1. Introduction
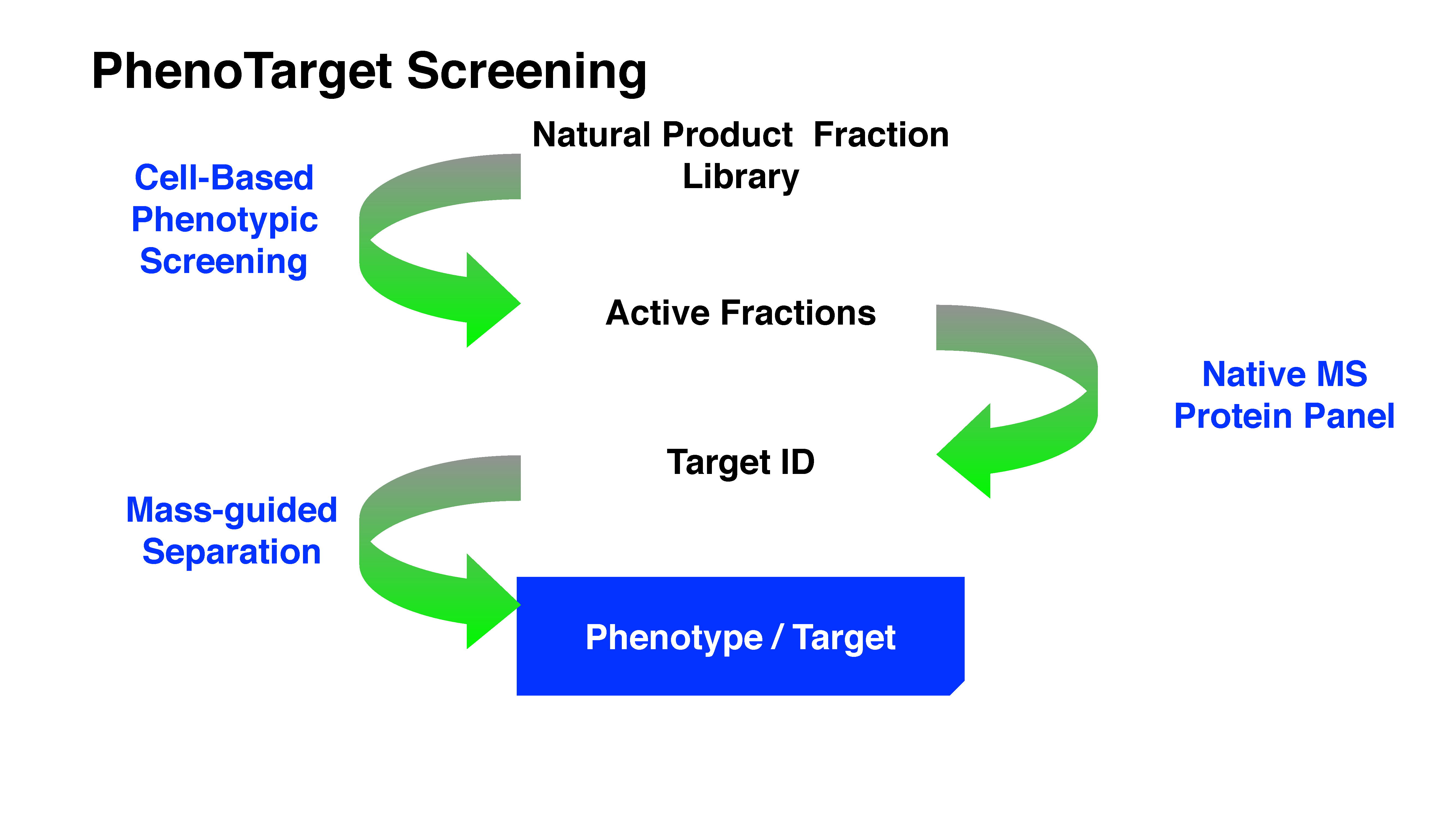
Drug discovery research and development has experienced two periods with different centric strategies, namely phenotypic-based drug discovery (PDD) and target-based drug discovery (TDD) (
Figure 1). Commonly PDD refers to an approach without prior knowledge of the target. In phenotypic-based screening, compounds that modify a phenotype to generate a positive outcome in cell culture or in a whole organism are identified. TDD examines a specific drug target which is hypothesized to play an important role in disease.
Before the 1980s, the era without recombinant DNA technology, PDD was the primary approach in drug discovery. Most drugs at that time were discovered serendipitously by phenotypic assays in live animals or isolated tissues [
3]. Examples are penicillin isolated from a
Penicillium species in 1928 [
2,
4] and ivermectin isolated from
Streptomyces avermitilis in 1975 [
5]. From the 1990s, the development of genomics allowed the identification of drug target proteins. The advent of high-throughput screening and combinatorial libraries enabled the screening of target proteins in high throughput. Due to the development of technologies including X-ray crystallography, computational modeling and screening (virtual docking), visualization of the interaction of target protein and compound greatly facilitated the later stage of structure-based development. The development of these technologies appealed to the pharmaceutical industry and academic researchers who then switched to focus on TDD during the last three decades.
In the era that mainly focused on TDD, the total number of new molecular entities (NMEs) and new biologics approved by the Food and Drug Administration was far below expectations [
6]. The timeline of cumulative NME approvals from 1950 to 2008 was contributed to by the three most productive companies in the industry and showed almost straight lines, indicating that productivity continued at a constant rate for almost 60 years [
1]. The introduction of new molecular biology tools such as recombinant DNA technology, deep sequencing, mining of Expressed Sequence Tagged (cDNA) libraries and the draft human genome did not facilitate drug innovation as expected. This was also indicated in a later NMEs analysis with a timeline spanning over five years to 2013 [
7]. More dishearteningly, while the number of NMEs per year has remained relatively constant for the past four decades, the investment in pharmaceutical research and development (R&D) has increased dramatically to over 50 billion USD per year. Today the number of NMEs launched per billion dollars of investment is well below the return for an equivalent billion-dollar investment 50 years ago [
1]. The asymmetrical output raises questions about the limitation of the popular target centric strategy of R&D in recent decades.
Consequently, there is a revival of interest in PDD. A significant analysis by Swinney demonstrated that the majority of NMEs approved by the FDA during the 10-year period between 1999 and 2008 were discovered using phenotypic assays, where 28 came from phenotypic screening approaches and 17 came from target-based approaches [
8]. It was suggested that the lack of successful NMEs in the post-genomic era was mainly due to the limited use of phenotypic screening. Because an organism is a complex biological system, a simplified single protein assay may not efficiently represent the disease pathogenesis [
8] The effects on a single protein may not be translated to meaningful therapeutic efficacy. Conversely, phenotypic screening is an approach for unbiased targets. Phenotypic-based screening is, therefore, being reconsidered for screening compounds due to the realization that results for a single target protein may not fully correlate in the context of a complex biological process [
9]. Phenotypic screening also holds the unique promise to uncover new mechanisms of action for currently untreated diseases such as rare diseases and/or neglected diseases [
2].
The current phenotypic based approach should not be regarded as a step back to the classical phenotypic screening but as a new discipline [
7]. Currently, in vivo and in vitro approaches are involved in phenotypic screening, of which in vitro cell-based phenotypic assays can be easily adapted to a high-throughput format for automated phenotypic analysis. New technologies such as gene expression, genetic modifier screening, resistance mutation and computational inference are increasingly being applied in phenotypic screening. These sophisticated phenotypic screening methodologies enhance the identification of novel compounds as well as their mechanism of action [
10].
Target identification is a crucial part of drug discovery. Most large pharmaceutical companies strongly recommend target identification because failing to assign the mechanism of action is frequently regarded as a major risk factor for clinical development and regulatory approval [
8]. Even with the most advanced phenotypic screens, in most cases, it is still difficult to determine the mechanism of action. Target identification is the key value of TDD [
6]. With the target protein in hand, detailed drug-protein characterization is possible and this provides a better understanding of structure-activity relationships, which is a challenge in phenotypic screening without a known target [
11].
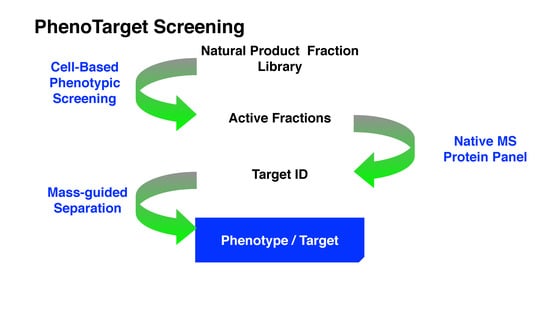
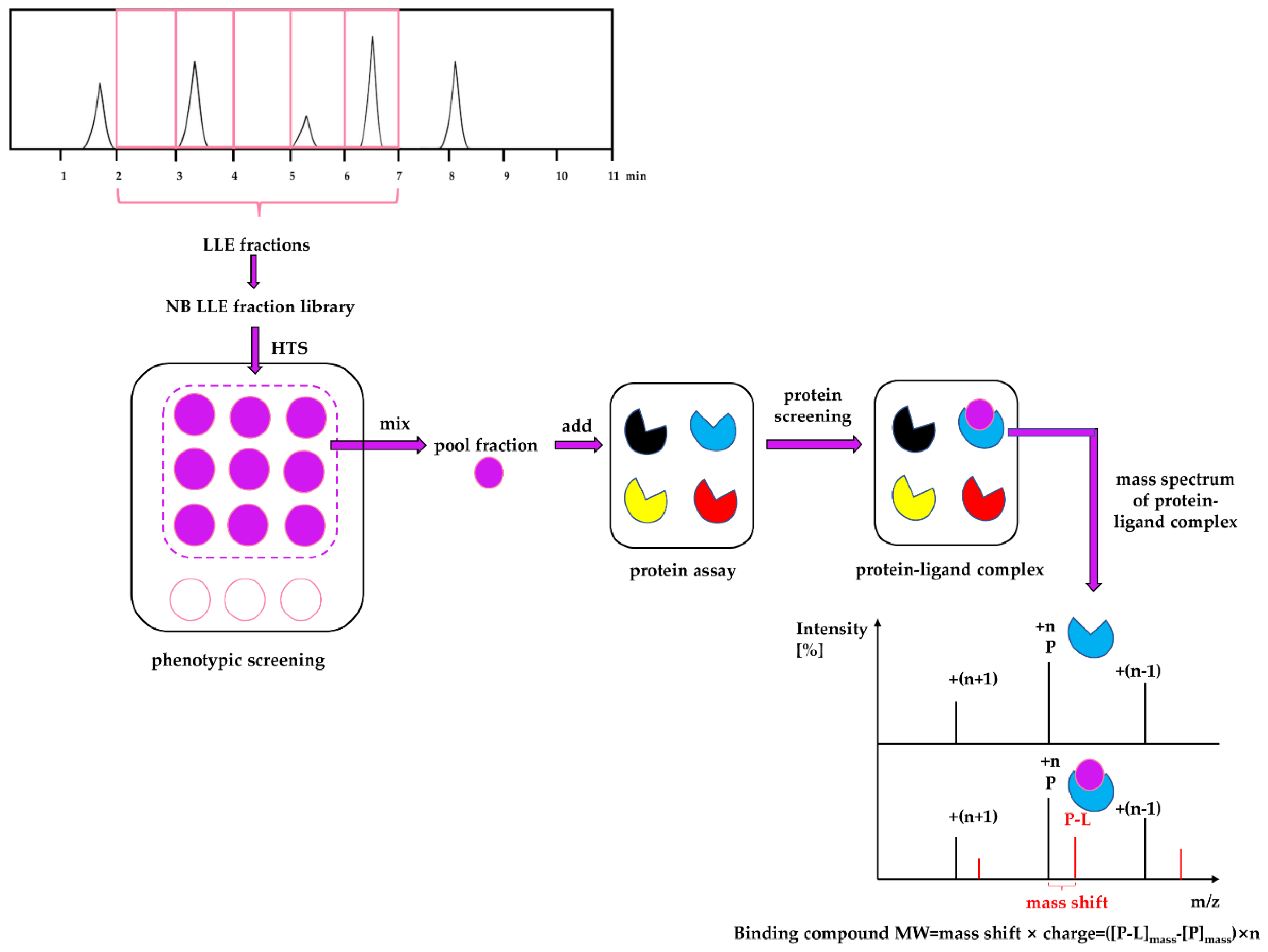
Considering the strengths and weaknesses of PDD and TDD, these two approaches should not be treated monolithically but as complementary approaches that can work together to increase the productivity of drug discovery and development. This article describes an approach that combines PDD and TDD: PhenoTarget screening to identify active compounds from natural resources. As a proof of principle, we apply this combination approach to probe fractions of natural product libraries for compounds active against Mycobacterium tuberculosis H37Rv, the etiological agent responsible for tuberculosis (TB). The active fraction was then screened against a panel of 37 unique mycobacterial proteins to identify the potential compound and protein responsible for the activity against M. tuberculosis.
3. Discussion
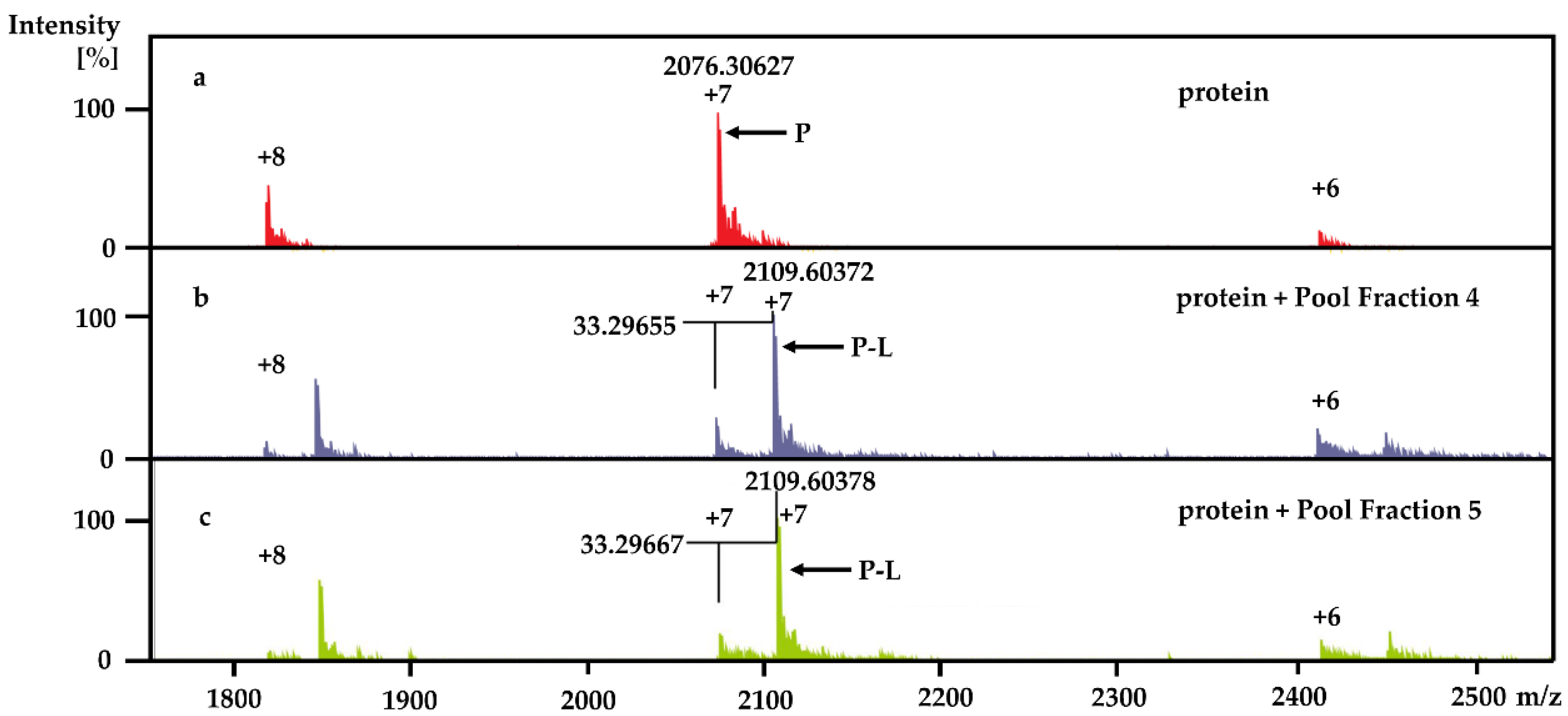
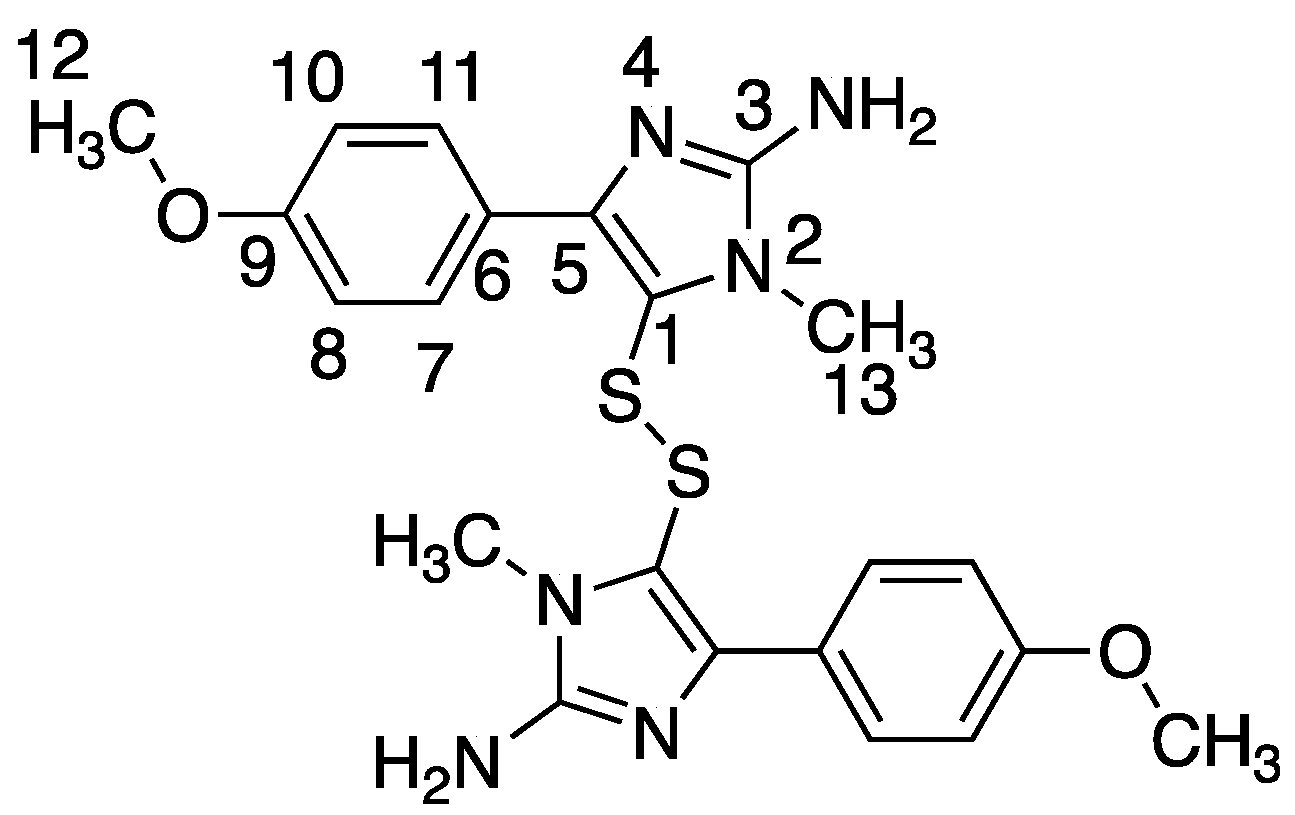
Using the PhenoTarget approach, a ligand from a natural product fraction library was identified that bound to the
M. tuberculosis protein Rv1466. The ligand molecular weight from the native MRMS spectrum was 233 Da. The compound was shown to be polycarpine with a molecular mass of 468 Da. The ligand MW of 233 Da was half the molecular weight of polycarpine less one proton, indicating that polycarpine formed a covalent bond with Rv1466. Under mild silica chromatography, the polycarpine disulfide bond has been shown to be cleaved [
15,
16]. Rv1466 has a single cysteine and the structure of Rv1466 (5IRD) indicates this residue is exposed on the surface of the protein so that it could attack the disulfide bond of polycarpine to form a covalent disulfide linked complex. The polycarpine-Rv1466 pseudo-
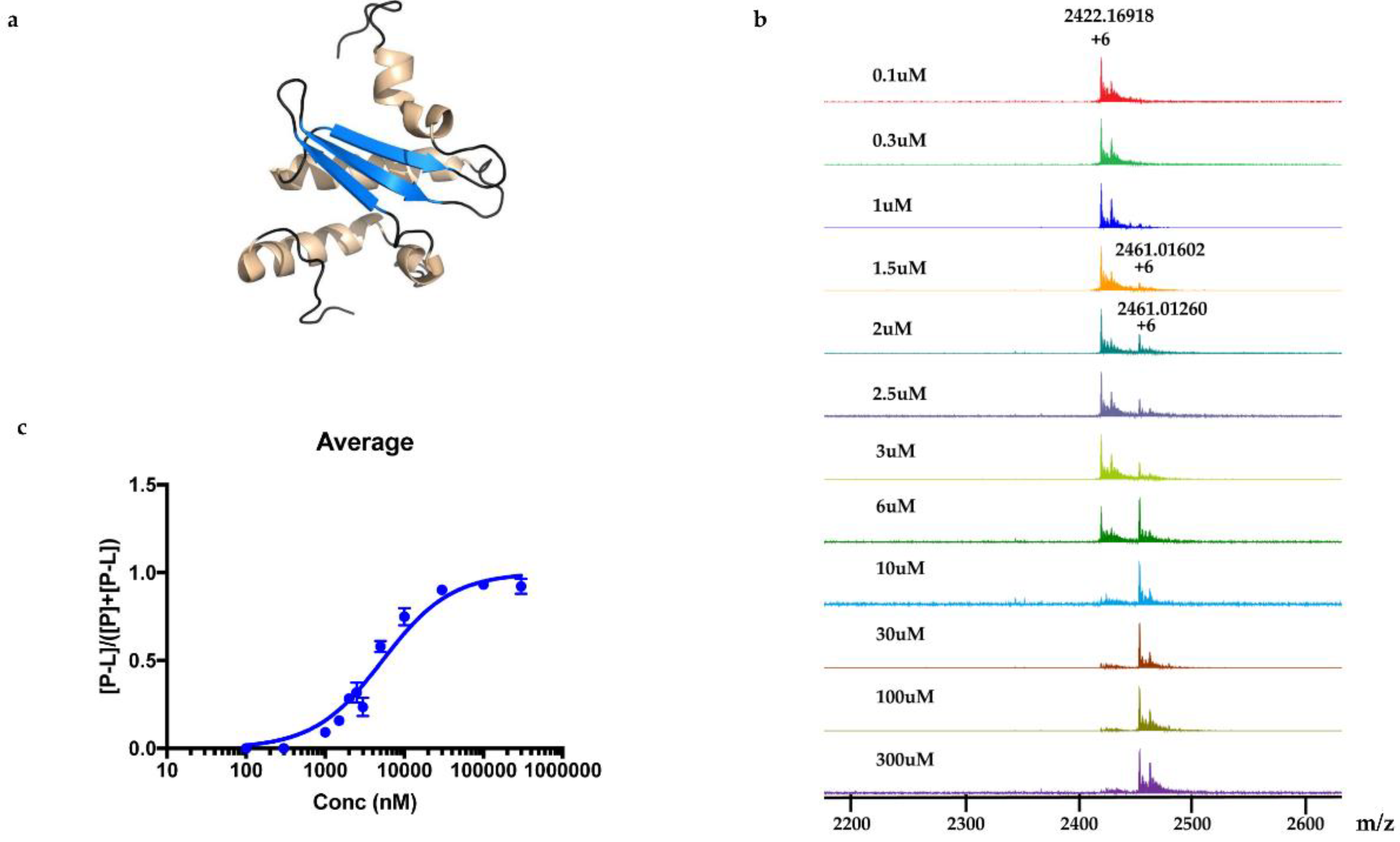
KD was determined to be 5.3 ± 0.4 μM.
P. aurata, a species of tunicate in the family Styelidae, is rich in alkaloids containing sulfur such as polycarpine, polycarpaurine A, polycarpaurine B and polycarpaurine C [
15,
16,
19,
20].
In the PhenoTarget approach, phenotypic screening is conducted initially to detect cellular active components. In our case a natural product fraction library was used, however, this could be equally applied to pure compound libraries. Mass spectrometry using a protein panel of putative targets provided sufficient throughput to analyze the phenotypic hits. Native mass spectrometry offers the further advantage that it can be used for cloned, expressed and purified proteins that lack structural information. This opens the option to explore the proteome of many species, in our case, the
Mycobacterium proteome. As well as its high sensitivity, high-throughput capability and in time observation of protein-ligand interaction, it can also provide exact mass information of the binding compound [
21]. The mass information is useful for isolating the compound from the fraction mixture using mass-guided isolation. In addition, mass spectrometry has been reported as an alternative technique for the quantitative characterization of protein–ligand interactions in solution, that is, the determination of equilibrium constants for protein–ligand association (Ka) or dissociation (Kd = 1/Ka) [
22,
23]. Among all the mass spectrometers, MRMS provides the highest accuracy and resolution. Nanoelectrospray was used for sample delivery. Compared to conventional electrospray, its benefits include an increased sensitivity signal, lower consumption of ligand and protein, and no sample-to-sample cross-contamination, a faster speed and a higher degree of nozzle-to-nozzle reproducibility [
24]. A high degree of nozzle-to-nozzle reproducibility is important for quantitative studies such as
KD determination. It has been proven that
KD results from chip-based electrospray are consistent with conventional techniques [
17].
While there are advantages in the phenotypic drug discovery approach it should be noted that there are also some disadvantages. Foremost is the procurement of pure protein panels for target identification. For example, the genome of
M. tuberculosis contains over 4000 genes [
25] and our exploratory panel contained only 37 different proteins. This bottleneck can be partly circumvented by using orthologous proteins from related species [
26]. Indeed, in addition to
M. tuberculosis (12 proteins), our mycobacterial panel contained proteins from
Mycobacterium smegmatis (5),
Mycobacterium abscessus (4),
Mycobacterium laprae (3),
Mycobacterium marinum (3),
Mycobacterium ulcerans (3),
Mycobacterium fotuitum (3),
Mycobacterium avium (2), and
Mycobacterium paratuberculosis (2). Advantages, on the other hand, lie in the sensitivity and specificity of MRMS for ligand identification.
In summary, while the PhenoTarget approach has some limitations, this article is an example of the powerful potential of the PhenoTarget approach in identifying new lead compounds, from a natural product fraction library, as well as the target protein of the lead compound. In the PhenoTarget approach, phenotypic screening is conducted initially to detect cellular active components. The hits are then screened against a panel of putative targets. This PhenoTarget protocol can be equally applied to pure compound libraries as well as natural product fractions and may prove to be a valuable alternative strategy for developing new intervention therapies.
4. Materials and Methods
4.1. General Experimental Procedures
NMR spectra were recorded in DMSO-d6 (δH 2.50 and δC 39.5), CD3OD (δH 3.31, 4.78) at 25 °C on the Bruker AVANCE III HDX 800 MHz NMR spectrometer (Fllanden, Zrich, Switzerland) equipped with a triple resonance cryoprobe. The low-resolution LC-MS was acquired using Ultimate 3000 RS UHPLC coupled to a Thermo Fisher MSQ Plus single quadruple ESI mass spectrometer (Waltham, MA, USA) with a Thermo Accucore C18 column (2.6 μm, 2.1 × 150 mm). High-resolution mass spectra (HRESIMS) were recorded on a Bruker maXis II ETD ESI- qTOF (Bremen, Germany). The HPLC system for LLE fractions for phenotypic screening included a Waters 600 pump (Milford, MA, USA) fitted with a 996-photodiode array detector and Gilson FC204 fraction collector (Middleton, WI, USA). The HPLC system for Fractions A~E from re-extraction by ethanol was the Thermo Ultimate 3000 system (Waltham, MA, USA). Semi-preparative HPLC was also programmed on Thermo Ultimate 3000 with a PDA detector. A Phenomenex C18 Monolithic column (5 μm, 4.6 × 100 mm) was used for analytical HPLC; a Thermo Electron Betasil C18 column (5 μm, 21.2 × 150 mm) was used for semi-preparative HPLC.
The fraction library (202,983 fractions) was from Nature Bank (Griffith Institute for Drug Discovery, Brisbane, Australia). All the solvents used for extraction, chromatography and MS were Lab-Scan HPLC grade, and the H2O was Millipore Milli-Q PF filtered.
Ammonium acetate was purchased from. Sigma-Aldrich (St Louis, O.M., USA). Rv1466 and the other 36 unique proteins in the target panel were supplied by the Seattle Structural Genomics Center for Infectious Diseases (SSGCID, Seattle, WA, USA). These were “well behaved” proteins whose structures have been solved by SSGCID and are publicly available in the PDB. All proteins contained a non-natural, N-terminal tag for purification (MAHHHHHHMGTLEAQTQGPGS-). For protein buffer exchange, Nalgene NAP-5 size G25, from GE Healthcare (Parramatta, NSW, Australia) was used (purchased through Thermo Fisher Scientific Australia Pty Ltd., Scoresby, VIC, Australia). The screening of protein-ligand complexes was conducted using Bruker Daltonics SolariX 12 T Magnetic Resonance Mass Spectrometry (Bruker Daltonics Inc., Billerica, MA, USA) equipped with automatic Nanoelectrospray system (TriVersa NanoMate, Advion Biosciences, Ithaca, NY, USA).
4.2. Lead-Like Enhanced Fraction Library
The lead-like enhanced fraction library was prepared as previously described [
14].
4.3. Phenotypic Screening-Activity against Replicating M. tuberculosis H37Rv
Growth inhibition of M. tuberculosis was monitored using a 1 μL of fraction (250 μge/μL) dispensed into each well of a 384-well plate. To this, 40 µL of M. tuberculosis ATCC 27294 H37Rv (3–5 × 105/mL in Middlebrook 7H9 broth with 0.05% Tween 80, 10 v/v ADC and Casamino acids) was added with a Multidrop dispenser. The plates were then incubated at 37 °C for 7 days. A 10 μL solution of Resazurin (20 mg/100 mL diluted 1:1 with 10% Tween 80) was added and incubated further for an additional 24 h at 37 °C for color development. Absorbance was monitored at two wavelengths (575 and 610 nm) using Spectramax and the ratios were determined to calculate the % inhibition. Growth controls in the absence of compound as well as media controls served as inhibition ~0% and −100% respectively.
Single point screen: samples (1 μL) which gave ≥80% inhibition were considered positive. MIC: the least concentration which gave ≥80% inhibition was considered as MIC (start conc. = 1 μL of fraction (250 μge/μL).
4.4. Re-extraction and Fractionation
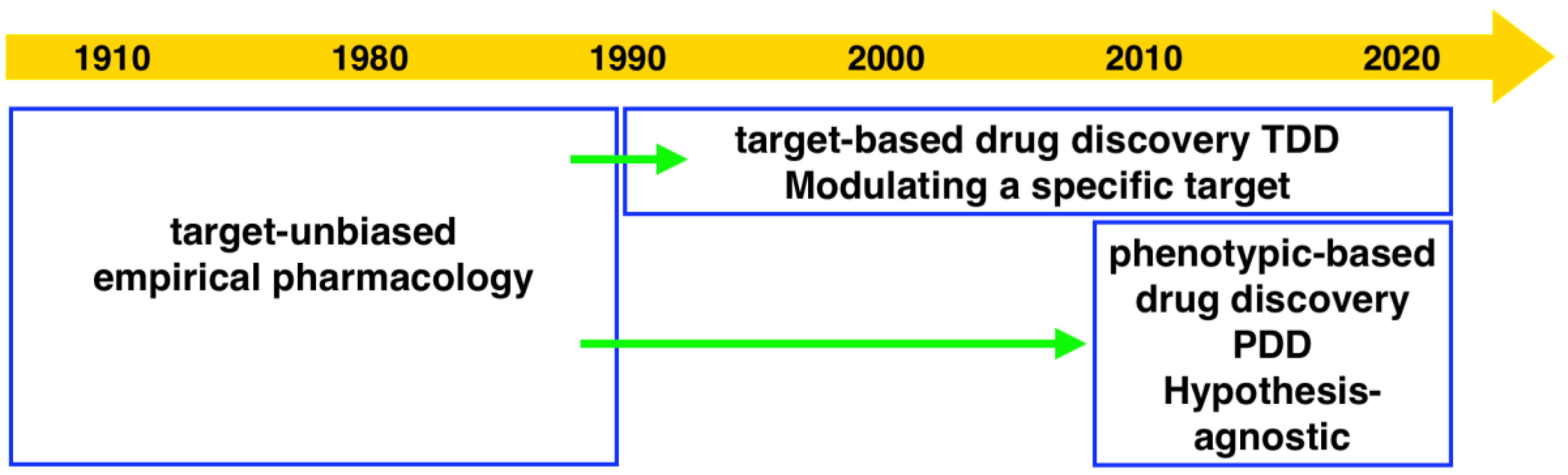
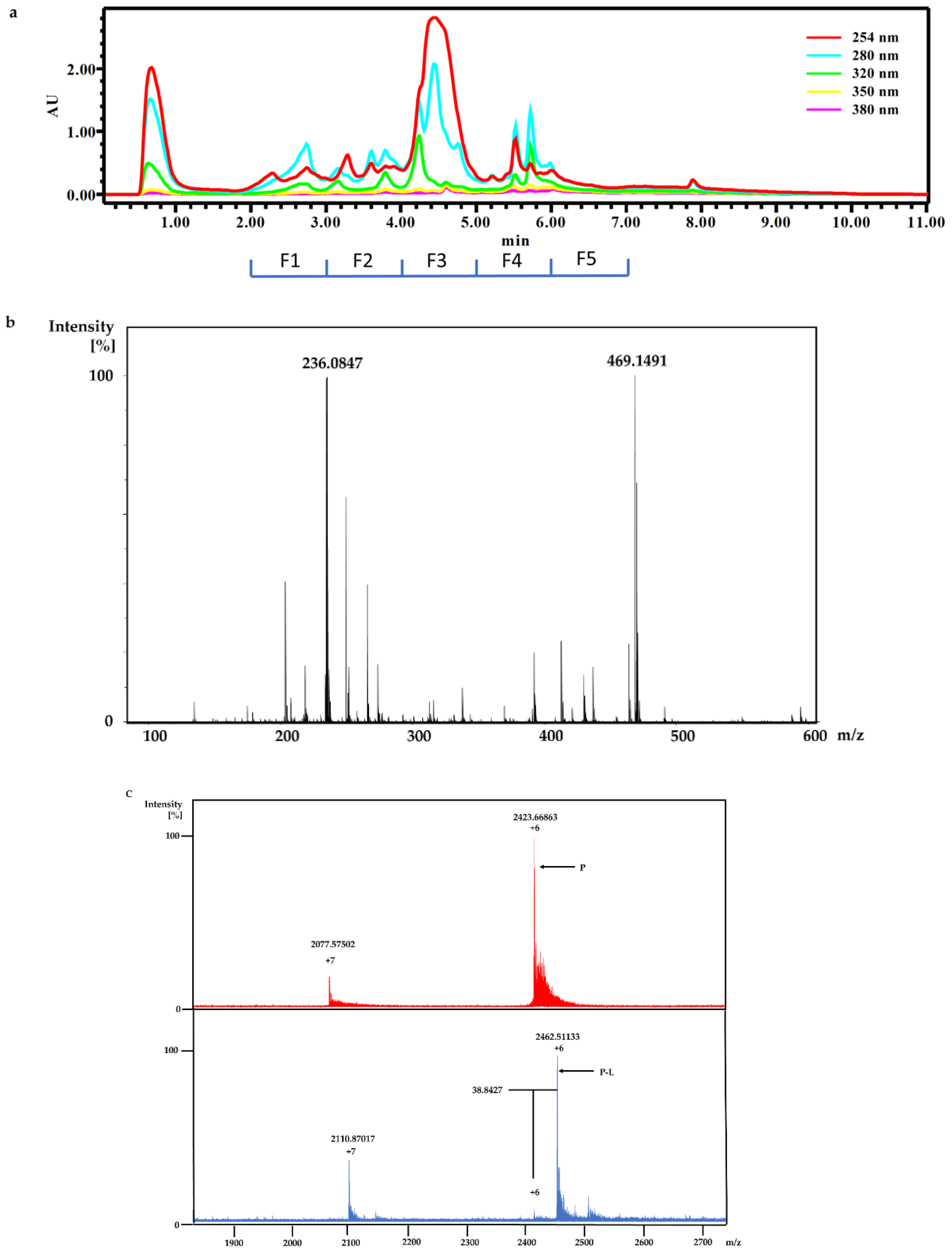
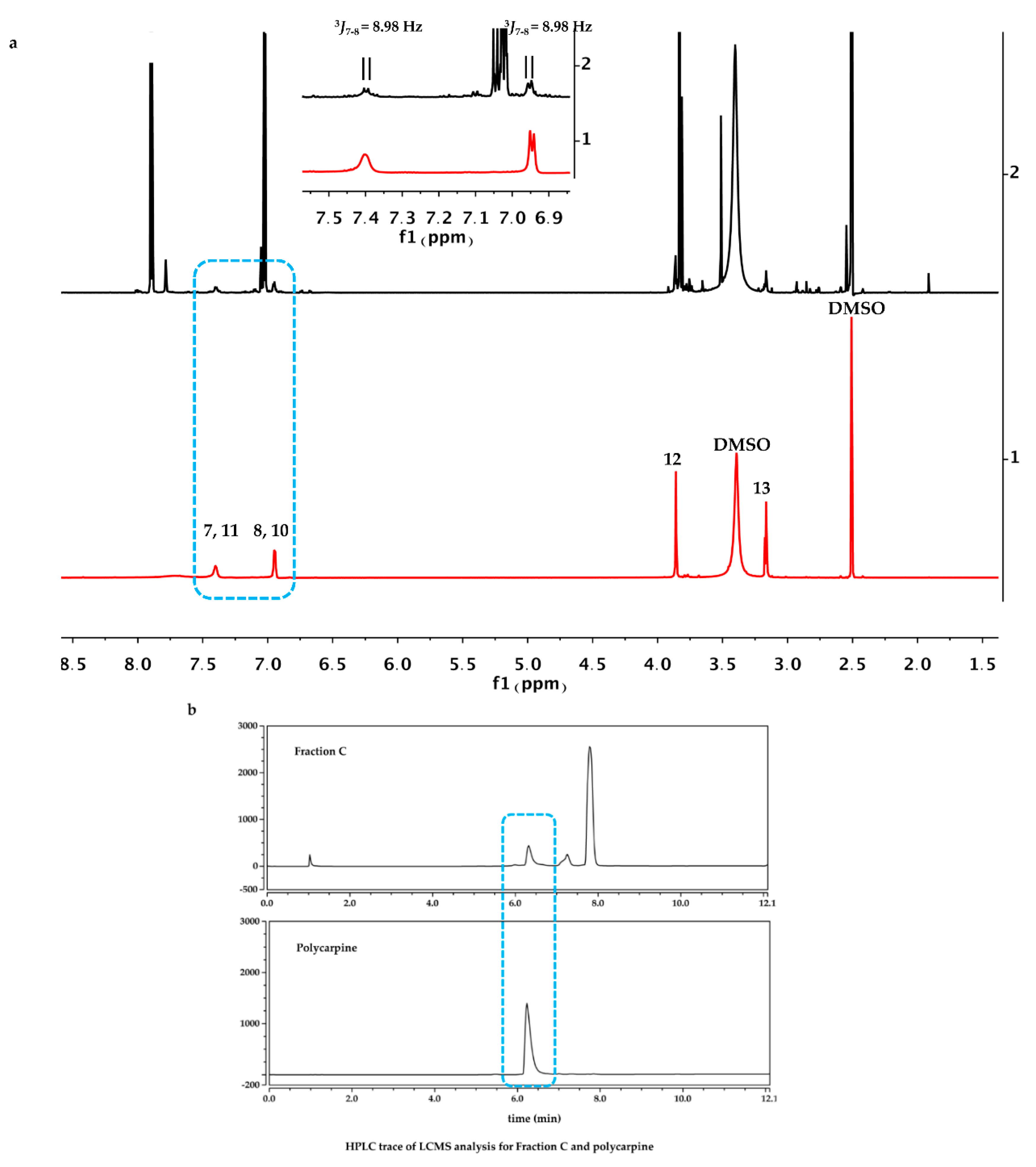
The dry powder of P. aurata (1.8 g) was extracted with 95% ethanol (3 × 150 mL) to afford a crude extract (1.48 g). A portion of the crude extract (40 mg) was dissolved in DMSO-d6 (600 μL). Twice injection (2 × 100 μL) was performed for each sample. HPLC separations were performed on a Phenomenex C18 Monolithic HPLC column (4.6 × 100 mm) using conditions that consisted of a linear gradient (curve 6) from 90% H2O (0.1% TFA)/10% MeOH (0.1% TFA) to 50% H2O (0.1% TFA)/50% MeOH (0.1% TFA) in 3 min at a flow rate of 4 mL/min; A convex gradient (curve 6) to 50% H2O (0.1% TFA)/50% MeOH (0.1% TFA) at a flow rate of 3 mL/min in 0.01 min, then a linear gradient (curve 5) to 100% MeOH (0.1% TFA) in 3.50 min; A held at 100% MeOH (0.1% TFA) (curve 6) for 1.50 min at a flow rate of 3 mL/min, then held at 100% MeOH (0.1% TFA) (curve 6) with a flow rate increasing to 4 mL/min in 0.01 min; A held at 100% MeOH (0.1% TFA) (curve 6) at a flow rate of 4 mL/min for a further 1 min; A linear gradient (curve 6) back to 90% H2O (0.1% TFA)/10% MeOH (0.1% TFA) in 1 min at a flow rate of 4 mL/min, then held at 90% H2O (0.1% TFA)/10% MeOH (0.1% TFA) (curve 6) for 2 min at a flow rate of 4 mL/min. The total run time for each injection was 11 min, and 5 fractions were collected between 2.0 and 7.0 min, i.e., Fraction A (time = 2.01-3.00 min), Fraction B (time = 3.01-4.00 min), Fraction C (time = 4.01-5.00 min), Fraction D (time = 5.01-6.00 min) and Fraction E (time = 6.01-7.00 min). Each fraction was dissolved in 200 μL of DMSO-d6 and was run for 1H NMR fingerprint in a 3 mm NMR tube on a Bruker AVANCE III HDX 800 MHz NMR instrument.
4.5. Automatic Chip-Based MRMS High Throughtput Screening
Proteins were buffer-exchanged into a suitable volatile buffer (ammonium acetate) under near physiological conditions using size exclusion chromatography prior to MRMS analysis. Depending on the protein, the buffer and its concentration were chosen to obtain the highest sensitivity in the mass spectrometer. Final concentration was 10 μM protein in 10 mM ammonium acetate after buffer change.
All screening samples were prepared fresh on the day for analyzing data to avoid the precipitation or decomposition in the sample.
Each 9 LLE fractions (9 × 1 μL) were combined as a Pool Fraction. Every Pool Fraction was dried, re-suspended in 9 μL MeOH. For the screening of Pool Fractions, 1 μL MeOH solution of pool fraction was mixed with 9 μL of 10 µM protein and then incubated for 1 h at room temperature before being analyzed by MRMS.
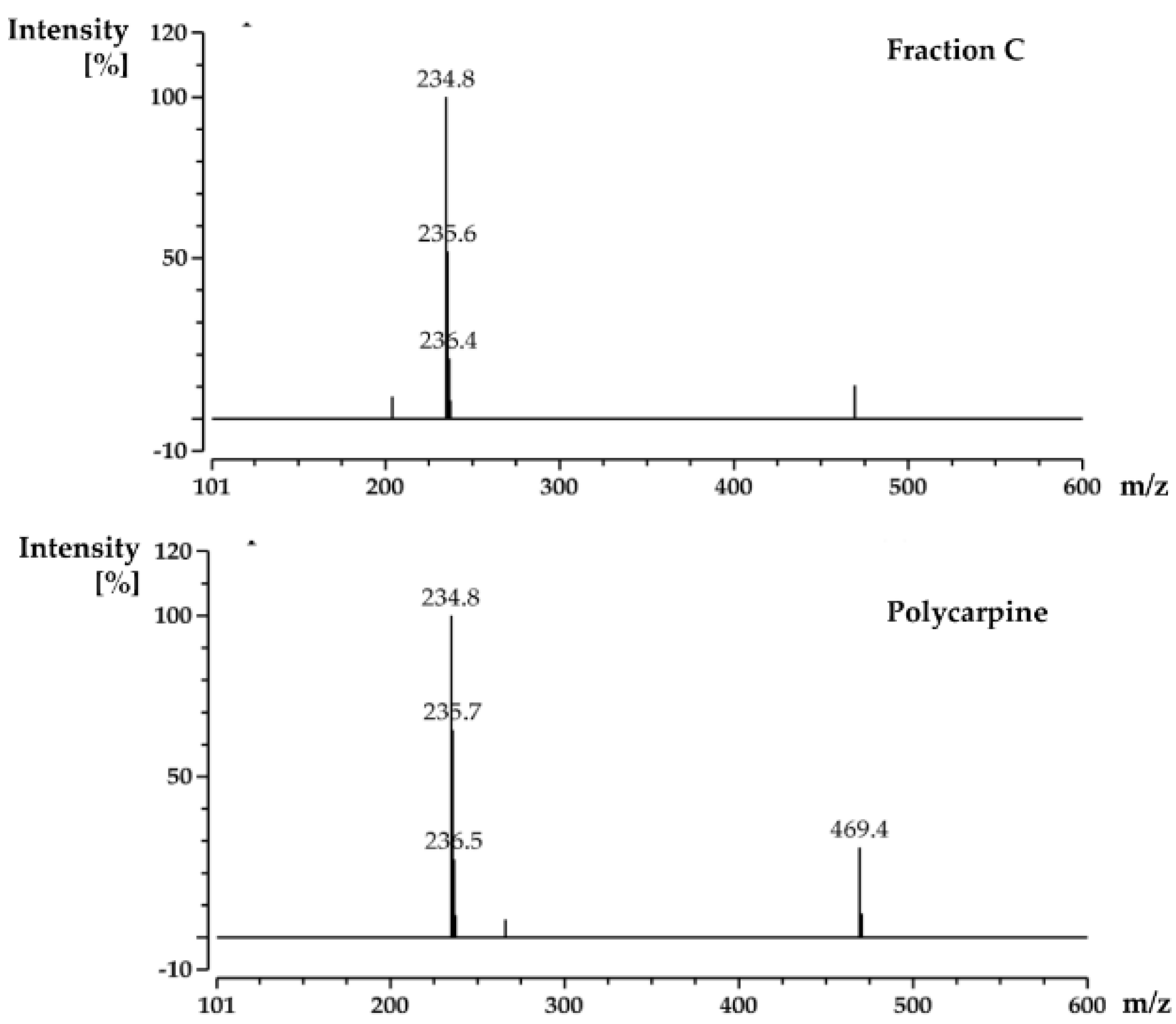
For screening the incubation of Fraction C of P. aurata with Rv1466-Putative uncharacterized protein, Fraction C was dried and re-suspended in 400 μL MeOH. An aliquot solution (1 μL) was added to protein (9 μL at 10 µM) for 15 min at room temperature and then analyzed by MRMS.
For the screening of pure compound-polycarpine, the stock solution of polycarpine, 3000 μM dissolved in DMSO-d6, was further diluted to additional varied concentrations between 1–1000 μM for dose-response measurement. Polycarpine solution in DMSO-d6 was dried and then dissolved in MeOH. A 40 μL sample of these polycarpine solutions was lyophilized and resuspended in 40 μL of MeOH. An aliquot (1 μL) of varied concentration of polycarpine in MeOH was incubated with 9μL of protein (10 µM) for 15 min at room temperature and then analyzed by MRMS.
Experiments were performed on a Bruker SolariX XR 12T MRMS (Bruker Daltonics Inc., Billerica, MA) equipped with an external automated chip-based nanoelectrospray. The ESI mass spectra were recorded in positive mode with a mass range from 294 to 10,000 m/z. Each spectrum was composed of 2 M data points. All the aspects of instrument parameter and data acquisition were controlled by Solarix control software under Windows operating system. Instrument parameters were tuned as follows for Pool Fraction screening: sample flow rate 120 µL/min, nebulizer gas (N2) pressure 3, end plate offset voltage 0 V, capillary voltage 1000 V, drying gas (N2) flow rate 1.5 L/min, drying gas temperature 100 °C, capillary exit voltage 220 V, skimmer 1 voltage 60 V, collision voltage −5, time of flight 2.1 s, scan 8 and accumulation time 0.5 s. For the screening of Rv1466-Putative uncharacterized protein with Fraction C and polycarpine, six parameters change including skimmer 1 voltage (30 V), collision voltage (−10 V), time of flight (1.8 s), scan (32) and accumulation time (2 s).
All sample solutions were injected by fully automated chip-based nanoelectrospray, which was mounted to the mass spectrometer. A Triversa NanoMate incorporating ESI chip technology (Advion Biosciences, Ithaca, NY) was used as an automatic chip system. The automated chip system consists of a 384-well plate, a rack of 96 disposable, conductive pipette tips, and the chip, which was positioned a few millimeters from the sampling cone. The system was programmed by ChipSoft software (Version 8.3.1, Advion BioSciences), which sequentially picks up a new pipette tip, aspirates 3 μL of sample from the 384-well plate followed by 1.5 µL of air and then delivers to the inlet side of ESI chip. Nanoelectrospray was carried out by applying a 1.60 kV spray voltage and a 1.0 psi gas pressure to the sample in the pipette tip. Sample solutions for screening were transferred into the 384-well plate. For every sample, a fresh tip and nozzle were used, thus preventing cross-contamination of samples. Following sample infusion and MS analysis, the pipet tip was disposed.
When the protein-ligand complex was found, the molecular weight of the binding ligand was estimated from the spectrum using the following equation: MW ligand = Δ m/z × z.
4.6. Isolation
Crude ethanolic extract of P. aurata (200 mg) was dissolved in MeOH. HPLC separation was conducted on a Thermo Electron Betasil C18 column (5 μm, 21.2 × 150 mm) at a flow rate of 9 mL/min: using gradient solvent system (10–100% MeOH in 50 min, 100% MeOH from 50–60min) and 60 fractions were collected by minutes. Fraction 33 and 34 were further purified on the same column using gradient solvent from 30–80% MeOH to afford polycarpine.
Polycarpine was obtained as yellow powder. HRMS (positive mode): m/z 469 [M + H] + (calculated for C22H25N6O2S2, 469.1475). 1H NMR (CD3OD, 800MHz) δ 7.49 (2H, d, J = 8.98Hz, H-7/11), 7.03 (2H, d, J = 8.98Hz, H-8/10), 3.91 (3H, s, 12-CH3), 3.25 (3H, s, 13-CH3). Carbon chemical shift was confirmed from 2D NMR, δ 109.8 (C-1), 148.1 (C-3), 137.6 (C-5), 117.6 (C-6), 127.7 (C-7/11), 113.7 (C-8/10), 161.4 (C-9), 54.4 (C-12) and 27.8 (C-13).
4.7. Dose-Response Data Analysis
The relative abundances of protein-ligand complex to total protein in the mass spectra correlated to the relative equilibrium concentrations of ligand to total protein in solution. The pseudo
KD of polycarpine with Rv1466 was determined using the following equations [
18]:
Experimental relative ratios of protein-ligand complex and total protein ion abundances were plotted against the total concentration of ligand. The pseudo KD could be obtained as a parameter of a nonlinear least-squares curve fitting.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







