CYC31, A Natural Bromophenol PTP1B Inhibitor, Activates Insulin Signaling and Improves Long Chain-Fatty Acid Oxidation in C2C12 Myotubes


Abstract

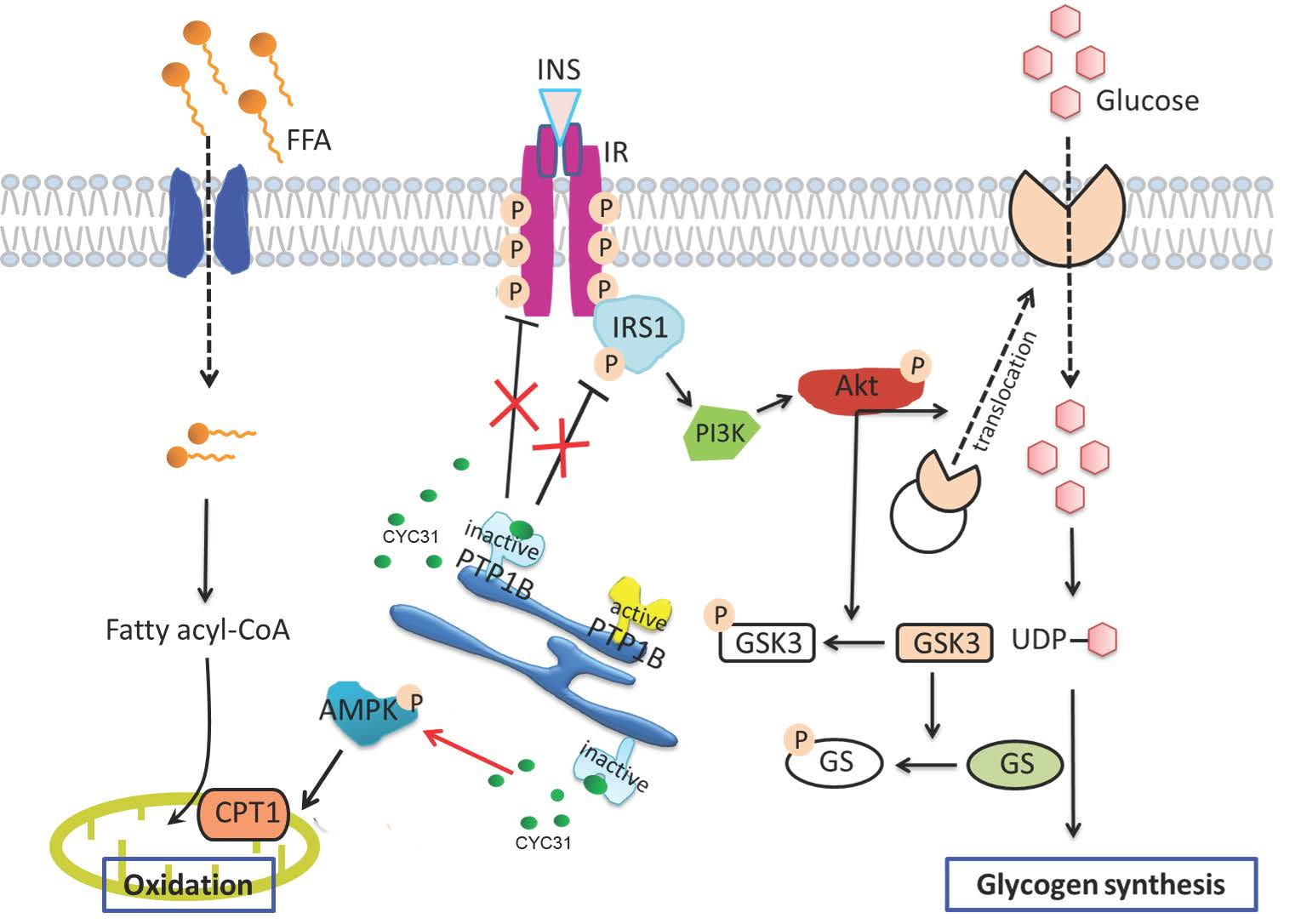{kind=link}
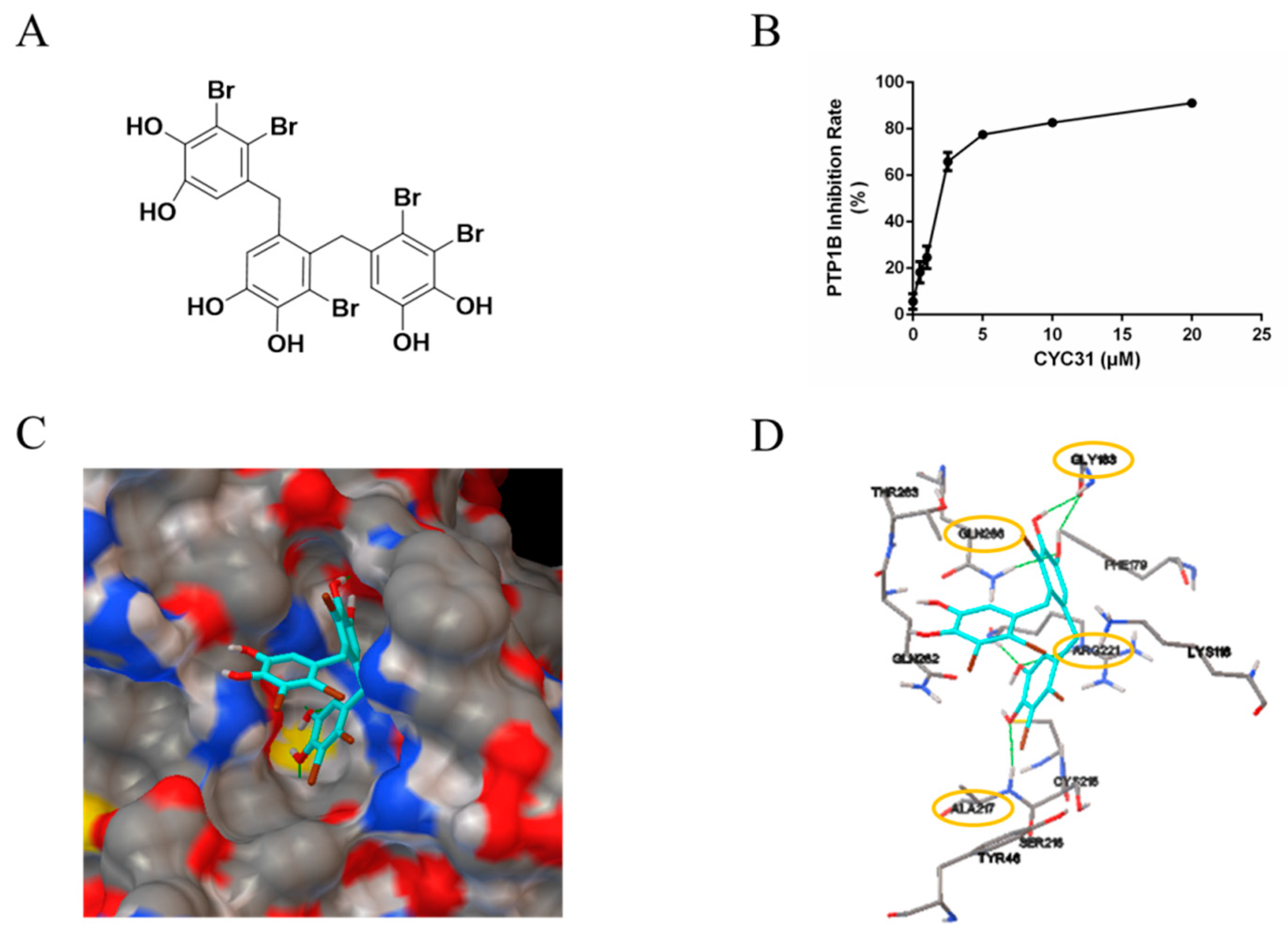{kind=link}
{kind=link}
{kind=link}
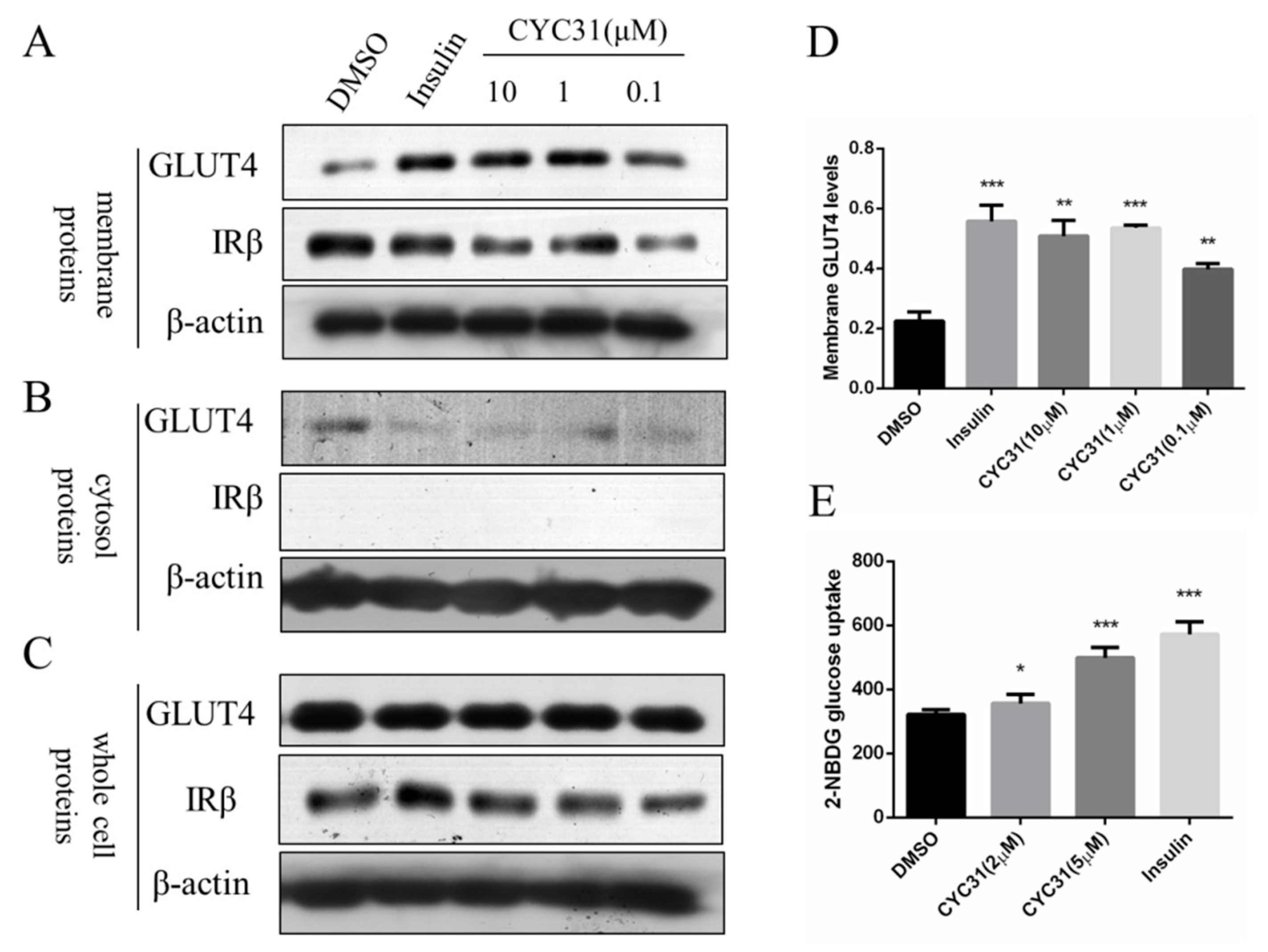{kind=link}
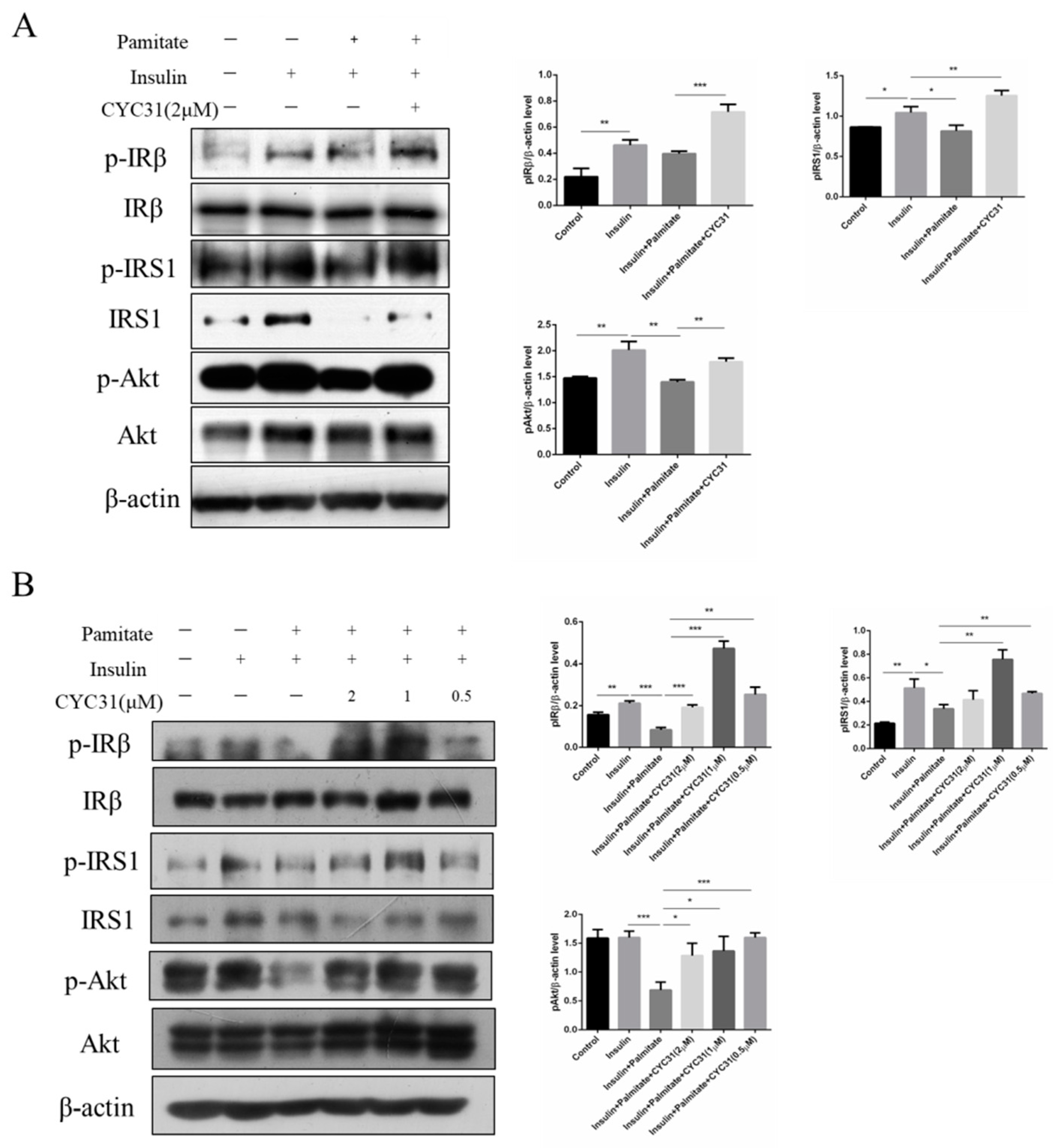{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CYC31 Embeds Into the Catalytic Pocket of PTP1B
2.2. CYC31 Increases the Activity of Insulin Signaling in C2C12 Myotubes
2.3. CYC31 Enhances the Glycogen Synthase Signaling
2.4. CYC31 Promotes Glucose Uptake Via GLUT4 Translocation
2.5. CYC31 Prevents Palmitate-Induced Insulin Resistance
2.6. CYC31 Increases the Fatty Acid Oxidation Signal in Palmitate-Exposed C2C12 Myotubes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. PTP1B Enzymatic Assay
4.3. Molecular Docking
4.4. Cell Culture and Viability Assay
4.5. Immunoblotting
4.6. Measurements of mRNA
4.7. 2-NBDG Uptake
4.8. Membrane and Cytosol Protein Extraction
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A. Role of beta-cell dysfunction, ectopic fat accumulation and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2011, 93, S60–S65. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Amati, F.; Dube, J.J.; Coen, P.M.; Stefanovic-Racic, M.; Toledo, F.G.; Goodpaster, B.H. Physical inactivity and obesity underlie the insulin resistance of aging. Diabetes Care 2009, 32, 1547–1549. [Google Scholar] [CrossRef]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic review series: Adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef]
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Investig. 2002, 32 (Suppl. 3), 14–23. [Google Scholar] [CrossRef]
- Lark, D.S.; Fisher-Wellman, K.H.; Neufer, P.D. High-fat load: Mechanism(s) of insulin resistance in skeletal muscle. Int. J. Obes. Suppl. 2012, 2, S31–S36. [Google Scholar] [CrossRef]
- Yazici, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef]
- Seely, B.L.; Staubs, P.A.; Reichart, D.R.; Berhanu, P.; Milarski, K.L.; Saltiel, A.R.; Kusari, J.; Olefsky, J.M. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes 1996, 45, 1379–1385. [Google Scholar] [CrossRef]
- Byon, J.C.H.; Kusari, A.B.; Kusari, J. Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Mol. Cell Biochem. 1998, 182, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wertheimer, S.J.; Lin, C.H.; Katz, S.L.; Amrein, K.E.; Burn, P.; Quon, M.J. Protein-tyrosine phosphatases PTP1B and Syp are modulators of insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. J. Biol. Chem. 1997, 272, 8026–8031. [Google Scholar] [CrossRef]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.-B.; Sharpe, A.H. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Owen, C.; Lees, E.; Grant, L.; Zimmer, D.; Mody, N.; Bence, K.; Delibegović, M. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia 2013, 56, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.-G.; Cho, Y.-R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Bence, K.K.; Mody, N.; Hong, E.-G.; Ko, H.J.; Kim, J.K.; Kahn, B.B.; Neel, B.G. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol. Cell Biol. 2007, 27, 7727–7734. [Google Scholar] [CrossRef]
- Luo, J.; Xu, Q.; Jiang, B.; Zhang, R.; Jia, X.; Li, X.; Wang, L.; Guo, C.; Wu, N.; Shi, D. Selectivity, cell permeability and oral availability studies of novel bromophenol derivative HPN as protein tyrosine phosphatase 1B inhibitor. Br. J. Pharmacol. 2018, 175, 140–153. [Google Scholar] [CrossRef]
- Zhang, X.; Tian, J.; Li, J.; Huang, L.; Wu, S.; Liang, W.; Zhong, L.; Ye, J.; Ye, F. A novel protein tyrosine phosphatase 1B inhibitor with therapeutic potential for insulin resistance. Br. J. Pharmacol. 2016, 173, 1939–1949. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, Y.; Xu, C.; Hou, D.; Li, J.; Zhang, Y.; Peng, W.; Zen, K.; Zhang, C.Y.; Jiang, X. Norathyriol reverses obesity- and high-fat-diet-induced insulin resistance in mice through inhibition of PTP1B. Diabetologia 2014, 57, 2145–2154. [Google Scholar] [CrossRef]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Shi, D.; Cui, Y.; Guo, S. Design, synthesis, and biological evaluation of bromophenol derivatives as protein tyrosine phosphatase 1B inhibitors. Arch. Pharm. (Weinheim) 2012, 345, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Xu, F.; He, J.; Li, J.; Fan, X.; Han, L. Inhibition of bromophenols against PTP1B and anti-hyperglycemic effect of Rhodomela confervoides extract in diabetic rats. Chin. Sci. Bull. 2008, 53, 2476–2479. [Google Scholar] [CrossRef]
- Thareja, S.; Aggarwal, S.; Bhardwaj, T.R.; Kumar, M. Protein tyrosine phosphatase 1B inhibitors: A molecular level legitimate approach for the management of diabetes mellitus. Med. Res. Rev. 2012, 32, 459–517. [Google Scholar] [CrossRef]
- Li, C.; Luo, J.; Guo, S.; Jia, X.; Guo, C.; Li, X.; Xu, Q.; Shi, D. Highly Selective Protein Tyrosine Phosphatase Inhibitor, 2,2′,3,3′-Tetrabromo-4,4′,5,5′-tetrahydroxydiphenylmethane, Ameliorates Type 2 Diabetes Mellitus in BKS db Mice. Mol. Pharm. 2019, 16, 1839–1850. [Google Scholar] [CrossRef]
- Maeda, A.; Kai, K.; Ishii, M.; Ishii, T.; Akagawa, M. Safranal, a novel protein tyrosine phosphatase 1B inhibitor, activates insulin signaling in C2C12 myotubes and improves glucose tolerance in diabetic KK-Ay mice. Mol. Nutr. Food Res. 2014, 58, 1177–1189. [Google Scholar] [CrossRef]
- Fukuda, S.; Ohta, T.; Sakata, S.; Morinaga, H.; Ito, M.; Nakagawa, Y.; Tanaka, M.; Matsushita, M. Pharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551. Diabetes Obes. Metab. 2010, 12, 299–306. [Google Scholar] [CrossRef]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef]
- Sebastian, D.; Herrero, L.; Serra, D.; Asins, G.; Hegardt, F.G. CPT I overexpression protects L6E9 muscle cells from fatty acid-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E677–E686. [Google Scholar] [CrossRef]
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 2009, 58, 550–558. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Wang, Y.; Shen, Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J. Biochem. Biophys. Methods 2005, 64, 207–215. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Hou, Y.; Xie, M.; Ma, W.; Shi, D.; Jiang, B. CYC31, A Natural Bromophenol PTP1B Inhibitor, Activates Insulin Signaling and Improves Long Chain-Fatty Acid Oxidation in C2C12 Myotubes. Mar. Drugs 2020, 18, 267. https://doi.org/10.3390/md18050267
Luo J, Hou Y, Xie M, Ma W, Shi D, Jiang B. CYC31, A Natural Bromophenol PTP1B Inhibitor, Activates Insulin Signaling and Improves Long Chain-Fatty Acid Oxidation in C2C12 Myotubes. Marine Drugs. 2020; 18(5):267. https://doi.org/10.3390/md18050267
Chicago/Turabian StyleLuo, Jiao, Yufei Hou, Mengyue Xie, Wanli Ma, Dayong Shi, and Bo Jiang. 2020. "CYC31, A Natural Bromophenol PTP1B Inhibitor, Activates Insulin Signaling and Improves Long Chain-Fatty Acid Oxidation in C2C12 Myotubes" Marine Drugs 18, no. 5: 267. https://doi.org/10.3390/md18050267
APA StyleLuo, J., Hou, Y., Xie, M., Ma, W., Shi, D., & Jiang, B. (2020). CYC31, A Natural Bromophenol PTP1B Inhibitor, Activates Insulin Signaling and Improves Long Chain-Fatty Acid Oxidation in C2C12 Myotubes. Marine Drugs, 18(5), 267. https://doi.org/10.3390/md18050267