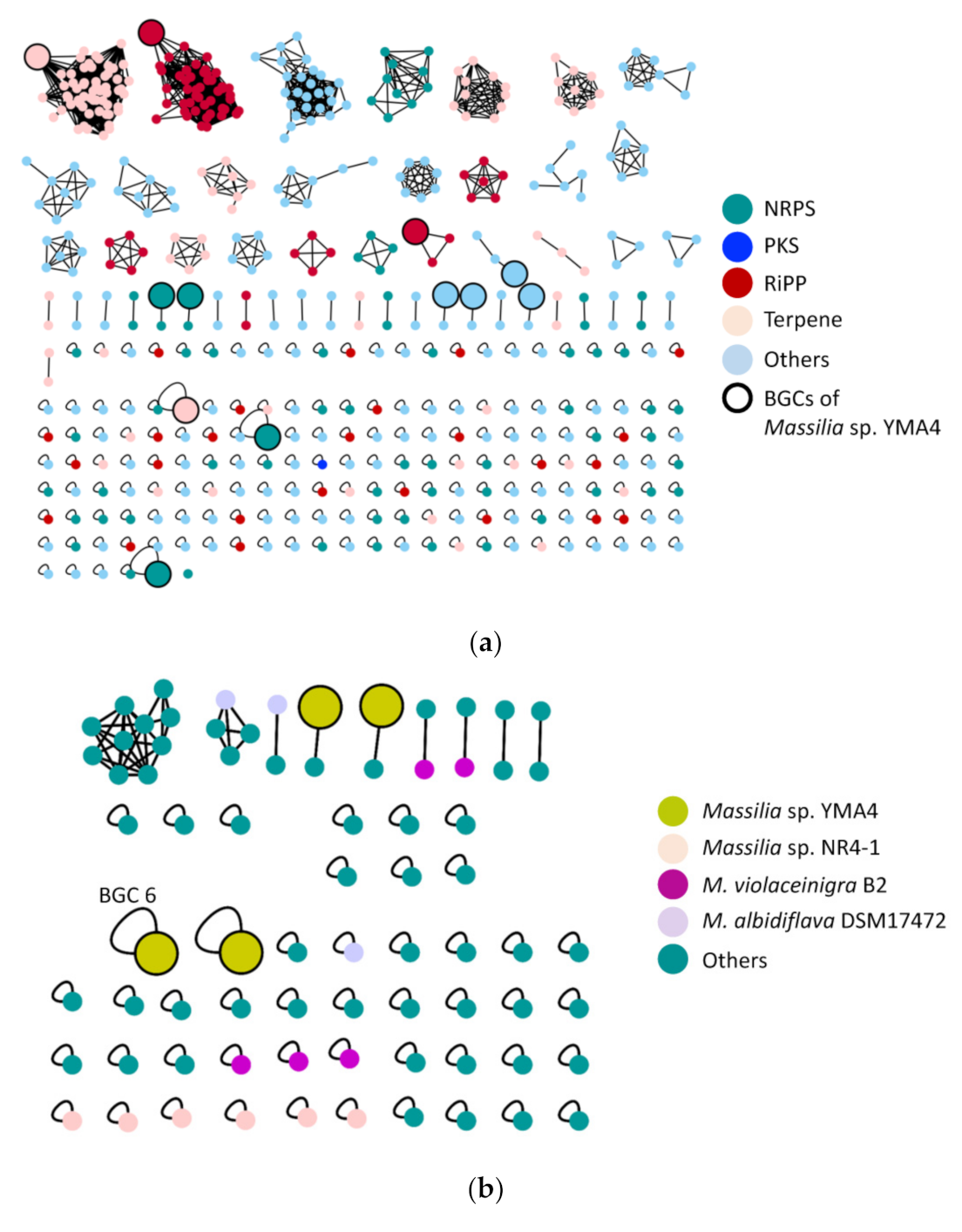
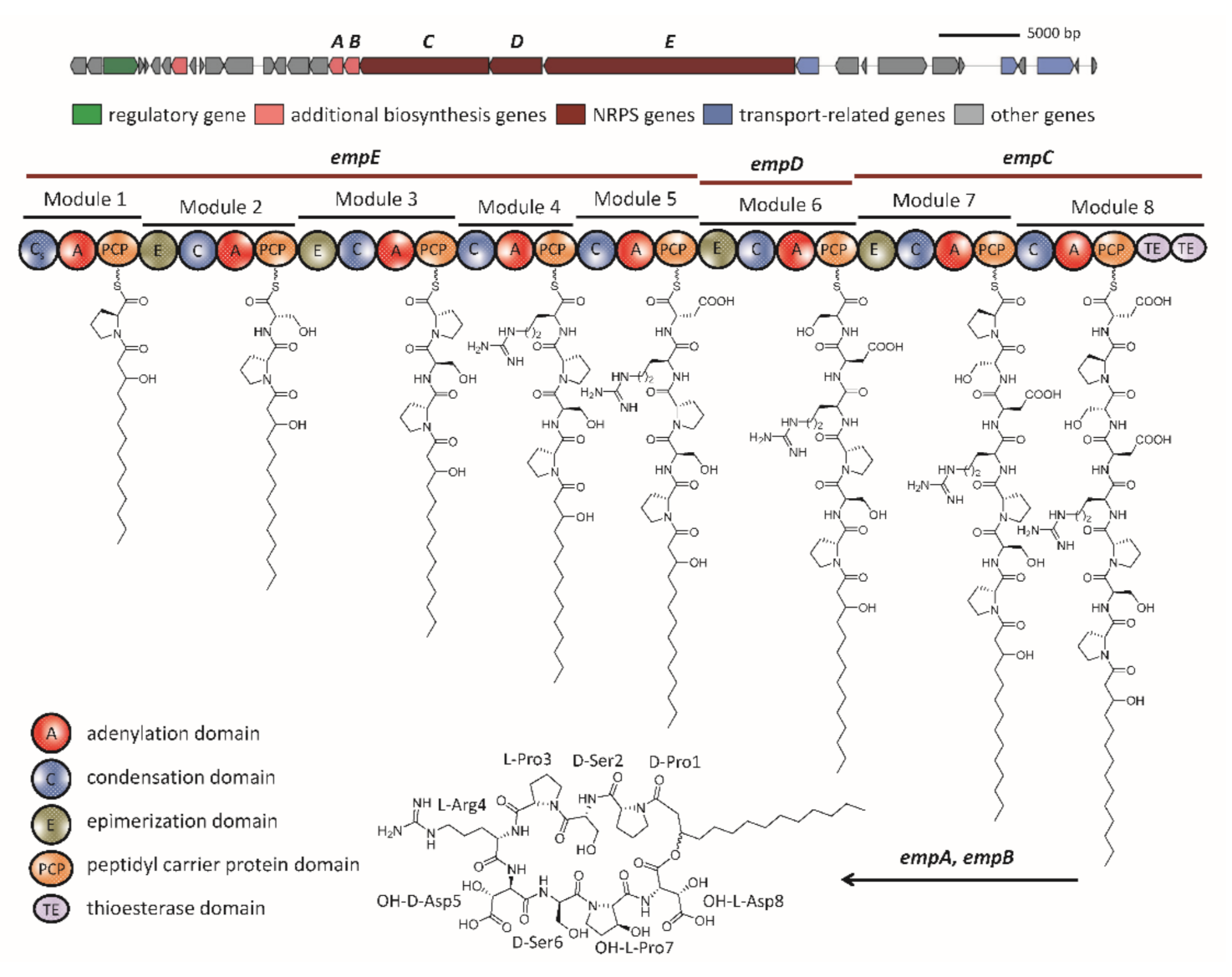
2.2. The Biosynthetic Potential of Secondary Metabolites of the Massilia Strains and the Discovery of the Empedopeptin Biosynthesis Gene Cluster
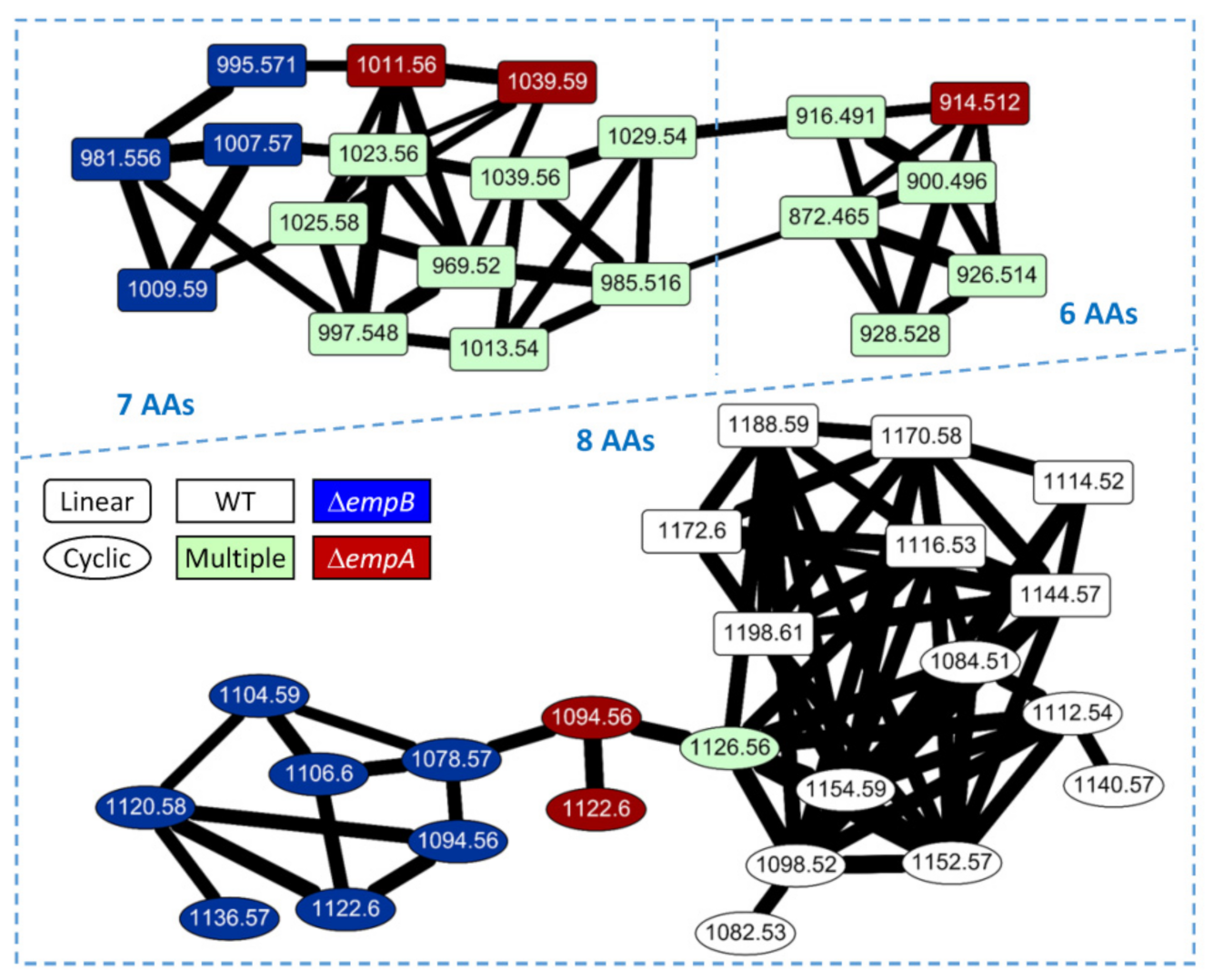
2.3. Molecular Networking of Empedopeptin and Its Analogs from Massilia sp. YMA4 Wild-Type and Mutant Strains
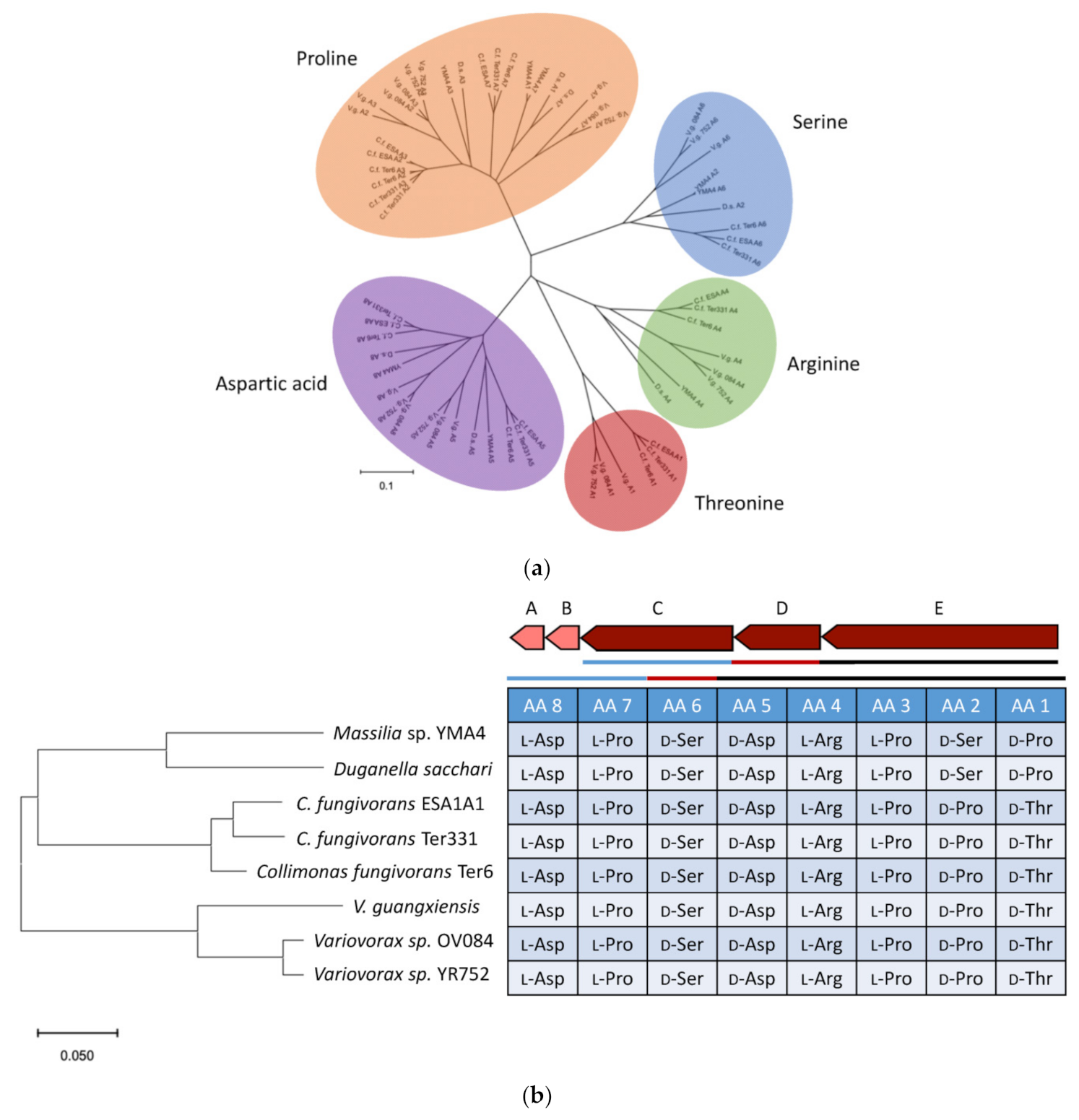
2.4. Genome Mining for Discovering Other Empedopeptin-Like Compound Producing Bacteria
3. Materials and Methods
3.1. Bacterial Culture Conditions and Antagonistic Assay
3.2. DNA Sample Preparation, Whole-Genome Sequencing, Assembly, and Annotation
3.3. Genome Mining and Bioinformatic Analysis
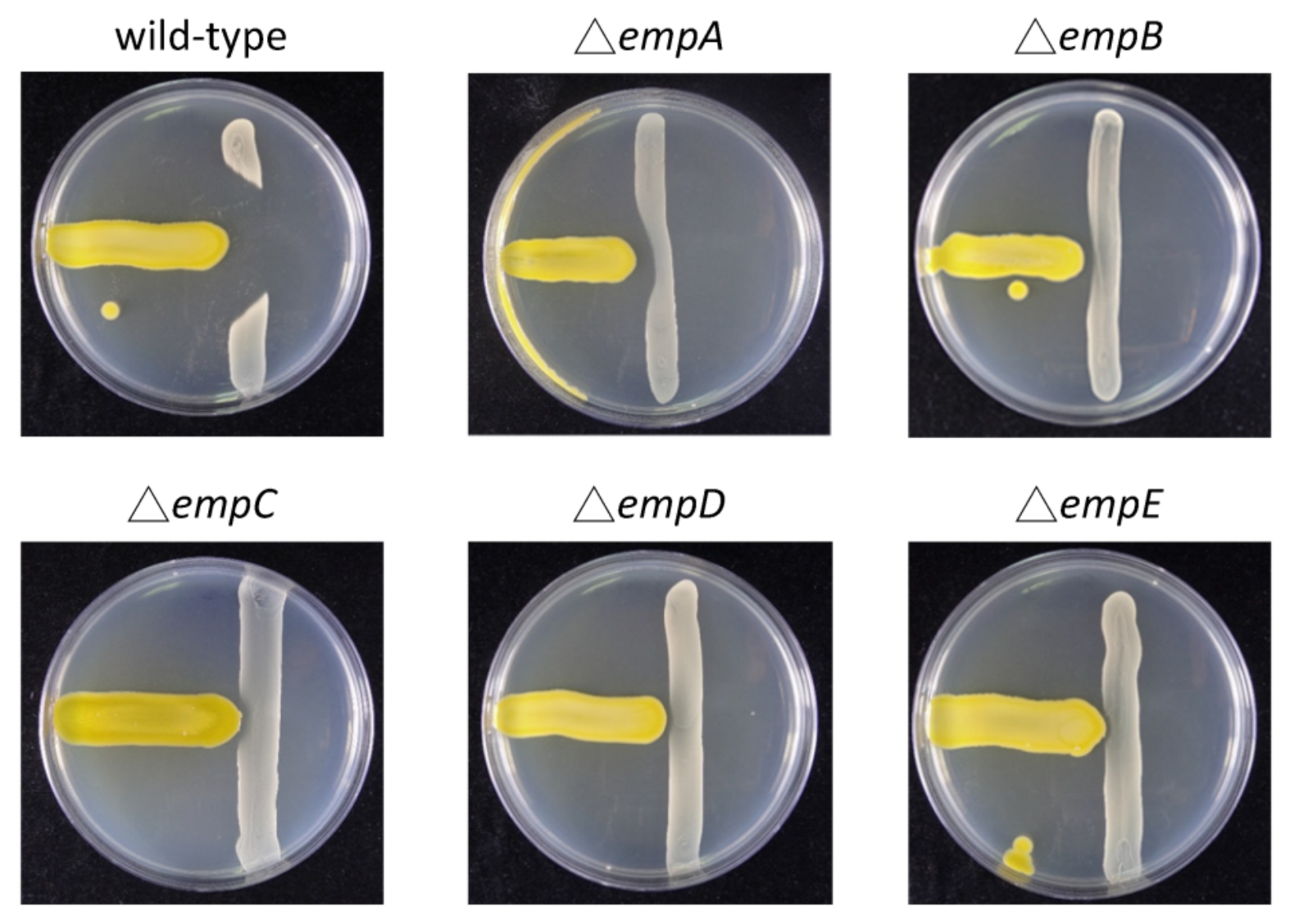
3.4. Construction of Empedopeptin Biosynthetic Gene-Null Mutant Strains
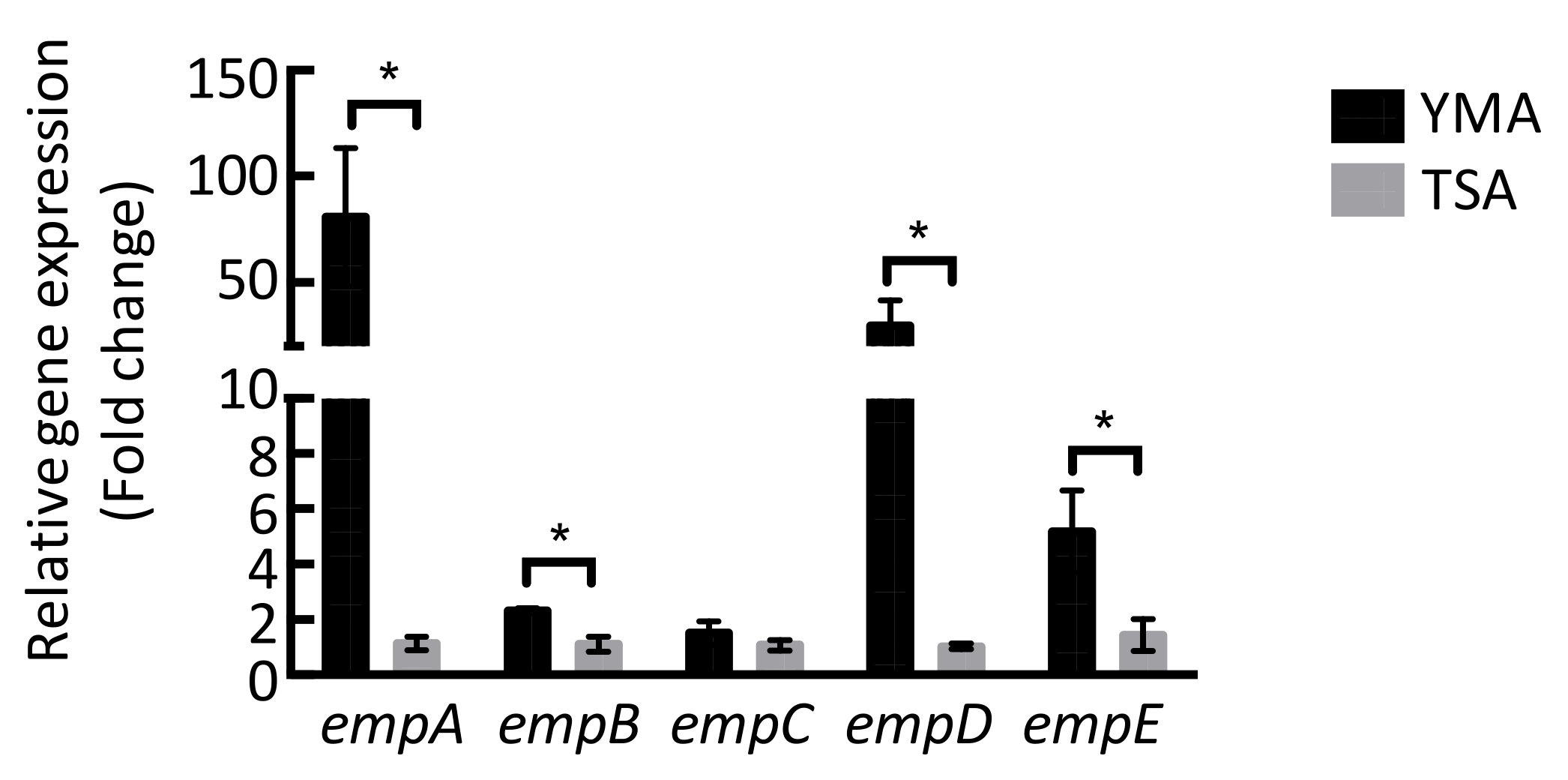
3.5. Quantitative Reverse Transcription PCR (qRT-PCR)
3.6. Comparative Metabolomics and Molecular Networking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffmeister, D.; Keller, N.P. Natural products of filamentous fungi: Enzymes, genes, and their regulation. Nat. Prod. Rep. 2007, 24, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.N.; Yen, M.R.; Chiang, C.Y.; Lin, H.C.; Chen, P.Y. Detecting and prioritizing biosynthetic gene clusters for bioactive compounds in bacteria and fungi. Appl. Microbiol. Biotechnol. 2019, 103, 3277–3287. [Google Scholar] [CrossRef] [PubMed]
- Atencio, L.A.; Boya, P.C.; Martin, H.C.; Mejia, L.C.; Dorrestein, P.C.; Gutierrez, M. Genome mining, microbial interactions, and molecular networking reveals new dibromoalterochromides from strains of Pseudoalteromonas of Coiba National Park-Panama. Mar. Drugs 2020, 18, 456. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Tomoda, H. MS network-based screening for new antibiotics discovery. J. Antibiot. 2019, 72, 54–56. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Wiese, J.; Imhoff, J.F. Marine bacteria and fungi as promising source for new antibiotics. Drug Dev. Res. 2019, 80, 24–27. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- La Scola, B.; Birtles, R.J.; Mallet, M.N.; Raoult, D. Massilia timonae gen. nov., sp. nov., isolated from blood of an immunocompromised patient with cerebellar lesions. J. Clin. Microbiol. 1998, 36, 2847–2852. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef]
- Agematu, H.; Suzuki, K.; Tsuya, H. Massilia sp. BS-1, a novel violacein-producing bacterium isolated from soil. Biosci. Biotechnol. Biochem. 2011, 75, 2008–2010. [Google Scholar] [CrossRef][Green Version]
- Myeong, N.R.; Seong, H.J.; Kim, H.J.; Sul, W.J. Complete genome sequence of antibiotic and anticancer agent violacein producing Massilia sp. strain NR 4-1. J. Biotechnol. 2016, 223, 36–37. [Google Scholar] [CrossRef]
- Miess, H.; Arlt, P.; Apel, A.K.; Weber, T.; Nieselt, K.; Hanssen, F.; Czemmel, S.; Nahnsen, S.; Gross, H. The draft whole-genome sequence of the antibiotic producer Empedobacter haloabium ATCC 31962 provides indications for its taxonomic reclassification. Microbiol. Resour. Announc. 2019, 8, 45. [Google Scholar] [CrossRef]
- Konishi, M.; Sugawara, K.; Hanada, M.; Tomita, K.; Tomatsu, K.; Miyaki, T.; Kawaguchi, H.; Buck, R.E.; More, C.; Rossomano, V.Z. Empedopeptin (BMY-28117), a new depsipeptide antibiotic. I. Production, isolation and properties. J. Antibiot. 1984, 37, 949–957. [Google Scholar] [CrossRef][Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Navarro-Munoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Rottig, M.; Medema, M.H.; Blin, K.; Weber, T.; Rausch, C.; Kohlbacher, O. NRPSpredictor2-a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011, 39, W362–W367. [Google Scholar] [CrossRef]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar]
- Girard, L.; Hofte, M.; Mot, R. Lipopeptide families at the interface between pathogenic and beneficial Pseudomonas-plant interactions. Crit. Rev. Microbiol. 2020, 46, 397–419. [Google Scholar] [CrossRef]
- Gross, H.; Loper, J.E. Genomics of secondary metabolite production by Pseudomonas spp. Nat. Prod. Rep. 2009, 26, 1408–1446. [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Burholt, S.; Hamley, I.W. Peptide hormones and lipopeptides: From self-assembly to therapeutic applications. J. Pept. Sci. 2017, 23, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Sussmuth, R.D.; Mainz, A. Nonribosomal peptide synthesis-principles and prospects. Angew. Chem. Int. Ed. Engl. 2017, 56, 3770–3821. [Google Scholar] [CrossRef] [PubMed]
- Loper, J.E.; Hassan, K.A.; Mavrodi, D.V.; Davis, E.W., 2nd; Lim, C.K.; Shaffer, B.T.; Elbourne, L.D.; Stockwell, V.O.; Hartney, S.L.; Breakwell, K.; et al. Comparative genomics of plant-associated Pseudomonas spp.: Insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 2012, 8, e1002784. [Google Scholar] [CrossRef] [PubMed]
- Jahanshah, G.; Yan, Q.; Gerhardt, H.; Pataj, Z.; Lammerhofer, M.; Pianet, I.; Josten, M.; Sahl, H.G.; Silby, M.W.; Loper, J.E.; et al. Discovery of the cyclic lipopeptide gacamide A by genome mining and repair of the defective GacA regulator in Pseudomonas fluorescens Pf0-1. J. Nat. Prod. 2019, 82, 301–308. [Google Scholar] [CrossRef]
- Strieker, M.; Tanovic, A.; Marahiel, M.A. Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 2010, 20, 234–240. [Google Scholar] [CrossRef]
- Medema, M.H.; Paalvast, Y.; Nguyen, D.D.; Melnik, A.; Dorrestein, P.C.; Takano, E.; Breitling, R. Pep2Path: Automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput. Biol. 2014, 10, e1003822. [Google Scholar] [CrossRef]
- Sanchez, S.; Chavez, A.; Forero, A.; Garcia-Huante, Y.; Romero, A.; Sanchez, M.; Rocha, D.; Sanchez, B.; Avalos, M.; Guzman-Trampe, S.; et al. Carbon source regulation of antibiotic production. J. Antibiot. 2010, 63, 442–459. [Google Scholar] [CrossRef]
- Muller, A.; Munch, D.; Schmidt, Y.; Reder-Christ, K.; Schiffer, G.; Bendas, G.; Gross, H.; Sahl, H.G.; Schneider, T.; Brotz-Oesterhelt, H. Lipodepsipeptide empedopeptin inhibits cell wall biosynthesis through Ca2+-dependent complex formation with peptidoglycan precursors. J. Biol. Chem. 2012, 287, 20270–20280. [Google Scholar] [CrossRef]
- Hover, B.M.; Kim, S.H.; Katz, M.; Charlop-Powers, Z.; Owen, J.G.; Ternei, M.A.; Maniko, J.; Estrela, A.B.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef]
- Medema, M.H.; Takano, E.; Breitling, R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 2013, 30, 1218–1223. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant. Genomics 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M. The SEED and the rapid rnnotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2013, 42, D206–D214. [Google Scholar] [CrossRef]
- Bertels, F.; Silander, O.K.; Pachkov, M.; Rainey, P.B.; van Nimwegen, E. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 2014, 31, 1077–1088. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Ceniceros, A.; Dijkhuizen, L.; Petrusma, M.; Medema, M.H. Genome-based exploration of the specialized metabolic capacities of the genus Rhodococcus. BMC Genom. 2017, 18, 1–16. [Google Scholar] [CrossRef]
- Yeong, M. BiG-SCAPE: Exploring Biosynthetic Diversity through Gene Cluster Similarity Networks; Wageningen University: Wageningen, The Netherlands, 2016. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, S.-T.; Ho, Y.-N.; Lin, C.; Hsu, W.-C.; Lee, H.-J.; Peng, C.-C.; Cheng, H.-T.; Yang, Y.-L. Integrated Omics Strategy Reveals Cyclic Lipopeptides Empedopeptins from Massilia sp. YMA4 and Their Biosynthetic Pathway. Mar. Drugs 2021, 19, 209. https://doi.org/10.3390/md19040209
Ho S-T, Ho Y-N, Lin C, Hsu W-C, Lee H-J, Peng C-C, Cheng H-T, Yang Y-L. Integrated Omics Strategy Reveals Cyclic Lipopeptides Empedopeptins from Massilia sp. YMA4 and Their Biosynthetic Pathway. Marine Drugs. 2021; 19(4):209. https://doi.org/10.3390/md19040209
Chicago/Turabian StyleHo, Shang-Tse, Ying-Ning Ho, Chih Lin, Wei-Chen Hsu, Han-Jung Lee, Chia-Chi Peng, Han-Tan Cheng, and Yu-Liang Yang. 2021. "Integrated Omics Strategy Reveals Cyclic Lipopeptides Empedopeptins from Massilia sp. YMA4 and Their Biosynthetic Pathway" Marine Drugs 19, no. 4: 209. https://doi.org/10.3390/md19040209
APA StyleHo, S.-T., Ho, Y.-N., Lin, C., Hsu, W.-C., Lee, H.-J., Peng, C.-C., Cheng, H.-T., & Yang, Y.-L. (2021). Integrated Omics Strategy Reveals Cyclic Lipopeptides Empedopeptins from Massilia sp. YMA4 and Their Biosynthetic Pathway. Marine Drugs, 19(4), 209. https://doi.org/10.3390/md19040209