Endolysins from Antarctic Pseudomonas Display Lysozyme Activity at Low Temperature



Abstract
1. Introduction
2. Results
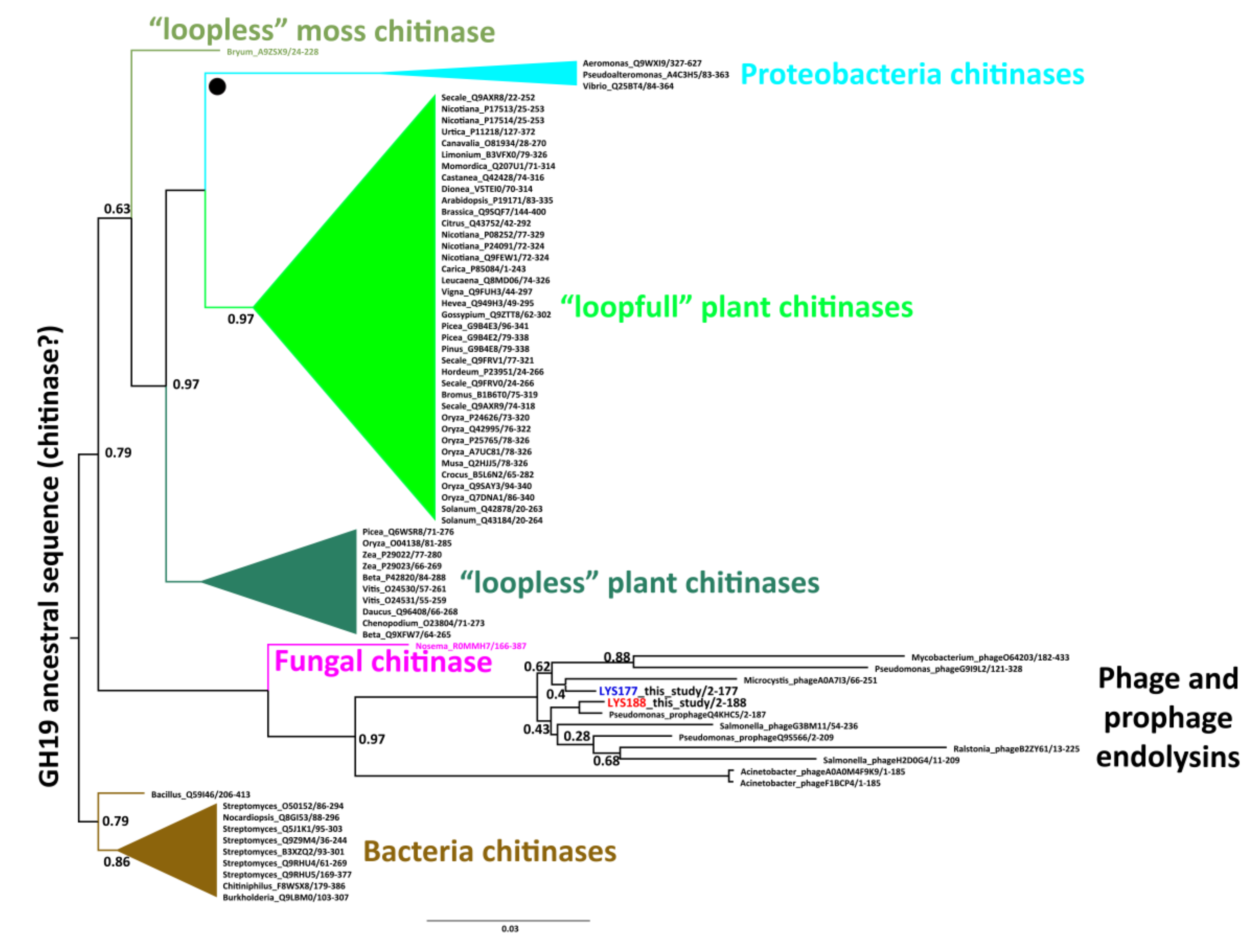
2.1. Mining and Molecular Evolution of Glycoside Hydrolases from the Bacterial Isolate
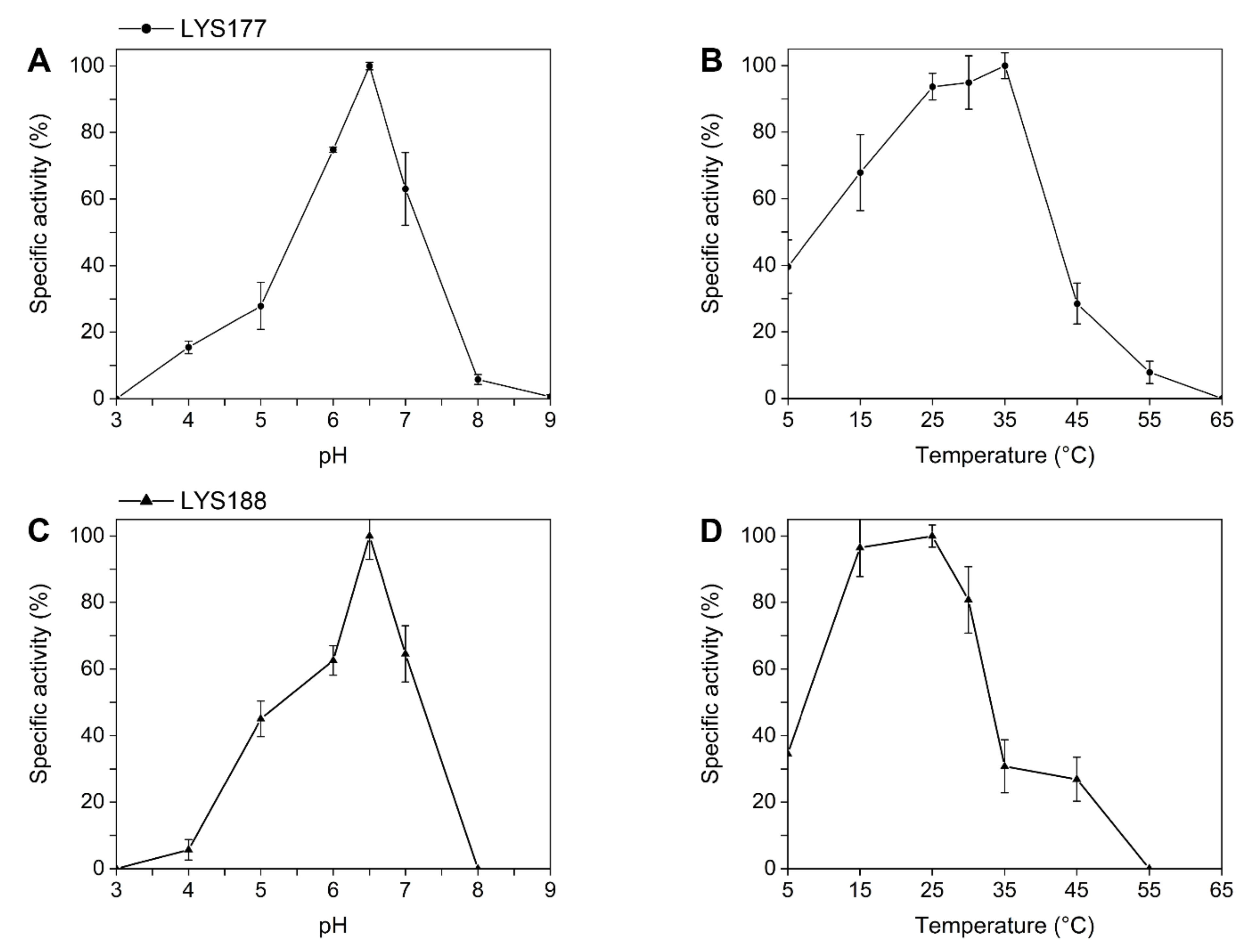
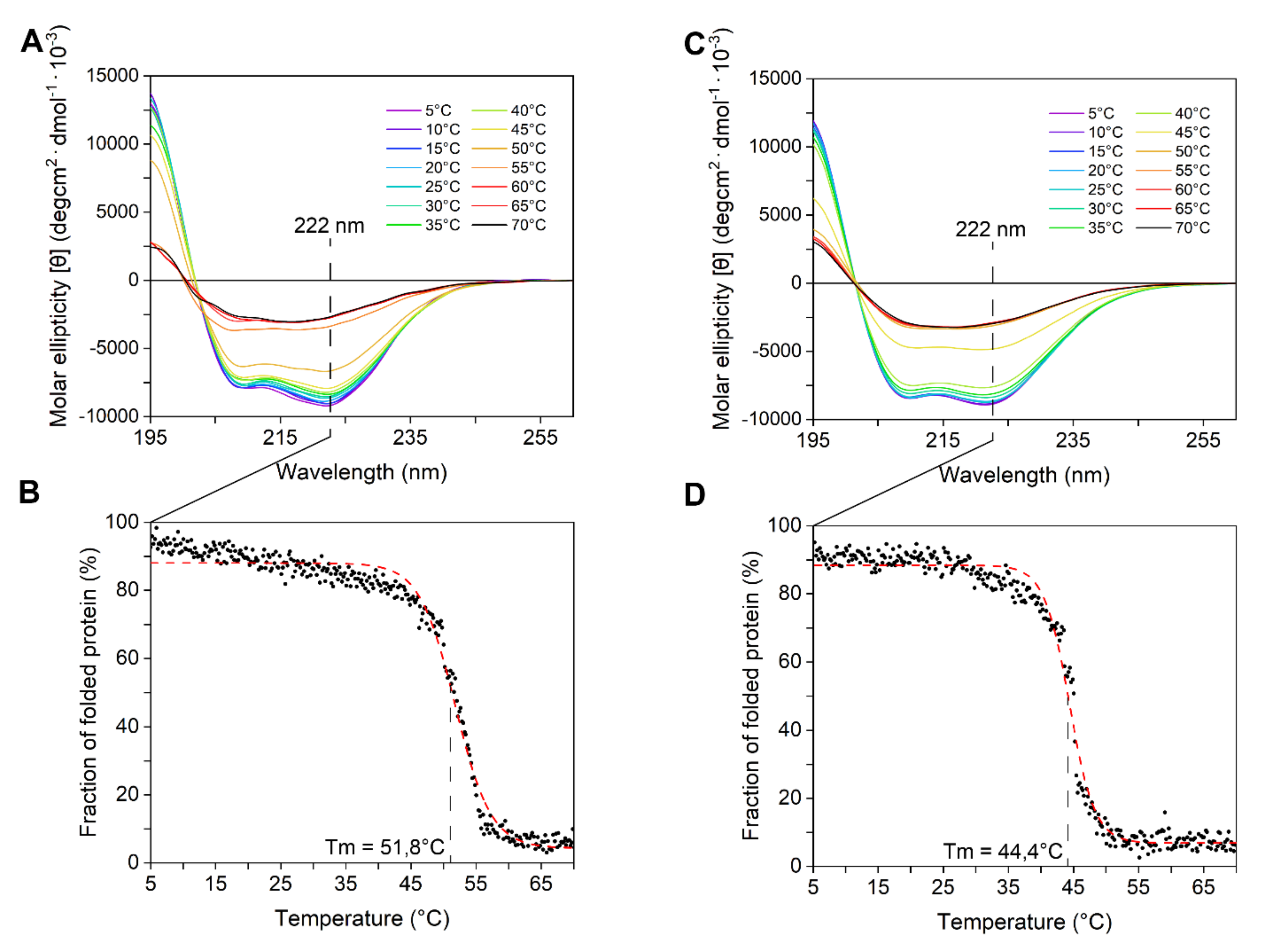
2.2. LYS177 and LYS188 are Cold-Active Thermolabile Glycosidases with Lysozyme Activity
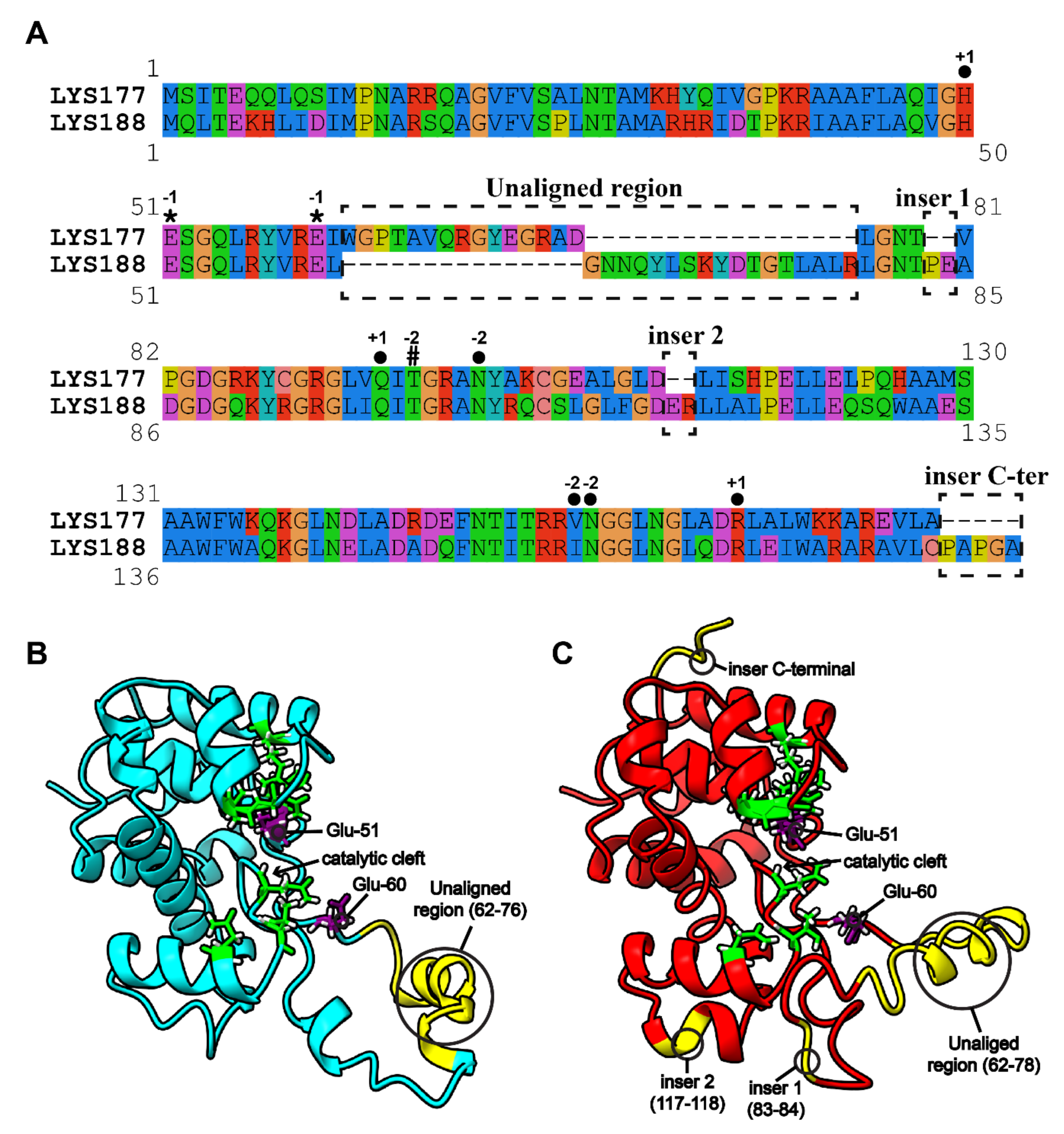
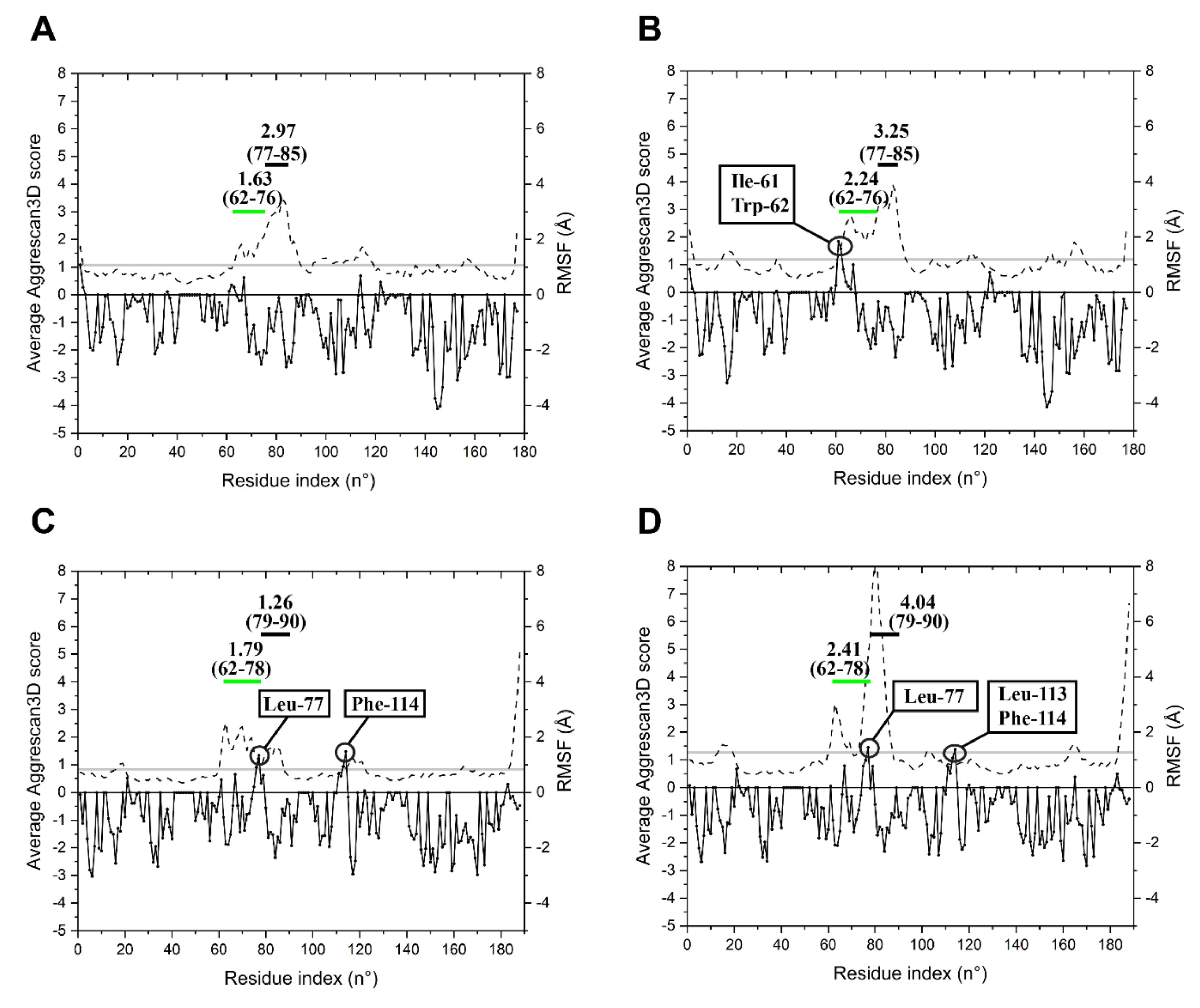
2.3. Specific Sequence Signatures May Account for Differences in Heat Stability
3. Discussion
4. Materials and Methods
4.1. Identification of the Bacterial Strain and of Glycoside Hydrolases Sequences
4.2. Enzymes Expression and Purification
4.3. Enzymes Characterization
4.3.1. Lysozyme Activity Assay
4.3.2. Chitinase Assay
4.3.3. Circular Dichroism (CD) Spectroscopy
4.3.4. Thermal Stability
4.4. In Silico Analysis
4.4.1. Phylogenetic Analysis
4.4.2. Alignment and Annotation of Close Homologues of LYS177 and LYS188
4.4.3. Modelling and Molecular Dynamics (MD) Simulations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D‘Amico, S.; Collins, T.; Marx, J.C.; Feller, G.; Gerday, C. Psychrophilic microorganisms: Challenges for life. EMBO Rep. 2006, 7, 385–389. [Google Scholar] [CrossRef] [PubMed]
- De Maayer, P.; Anderson, D.; Cary, C.; Cowan, D.A. Some like it cold: Understanding the survival strategies of psychrophiles. EMBO Rep. 2014, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 2003, 1, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.S.; Cavicchioli, R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef]
- D‘Amico, S.; Marx, J.-C.; Gerday, C.; Feller, G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem. 2003, 278, 7891–7896. [Google Scholar] [CrossRef]
- Siddiqui, K.S.; Feller, G.; D‘Amico, S.; Gerday, C.; Giaquinto, L.; Cavicchioli, R. The active site is the least stable structure in the unfolding pathway of a multidomain cold-adapted α-amylase. J. Bacteriol. 2005, 187, 6197–6205. [Google Scholar] [CrossRef]
- Mangiagalli, M.; Lapi, M.; Maione, S.; Orlando, M.; Brocca, S.; Pesce, A.; Barbiroli, A.; Camilloni, C.; Pucciarelli, S.; Lotti, M. The co-existence of cold activity and thermal stability in an Antarctic GH42 β-galactosidase relies on its hexameric quaternary arrangement. FEBS J. 2020. [Google Scholar] [CrossRef]
- Santiago, M.; Ramírez-Sarmiento, C.A.; Zamora, R.A.; Parra, L.P. Discovery, molecular mechanisms, and industrial applications of cold-active enzymes. Front. Microbiol. 2016, 7, 1408. [Google Scholar] [CrossRef]
- Mangiagalli, M.; Brocca, S.; Orlando, M.; Lotti, M. The “cold revolution”. Present and future applications of cold-active enzymes and ice-binding proteins. New Biotechnol. 2020, 55, 5–11. [Google Scholar] [CrossRef]
- Collins, T.; Margesin, R. Psychrophilic lifestyles: Mechanisms of adaptation and biotechnological tools. Appl. Microbiol. Biotechnol. 2019, 103, 2857–2871. [Google Scholar] [CrossRef]
- Yang, G.; Aprile, L.; Turturo, V.; Pucciarelli, S.; Pucciarelli, S.; Miceli, C. Characterization and comparative analysis of psychrophilic and mesophilic alpha-amylases from Euplotes species: A contribution to the understanding of enzyme thermal adaptation. Biochem. Biophys. Res. Commun. 2013, 438, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Perfumo, A.; Banat, I.M.; Marchant, R. Going green and cold: Biosurfactants from low-temperature environments to biotechnology applications. Trends Biotechnol. 2018, 36, 277–289. [Google Scholar] [CrossRef] [PubMed]
- John, M.S.; Nagoth, J.A.; Ramasamy, K.P.; Ballarini, P.; Mozzicafreddo, M.; Mancini, A.; Telatin, A.; Liò, P.; Giuli, G.; Natalello, A. Horizontal gene transfer and silver nanoparticles production in a new Marinomonas strain isolated from the Antarctic psychrophilic ciliate Euplotes focardii. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vallesi, A.; Pucciarelli, S.; Buonanno, F.; Fontana, A.; Mangiagalli, M. Bioactive molecules from protists: Perspectives in biotechnology. Eur. J. Protistol. 2020, 125720. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Pons, L.; Shilling, A.; Verde, C.; Baker, B.J.; Giordano, D. Marine Terpenoids from Polar Latitudes and Their Potential Applications in Biotechnology. Mar. Drugs 2020, 18, 401. [Google Scholar] [CrossRef]
- Mangiagalli, M.; Bar-Dolev, M.; Tedesco, P.; Natalello, A.; Kaleda, A.; Brocca, S.; Pascale, D.; Pucciarelli, S.; Miceli, C.; Braslavsky, I. Cryo-protective effect of an ice-binding protein derived from Antarctic bacteria. FEBS J. 2017, 284, 163–177. [Google Scholar] [CrossRef]
- Pischedda, A.; Ramasamy, K.P.; Mangiagalli, M.; Chiappori, F.; Milanesi, L.; Miceli, C.; Pucciarelli, S.; Lotti, M. Antarctic marine ciliates under stress: Superoxide dismutases from the psychrophilic Euplotes focardii are cold-active yet heat tolerant enzymes. Sci. Rep. 2018, 8, 14721. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef]
- Brameld, K.A.; Goddard, W.A. The role of enzyme distortion in the single displacement mechanism of family 19 chitinases. Proc. Natl. Acad. Sci. USA 1998, 95, 4276–4281. [Google Scholar] [CrossRef]
- Oliveira, H.; Melo, L.D.; Santos, S.B.; Nóbrega, F.L.; Ferreira, E.C.; Cerca, N.; Azeredo, J.; Kluskens, L.D. Molecular aspects and comparative genomics of bacteriophage endolysins. J. Virol. 2013, 87, 4558–4570. [Google Scholar] [CrossRef]
- Vidová, B.; Šramková, Z.; Tišáková, L.; Oravkinová, M.; Godány, A. Bioinformatics analysis of bacteriophage and prophage endolysin domains. Biologia 2014, 69, 541–556. [Google Scholar] [CrossRef]
- Lai, M.-J.; Lin, N.-T.; Hu, A.; Soo, P.-C.; Chen, L.-K.; Chen, L.-H.; Chang, K.-C. Antibacterial activity of Acinetobacter baumannii phage ϕAB2 endolysin (LysAB2) against both gram-positive and gram-negative bacteria. Appl. Microbiol. Biotechnol. 2011, 90, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, N.; Kurokawa, Y.; Sako, Y.; Nagasaki, K.; Yoshida, T.; Hiroishi, S. The functional effect of Gly209 and Ile213 substitutions on lysozyme activity of family 19 chitinase encoded by cyanophage Ma-LMM01. Fish. Sci. 2011, 77, 665–670. [Google Scholar] [CrossRef]
- Walmagh, M.; Briers, Y.; Dos Santos, S.B.; Azeredo, J.; Lavigne, R. Characterization of modular bacteriophage endolysins from Myoviridae phages OBP, 201φ2–1 and PVP-SE1. PLoS ONE 2012, 7, e36991. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-A.; Shin, H.; Kang, D.-H.; Ryu, S. Characterization of endolysin from a Salmonella Typhimurium-infecting bacteriophage SPN1S. Res. Microbiol. 2012, 163, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Pohane, A.A.; Joshi, H.; Jain, V. Molecular Dissection of Phage Endolysin an interdomain interaction confers host specificity in lysin a of mycobacterium phage D29. J. Biol. Chem. 2014, 289, 12085–12095. [Google Scholar] [CrossRef]
- Oliveira, H.; Vilas Boas, D.; Mesnage, S.; Kluskens, L.D.; Lavigne, R.; Sillankorva, S.; Secundo, F.; Azeredo, J. Structural and enzymatic characterization of ABgp46, a novel phage endolysin with broad anti-Gram-negative bacterial activity. Front. Microbiol. 2016, 7, 208. [Google Scholar] [CrossRef]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef]
- Gerstmans, H.; Rodriguez-Rubio, L.; Lavigne, R.; Briers, Y. From endolysins to Artilysins®: Novel enzyme-based approaches to kill drug-resistant bacteria. Biochem. Soc. Trans. 2016, 44, 123–128. [Google Scholar] [CrossRef]
- Ramasamy, K.P.; Telatin, A.; Mozzicafreddo, M.; Miceli, C.; Pucciarelli, S. Draft Genome Sequence of a New Pseudomonas sp. Strain, ef1, Associated with the Psychrophilic Antarctic Ciliate Euplotes focardii. Microbiol. Resour. Announc. 2019, 8, e00867-19. [Google Scholar] [CrossRef]
- John, M.S.; Nagoth, J.A.; Ramasamy, K.P.; Mancini, A.; Giuli, G.; Natalello, A.; Ballarini, P.; Miceli, C.; Pucciarelli, S. Synthesis of Bioactive Silver Nanoparticles by a Pseudomonas Strain Associated with the Antarctic Psychrophilic Protozoon Euplotes focardii. Mar. Drugs 2020, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Pucciarelli, S.; Devaraj, R.R.; Mancini, A.; Ballarini, P.; Castelli, M.; Schrallhammer, M.; Petroni, G.; Miceli, C. Microbial consortium associated with the Antarctic marine ciliate Euplotes focardii: An investigation from genomic sequences. Microb. Ecol. 2015, 70, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.W.; Kim, J.S.; Park, I.C.; Yoon, S.H.; Park, D.H.; Lim, C.K.; Go, S.J. Pseudomonas koreensis sp. nov., Pseudomonas umsongensis sp. nov. and Pseudomonas jinjuensis sp. nov., novel species from farm soils in Korea. Int. J. Syst. 2003, 53, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Ohnuma, T.; Fukamizo, T. Insertion of a Loop Structure into the “Loopless” GH19 Chitinase from Bryum coronatum. J. Appl. Glycosci. 2017, 64, 39–42. [Google Scholar] [CrossRef]
- Boller, T.; Gehri, A.; Mauch, F.; Vögeli, U. Chitinase in bean leaves: Induction by ethylene, purification, properties, and possible function. Planta 1983, 157, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Levashov, P.A.; Sedov, S.A.; Shipovskov, S.; Belogurova, N.G.; Levashov, A.V. Quantitative turbidimetric assay of enzymatic gram-negative bacteria lysis. Anal. Chem. 2010, 82, 2161–2163. [Google Scholar] [CrossRef]
- Banerjee, S.K.; Holler, E.; Hess, G.P.; Rupley, J.A. Reaction of N-acetylglucosamine oligosaccharides with lysozyme. Temperature, pH, and solvent deuterium isotope effects; equilbrium, steady state, and pre-steady state measurements*. J. Biol. Chem. 1975, 250, 4355–4367. [Google Scholar]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Buchholz, P.C.; Vogel, C.; Reusch, W.; Pohl, M.; Rother, D.; Spieß, A.C.; Pleiss, J. BioCatNet: A database system for the integration of enzyme sequences and biocatalytic experiments. ChemBioChem 2016, 17, 2093–2098. [Google Scholar] [CrossRef]
- Davies, G.J.; Wilson, K.S.; Henrissat, B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K. Evolution of the Taebaeksan Basin, Korea: I, early Paleozoic sedimentation in an epeiric sea and break-up of the Sino-Korean Craton from Gondwana. Isl. Arc 2019, 28, e12275. [Google Scholar] [CrossRef]
- Barnes, D.K.; Clarke, A. Antarctic marine biology. Curr. Biol. 2011, 21, R451–R457. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.U.; Jayanthi, M.; Sabarinathan, R.; Kangueane, P.; Mathew, L.; Sekar, K. Evolution, homology conservation, and identification of unique sequence signatures in GH19 family chitinases. J. Mol. Evol. 2010, 70, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Isaksen, G.V.; Brandsdal, B.O. Computation of enzyme cold adaptation. Nat. Rev. Chem. 2017, 1, 51. [Google Scholar] [CrossRef]
- Briers, Y.; Lavigne, R. Breaking barriers: Expansion of the use of endolysins as novel antibacterials against Gram-negative bacteria. Future Microbiol. 2015, 10, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lim, J.A.; Kong, M.; Ryu, S.; Rhee, S. Structure of bacteriophage SPN 1 S endolysin reveals an unusual two-module fold for the peptidoglycan lytic and binding activity. Mol. Microbiol. 2014, 92, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. Bioessays 2011, 33, 43–51. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Brocca, S.; Ferrari, C.; Barbiroli, A.; Pesce, A.; Lotti, M.; Nardini, M. A bacterial acyl aminoacyl peptidase couples flexibility and stability as a result of cold adaptation. FEBS J. 2016, 283, 4310–4324. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Lavigne, R.; Volckaert, G.; Hertveldt, K. A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J. Biochem. Biophys. Meth. 2007, 70, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Redelings, B.D. BAli-Phy: Simultaneous Bayesian inference of alignment and phylogeny. Bioinformatics 2006, 22, 2047–2048. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Redelings, B.D.; Suchard, M.A. Incorporating indel information into phylogeny estimation for rapidly emerging pathogens. BMC Evol. Biol. 2007, 7, 40. [Google Scholar] [CrossRef]
- Bansal, M.S.; Kellis, M.; Kordi, M.; Kundu, S. RANGER-DTL 2.0: Rigorous Reconstruction of Gene-Family Evolution by Duplication, Transfer, and Loss. Bioinformatics 2018, 34, 3214–3216. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lindahl, E.R. Molecular dynamics simulations. Methods Mol. Biol. 2008, 443, 3–23. [Google Scholar] [CrossRef]
- Kuriata, A.; Iglesias, V.; Kurcinski, M.; Ventura, S.; Kmiecik, S. Aggrescan3D standalone package for structure-based prediction of protein aggregation properties. Bioinformatics 2019, 35, 3834–3835. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Pairwise Identity (%) to lys188 | Pairwise Identity (%) to lys177 | Pairwise Identity (%) to Pseudomonas Ef1 16S | RefSeq Genome Assembly (GenBank AN) |
|---|---|---|---|---|
| Pseudomonas Ef1 (paralogue gene) | 61.92 a | 61.92 a | 100 | GCF_007293365.1 |
| Pseudomonas fluorescens 90f12-2 | 93.65 | - | 99.93 | GCF_003732335.1 |
| Pseudomonas koreensis D26 | 100 | 100 | 99.87 | GCF_001605965.1 |
| Pseudomonas moravensis BS3668 b | 91.53 | 89.51 | 99.87 | GCF_900105805.1 |
| Pseudomonas koreensis IMBL1 | 94.53 | 87.27 | 99.8 | GCF_001856885.1 |
| Pseudomonas koreensis BS3658 b | 83.6 | - | 99.8 | GCF_900101415.1 |
| Pseudomonas koreensis CRS05-R5 | 83.6 | - | 99.8 | GCF_001654515.1 |
| Pseudomonas sp RIT288 b | 84.83 | - | 99.67 | GCF_000631985.1 |
| Pseudomonas granadiensis LMG 27940 | 84.13 | - | 99.54 | GCF_900105485.1 |
| Pseudomonas reinekei MT1 | 78.48 | - | 99.54 | GCF_001945365.1 |
| Pseudomonas koreensis CI12 | 82.19 | - | 98.96 | GCF_002003425.1 |
| Pseudomonas koreensis P2 | 68.08 | - | 98.24 | GCF_002177125.1 |
| Pseudomonas putida NBRC 14164 | - | - | 97.98 | GCF_000412675.1 |
| Pseudomonas viridiflava | - | - | 96.54 | GCF_900184295.1 |
| Pseudomonas aeruginosa PA96 | - | - | 95.43 | GCF_000626655.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlando, M.; Pucciarelli, S.; Lotti, M. Endolysins from Antarctic Pseudomonas Display Lysozyme Activity at Low Temperature. Mar. Drugs 2020, 18, 579. https://doi.org/10.3390/md18110579
Orlando M, Pucciarelli S, Lotti M. Endolysins from Antarctic Pseudomonas Display Lysozyme Activity at Low Temperature. Marine Drugs. 2020; 18(11):579. https://doi.org/10.3390/md18110579
Chicago/Turabian StyleOrlando, Marco, Sandra Pucciarelli, and Marina Lotti. 2020. "Endolysins from Antarctic Pseudomonas Display Lysozyme Activity at Low Temperature" Marine Drugs 18, no. 11: 579. https://doi.org/10.3390/md18110579
APA StyleOrlando, M., Pucciarelli, S., & Lotti, M. (2020). Endolysins from Antarctic Pseudomonas Display Lysozyme Activity at Low Temperature. Marine Drugs, 18(11), 579. https://doi.org/10.3390/md18110579