Osirisynes G-I, New Long-Chain Highly Oxygenated Polyacetylenes from the Mayotte Marine Sponge Haliclona sp.

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
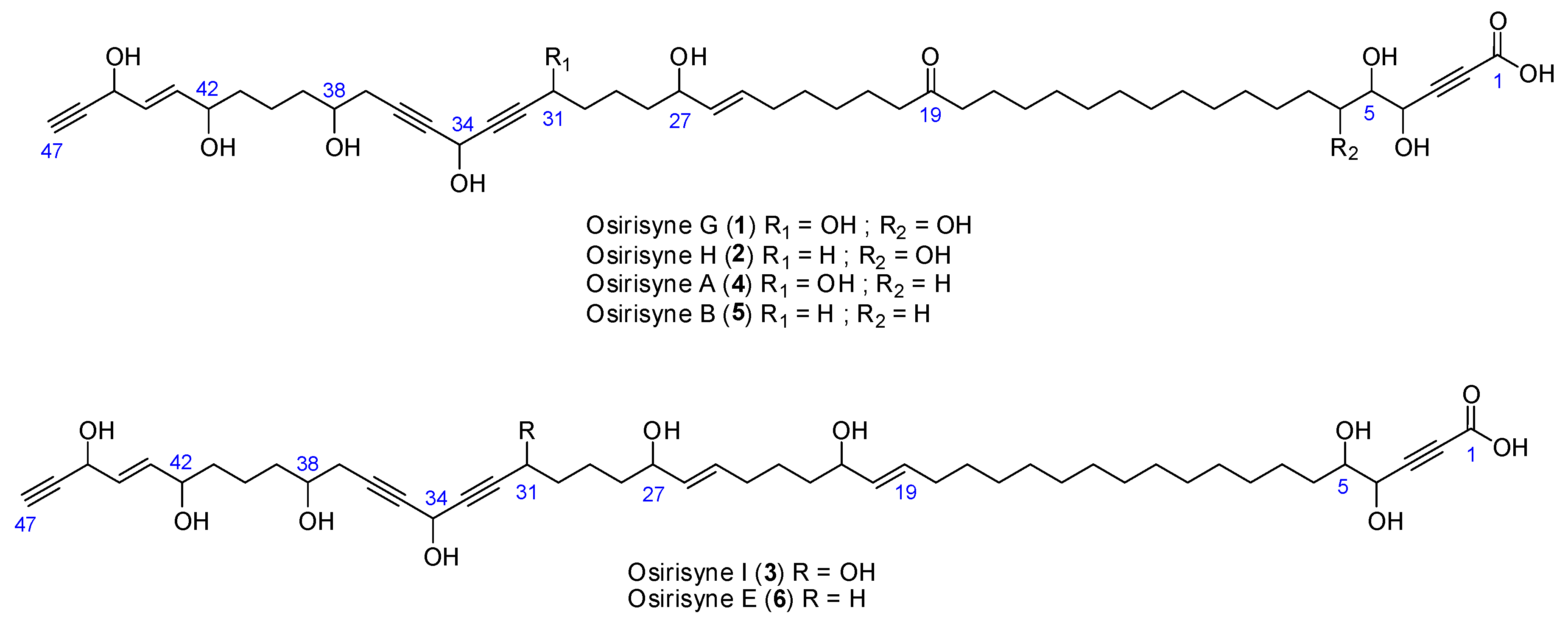
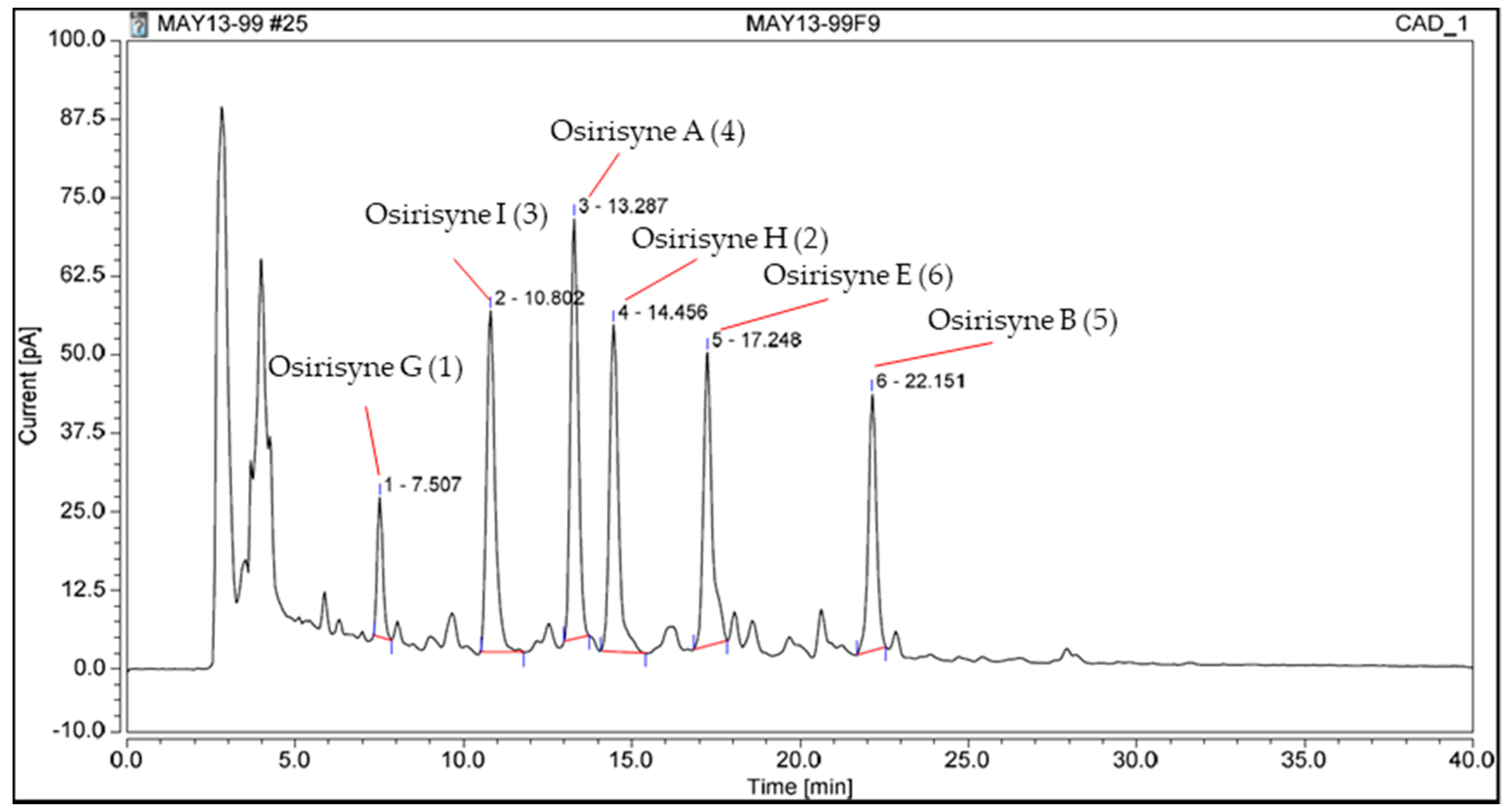
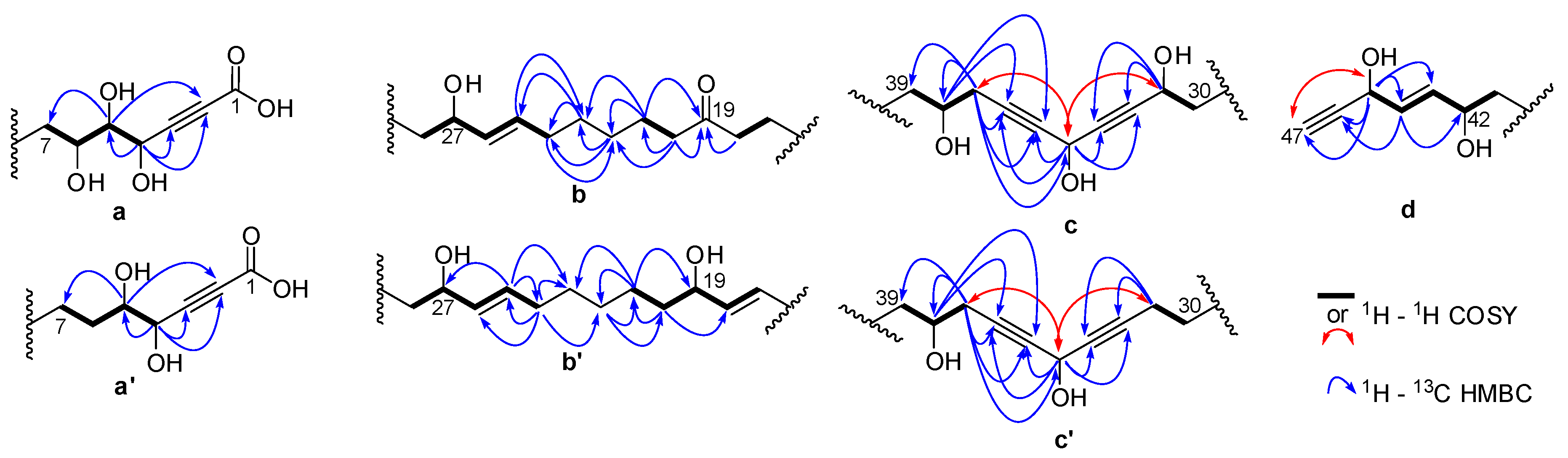
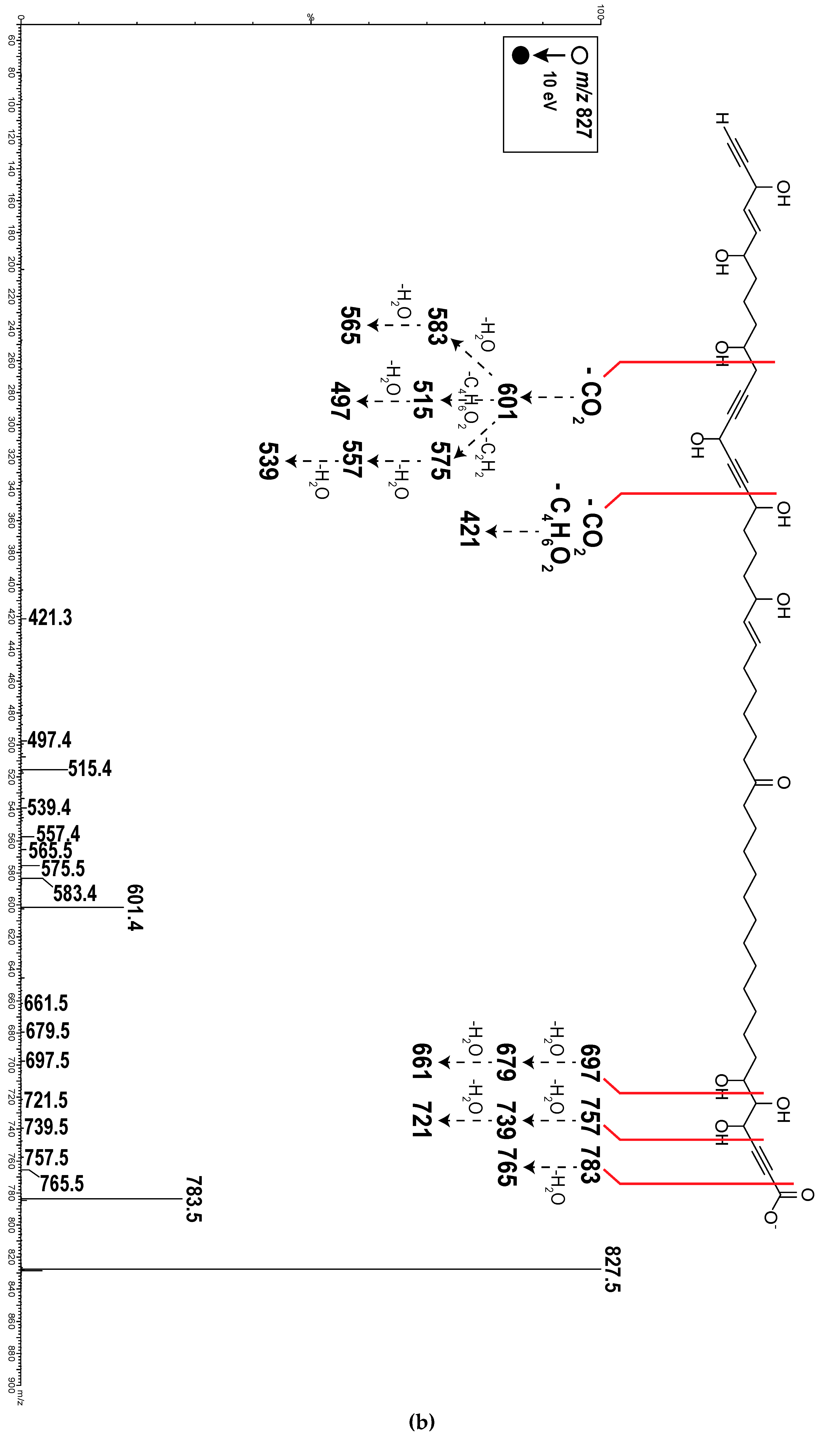
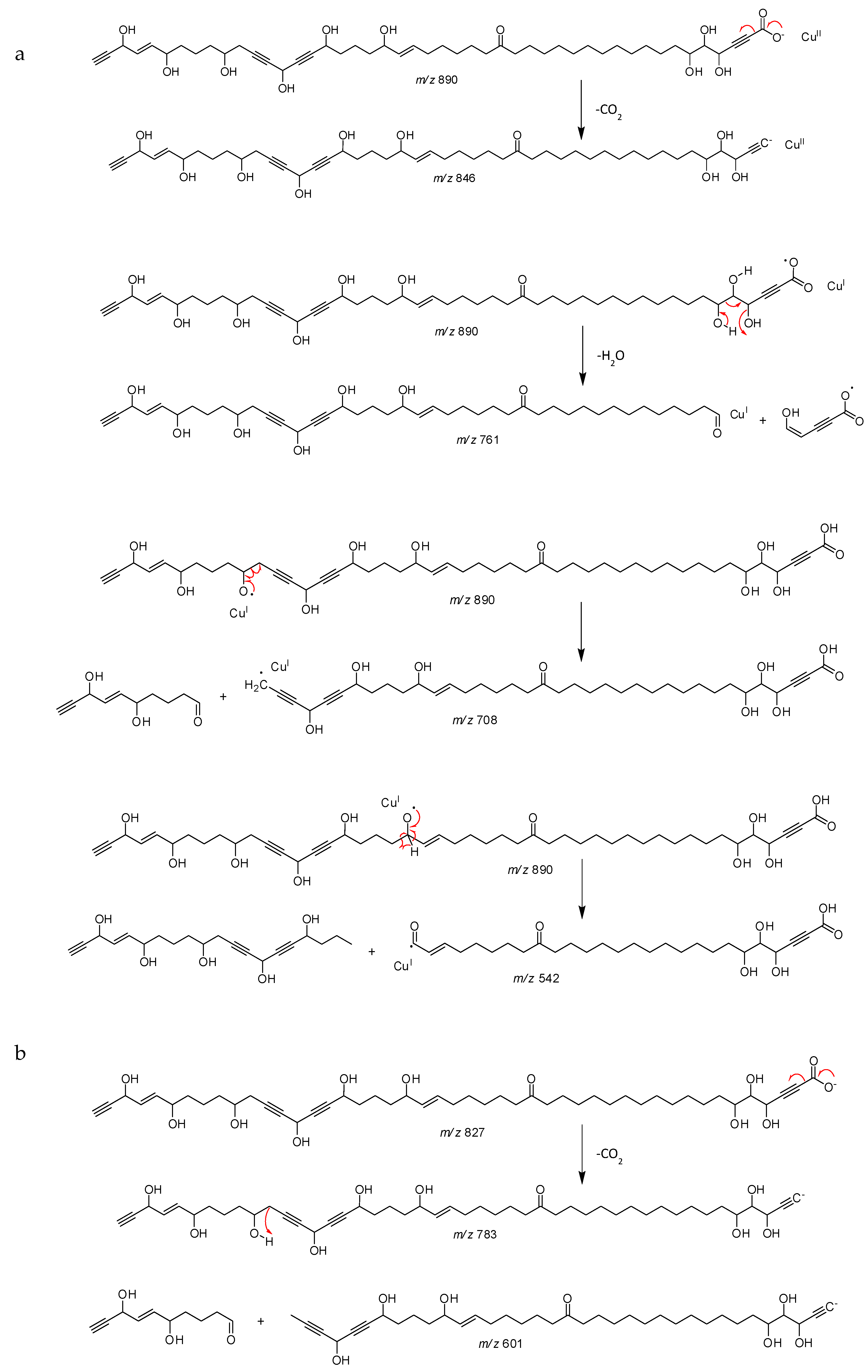
2.1. Chemistry
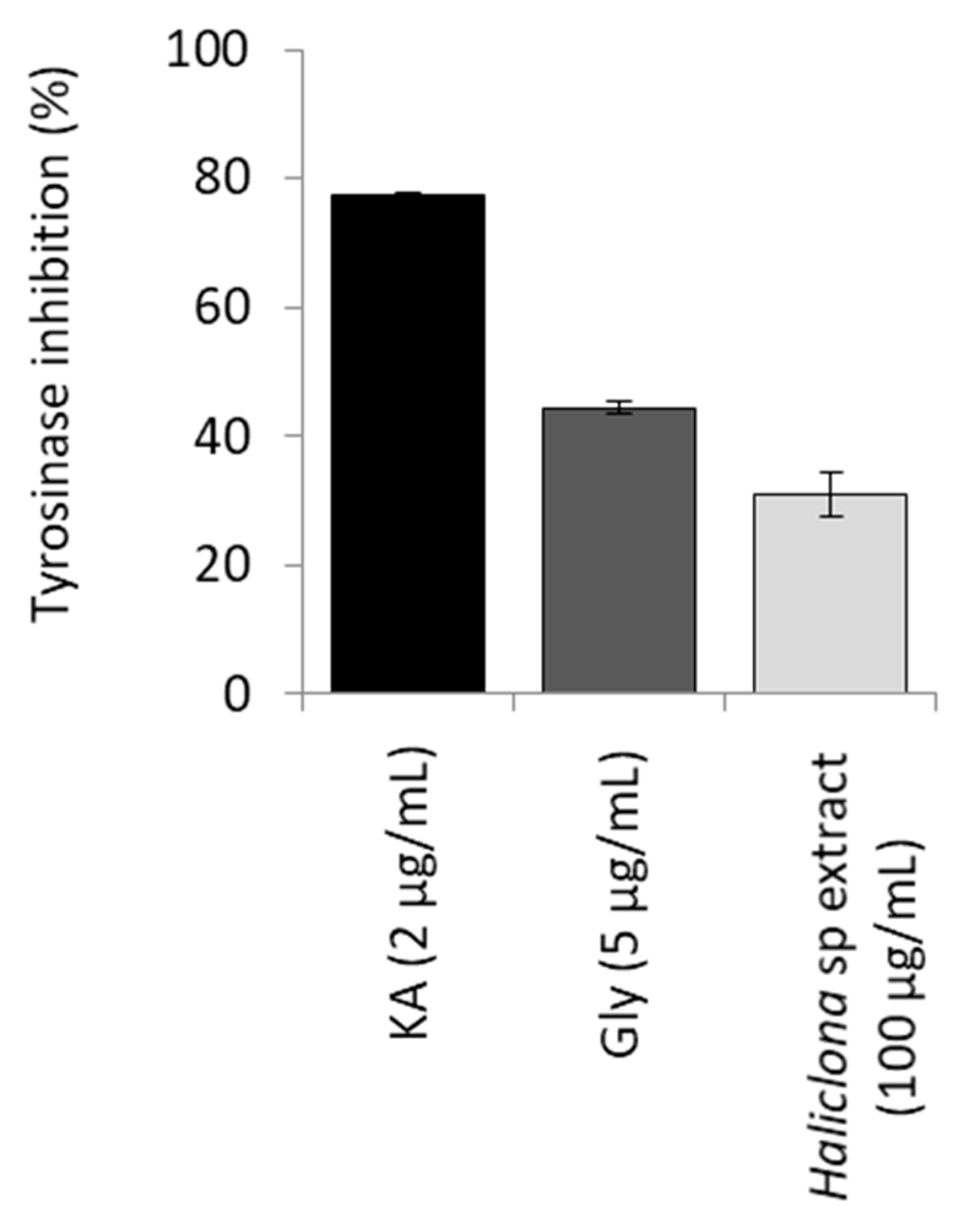
2.2. Biological Activity
3. Materials and Methods
3.1. General Experiment Procedures
- ESI+: capillary voltage: 2.8 kV, cone voltage: 20 V, gas flow (N2): 100 L/h, collision gas: Ar.
- ESI−: capillary voltage: −2.27 kV, cone voltage: −20 V, gas flow (N2): 100 L/h, collision gas: Ar.
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Catalase Activation Assays
3.5. Sirtuin 1 Activation Assays
3.6. CDK7 Inhibition Assays
3.7. Proteasome Inhibition Assays
3.8. FynB Kinase Inhibition Assays
3.9. Tyrosinase Inhibition Assays
3.10. Elastase Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase Inhibitors from Marine Sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Imada, C. Enzyme Inhibitors of Marine Microbial Origin with Pharmaceutical Importance. Mar. Biotechnol. 2004, 6, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Minto, R.E.; Blacklock, B.J. Biosynthesis and Function of Polyacetylenes and Allied Natural Products. Prog. Lipid Res. 2008, 47, 233–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, S.; Kashman, Y.; Loya, S.; Hizi, A.; Loya, Y. Petrosynol and Petrosolic Acid, Two Novel Natural Inhibitors of the Reverse Transcriptase of Human Immunodeficiency Virus from Petrosia sp. Tetrahedron 1993, 49, 10435–10438. [Google Scholar] [CrossRef]
- Li, H.-Y.; Matsunaga, S.; Fusetani, N. Corticatic Acids A-C, Antifungal Acetylenic Acids from the Marine Sponge, Petrosia Corticata. J. Nat. Prod. 1994, 57, 1464–1467. [Google Scholar] [CrossRef]
- Fusetani, N.; Shiragaki, T.; Matsunaga, S.; Hashimoto, K. Bioactive Marine Metabolites XX. Petrosynol and Petrosynone, Antimicrobial C30 Polyacetylenes from the Marine Sponge Petrosia sp.: Determination of the Absolute Configuration. Tetrahedron Lett. 1987, 28, 4313–4314. [Google Scholar] [CrossRef]
- Ayyad, S.-E.N.; Katoua, D.F.; Alarif, W.M.; Sobahi, T.R.; Aly, M.M.; Shaala, L.A.; Ghandourah, M.A. Two New Polyacetylene Derivatives from the Red Sea Sponge Xestospongia sp. Z. Naturforsch. C 2015, 70, 297–303. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Cardellina, J.H.; Balaschak, M.S.; Alexander, M.R.; Prather, T.R.; Shoemaker, R.H.; Boyd, M.R. Antitumor Activity and Stereochemistry of Acetylenic Alcohols from the Sponge Cribrochalina vasculum. J. Nat. Prod. 1995, 58, 1801–1807. [Google Scholar] [CrossRef]
- Nuzzo, G.; Ciavatta, M.L.; Villani, G.; Manzo, E.; Zanfardino, A.; Varcamonti, M.; Gavagnin, M. Fulvynes, Antimicrobial Polyoxygenated Acetylenes from the Mediterranean Sponge Haliclona fulva. Tetrahedron 2012, 68, 754–760. [Google Scholar] [CrossRef]
- Shin, J.; Seo, Y.; Cho, K.W.; Rho, J.-R.; Paul, V.J. Osirisynes A-F, Highly Oxygenated Polyacetylenes from the Sponge Haliclona osiris. Tetrahedron 1998, 54, 8711–8720. [Google Scholar] [CrossRef]
- Fusetani, N.; Sugano, M.; Matsunaga, S.; Hashimoto, K. H,K-ATPase Inhibitors from the Marine Sponge Siphonochalina truncata: Absolute Configuration of Siphonodiol and Two Related Metabolites. Tetrahedron Lett. 1987, 28, 4311–4312. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Yoshida, W.Y.; Kelly, M.; Scheuer, P.J. Polyacetylenes from a Red Sea Sponge Callyspongia Species. J. Nat. Prod. 2000, 63, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.P. Chapter 20—Bioactivity of Polyacetylenes in Food Plants. In Bioactive Foods in Promoting Health; Watson, R.R., Preedy, V.R., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 285–306. [Google Scholar]
- Wade Harper, J.; Elledge, S.J. Cdk Inhibitors in Development and Cancer. Curr. Opin. Genet. Dev. 1996, 6, 56–64. [Google Scholar] [CrossRef]
- Geutjes, E.-J.; Bajpe, P.K.; Bernards, R. Targeting the Epigenome for Treatment of Cancer. Oncogene 2012, 31, 3827–3844. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn Kinase Inhibition as a Novel Therapy for Alzheimer’s Disease. Alzheimers Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Strooper, B.D.; Vassar, R.; Golde, T. The Secretases: Enzymes with Therapeutic Potential in Alzheimer Disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, N.; Costantini, S.; Palumbo, F.; Costantini, M. Marine Sponges and Bacteria as Challenging Sources of Enzyme Inhibitors for Pharmacological Applications. Mar. Drugs 2017, 15, 173. [Google Scholar] [CrossRef] [Green Version]
- Gianelli, L.; Amendola, V.; Fabbrizzi, L.; Pallavicini, P.; Mellerio, G.G. Investigation of reduction of Cu(II) complexes in positive-ion mode electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2001, 15, 2347–2353. [Google Scholar] [CrossRef]
- Afonso, C.; Riu, A.; Xu, Y.; Fournier, F.; Tabet, J.-C. Structural characterization of fatty acids cationized with copper by electrospray ionization mass spectrometry under low-energy collision-induced dissociation. J. Mass. Spectrom. 2005, 40, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Mistry, N.; Wood, C.; Herbert, K.E.; Lunec, J. Immunogenicity of DNA damaged by reactive oxygen species—Implications for anti-DNA antibodies in lupus. Free Radic. Biol. Med. 1997, 22, 151–159. [Google Scholar] [CrossRef]
- Sun, C.; Shan, C.Y.; Gao, X.D.; Tan, R.X. Protection of PC12 cells from hydrogen peroxide-induced injury by EPS2, an exopolysaccharide from a marine filamentous fungus Keissleriella sp. YS4108. J. Biotechnol. 2005, 115, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Wątroba, M.; Szukiewicz, D. The role of sirtuins in aging and age-related diseases. Adv. Med. Sci. 2016, 61, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kelso, T.W.R.; Baumgart, K.; Eickhoff, J.; Albert, T.; Antrecht, C.; Lemcke, S.; Klebl, B.; Meisterernst, M. Cyclin-dependent kinase 7 controls mRNA synthesis by affecting stability of preinitiation complexes, leading to altered gene expression, cell cycle progression, and survival of tumor cells. Mol. Cell. Biol. 2014, 34, 3675–3688. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef] [Green Version]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef]
- Resh, M.D. Fyn, a Src family tyrosine kinase. Int. J. Biochem. Cell Biol. 1998, 30, 1159–1162. [Google Scholar] [CrossRef]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A novel molecular target in cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Noh, S.G.; Park, Y.; Kang, D.; Chun, P.; Chung, H.Y.; Moon, H.R. In vitro and in silico insights into tyrosinase inhibitors with (E)-benzylidene-1-indanone derivatives. Comput. Struct. Biotechnol. J. 2019, 17, 1255–1264. [Google Scholar] [CrossRef]
- Azam, M.S.; Choi, J.; Lee, M.-S.; Kim, H.-R. Hypopigmenting Effects of Brown Algae-Derived Phytochemicals: A Review on Molecular Mechanisms. Mar. Drugs 2017, 15, 297. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Yang, X.; Cao, Y. Study of Ellagic Acid as a Natural Elastase Inhibitor by Spectroscopic Methods. J. Appl. Spectrosc. 2016, 83, 149–155. [Google Scholar] [CrossRef]
- Andrés-Ramos, I.; Alegría-Landa, V.; Gimeno, I.; Pérez-Plaza, A.; Rütten, A.; Kutzner, H.; Requena, L. Cutaneous Elastic Tissue Anomalies. Am. J. Dermatopathol. 2019, 41, 85–117. [Google Scholar] [CrossRef]
- Teicher, B.A.; Ara, G.; Herbst, R.; Palombella, V.J.; Adams, J. The Proteasome Inhibitor PS-341 in Cancer Therapy. Clin. Cancer Res. 1999, 5, 2638–2645. [Google Scholar]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. BBA Mol. Cell Res. 2014, 1843, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Summers, F.A.; Mason, R.P. Photooxidation of Amplex Red to Resorufin: Implications of Exposing the Amplex Red Assay to Light. Free Radic. Biol. Med. 2012, 53, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Totoritis, R.D.; Lor, L.A.; Schwartz, B.; Caprioli, P.; Jurewicz, A.J.; Zhang, G. Evaluation of an Antibody-Free ADP Detection Assay: ADP-Glo. ASSAY Drug Dev. Technol. 2009, 7, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Odaka, Y.; Ogawa, N.; Nakamoto, K.; Kuninaga, H. Identification of geranic acid, a tyrosinase inhibitor in lemongrass (Cymbopogon citratus). J. Agric. Food Chem. 2008, 56, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Angelis, A.; Aligiannis, N.; Rosalia, M.; Abedini, A.; Bakiri, A.; Reynaud, R.; Nuzillard, J.M.; Gangloff, S.C.; Skaltsounis, A.L.; et al. In Vitro Dermo-Cosmetic Evaluation of Bark Extracts from Common Temperate Trees. Planta Med. 2016, 82, 1351–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | δC, Type | δH, mult. (J in Hz) | COSY (1H-1H) | HMBC (1H-13C) |
|---|---|---|---|---|
| 1 | 161.3, C | |||
| 2 | 79.9, C | |||
| 3 | 83.4, C | |||
| 4 | 65.0, CH | 4.60, 1H, d (4.2) | 5 | 2, 3, 5 |
| 5 | 78.5, CH | 3.43, 1H, dd (8.1, 4.3) | 4, 6 | 2, 4, 6, 7 |
| 6 | 72.7, CH | 3.61, 1H, td (8.6, 2.5) | 5, 7 | 4, 8 |
| 7 | 34.3, CH2 | 1.78, 1H, m; 1.38, 1H, m | 6, 8 | |
| 8 | 24.0, CH2 | 1.57, 2H, m | ||
| 9–16 | 30.8, CH2 | 1.40–1.30, 16H, m | ||
| 17 | 24.8, CH2 | 1.54, 2H, m | 18 | |
| 18 | 43.4, CH2 | 2.46, 2H, t (7.4) | 17 | 16, 17, 19 |
| 19 | 214.5, C | |||
| 20 | 43.5, CH2 | 2.44, 2H, t (8.1) | 21 | 19, 21, 22 |
| 21 | 24.8, CH2 | 1.54, 2H, m | 20 | 19, 20, 22, 23 |
| 22 | 30.2, CH2 | 1.40, 2H, m | 21 | 21, 23, 24 |
| 23 | 30.2, CH2 | 1.48, 1H, m; 1.38, 1H, m | 24 | 21, 22, 24, 25 |
| 24 | 33.1, CH2 | 2.05, 2H, q (7.1) | 23, 25 | 22, 23, 25, 26 |
| 25 | 132.3, CH | 5.62, 1H, dtd (15.3, 6.5, 0.8) | 24, 26 | 23, 24, 26, 27 |
| 26 | 134.3, CH | 5.43, 1H, ddt (15.3, 7.1, 1.4) | 25, 27 | 24, 27 |
| 27 | 73.5, CH | 3.97, 1H, m | 26, 28 | 25, 26, 28, 29 |
| 28 | 38.1, CH2 | 1.56, 1H, m, 1.49, 1H, m | 27 | 27 |
| 29 | 22.4, CH2 | 1.50, 2H, m | 27, 31 | |
| 30 | 38.5, CH2 | 1.67, 2H, m | 31 | 31, 32 |
| 31 | 62.5, CH | 4.33, 1H, td (6.7, 1.6) | 30, 34 | 29, 30, 32, 33 |
| 32 | 85.0, C | |||
| 33 | 83.8, C | |||
| 34 | 52.3, CH | 5.11, 1H, m | 31, 37 | 32, 33, 35, 36 |
| 35 | 80.9, C | |||
| 36 | 81.9, C | |||
| 37 | 28.0, CH2 | 2.38, 2H, dd (5.9, 1.9) | 34, 38 | 35, 36, 38, 39 |
| 38 | 70.7, CH | 3.69, 1H, tt (10.9, 6.3) | 37, 39 | 36, 37, 39, 40 |
| 39 | 37.0, CH2 | 1.50, 2H, m | 38 | 37, 38 |
| 1.64, 2H, m | ||||
| 40 | 22.6, CH2 | 1.49, 2H, m | 38, 42 | |
| 41 | 38.1 CH2 | 1.58, 1H, m; 1.50, 1H, m | 42 | 42, 43 |
| 42 | 72.2, CH | 4.09, 1H, q (6.0) | 41, 43 | 40, 41, 43 |
| 43 | 136.0 CH | 5.88, 1H, ddd (15.4, 6.2, 1.3) | 42, 44 | |
| 44 | 130.3, CH | 5.76, 1H, ddd (15.4, 5.7, 1.1) | 43, 45 | 42, 43, 45, 46 |
| 45 | 62.5, CH | 4.82, 1H, dm (5.7) | 44, 47 | 43, 44, 46, 47 |
| 46 | 84.7 C | |||
| 47 | 74.9, CH | 2.92, 1H, d (2.2) | 45 | 45 |
| No. | Osirisyne H (2) | Osirisyne I (3) | ||
|---|---|---|---|---|
| δC, Type | δH, mult. (J in Hz) | δC, Type | δH, mult. (J in Hz) | |
| 1 | 158.6, C | 161.5, C | ||
| 2 | 80.0, C | 80.7, C | ||
| 3 | 83.3, C | 83.3, C | ||
| 4 | 65.0, CH | 4.60, 1H, d (4.3) | 67.2, CH | 4.19, 1H, d (5.2) |
| 5 | 78.5, CH | 3.43, 1H, dd (8.1, 4.3) | 75.5, CH | 3.56, 1H, ddd (8.6, 5.0, 3.4) |
| 6 | 72.9, CH | 3.62, 1H, td (8.8, 2.3) | 33.3, CH2 | 1.67, 1H, m; 1.47, 1H, m |
| 7 | 34.1, CH2 | 1.77, 1H, m; 1.38, 1H, m | 26.9, CH2 | 1.54, 1H, m; 1.35, 1H, m |
| 8 | 24.7, CH2 | 1.56, 2H, m | 30.8, CH2 | 1.40–1.30, 2H, m |
| 9–16 | 30.8, CH2 | 1.40–1.30, 16H, m | 30.8, CH2 | 1.40–1.30, 16H, m |
| 17 | 24.3, CH2 | 1.54, 2H, m | 30.5, CH2 | 1.39, 2H, m |
| 18 | 43.3, CH2 | 2.45, 2H, t (7,4) | 33.3, CH2 | 2.05, 2H, m |
| 19 | 214.5, C | 132.5, CH | 5.62, 1H, dq (14.5, 7.0) | |
| 20 | 43.3, CH2 | 2.45, 2H, t (7.4) | 134.5, CH | 5.43, 1H, ddt (15.3, 7.1, 1.3) |
| 21 | 24.3, CH2 | 1.54, 2H, m | 73.6, CH | 3.97, 1H, q (6.3) |
| 22 | 30.1, CH2 | 1.41, 2H, m | 37.9, CH2 | 1.56, 1H, m; 1.45, 1H, m |
| 23 | 30.1, CH2 | 1.40, 2H, m | 26.3, CH2 | 1.39, 2H, m |
| 24 | 33.0, CH2 | 2.05, 2H, q (7.1) | 33.2, CH2 | 2.05, 2H, m |
| 25 | 132.2, CH | 5.61, 1H, dtd (15.3, 6.7, 0.6) | 132.3, CH | 5.62, 1H, dq (14.5, 7.0) |
| 26 | 134.3, CH | 5.42, 1H, ddt (15.3, 7.1, 1.3) | 134.4, CH | 5.43, 1H, ddt (15.3, 7.1, 1.3) |
| 27 | 73.4, CH | 3.97, 1H, q (6.3) | 73.6, CH | 3.98, 1H, q (6.3) |
| 28 | 37.5, CH2 | 1.51, 2H, m | 38.1, CH2 | 1.50, 2H, m |
| 29 | 26.2, CH2 | 1.43, 2H, m | 22.5, CH2 | 1.47, 2H, m |
| 30 | 29.2, CH2 | 1.52, 2H, m | 38.8, CH2 | 1.68, 2H, m |
| 31 | 19.1, CH2 | 2.23, 2H, td (6.9, 2.0) | 62.6, CH | 4.33, 1H, td (6.7, 1.6) |
| 32 | 79.3, C | 85.5, C | ||
| 33 | 84.3, C | 84.4, C | ||
| 34 | 52.4, CH | 5.04, 1H, quint (1.8) | 52.5, CH | 5.11, 1H, q (1.7) |
| 35 | 81.1, C | 80.7, C | ||
| 36 | 81.5, C | 82.1, C | ||
| 37 | 28.0, CH2 | 2.38, 2H, dq (5.8, 1.0) | 28.1, CH2 | 2.38, 2H, dd (5.9, 2.0) |
| 38 | 70.6, CH | 3.69, 1H, m | 70.9, CH | 3.69, 1H, m |
| 39 | 36.6, CH2 | 1.50, 1H, m; 1.65, 1H, m | 37.0, CH2 | 1.50, 1H, m; 1.64, 1H, m |
| 40 | 22.3, CH2 | 1.49, 2H, m | 22.5, CH2 | 1.49, 2H, m |
| 41 | 37.8 CH2 | 1.58, 1H, m; 1.50, 1H, m | 38.1 CH2 | 1.60, 1H, m; 1.51, 1H, m |
| 42 | 72.2, CH | 4.09, 1H, q (6.1) | 72.4, CH | 4.09, 1H, q (6.1) |
| 43 | 136.0 CH | 5.88, 1H, ddd (15.4, 6.2, 1.3) | 136.2 CH | 5.88, 1H, ddd (15.3, 6.2, 1.2) |
| 44 | 130.4, CH | 5.76, 1H, ddd (15.4, 5.7, 1.1) | 130.5, CH | 5.76, 1H, ddd (15.4, 5.7, 1.1) |
| 45 | 62.4, CH | 4.82, 1H, dm (5.7) | 62.6, CH | 4.82, 1H, dm (5.7) |
| 46 | 84.0 C | 84.4 C | ||
| 47 | 74.5, CH | 2.92, 1H, d (2.2) | 74.9, CH | 2.91, 1H, d (2.2) |
| Compound | FynB Kinase Inhibition | CDK7 Kinase Inhibition | Proteasome Inhibition |
|---|---|---|---|
| Osirisyne G (1) | >40.20 | >40.20 | |
| Osirisyne H (2) | >40.99 | ||
| Osirisyne I (3) | >40.99 | >40.99 | |
| Osirisyne A (4) | >40.99 | >40.99 | |
| Osirisyne B (5) | 18.44 | 9.13 | 0.26 |
| Osirisyne E (6) | >41.81 | >41.81 | >41.81 |
| Staurosporine | 0.137 | 0.172 | |
| ONX-0914 | 4.63 × 10−5 |
| protein | 300 nM CDK7 (Crelux construct CZY-3, PC09891) |
| substrate | 30 μM CDKtide |
| ATP | 125 μM Ultra Pure ATP (Sigma) |
| buffer | 20 mM Hepes pH 7.5, 150 mM NaCl, 10 mM MgCl2 |
| extracts | stock conc. 10 mg/mL in DMSO |
| instrument | TECAN INFINITE M1000 PRO |
| settings | Mode: Luminescence; Integration time: 100 ms |
| Compound plates | 1 mg/mL in DMSO |
| Protein | Yeast proteasome (TUM Groll group) |
| Storage buffer | 20 mM Tris/HCl pH 7.5 |
| substrate | Suc-Leu-Leu-Val-Tyr-AMC (Enzo Lifesciences, BML-P802-0005) |
| assay buffer | 100 mM Tris pH 7.5, 1 mM MgCl2 |
| instrument | TECAN INFINITE M1000 PRO |
| settings | mode: Fluorescence Intensity; ex 380 nm, em 460 nm; bandwidth: +/−10 nm |
| Compound plates | 1 mg/mL in DMSO |
| protein | 200 nM FynB wt (Crelux construct CTX4, PC09815-1) |
| substrate | 10 μM Fyn substrate (Enzo, P-215) |
| ATP | 100 μM Ultra Pure ATP (Promega) |
| buffer | 20 mM Tris pH 8.0, 170 mM NaCl, 10 mM MgCl2 |
| instrument | TECAN INFINITE M1000 PRO |
| settings | Mode: Luminescence; Integration time: 100 ms |
| Compound plates | 1 mg/mL in DMSO |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, P.-E.; Herbette, G.; Chendo, C.; Clerc, P.; Tintillier, F.; de Voogd, N.J.; Papanagnou, E.-D.; Trougakos, I.P.; Jerabek, M.; Bignon, J.; et al. Osirisynes G-I, New Long-Chain Highly Oxygenated Polyacetylenes from the Mayotte Marine Sponge Haliclona sp. Mar. Drugs 2020, 18, 350. https://doi.org/10.3390/md18070350
Campos P-E, Herbette G, Chendo C, Clerc P, Tintillier F, de Voogd NJ, Papanagnou E-D, Trougakos IP, Jerabek M, Bignon J, et al. Osirisynes G-I, New Long-Chain Highly Oxygenated Polyacetylenes from the Mayotte Marine Sponge Haliclona sp. Marine Drugs. 2020; 18(7):350. https://doi.org/10.3390/md18070350
Chicago/Turabian StyleCampos, Pierre-Eric, Gaëtan Herbette, Christophe Chendo, Patricia Clerc, Florent Tintillier, Nicole J. de Voogd, Eleni-Dimitra Papanagnou, Ioannis P. Trougakos, Moran Jerabek, Jérôme Bignon, and et al. 2020. "Osirisynes G-I, New Long-Chain Highly Oxygenated Polyacetylenes from the Mayotte Marine Sponge Haliclona sp." Marine Drugs 18, no. 7: 350. https://doi.org/10.3390/md18070350
APA StyleCampos, P.-E., Herbette, G., Chendo, C., Clerc, P., Tintillier, F., de Voogd, N. J., Papanagnou, E.-D., Trougakos, I. P., Jerabek, M., Bignon, J., Le Goff, G., Ouazzani, J., & Gauvin-Bialecki, A. (2020). Osirisynes G-I, New Long-Chain Highly Oxygenated Polyacetylenes from the Mayotte Marine Sponge Haliclona sp. Marine Drugs, 18(7), 350. https://doi.org/10.3390/md18070350