Characteristics, Cryoprotection Evaluation and In Vitro Release of BSA-Loaded Chitosan Nanoparticles

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. NPs Preparation and Characterization
2.2. Preparation of the NPs through DHPM
2.3. NPs Lyophilization
2.4. Microstructure of NPs
2.4.1. Fluorescence Quenching of BSA
2.4.2. Interaction Parameter and the Combination Types
2.4.3. Fourier Transform Infrared Spectroscopy (FT-IR)
2.4.4. CD Spectra Studies
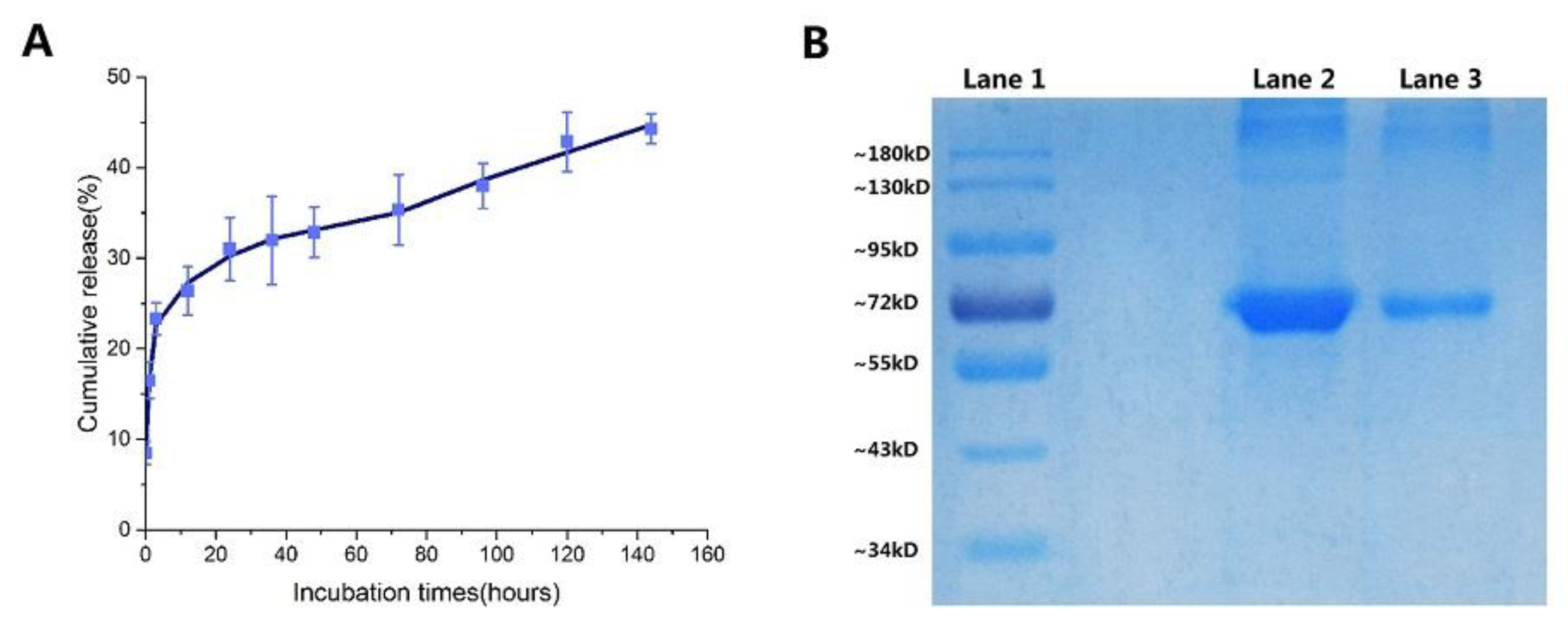
2.5. In Vitro Release of NPs
2.6. Cytotoxicity Analysis
3. Materials and Methods
3.1. Materials
3.2. Preparation of the NPs
3.3. NPs Recovery
3.4. Physicochemical Characteristics of NPs
3.4.1. Particle Size and Morphology
3.4.2. The Encapsulation Efficiency and Loading Capacity of NPs
3.5. NPs Release and Stability Studies
3.5.1. In Vitro BSA Release Studies
3.5.2. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis
3.6. Multi-Spectroscopic Investigation of NPs
3.6.1. Fluorescence Spectroscopy Measurements
3.6.2. FT-IR Measurements
3.6.3. CD Spectra Measurements
3.7. Cytotoxicity Assay
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CS-NPs | Chitosan nanoparticles |
| TPP | Tripolyphosphate pentasodium |
| HMWC | High molecular weight chitosan |
| MMWC | Medium molecular weight chitosan |
| LMWC | Low molecular weight chitosan |
| BSA | Bovine serum albumin |
| FITC | Fluorescein 5(6)-isothiocyanate |
| DLS | Dynamic light scattering |
| MHD | Mean hydrodynamic diameter |
| SD | Standard deviation |
| PDI | Polydispersity index |
| TEM | Transmission electron microscope |
| EE | Encapsulation efficiency |
| LC | Loading capacity |
| Trp | Tryptophan |
| FT-IR | Fourier transform infrared |
| CD | Circular dichroism |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| DHPM | Dynamic high pressure microfluidization |
| OVA | Ovalbumin |
References
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Chin. J. Process. Eng. 2014, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Pechenov, S.; Anand Subramony, J. Oral peptide delivery: Translational challenges due to physiological effects. J. Control. Release 2018, 287, 167–176. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Encapsulation, protection, and delivery of bioactive proteins and peptides using nanoparticle and microparticle systems: A review. Adv. Colloid Interface Sci. 2018, 253, 1–22. [Google Scholar] [CrossRef]
- Li, L.; Yang, L.; Li, M.; Zhang, L. A cell-penetrating peptide mediated chitosan nanocarriers for improving intestinal insulin delivery. Carbohydr. Polym. 2017, 174, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Razi, M.A.; Wakabayashi, R.; Goto, M.; Kamiya, N. Self-Assembled Reduced Albumin and Glycol Chitosan Nanoparticles for Paclitaxel Delivery. Langmuir 2019, 35, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fan, Q.; Hao, D.; Wu, J.; Ma, G.; Su, Z. Chitosan-based mucosal adjuvants: Sunrise on the ocean. Vaccine 2015, 33, 5997–6010. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.; Castro, P.M.; Madureira, A.R.; Sarmento, B.; Pintado, M. Recent insights in the use of nanocarriers for the oral delivery of bioactive proteins and peptides. Peptides 2018, 101, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.T.; Keller, S.; Manganiello, M.J.; Cheng, C.; Lee, C.C.; Opara, C.; Convertine, A.; Stayton, P.S. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 2013, 7, 3912–3925. [Google Scholar] [CrossRef]
- Bugnicourt, L.; Ladaviere, C. Interests of chitosan nanoparticles ionically cross-linked with tripolyphosphate for biomedical applications. Prog. Polym. Sci. 2016, 60, 1–17. [Google Scholar] [CrossRef]
- Grenha, A. Chitosan nanoparticles: A survey of preparation methods. J. Drug Target. 2012, 20, 291–300. [Google Scholar] [CrossRef]
- Almalik, A.; Alradwan, I.; Kalam, M.A.; Alshamsan, A. Effect of cryoprotection on particle size stability and preservation of chitosan nanoparticles with and without hyaluronate or alginate coating. Saudi. Pharm. J. 2017, 25, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Sims, L.B.; Curry, K.C.; Parupalli, S.; Horner, G.; Frieboes, H.B.; Steinbach-Rankins, J.M. Efficacy of Surface-Modified PLGA Nanoparticles as a Function of Cervical Cancer Type. Pharm. Res. 2019, 36, 66. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yang, H.; Su, L.; Yang, Z.; Tang, X. Effect of size on the in vitro/in vivo drug release and degradation of exenatide-loaded PLGA microspheres. J. Drug Deliv. Sci. Technol. 2018, 45, 346–356. [Google Scholar] [CrossRef]
- Xu, Y.; Du, Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int. J. Pharm. 2003, 250, 215–226. [Google Scholar] [CrossRef]
- Bulmer, C.; Margaritis, A.; Xenocostas, A. Production and characterization of novel chitosan nanoparticles for controlled release of rHu-Erythropoietin. Biochem. Eng. J. 2012, 68, 61–69. [Google Scholar] [CrossRef]
- Calvo, P.; Remunan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Ferreira, D. Development and characterization of new insulin containing polysaccharide nanoparticles. Colloids Surf. B Biointerfaces 2006, 53, 193–202. [Google Scholar] [CrossRef]
- Zhang, H.L.; Wu, S.H.; Tao, Y.; Zang, L.Q.; Su, Z.Q. Preparation and characterization of water-soluble chitosan nanoparticles as protein delivery system. J. Nanomater. 2009, 2010. [Google Scholar] [CrossRef]
- Gan, Q.; Wang, T. Chitosan nanoparticle as protein delivery carrier—Systematic examination of fabrication conditions for efficient loading and release. Colloids Surf. B Biointerfaces 2007, 59, 24–34. [Google Scholar] [CrossRef]
- Tokeshi, M.; Sato, K. Micro/Nano Devices for Chemical Analysis. Micromachine 2016, 7, 164. [Google Scholar] [CrossRef]
- Bhise, N.S.; Ribas, J.; Manoharan, V.; Zhang, Y.S.; Polini, A.; Massa, S.; Dokmeci, M.R.; Khademhosseini, A. Organ-on-a-chip platforms for studying drug delivery systems. J. Control. Release 2014, 190, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Kaasalainen, M.; Aseyev, V.; von Haartman, E.; Karaman, D.S.; Makila, E.; Tenhu, H.; Rosenholm, J.; Salonen, J. Size, Stability, and Porosity of Mesoporous Nanoparticles Characterized with Light Scattering. Nanoscale Res. Lett. 2017, 12, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Wang, T.; Cochrane, C.; Mccarron, P. Modulation of surface charge, particle size and morphological properties of chitosan–TPP nanoparticles intended for gene delivery. Colloids Surf. B Biointerfaces 2005, 44, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef] [PubMed]
- Rampino, A.; Borgogna, M.; Blasi, P.; Bellich, B.; Cesàro, A. Chitosan nanoparticles: Preparation, size evolution and stability. Int. J. Pharm. 2013, 455, 219–228. [Google Scholar] [CrossRef]
- Muthu, M.S.; Feng, S.S. Pharmaceutical stability aspects of nanomedicines. Nanomedicine 2009, 4, 857–860. [Google Scholar] [CrossRef]
- Beck-Broichsitterab, M.; Schmehl, T.; Betz, T.; Bakowsky, U.; Kissel, T.; Seeger, W. Impact of lyoprotectants for the stabilization of biodegradable nanoparticles on the performance of air-jet, ultrasonic, and vibrating-mesh nebulizers. Eur. J. Pharm. Biopharm. 2012, 82, 272–280. [Google Scholar] [CrossRef]
- Chishti, N.; Jagwani, S.; Dhamecha, D.; Jalalpure, S.; Dehghan, M.H. Preparation, Optimization, and In Vivo Evaluation of Nanoparticle-Based Formulation for Pulmonary Delivery of Anticancer Drug. Medicina 2019, 55, 294. [Google Scholar] [CrossRef]
- Fonte, P.; Reis, S.; Sarmento, B. Facts and evidences on the lyophilization of polymeric nanoparticles for drug delivery. J. Control. Release 2016, 225, 75–86. [Google Scholar] [CrossRef]
- Abdelwahed, W.; Degobert, G.; Fessi, H. Investigation of nanocapsules stabilization by amorphous excipients during freeze-drying and storage. Eur. J. Pharm. Biopharm. 2006, 63, 87–94. [Google Scholar] [CrossRef]
- Lins, R.D.; Pereira, C.S.; Hunenberger, P.H. Trehalose-protein interaction in aqueous solution. Proteins 2004, 55, 177–186. [Google Scholar] [CrossRef]
- Fonte, P.; Andrade, F.; Azevedo, C.; Pinto, J.; Seabra, V.; Van, D.W.M.; Reis, S.; Sarmento, B. Effect of the Freezing Step in the Stability and Bioactivity of Protein-Loaded PLGA Nanoparticles Upon Lyophilization. Pharm. Res. 2016, 33, 2777–2793. [Google Scholar] [CrossRef] [PubMed]
- Siri, M.; Grasselli, M.; Alonso, S.D. Albumin-based nanoparticle trehalose lyophilisation stress-down to preserve structure/function and enhanced binding. J. Pharm. Biomed. Anal. 2016, 126, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, C.R.; Jiang, M.; Zhu, Y.Y.; Wang, J.; Chen, J.; Shi, J.H. Binding interaction of atorvastatin with bovine serum albumin: Spectroscopic methods and molecular docking. Spectrochim. Acta A 2015, 156, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Bekale, L.; Agudelo, D.; Tajmirriahi, H.A. Effect of polymer molecular weight on chitosan-protein interaction. Colloids Surf. B. Biointerfaces 2015, 125, 309–317. [Google Scholar] [CrossRef]
- Chanphai, P.; Tajmir-Riahi, H.A. Encapsulation of testosterone by chitosan nanoparticles. Int. J. Biol. Macromol. 2017, 98, 535–541. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Zhou, K.L.; Lou, Y.Y.; Pan, D.; Shi, J.H. Investigation of the binding interaction between estazolam and bovine serum albumin: Multi-spectroscopic methods and molecular docking technique. J. Biomol. Struct. Dyn. 2016, 35, 3605–3614. [Google Scholar] [CrossRef]
- Lou, Y.Y.; Zhou, K.L.; Shi, J.H.; Pan, D.Q. Characterizing the binding interaction of fungicide boscalid with bovine serum albumin (BSA): A spectroscopic study in combination with molecular docking approach. J. Photochem. Photobiol. B 2017, 173, 589–597. [Google Scholar] [CrossRef]
- Hooshyar, Z.; Bardajee, G.R.; Shafagh, P.; Ghiasvand, S.; Khanjari, M.; Dianatnejad, N. In vitro study on the interaction of Mn (II)-DiAmsar with human serum albumin (HSA) and bovine serum albumin (BSA) by spectroscopic and molecular docking methods. J. Iran. Chem.Soc. 2015, 12, 715–725. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of macromolecular association reactions: Analysis of forces contributing to stabilization. Biophys. J. 1980, 32, 79–81. [Google Scholar] [CrossRef]
- Naveenraj, S.; Anandan, S. Binding of serum albumins with bioactive substances—Nanoparticles to drugs. J. Photochem. Photobiol. C Photochem. Rev. 2013, 14, 53–71. [Google Scholar] [CrossRef]
- Esfandiari, F.; Motazedian, M.H.; Asgari, Q.; Morowvat, M.H.; Molaei, H.; Heli, H. Paromomycin-loaded mannosylated chitosan nanoparticles: Synthesis, characterization and targeted drug delivery against leishmaniasis. Acta Trop. 2019, 197, 105045–105057. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zou, S.; Rao, Z.; Min, L.; Liu, M.; Liu, L.; Fan, L. Preparation and characterization of hydroxypropyl chitosan modified with nisin. Int. J. Biol. Macromol. 2017, 105, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Vandana, M.; Sahoo, S.K. Optimization of physicochemical parameters influencing the fabrication of protein-loaded chitosan nanoparticles. Nanomedicine 2009, 4, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, A.; Asgari, A. In vitro release and biological activities of Carum copticum essential oil (CEO) loaded chitosan nanoparticles. Int. J. Biol. Macromol. 2015, 81, 283–290. [Google Scholar] [CrossRef]
- Luo, Y.C.; Zhang, B.; Cheng, W.H.; Wang, Q. Preparation, characterization and evaluation of selenite-loaded chitosan/TPP nanoparticles with or without zein coating. Carbohydr. Polym. 2010, 82, 942–951. [Google Scholar] [CrossRef]
- Bahreini, E.; Aghaiypour, K.; Abbasalipourkabir, R.; Mokarram, A.R.; Goodarzi, M.T.; Saidijam, M. Preparation and nanoencapsulation of l-asparaginase II in chitosan-tripolyphosphate nanoparticles and in vitro release study. Nanoscale Res. Lett. 2014, 9, 340. [Google Scholar] [CrossRef]
- Wu, J.; Bi, S.Y.; Sun, X.Y.; Zhao, R.; Wang, J.H.; Zhou, H.F. Study on the interaction of fisetholz with BSA/HSA by multi-spectroscopic, cyclic voltammetric, and molecular docking technique. J. Biomol. Struct. Dyn. 2019, 37, 3496–3505. [Google Scholar] [CrossRef]
- Shi, J.H.; Lou, Y.Y.; Zhou, K.L.; Pan, D.Q. Probing the behavior of calf thymus DNA upon binding to a carboxamide fungicide boscalid: Insights from spectroscopic and molecular docking approaches. J. Biomol. Struct. Dyn. 2018, 36, 2738–2745. [Google Scholar] [CrossRef]
- Norde, W.; Giacomelli, C.E. BSA structural changes during homomolecular exchange between the adsorbed and the dissolved states. J. Biotechnol. 2000, 79, 259–268. [Google Scholar] [CrossRef]
- Xiao, J.T.; Qiu, F.Z.; Kai, L.; Yi, C.C.; Wen, Z.Y.; Fen, F.G.; Gang, G.S. Effect of pullulan nanoparticle surface charges on HSA complexation and drug release behavior of HSA-bound nanoparticles. PLoS ONE 2012, 7, e49304. [Google Scholar]
- Fernandez, D.I.; Sani, M.A.; Miles, A.J.; Wallace, B.A.; Separovic, F. Membrane defects enhance the interaction of antimicrobial peptides, aurein 1.2 versus caerin 1.1. Biochim. Et Biophys. Acta Biomembr. 2013, 1828, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Xian, W.; Minquan, X.; Yuanhua, Z.; Limei, W.; Xiaolong, F.; Kun, Y.; Jing, M.; Zhenshun, L.; Lan, W.; Weiqing, S. Gel properties of myofibrillar proteins heated at different heating rates under a low-frequency magnetic field. Food Chem. 2020, 321, 126728–126735. [Google Scholar]
- Emanuele, C.; Salvatore, M. Direct spectroscopic evidence for competition between thermal molecular agitation and magnetic field in a tetrameric protein in aqueous solution. Phys. Lett. A 2018, 382, 1389–1394. [Google Scholar]
- You, G.; Niu, G.; Long, H.; Zhang, C.; Liu, X. Elucidation of interactions between gelatin aggregates and hsian-tsao gum in aqueous solutions. Food Chem. 2020, 319, 126532–126543. [Google Scholar] [CrossRef]
- Koppolu, B.P.; Smith, S.G.; Ravindranathan, S.; Jayanthi, S.; Suresh Kumar, T.K.; Zaharoff, D.A. Controlling chitosan-based encapsulation for protein and vaccine delivery. Biomaterials 2014, 35, 4382–4389. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, W.; Wang, C.; Hu, J.; Fu, S. Chitosan nanoparticles as a novel delivery system for ammonium glycyrrhizinate. Int. J. Pharm. 2005, 295, 235–245. [Google Scholar] [CrossRef]
- Pawar, D.; Jaganathan, K.S. Mucoadhesive glycol chitosan nanoparticles for intranasal delivery of hepatitis B vaccine: Enhancement of mucosal and systemic immune response. Drug Deliv. 2016, 23, 185–194. [Google Scholar] [CrossRef]
- Hajji, S.; Hamdi, M.; Boufi, S.; Li, S.; Nasri, M. Suitability of chitosan nanoparticles as cryoprotectant on shelf life of restructured fish surimi during chilled storage. Cellulose 2019, 26, 6825–6847. [Google Scholar] [CrossRef]
- Garg, P.; Kaur, G.; Chaudhary Ganga, R.; Gawali Santosh, L.; Hassan, P.A. Structural and SAXS analysis of protein folding/unfolding with cationic single chain metallosurfactants. J. Mol. Liq. 2018, 271, 157–165. [Google Scholar] [CrossRef]
- Saha, D.; Diamai, S.; Warjri, W.; Negi, D.P.S. Effect of pH on the interaction of hypoxanthine and guanine with colloidal ZnS nanoparticles: A spectroscopic approach. J. Mol. Liq. 2019, 278, 460–466. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CS: TPP (w/w) | MHD ± SD (nm) | PDI | ζ-Pot ± SD (mv) | EE ± SD (%) | LC (%) |
|---|---|---|---|---|---|
| 3:1 | 92 ± 2 | 0.14 | 16 ± 0.4 | 53 ± 5 | 67 ± 1 |
| 4:1 | 99 ± 2 | 0.21 | 20 ± 0.5 | 34 ± 3 | 56 ± 2 |
| 5:1 | 118 ± 2 | 0.27 | 25 ± 0.2 | 28 ± 1 | 47 ± 1 |
| 6:1 | 120 ± 1 | 0.26 | 26 ± 0.2 | 20 ± 2 | / |
| blank NPs (3:1) | 112 ± 4 | 0.23 | 19 ± 0.5 | / | / |
| Pressure (psi) | MHD ± SD (nm) | PDI | ζ-pot ± SD (mv) | EE ± SD (%) | LC (%) |
|---|---|---|---|---|---|
| 10 | 98 ± 2 | 0.14 | 18 ± 0.3 | 82 ± 1 | 9 ± 1 |
| 20 | 89 ± 2 | 0.12 | 19 ± 0.2 | 82 ± 1 | 18 ± 17 |
| 30 | 89 ± 2 | 0.10 | 21 ± 0.5 | 83 ± 1 | 33 ± 2 |
| Coarse NPs | 92 ± 2 | 0.14 | 16 ± 0.4 | 53 ± 5 | 67 ± 2 * |
| Cycle Times(min) | MHD ± SD (nm) | PDI | ζ-pot ± SD (mv) | EE ± SD (%) | LC (%) |
|---|---|---|---|---|---|
| 2 | 89 ± 2 | 0.10 | 21 ± 0.5 | 93 ± 1 | 33 ± 1 |
| 4 | 88 ± 2 | 0.11 | 22 ± 0.1 | 85 ± 2 | 36 ± 1 |
| 6 | 90 ± 2 | 0.10 | 21 ± 0.1 | 84 ± 1 | 37 ± 2 |
| 8 | 89 ± 0.2 | 0.12 | 20 ± 0.2 | 87 ± 1 | 40 ± 1 |
| 10 | 92 ± 2 | 0.13 | 19 ± 0.2 | 85 ± 13 | 38 ± 1 |
| T(K) | Slope | Intercept | r2 a | Ksv × 105 (M−1) | Kq × 1013 (M−1 s−1) |
|---|---|---|---|---|---|
| 298 | 0.21 | 1.00 | 0.9922 | 2.13 | 3.61 |
| 303 | 0.22 | 1.00 | 0.9994 | 2.24 | 3.80 |
| 308 | 0.30 | 1.00 | 0.9955 | 2.95 | 5.00 |
| T(K) | LogKb | n | r2 a | Kb(M−1) | ΔG (kJ.mol−1) | ΔH (kJ.mol−1) | ΔS (kJ mol−1 K−1) |
|---|---|---|---|---|---|---|---|
| 298 | 3.4624 | 0.9084 | 0.9984 | 2.900 × 103 | −18.74 | 407.40 | 1.43 |
| 303 | 4.9684 | 0.9398 | 0.9954 | 9.298 × 104 | −25.89 | ||
| 308 | 5.7773 | 1.0673 | 0.9969 | 5.988 × 105 | −30.44 |
| Sample | BSA Concentration (µg/mL) | Cell Viability (%) |
|---|---|---|
| lyophilized BSA-loaded NPs | 100 | 82 ± 2 |
| 50 | 90 ± 2 | |
| 10 | 93 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Q.; Weng, J.; Wu, X.; Wang, W.; Yang, Q.; Guo, F.; Wu, D.; Song, Y.; Chen, F.; Yang, G. Characteristics, Cryoprotection Evaluation and In Vitro Release of BSA-Loaded Chitosan Nanoparticles. Mar. Drugs 2020, 18, 315. https://doi.org/10.3390/md18060315
Yan Q, Weng J, Wu X, Wang W, Yang Q, Guo F, Wu D, Song Y, Chen F, Yang G. Characteristics, Cryoprotection Evaluation and In Vitro Release of BSA-Loaded Chitosan Nanoparticles. Marine Drugs. 2020; 18(6):315. https://doi.org/10.3390/md18060315
Chicago/Turabian StyleYan, Qinying, Jiaqi Weng, Xieqi Wu, Weiwei Wang, Qingliang Yang, Fangyuan Guo, Danjun Wu, Ying Song, Fan Chen, and Gensheng Yang. 2020. "Characteristics, Cryoprotection Evaluation and In Vitro Release of BSA-Loaded Chitosan Nanoparticles" Marine Drugs 18, no. 6: 315. https://doi.org/10.3390/md18060315
APA StyleYan, Q., Weng, J., Wu, X., Wang, W., Yang, Q., Guo, F., Wu, D., Song, Y., Chen, F., & Yang, G. (2020). Characteristics, Cryoprotection Evaluation and In Vitro Release of BSA-Loaded Chitosan Nanoparticles. Marine Drugs, 18(6), 315. https://doi.org/10.3390/md18060315