
Comparative Metabologenomics Analysis of Polar Actinomycetes

 ,
,  ,
,  , and
, and 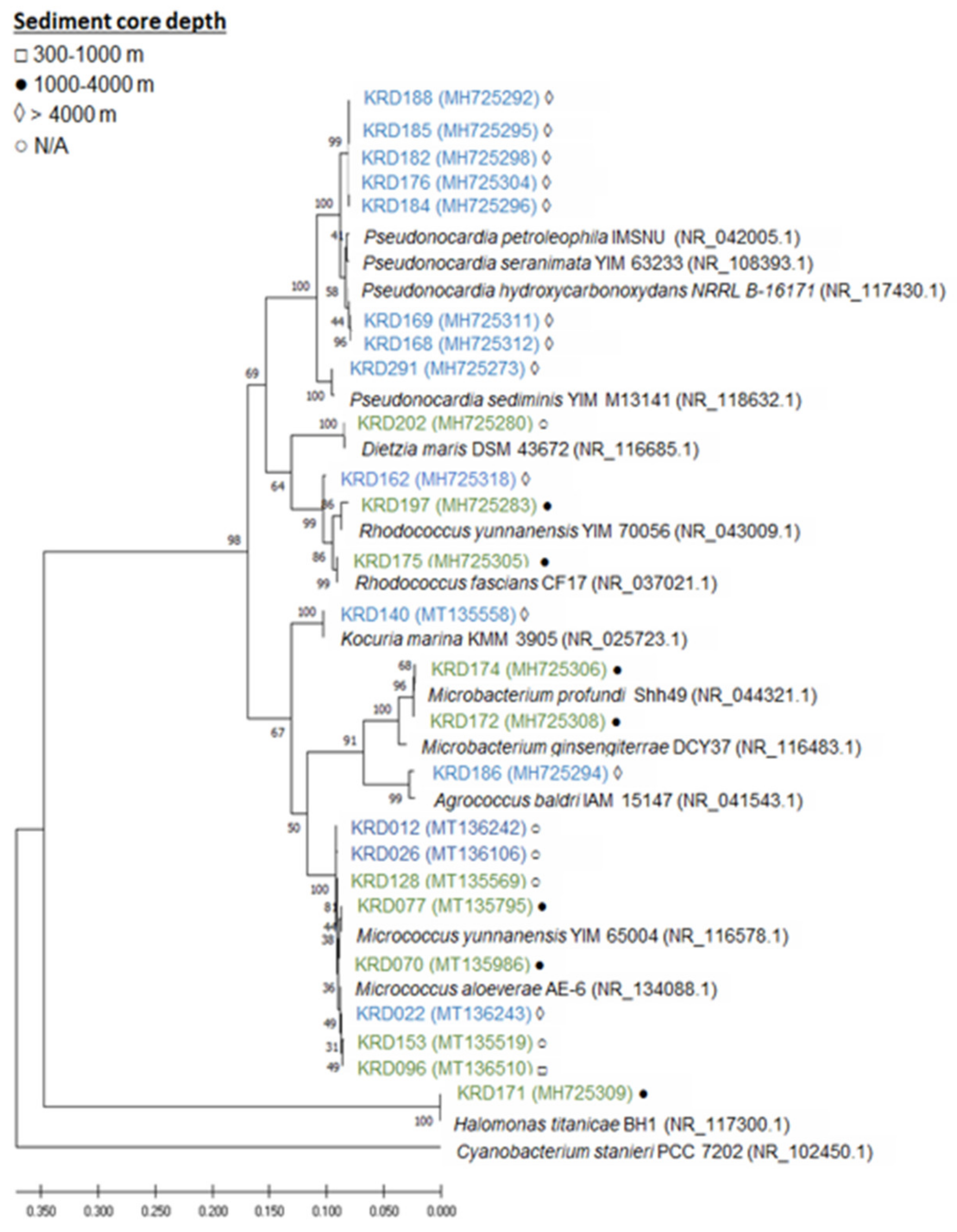{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
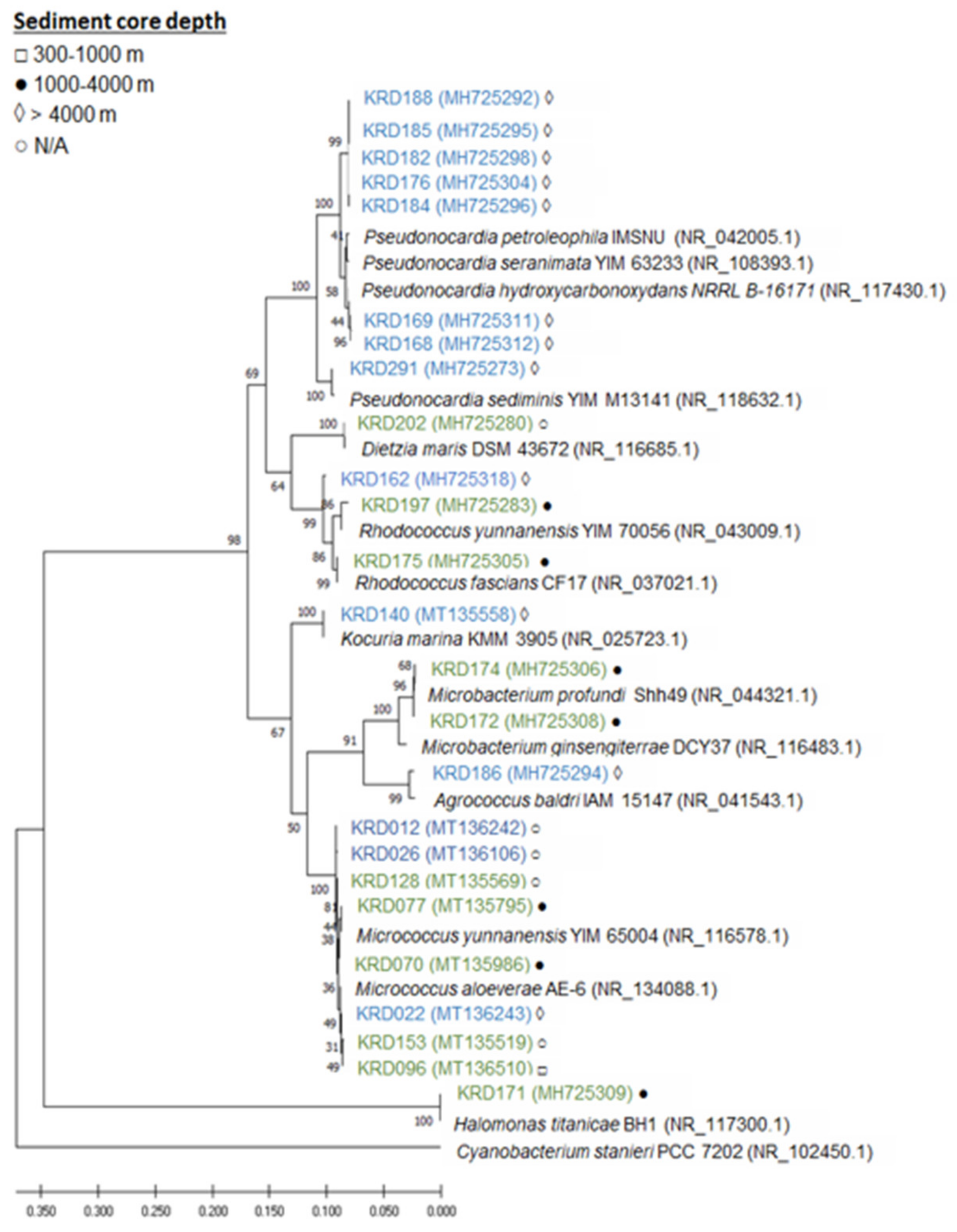
2.1. Phylogenetic Analysis
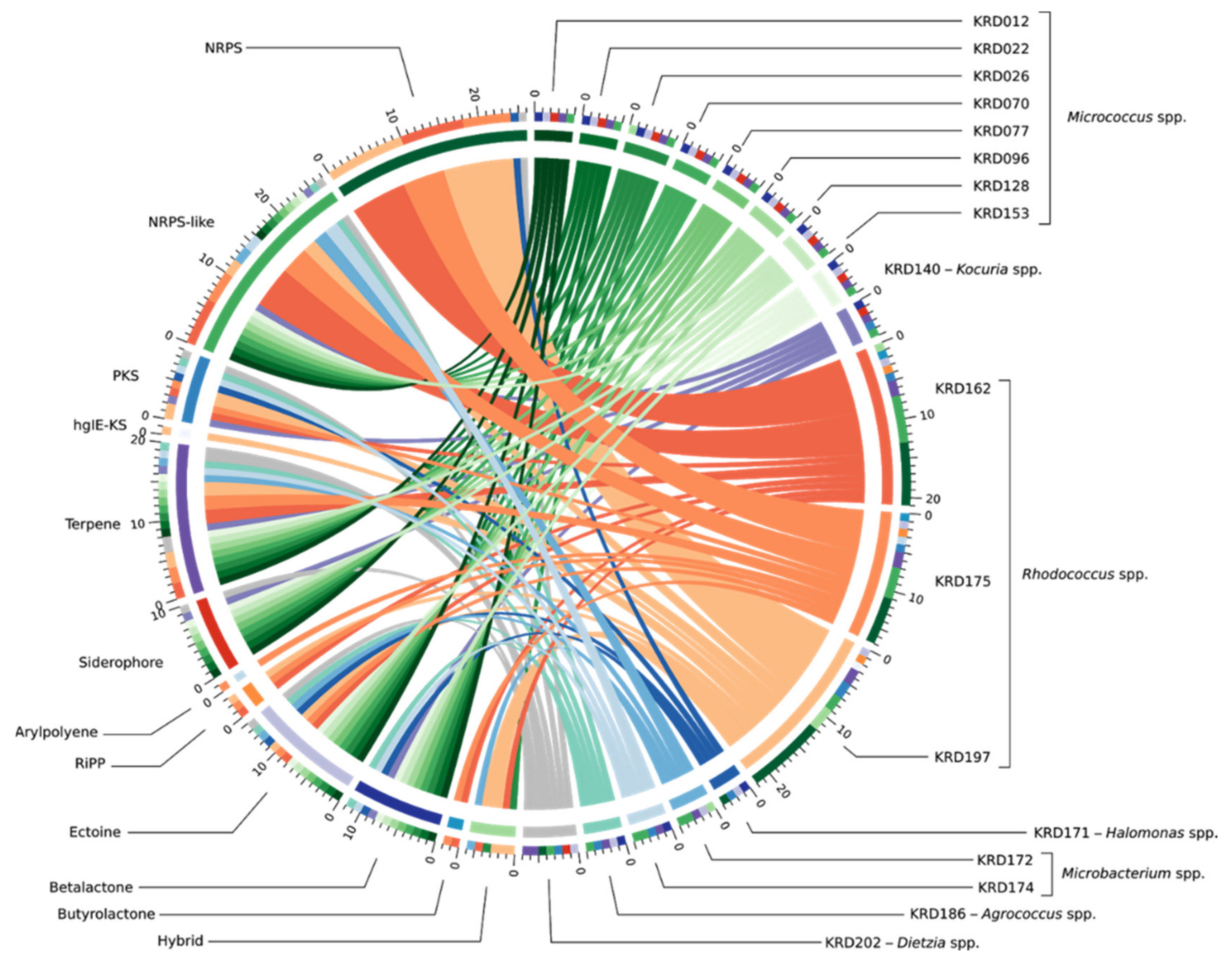
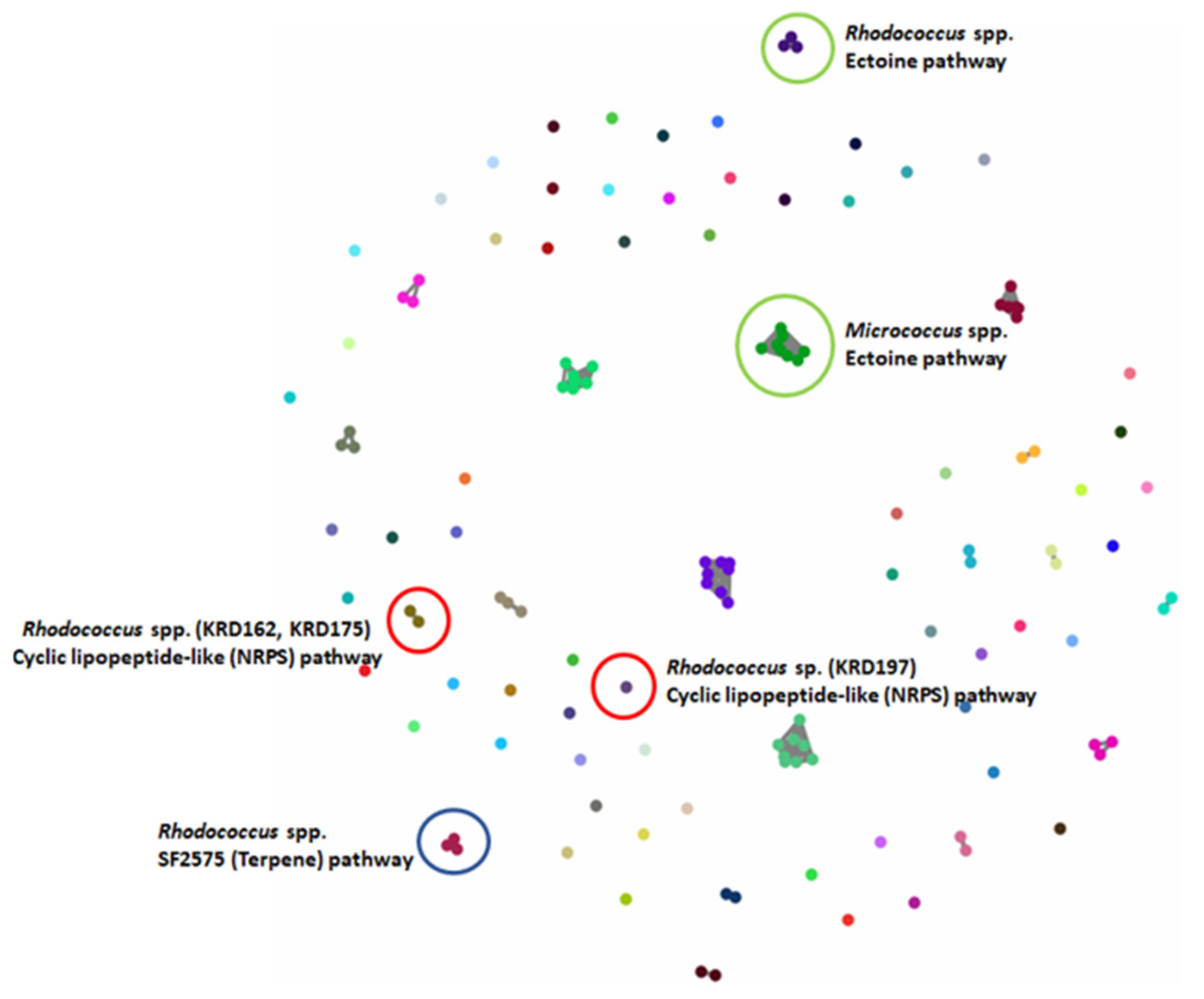
2.2. Genome Mining
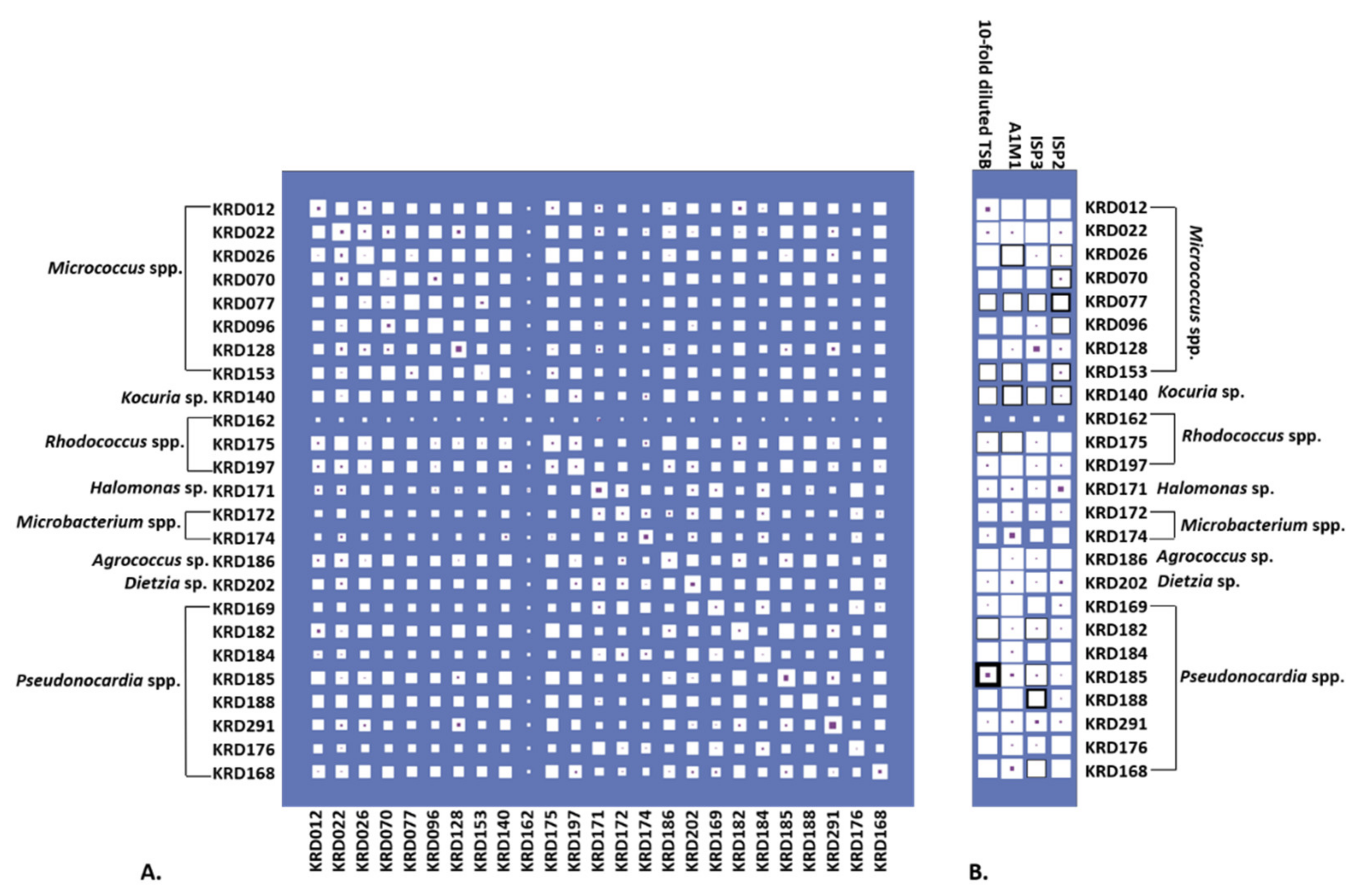
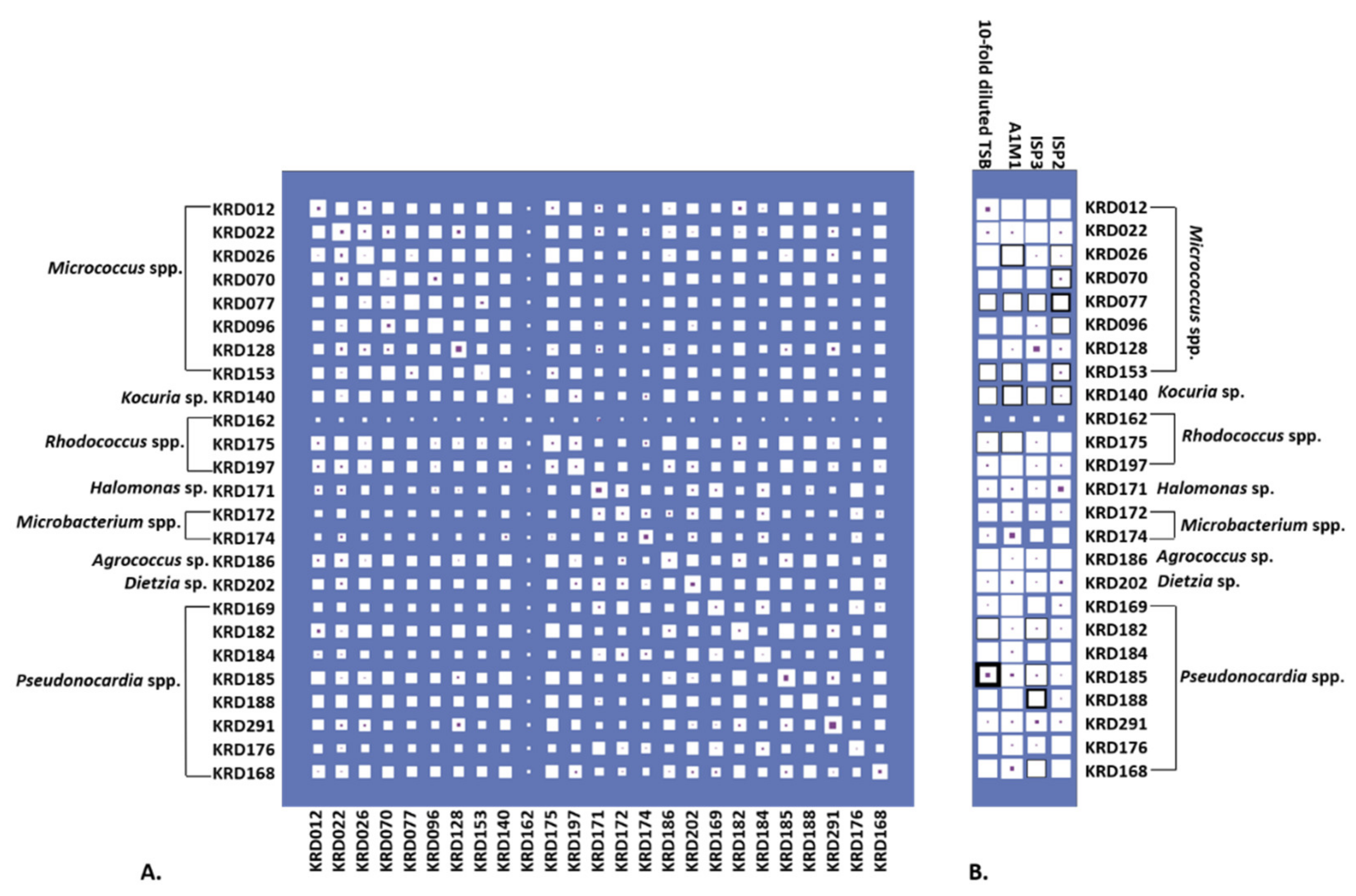
2.3. Antibacterial Activity and Parent Ion Distribution
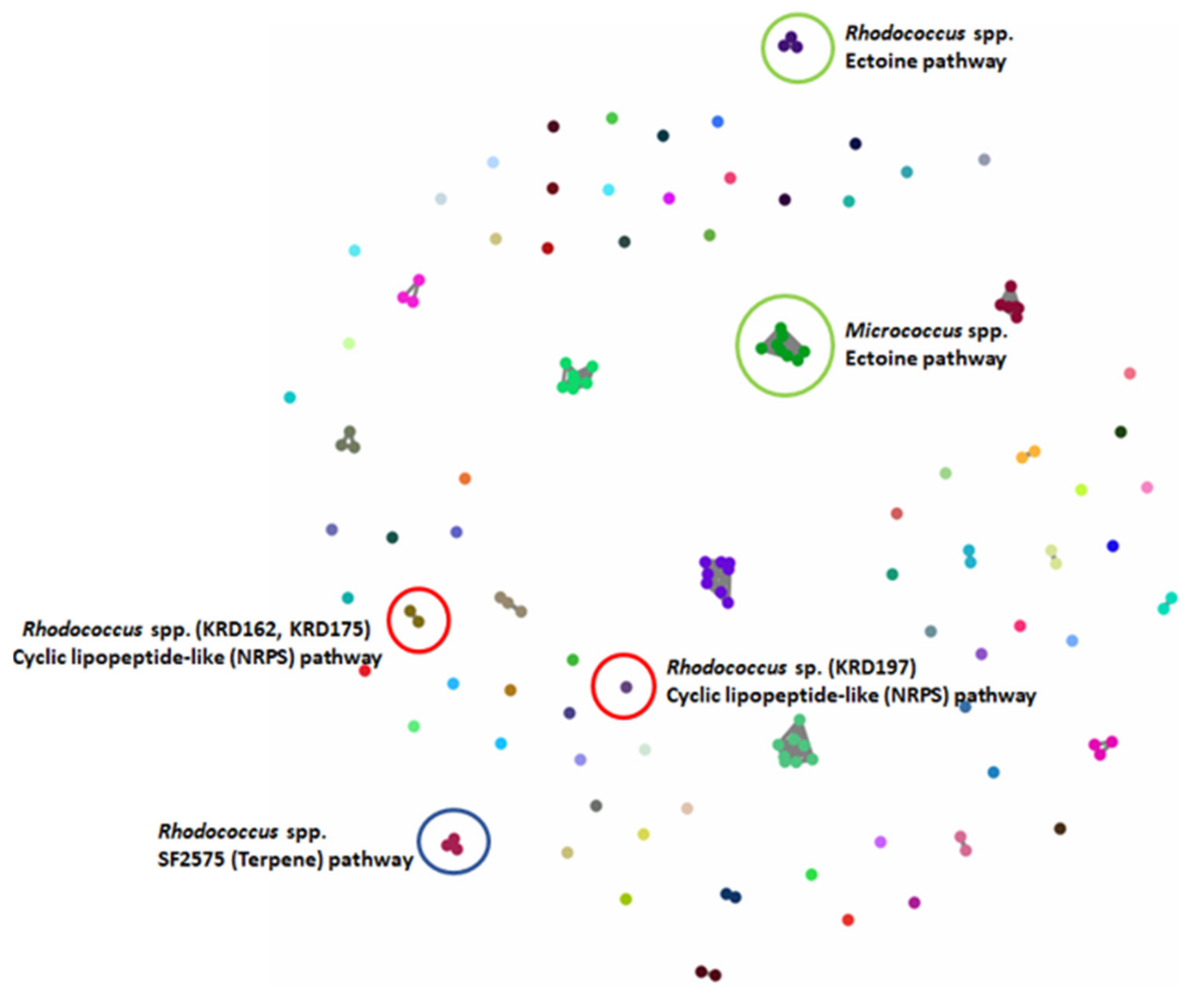
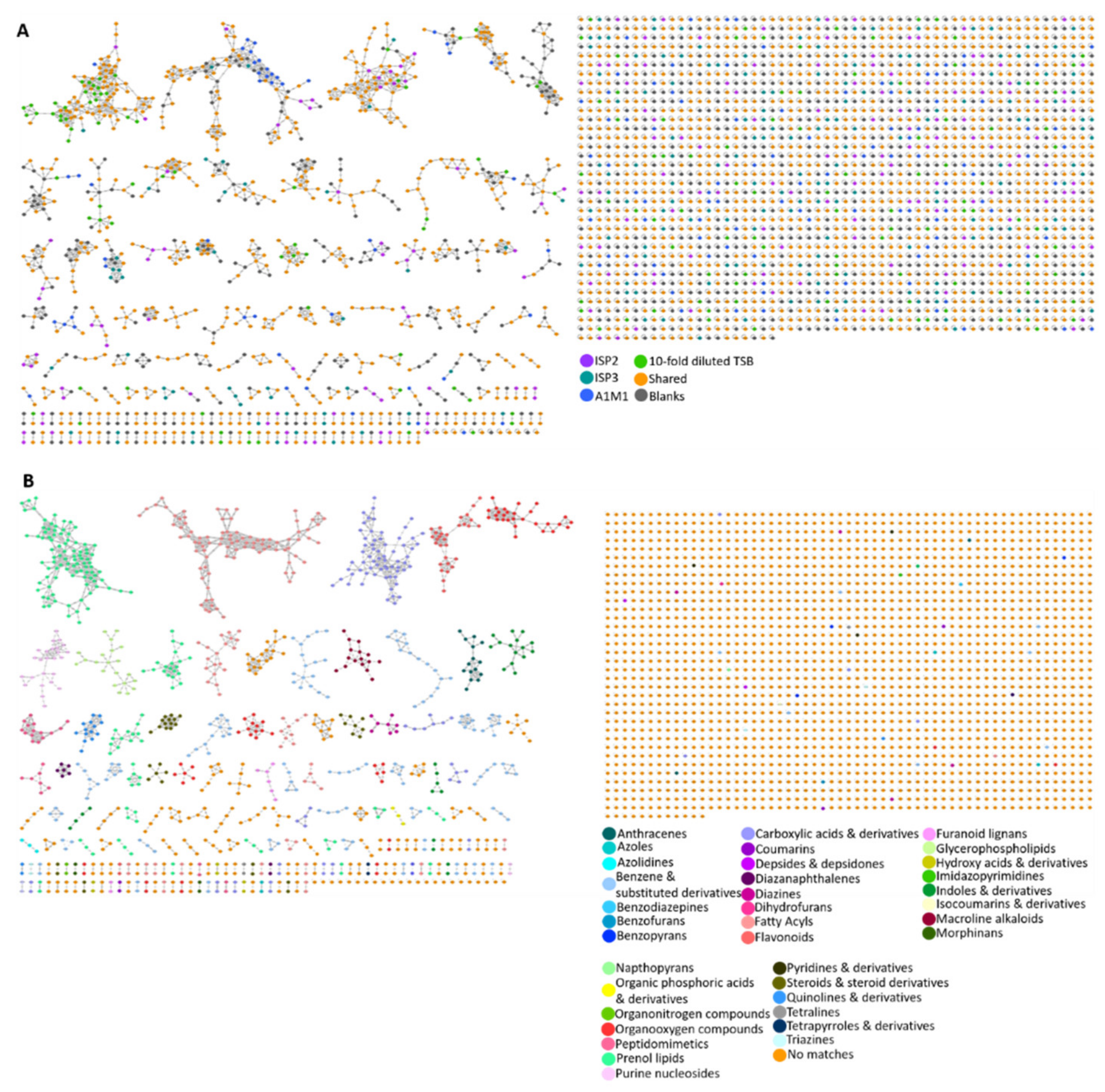
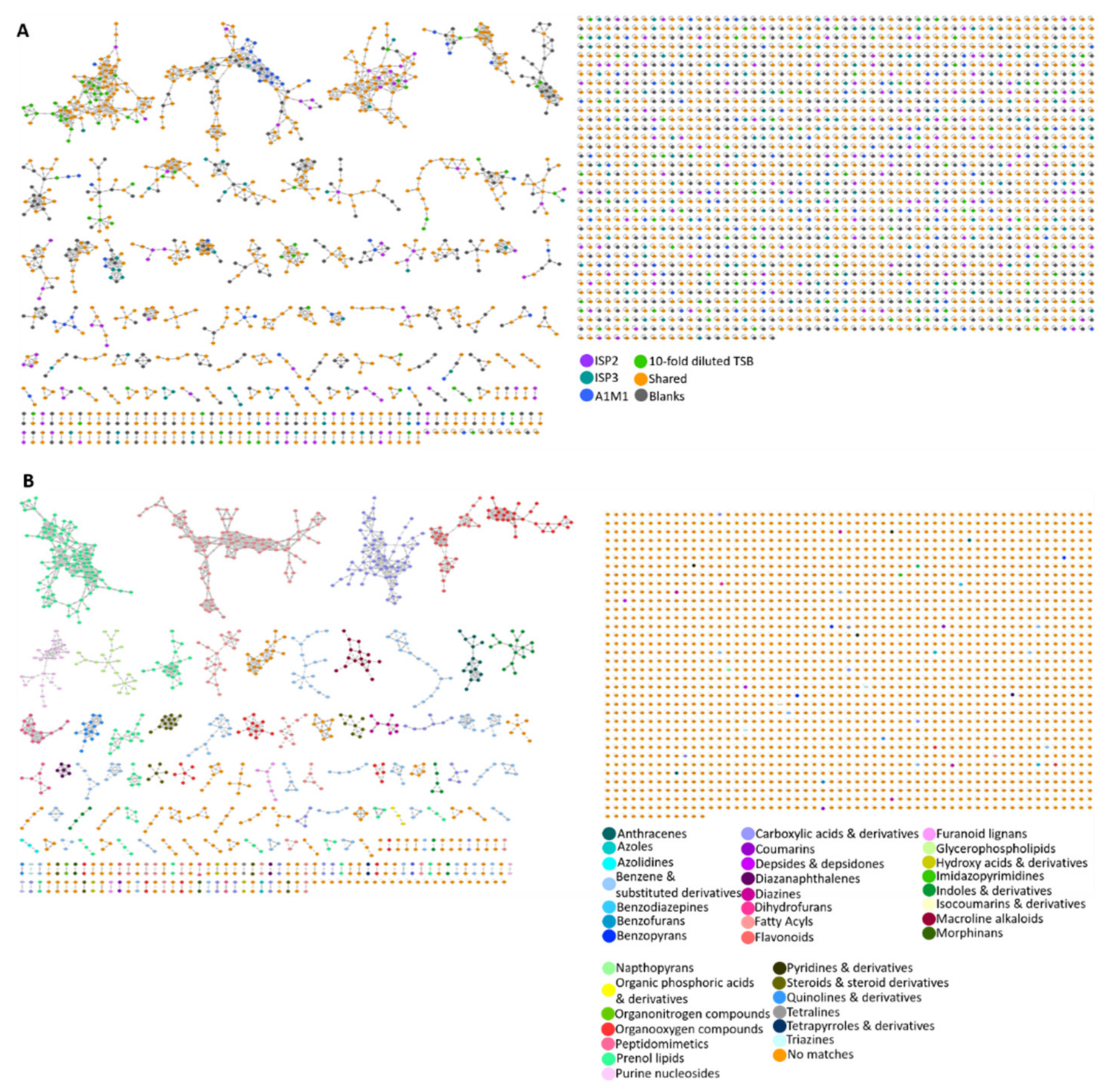
2.4. Molecular Networking
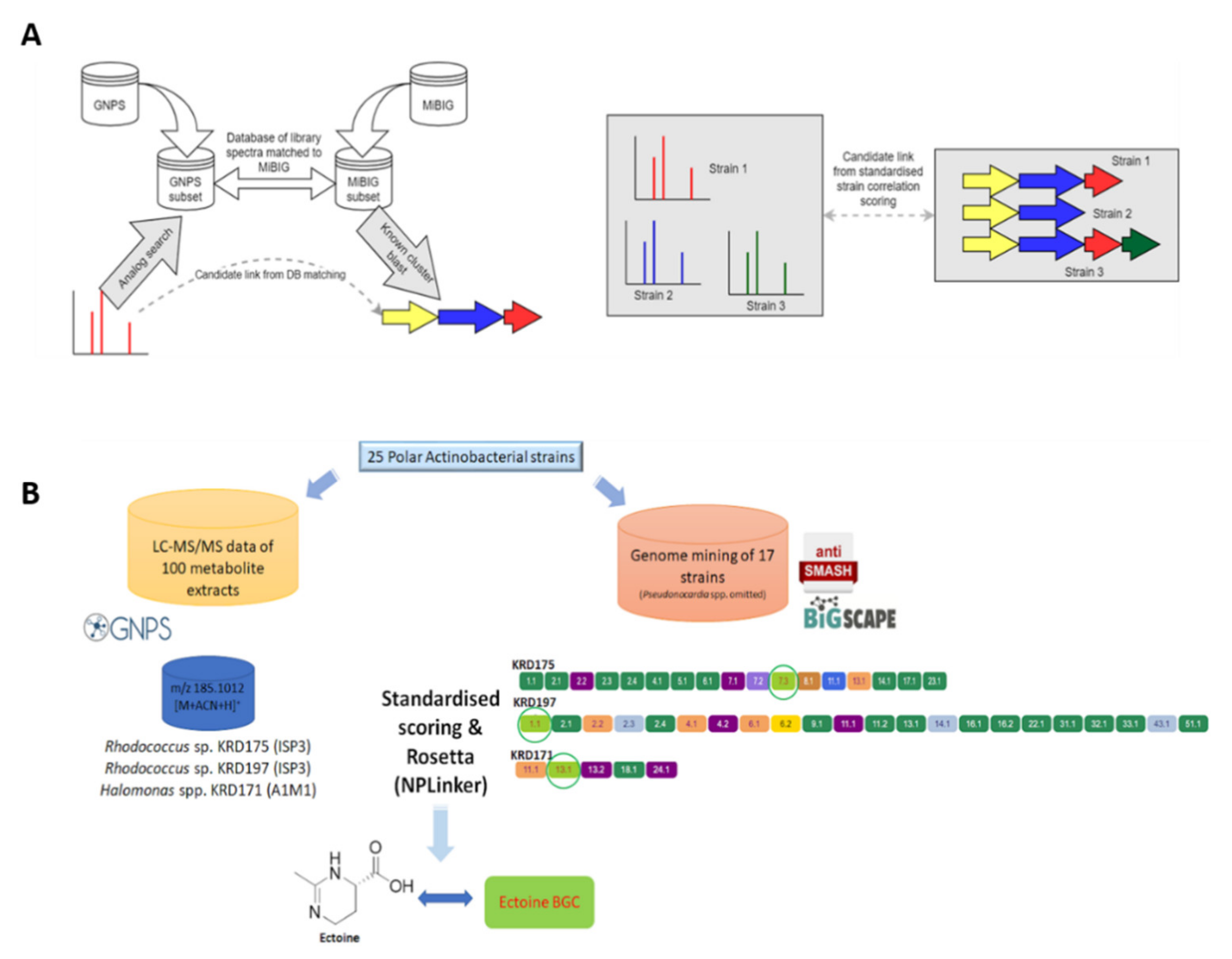
2.5. Computational Pattern Matching
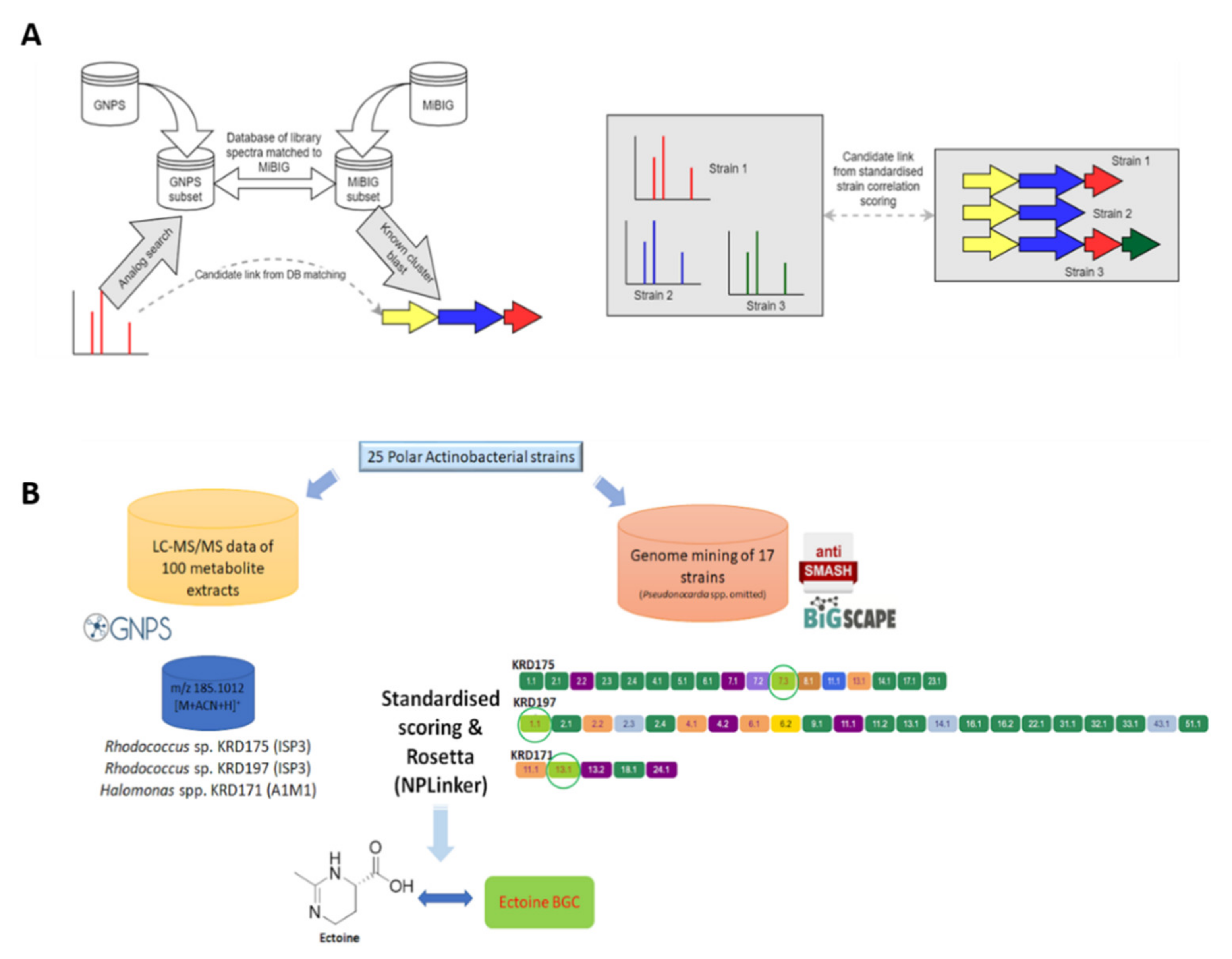
2.5.1. Computational Pattern Matching Using the Standardised Strain Correlation Scoring Method
2.5.2. Computational Pattern Matching Using Standardised Strain Correlation Scoring and the Rosetta Method
3. Discussion
4. Materials and Methods
4.1. Phylogenetic Analysis
4.2. Fermentation and Metabolite Extraction
4.3. Bioactivity Disc Diffusion Assay
4.4. Bioactivity Agar Plug Assay
4.5. Mass Spectral Data Acquisition
4.6. Mass Spectral Data Processing
4.7. Molecular Networking
4.8. MolNetEnhancer Workflow Description for Chemical Class Annotation of Molecular Networks
4.9. Genomic DNA Extraction
4.10. Genome Sequencing and Alignment
4.11. Biosynthetic Gene Cluster Mining and Comparison
4.12. Computational Pattern Matching
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- O’ Neil, J. Review on Antibiotic Resisitance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; The Wellcome Trust and the UK Department of Health: London, UK, 2014. [Google Scholar]
- O’ Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; The Wellcome Trust and the UK Department of Health: London, UK, 2016. [Google Scholar]
- Jackson, S.A.; Crossman, L.; Almeida, E.L.; Margassery, L.M.; Kennedy, J.; Dobson, A.D.W. Diverse and Abundant Secondary Metabolism Biosynthetic Gene Clusters in the Genomes of Marine Sponge Derived Streptomyces spp. Isolates. Mar. Drugs 2018, 16, 67. [Google Scholar] [CrossRef] [Green Version]
- Baltz, R.H. Renaissance in Antibacterial Discovery from Actinomycetes. Curr. Opin. Pharmacol. 2008, 8, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Lewin, G.R.; Carlos, C.; Chevrette, M.G.; Horn, H.A.; McDonald, B.R.; Stankey, R.J.; Fox, B.G.; Currie, C.R. Evolution and Ecology of Actinobacteria and Their Bioenergy Applications. Annu. Rev. Microbiol. 2016, 70, 235–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltz, R.H. Gifted Microbes for Genome Mining and Natural Product Discovery. J. Ind. Microbiol. Biotechnol. 2017, 44, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Schorn, M.A.; Alanjary, M.M.; Aguinaldo, K.; Korobeynikov, A.; Podell, S.; Patin, N.; Lincecum, T.; Jensen, P.R.; Ziemert, N.; Moore, B.S. Sequencing Rare Marine Actinomycete Genomes Reveals High Density of Unique Natural Product Biosynthetic Gene Clusters. Microbiology 2016, 162, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Letzel, A.-C.; Natalie, M.-A.; Amos, G.C.; Millán-Aguiñaga, N.; Ginigini, J.; Abdelmohsen, U.R.; Gaudêncio, S.P.; Ziemert, N.; Moore, B.S.; Jensen, P.R. Genomic Insights into Specialized Metabolism in the Marine Actinomycete salinispora. Environ. Microbiol. 2017, 19, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-Z.; Han, X.-B.; Zhang, M.-X.; Zhou, Y.; Chen, M.; Yao, Q.; Zhu, H.-H. Culture-Dependent and -Independent Analyses Reveal the Diversity, Structure, and Assembly Mechanism of Benthic Bacterial Community in the Ross Sea, Antarctica. Front. Microbiol. 2019, 10, 2523. [Google Scholar] [CrossRef] [Green Version]
- Millán-Aguiñaga, N.; Soldatou, S.; Brozio, S.; Munnoch, J.T.; Howe, J.A.; Hoskisson, P.A.; Duncan, K.R. Awakening Ancient Polar Actinobacteria: Diversity, Evolution and Specialized Metabolite Potential. Microbiology 2019, 165, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Purves, K.; Macintyre, L.; Brennan, D.; Hreggviðsson, G.; Kuttner, E.; Ásgeirsdóttir, M.E.; Young, L.C.; Green, D.H.; Edrada-Ebel, R.; Duncan, K.R. Using Molecular Networking for Microbial Secondary Metabolite Bioprospecting. Metabolites 2016, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Taglialatela-Scafati, O.; Zhao, F. Secondary Metabolites from Polar Organisms. Mar. Drugs 2017, 15, 28. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lu, Y.; Xing, Y.; Ma, Y.; Lu, J.; Bao, W.; Wang, Y.; Xi, T. A Novel Anticancer and Antifungus Phenazine Derivative from a Marine Actinomycete BM-17. Microbiol. Res. 2012, 167, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Saurav, K.; Yu, Z.; Mándi, A.; Kurtán, T.; Li, J.; Tian, X.; Zhang, Q.; Zhang, W.; Zhang, C. α-Pyrones with Diverse Hydroxy Substitutions from Three Marine-Derived Nocardiopsis Strains. J. Nat. Prod. 2016, 79, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Mondol, M.M.; Yu, T.K.; Lee, H.-S.; Lee, Y.-J.; Jung, H.J.; Kim, J.H.; Kwon, H.J. An Angiogenesis Inhibitor Isolated from a Marine-Derived Actinomycete, Nocardiopsis sp. 03N67. Phytochem. Lett. 2010, 3, 194–197. [Google Scholar] [CrossRef]
- Hoskisson, P.A.; Seipke, R.F. Cryptic or Silent? The Known Unknowns, Unknown Knowns, and Unknown Unknowns of Secondary Metabolism. mBio 2020, 11, 02642–20. [Google Scholar] [CrossRef]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “One Strain Many Compounds” (Osmac) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef] [Green Version]
- Rateb, M.E.; Houssen, W.E.; Harrison, W.T.A.; Deng, H.; Okoro, C.K.; Asenjo, J.A.; Andrews, B.A.; Bull, A.T.; Goodfellow, M.; Ebel, R.; et al. Diverse Metabolic Profiles of aStreptomycesStrain Isolated from a Hyper-Arid Environment. J. Nat. Prod. 2011, 74, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Che, Q.; Li, J.; Li, D.; Gu, Q.; Zhu, T. Structure and Absolute Configuration of Drimentine I, an Alkaloid from Streptomyces sp. CHQ-64. J. Antibiot. 2016, 69, 467–469. [Google Scholar] [CrossRef]
- Che, Q.; Zhu, T.; Qi, X.; Mándi, A.; Kurtán, T.; Mo, X.; Li, J.; Gu, Q.; Li, D. Hybrid Isoprenoids from a Reeds Rhizosphere Soil Derived Actinomycete Streptomyces sp. CHQ-64. Org. Lett. 2012, 14, 3438–3441. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big Effects from Small Changes: Possible Ways to Explore Nature’s Chemical Diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Quinn, R.A.; Nothias, L.F.; Vining, O.; Meehan, M.; Esquenazi, E.; Dorrestein, P.C. Molecular Networking As a Drug Discovery, Drug Metabolism, and Precision Medicine Strategy. Trends Pharmacol. Sci. 2017, 38, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; De Felicio, R.; Fenner, A.; et al. Molecular Networking as a Dereplication Strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [Green Version]
- Audoin, C.; Zampalégré, A.; Blanchet, N.; Giuliani, A.; Roulland, E.; Laprévote, O.; Genta-Jouve, G. MS/MS-Guided Isolation of Clarinoside, a New Anti-Inflammatory Pentalogin Derivative. Molecules 2018, 23, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, K.R.; Crüsemann, M.; Lechner, A.; Sarkar, A.; Li, J.; Ziemert, N.; Wang, M.; Bandeira, N.; Moore, B.S.; Dorrestein, P.C.; et al. Molecular Networking and Pattern-Based Genome Mining Improves Discovery of Biosynthetic Gene Clusters and their Products from Salinispora Species. Chem. Biol. 2015, 22, 460–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothias, L.-F.; Nothias-Esposito, M.; Da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-Based Molecular Networking for the Discovery of Drug Leads in Natural Product Bioassay-Guided Fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Hooft, V.D.J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.V.; Rogers, S. Topic Modeling for Untargeted Substructure Exploration in Metabolomics. Proc. Natl. Acad. Sci.USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [Green Version]
- Marchisio, M.A. In Silico Implementation of Synthetic Gene Networks BT-Synthetic Gene Networks: Methods and Protocols. Methods Mol. Biol. 2012, 813, 3–21. [Google Scholar]
- Ernst, M.; Bin Kang, K.; Caraballo-Rodríguez, A.M.; Nothias, L.-F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.-M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.; Kieser, H.M.; Harper, D.P.; et al. Complete Genome Sequence of the Model Actinomycete Streptomyces Coelicolor A3(2). Nat. Cell Biol. 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Machado, H.; Tuttle, R.N.; Jensen, P.R. Omics-Based Natural Product Discovery and the Lexicon of Genome Mining. Curr. Opin. Microbiol. 2017, 39, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemert, N.; Alanjary, M.; Weber, T. The Evolution of Genome Mining in Microbes—A Review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, M.H.; Fischbach, M.A. Computational Approaches to Natural Product Discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The AntiSMASH Database, a Comprehensive Database of MI-Crobial Secondary Metabolite Biosynthetic Gene Clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanjary, M.; Kronmiller, B.; Adamek, M.; Blin, K.; Weber, T.; Huson, D.H.; Philmus, B.; Ziemert, N. The Antibiotic Resistant Target Seeker (ARTS), An Exploration Engine for Antibiotic Cluster Prioritization and Novel Drug Target Discovery. Nucleic Acids Res. 2017, 45, W42–W48. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; Santos, E.L.C.D.L.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A Computational Framework to Explore Large-Scale Biosynthetic Diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Soldatou, S.; Eldjarn, G.H.; Huerta-Uribe, A.; Rogers, S.; Duncan, K.R. Linking Biosynthetic and Chemical Space to Accelerate Microbial Secondary Metabolite Discovery. FEMS Microbiol. Lett. 2019, 366, 142. [Google Scholar] [CrossRef]
- Goering, A.W.; McClure, R.A.; Doroghazi, J.R.; Albright, J.C.; Haverland, N.A.; Zhang, Y.; Ju, K.S.; Thomson, R.J.; Metcalf, W.W.; Kelleher, N.L. Metabologenomics: Correlation of Microbial Gene Clusters with Metabolites Drives Discovery of a Non-Ribosomal Peptide with an Unusual Amino Acid Monomer. ACS Cent. Sci. 2016, 2, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. Aroadmap for Natural Product Discovery Based on Large-Scale Genomics and Metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef]
- Eldjárn, G.H.; Ramsay, A.; van der Hooft, J.J.J.; Duncan, K.R.; Soldatou, S.; Rousu, J.; Daly, R.; Wandy, J.; Rogers, S. Ranking Microbial Metabolomic and Genomic Links in the NPLinker Framework Using Complementary Scoring Functions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R. Developing a New Resource for Drug Discovery: Marine Actinomycete Bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Pickens, L.B.; Kim, W.; Wang, P.; Zhou, H.; Watanabe, K.; Gomi, S.; Tang, Y. Biochemical Analysis of the Biosynthetic Pathway of an Anticancer Tetracycline SF2575. J. Am. Chem. Soc. 2009, 131, 17677–17689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatsu, M.; Sasaki, T.; Watabe, H.-O.; Miyadoh, S.; Nagasawa, M.; Shomura, T.; Sezaki, M.; Inouye, S.; Kondo, S. A New Tetracycline Antibiotic with Antitumor Activity. I. Taxonomy and Fermentation of the Producing Strain, Isolation and Characterization of SF2575. J. Antibiot. 1992, 45, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Martínez, L.T.; Borsetto, C.; Gomez-Escribano, J.P.; Bibb, M.J.; Al-Bassam, M.M.; Chandra, G.; Bibb, M. New Insights into Chloramphenicol Biosynthesis in Streptomyces venezuelae ATCC 10712. Antimicrob. Agents Chemother. 2014, 58, 7441–7450. [Google Scholar] [CrossRef] [Green Version]
- Kleijn, L.H.J.; Martin, N.I. The Cyclic Lipopeptide Antibiotics. In Antibacterials. Topics in Medicinal Chemistry; Fisher, J.F., Mobashery, S., Miller, M.J., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 27–53. ISBN 978-3-319-70839-3. [Google Scholar]
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Biosynthetic Pathway for High Structural Diversity of a Common Dilactone Core in Antimycin Production. Org. Lett. 2012, 14, 4142–4145. [Google Scholar] [CrossRef]
- Westley, J.W.; Liu, C.-M.; Evans, R.H.; Blount, J.F. Conglobatin, a Novel Macrolide Dilactone from Streptomyces conglobatus ATCC 31005. J. Antibiot. 1979, 32, 874–877. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Murphy, A.C.; Samborskyy, M.; Prediger, P.; Dias, L.C.; Leadlay, P.F. Iterative Mechanism of Macrodiolide Formation in the Anticancer Compound Conglobatin. Chem. Biol. 2015, 22, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebstock, M.C.; Crooks, H.M.; Controulis, J.; Bartz, Q.R. Chloramphenicol (Chloromycetin).1IV.1aChemical Studies. J. Am. Chem. Soc. 1949, 71, 2458–2462. [Google Scholar] [CrossRef]
- Van Der Hooft, J.J.J.; Mohimani, H.; Bauermeister, A.; Dorrestein, P.C.; Duncan, K.R.; Medema, M.H. Linking Genomics and Metabolomics to Chart Specialized Metabolic Diversity. Chem. Soc. Rev. 2020, 49, 3297–3314. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid Identification, Annotation and Analysis of Secondary Metabolite Biosynthesis Gene Clusters in Bacterial and Fungal GE-Nome Sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Klassen, J.L.; Currie, C.R. Gene Fragmentation in Bacterial Draft Genomes: Extent, Consequences and Mitigation. BMC Genom. 2012, 13, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of Strategies for the Assembly of Diverse Bacterial Genomes Using Minion Long-Read Sequencing. BMC Genom. 2019, 20, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltz, R.H. Molecular Beacons to Identify Gifted Microbes for Genome Mining. J. Antibiot. 2017, 70, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, T.H.M. The Importance of Genome Sequence Quality to Microbial Comparative Genomics. BMC Genom. 2019, 20, 1–4. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene Synthases Are Widely Dis-Tributed in Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Blodgett, J.A.V.; Oh, D.-C.; Cao, S.; Currie, C.R.; Kolter, R.; Clardy, J. Common Biosynthetic Origins for Polycyclic Tetramate Macrolactams from Phylogenetically Diverse Bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 11692–11697. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, Y.; Lu, C.; Zhang, J.; Zhu, J.; Wang, H.; Shen, Y. Activating a Cryptic Ansamycin Biosynthetic Gene Cluster To Produce Three New Naphthalenic Octaketide Ansamycins with n-Pentyl and n-Butyl Side Chains. Org. Lett. 2015, 17, 3706–3709. [Google Scholar] [CrossRef]
- Eltamany, E.E.; Abdelmohsen, U.R.; Ibrahim, A.K.; Hassanean, H.A.; Hentschel, U.; Ahmed, S.A. New Antibacterial Xan-Thone from the Marine Sponge-Derived Micrococcus sp. EG45. Bioorg. Med. Chem. Lett. 2014, 24, 4939–4942. [Google Scholar] [CrossRef]
- Homann, V.V.; Sandy, M.; Tincu, J.A.; Templeton, A.S.; Tebo, B.M.; Butler, A. Loihichelins A−F, a Suite of Amphiphilic Siderophores Produced by the Marine Bacterium Halomonas LOB-5. J. Nat. Prod. 2009, 72, 884–888. [Google Scholar] [CrossRef] [Green Version]
- Bitzer, J.; Grosse, T.; Wang, L.; Lang, S.; Beil, W.; Zeeck, A. New Aminophenoxazinones from a Marine Halomonas sp.: Fermentation, Structure Elucidation, and Biological Activity. J. Antibiot. 2006, 59, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, L.; Hermann, L.; Stöveken, N.; Richter, A.A.; Höppner, A.; Smits, S.H.J.; Heider, J.; Bremer, E. Role of the Extremolytes Ectoine and Hydroxyectoine as Stress Protectants and Nutrients: Genetics, Phylogenomics, Biochemistry, and Structural Analysis. Genes 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweiggert, R.; Carle, R. Carotenoid Deposition in Plant And Animal Foods and Its Impact on Bioavailability. Crit. Rev. Food Sci. Nutr. 2015, 57. [Google Scholar] [CrossRef] [PubMed]
- Iftime, D.; Kulik, A.; Härtner, T.; Rohrer, S.; Niedermeyer, T.H.J.; Stegmann, E.; Weber, T.; Wohlleben, W. Identification and Activation of Novel Biosynthetic Gene Clusters by Genome Mining in the Kirromycin Producer Streptomyces collinus Tü 365. J. Ind. Microbiol. Biotechnol. 2016, 43, 277–291. [Google Scholar] [CrossRef]
- Myronovskyi, M.; Tokovenko, B.; Brötz, E.; Rückert, C.; Kalinowski, J.; Luzhetskyy, A. Genome Rearrangements of Streptomyces Albus J1074 Lead to the Carotenoid Gene Cluster Activation. Appl. Microbiol. Biotechnol. 2014, 98, 795–806. [Google Scholar] [CrossRef]
- Debono, M.; Barnhart, M.; Carrell, C.B.; Hoffmann, J.A.; Occolowitz, J.L.; Abbott, B.J.; Fukuda, D.S.; Hamill, R.L.; Biemann, K.; Herlihy, W.C. A21978C, A Complex of New Acidic Peptide Antibiotics. Isolation, Chemistry, and Mass Spectral Structure Elucidation. J. Antibiot. 1987, 40, 761–777. [Google Scholar] [CrossRef] [Green Version]
- Kosmidis, C.; Levine, D.P. Daptomycin: Pharmacology and Clinical Use. Expert Opin. Pharmacother. 2010, 11, 615–625. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in Vitro Evaluating Antimicrobial Activity: A Review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Chapartegui-González, I.; Lázaro-Díez, M.; Bravo, Z.; Navas, J.; Icardo, J.M.; Ramos-Vivas, J. Acinetobacter Baumannii Maintains Its Virulence after Long-Time Starvation. PLoS ONE 2018, 13, e0201961. [Google Scholar] [CrossRef]
- Liaw, C.-C.; Chen, P.-C.; Shih, C.-J.; Tseng, S.-P.; Lai, Y.-M.; Hsu, C.-H.; Dorrestein, P.C.; Yang, Y. Vitroprocines, New Antibiotics against Acinetobacter Baumannii, Discovered from Marine Vibrio SP. QWI-06 Using Mass-Spectrometry-Based Metabolomics Approach. Sci. Rep. 2015, 5, 12856. [Google Scholar] [CrossRef] [Green Version]
- Vila-Farres, X.; Chu, J.; Ternei, M.A.; Lemetre, C.; Park, S.; Perlin, D.S.; Brady, S.F. An Optimized Synthetic-Bioinformatic Natural Product Antibiotic Sterilizes Multidrug-Resistant Acinetobacter baumannii-Infected Wounds. mSphere 2018, 3, e00528-17. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-S.; Cheng, W.-C.; Cheng, T.-J.R.; Wong, C.-H. Affinity-Based Screen for Inhibitors of Bacterial Transglycosylase. J. Am. Chem. Soc. 2018, 140, 2752–2755. [Google Scholar] [CrossRef]
- Zdouc, M.M.; Iorio, M.; Maffioli, S.I.; Crüsemann, M.; Donadio, S.; Sosio, M. Planomonospora: A Metabolomics Perspective on an Underexplored Actinobacteria Genus. J. Nat. Prod. 2021. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, J.A.; Kautsar, S.A.; Medema, M.H.; Linington, R.G. Microbial Natural Product Databases: Moving Forward in the Multiomics Era. Nat. Prod. Rep. 2021, 38, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Kurita, K.L.; Glassey, E.; Linington, R.G. Integration of High-Content Screening and Untargeted Metabolomics for ComPre-hensive Functional Annotation of Natural Product Libraries. Proc. Natl. Acad. Sci. USA 2015, 112, 11999–12004. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucle-Otide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Shirling, E.B.; Gottlieb, D. Methods for Characterization of Streptomyces Species. Int. J. Syst. Bacteriol. 1966, 16, 313–340. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, R.; Mallick, P. Data Conversion with Proteo Wizard msConvert. In Proteomics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 339–368. [Google Scholar]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of Biological Networks and Gene Expression Data Using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [Green Version]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. Classyfire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Maniatis, T.; Fritsch, E.F. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wood, E.; Salzberg, D.E.; Kraken, S.L. Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.-F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A Multi-Draft Based Scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soldatou, S.; Eldjárn, G.H.; Ramsay, A.; van der Hooft, J.J.J.; Hughes, A.H.; Rogers, S.; Duncan, K.R. Comparative Metabologenomics Analysis of Polar Actinomycetes. Mar. Drugs 2021, 19, 103. https://doi.org/10.3390/md19020103
Soldatou S, Eldjárn GH, Ramsay A, van der Hooft JJJ, Hughes AH, Rogers S, Duncan KR. Comparative Metabologenomics Analysis of Polar Actinomycetes. Marine Drugs. 2021; 19(2):103. https://doi.org/10.3390/md19020103
Chicago/Turabian StyleSoldatou, Sylvia, Grímur Hjörleifsson Eldjárn, Andrew Ramsay, Justin J. J. van der Hooft, Alison H. Hughes, Simon Rogers, and Katherine R. Duncan. 2021. "Comparative Metabologenomics Analysis of Polar Actinomycetes" Marine Drugs 19, no. 2: 103. https://doi.org/10.3390/md19020103
APA StyleSoldatou, S., Eldjárn, G. H., Ramsay, A., van der Hooft, J. J. J., Hughes, A. H., Rogers, S., & Duncan, K. R. (2021). Comparative Metabologenomics Analysis of Polar Actinomycetes. Marine Drugs, 19(2), 103. https://doi.org/10.3390/md19020103