Experimental and Computational Study to Reveal the Potential of Non-Polar Constituents from Hizikia fusiformis as Dual Protein Tyrosine Phosphatase 1B and α-Glucosidase Inhibitors

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
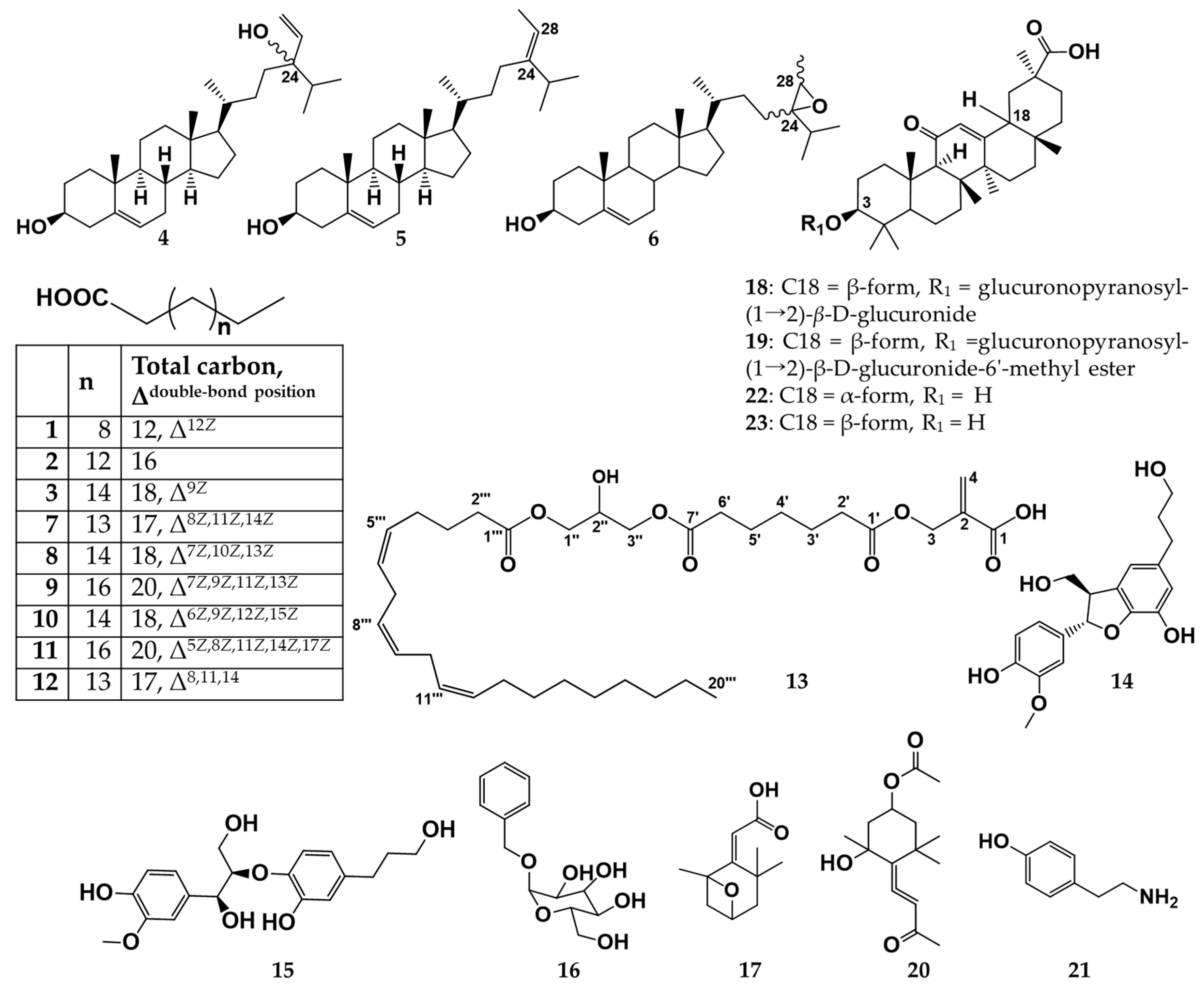
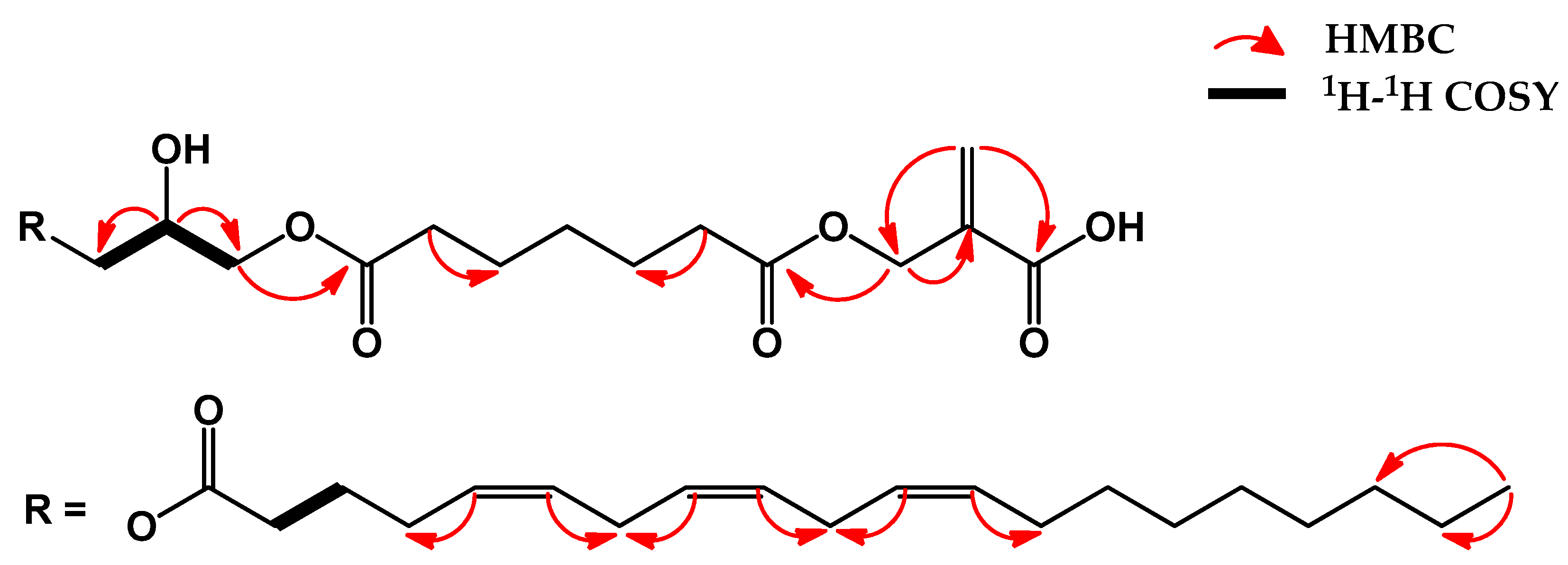
2.1. Structure Elucidation of Isolated Compounds
2.2. PTP1B and α-Glucosidase Inhibitory Activity of the Isolated Compounds from H. fusiformis
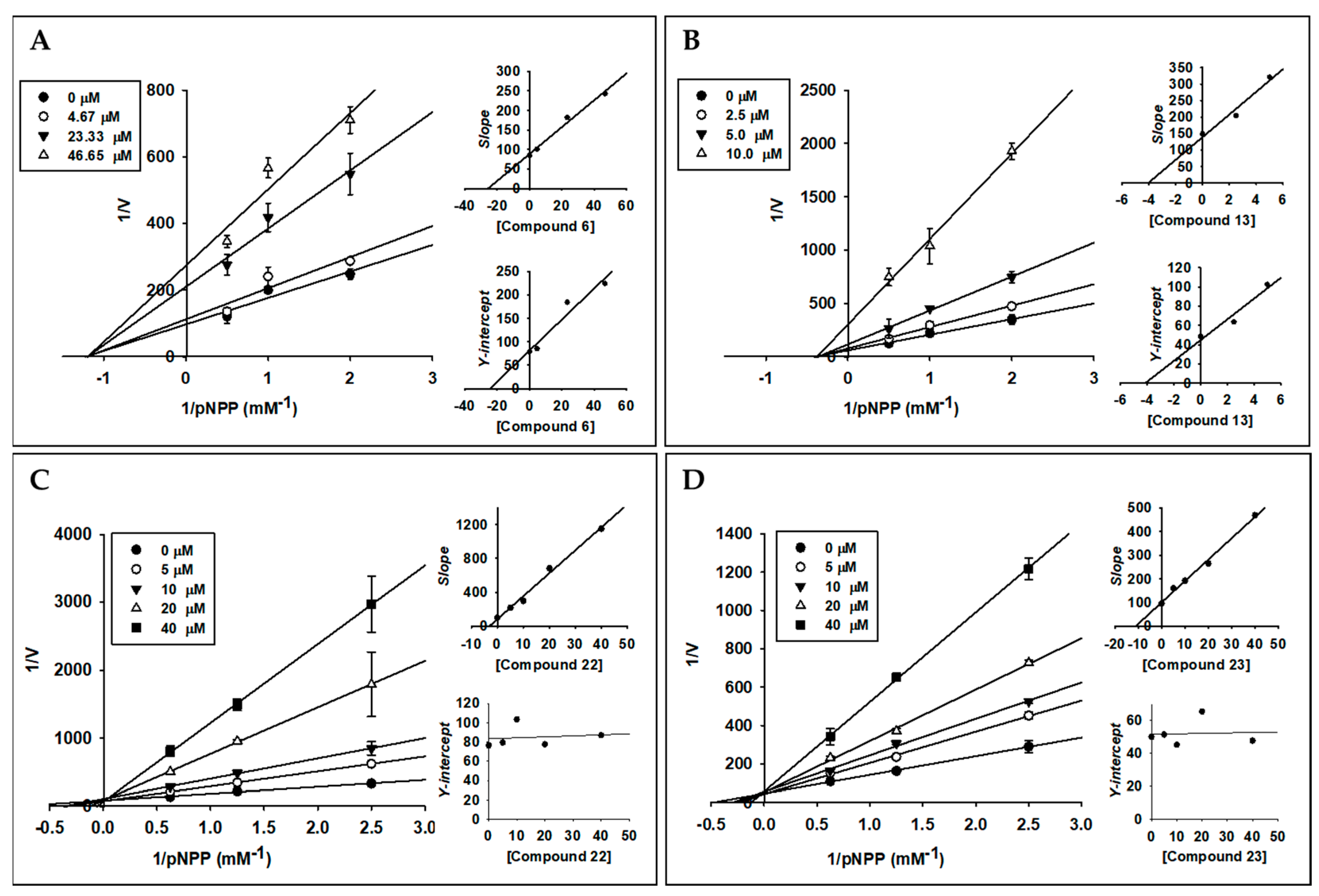
2.3. Enzyme Kinetic Analysis of Active Compounds with PTP1B
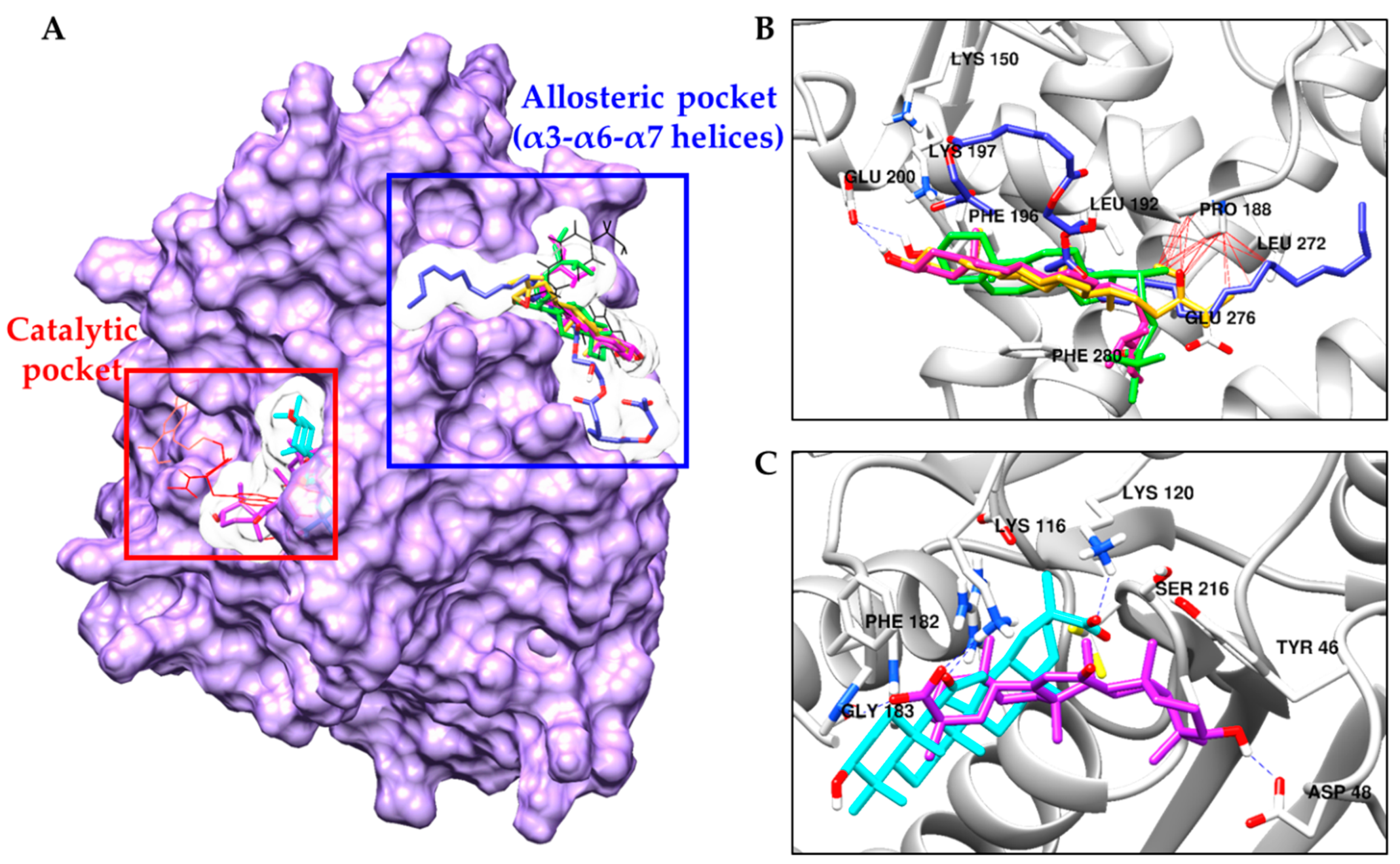
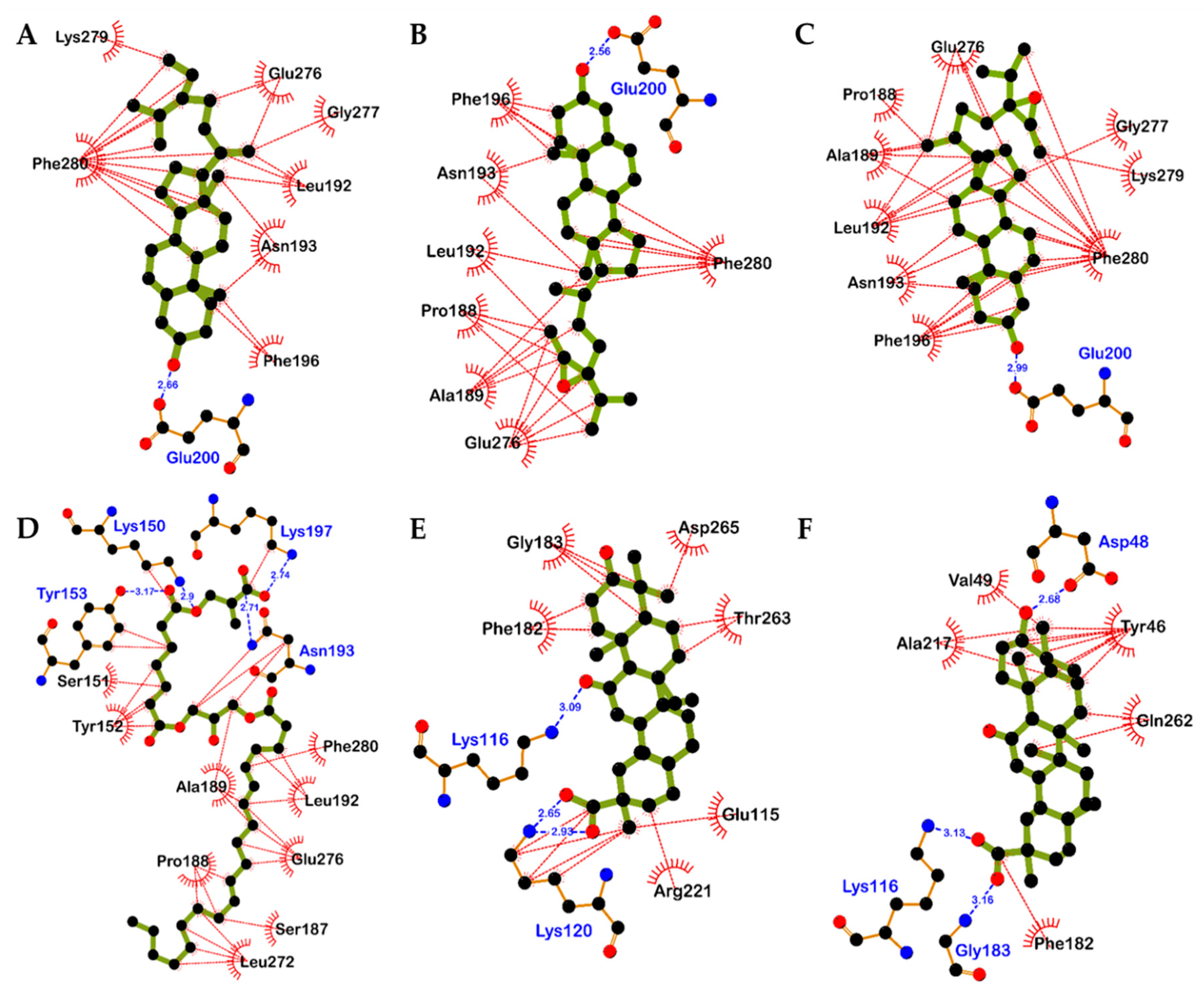
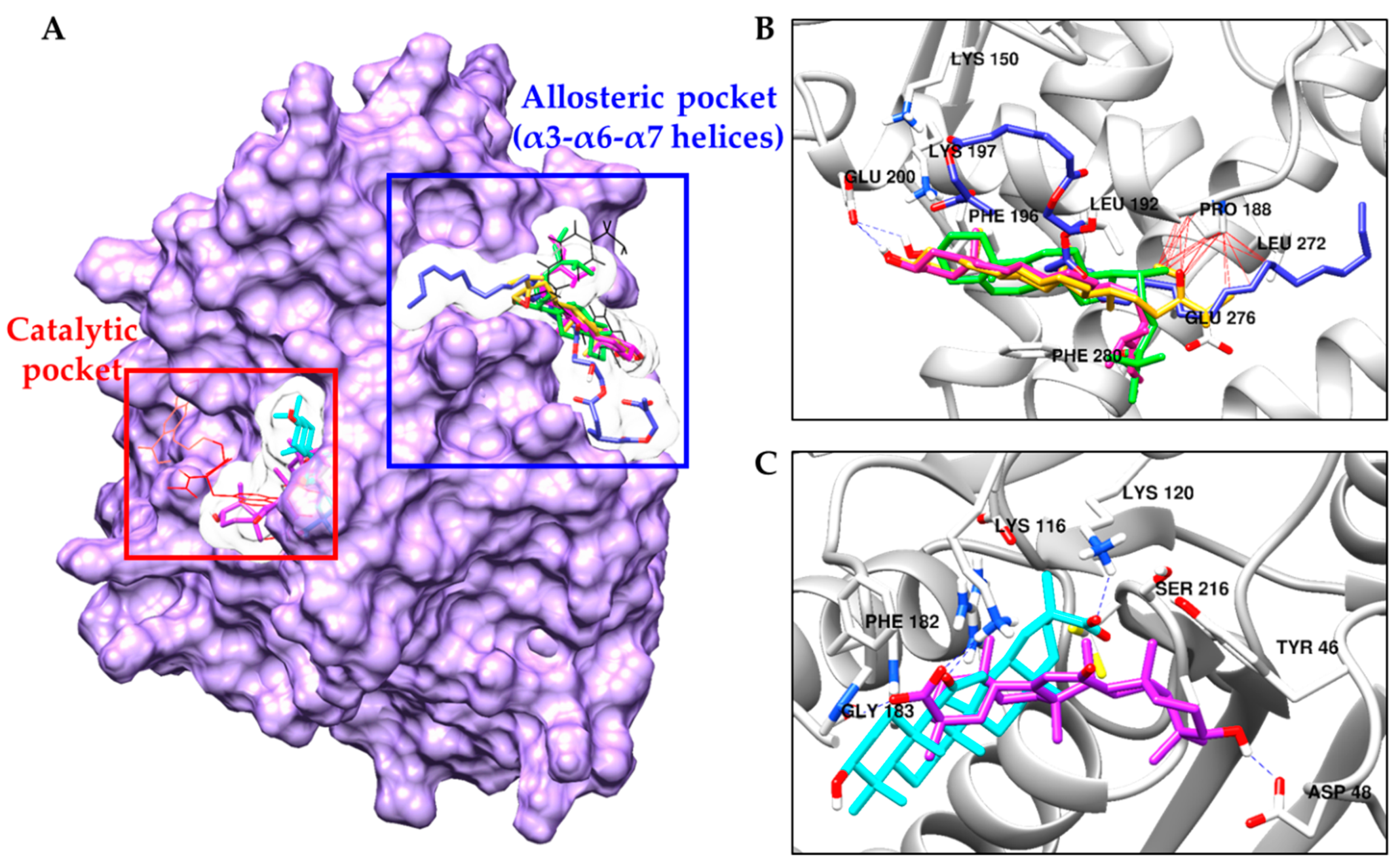
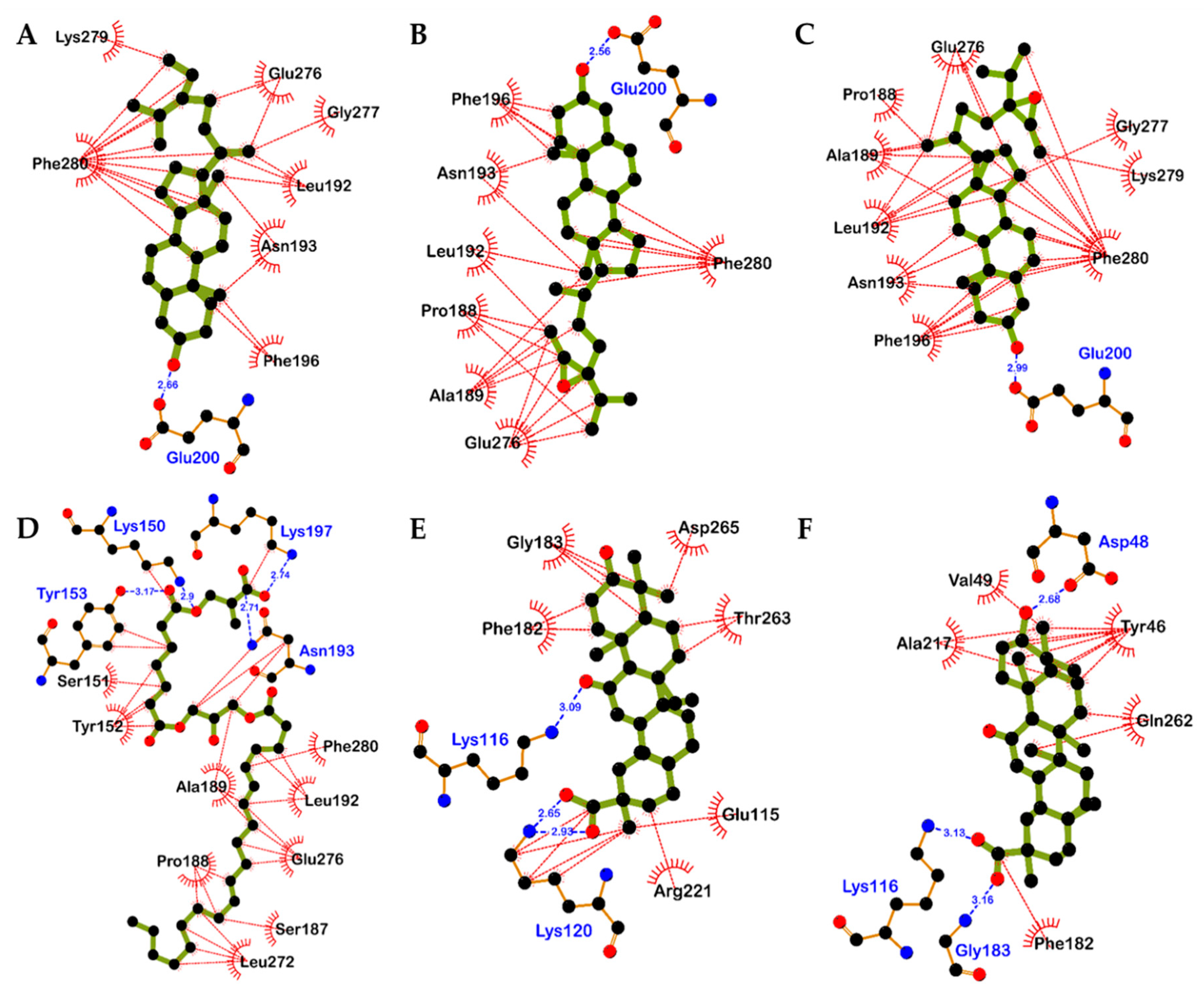
2.4. Molecular Docking Simulation in PTP1B Inhibition
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Chemicals and Reagents
4.3. Plant Material
4.4. Extraction, Fractionation, and Isolation
4.5. In Vitro α-Glucosidase Inhibitory Activity Assay
4.6. In Vitro PTP1B Inhibitory Activity Assay
4.7. Kinetic Parameters of Active Compounds towards PTP1B Inhibition
4.8. PTP1B Molecular Docking Simulations
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Diabetes Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 28 December 2018).
- Sun, J.; Cu, C.; Wang, Y.; Huang, H.; Zhang, M.; Li, H.; Zhang, Y.; Wang, Y.; Zou, W. PTP1B, a potential target of type 2 diabetes mellitus. Mol. Biol. 2016, 5, 1–6. [Google Scholar] [CrossRef]
- Shim, Y.J.; Doo, H.K.; Ahn, S.Y.; Kim, Y.S.; Seong, J.K.; Park, I.S.; Min, B.H. Inhibitory effect of aqueous extract from the gall of Rhus chinensis on alpha-glucosidase activity and postprandial blood glucose. J. Ethnopharmacol. 2013, 85, 283–287. [Google Scholar] [CrossRef]
- Seong, S.H.; Roy, A.; Jung, H.A.; Jung, H.J.; Choi, J.S. Protein tyrosine phosphatase 1B and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J. Ethnopharmacol. 2016, 194, 706–716. [Google Scholar] [CrossRef]
- Bischoff, H. The mechanism of alpha-glucosidase inhibition in the management of diabetes. Clin. Invest. Med. 1995, 18, 303–311. [Google Scholar] [PubMed]
- Dabhi, A.S.; Bhatt, N.R.; Shah, M.J. Voglibose: An alpha glucosidase inhibitor. J. Clin. Diagn. Res. 2013, 7, 3023–3027. [Google Scholar]
- Wubshet, S.G.; Tahtah, Y.; Heskes, A.M.; Kongstad, K.T.; Pateraki, I.; Hamberger, B.; Møller, B.M.; Staerk, D. Identification of PTP1B and α-glucosidase inhibitory serrulatanes from Eremophila spp. by combined use of dual high-resolution PTP1B and α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. J. Nat. Prod. 2016, 79, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696. [Google Scholar] [CrossRef]
- Song, Y.H.; Uddin, Z.; Jin, Y.M.; Li, Z.; Curtis-Long, M.S.; Kim, K.D.; Cho, J.K.; Park, K.H. Inhibition of protein tyrosine phosphatase (PTP1B) and α-glucosidase by geranylated flavonoids from Paulownia tomentosa. J. Enzyme Inhib. Med. Chem. 2017, 32, 1195–1202. [Google Scholar] [CrossRef]
- Ha, M.T.; Seong, S.H.; Nguyen, T.D.; Cho, W.K.; Ah, K.J.; Ma, J.Y.; Woo, M.H.; Choi, J.S.; Min, B.S. Chalcone derivatives from the root bark of Morus alba L. act as inhibitors of PTP1B and α-glucosidase. Phytochemistry 2018, 155, 114–125. [Google Scholar] [CrossRef]
- Ali, M.Y.; Kim, D.H.; Seong, S.H.; Kim, H.-R.; Jung, H.A.; Choi, J.S. α-Glucosidase and protein tyrosine phosphatase 1B inhibitory activity of plastoquinones from marine brown alga Sargassum serratifolium. Mar. Drugs 2017, 15, 368. [Google Scholar] [CrossRef]
- Ezzat, S.M.; Bishbishy, M.H.E.; Habtemariam, S.; Salehi, B.; Sharifi-Rad, M.; Martins, N.; Sharifi-Rad, J. Looking at marine-derived bioactive molecules as upcoming anti-diabetic agents: A special emphasis on PTP1B inhibitors. Molecules 2018, 23, 3334. [Google Scholar] [CrossRef]
- Ma, Z.; Wu, M.; Lin, L.; Thring, R.W.; Yu, H.; Zhang, X.; Zhao, M. Allelopathic interactions between the macroalga Hizikia fusiformis (Harvey) and the harmful blooms-forming dinoflagellate Karenia mikimotoi. Harmful Algae 2017, 65, 19–26. [Google Scholar] [CrossRef]
- Karawita, R.; Siriwardhana, N.; Lee, K.W.; Heo, M.S.; Yeo, I.K.; Lee, Y.D.; Jeon, Y.J. Reactive oxygen species scavenging, metal chelating, reducing power and lipid peroxidation inhibition properties of different solvent fractions from Hizikia fusiformis. Eur. Food Res. Technol. 2005, 220, 363–371. [Google Scholar] [CrossRef]
- Han, Y.R.; Ali, M.Y.; Woo, M.H.; Jung, H.A.; Choi, J.S. Anti-diabetic and anti-inflammatory potential of the edible brown alga Hizikia fusiformis. J. Food Biochem. 2015, 39, 417–428. [Google Scholar] [CrossRef]
- Wagle, A.; Seong, S.H.; Zhao, B.T.; Woo, M.H.; Jung, H.A.; Choi, J.S. Comparative study of selective in vitro and in silico BACE1 inhibitory potential of glycyrrhizin together with its metabolites, 18α- and 18β-glycyrrhetinic acid, isolated from Hizikia fusiformis. Arch. Pharm. Res. 2018, 41, 409–418. [Google Scholar] [CrossRef]
- Choi, E.Y.; Hwang, H.J.; Nam, T.J. Protective effect of a polysaccharide from Hizikia fusiformis against ethanol-induced cytotoxicity in IEC-6 cells. Toxicol. In Vitro 2010, 24, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Kim, E.; Kang, I.; Lee, M.; Lee, Y. Anti-diabetic effects and anti-inflammatory effects of Laminaria japonica and Hizikia fusiforme in skeletal muscle: in vitro and in vivo model. Nutrients 2018, 10, 491. [Google Scholar] [CrossRef]
- Hwang, H.J.; Kim, I.H.; Nam, T.J. Effect of a glycoprotein from Hizikia fusiformis on acetaminophen-induced liver injury. Food Chem. Toxicol. 2008, 46, 3475–3481. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.S.; Park, H.Y.; Nam, K.H. Whitening effects of 4-hydroxyphenethyl alcohol isolated from water boiled with Hizikia fusiformis. Food Sci. Biotechnol. 2014, 23, 555–560. [Google Scholar] [CrossRef]
- Zhao, B.T.; Nguyen, D.H.; Le, D.D.; Choi, J.S.; Min, B.S.; Woo, M.H. Protein tyrosine phosphatase 1B inhibitors from natural sources. Arch. Pharm. Res. 2018, 41, 130–161. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Jeong, S.Y.; Jung, H.A.; Choi, J.S.; Min, B.S.; Woo, M.H. Capsofulvesins A–C, cholinesterase inhibitors from Capsosiphon fulvescens. Chem. Pharm. Bull. 2012, 60, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Gunstone, F.D.; Pollard, M.R.; Scrimgeour, C.M.; Vedanayagam, H.S. 13C nuclear magnetic resonance studies of olefinic fatty acids and esters. Chem. Phys. Lipids 1977, 18, 115–129. [Google Scholar] [CrossRef]
- Jagannathan, N.R.; Rao, C.N.R. A 13C NMR spectroscopic study of the phase transitions of alkane dicarboxylic acids in the solid state. Chem. Phys. Lett. 1987, 140, 46–50. [Google Scholar] [CrossRef]
- Huh, G.W.; Lee, D.Y.; In, S.J.; Lee, D.G.; Park, S.Y.; Yi, T.H.; Kang, H.C.; Seo, W.D.; Baek, N.I. Fucosterols from Hizikia fusiformis and their proliferation activities on osteosarcoma-derived cell MG63. J. Korean Soc. Appl. Biol. Chem. 2012, 55, 551–555. [Google Scholar] [CrossRef]
- Catalan, C.A.N.; Kokke, W.C.M.C.; Duque, C.; Djerassi, C. Synthesis of (24R)- and (24S)-5,28-stigmastadien-3β-ol and determination of the stereochemistry of their 24-hydroxy analogs, the saringosterols. J. Org. Chem. 1983, 48, 5207–5214. [Google Scholar] [CrossRef]
- Manners, G.D.; Penn, D.D.; Jurd, L.; James, L.F. Chemistry of toxic range plants. Water-soluble lignols of Ponderosa pine needles. J. Agric. Food Chem. 1982, 30, 401–404. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Rastogi, R.P.; Osterdahl, B.G. 13C NMR spectral analysis of dihydrobenzofuran lignans. Org. Magn. Reson. 1983, 21, 119–121. [Google Scholar] [CrossRef]
- Seigler, D.S.; Pauli, G.F.; Nahrstedt, A.; Leen, R. Cyanogenic allosides and glucosides from Passiflora edulis and Carica papaya. Phytochemistry 2002, 60, 873–882. [Google Scholar] [CrossRef]
- Hoang, L.S.; Tran, M.H.; Nguyen, V.T.; Ngo, Q.M.T.; Lee, J.S.; Rho, S.S.; Kim, J.A.; Lee, J.H.; Woo, M.H.; Min, B.S. Isolation of a new homomonoterpene from Madhuca pasquieri and effect of isolated compounds on NO production. Nat. Prod. Commun. 2016, 11, 729–732. [Google Scholar] [CrossRef]
- Farag, M.A.; Porzel, A.; Wessjohann, L.A. Comparative metabolite profiling and fingerprinting of medicinal licorice roots using a multiplex approach of GC-MS, LC-MS and 1D NMR techniques. Phytochemistry 2012, 76, 60–72. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Qi, L.W.; Cui, X.B.; Peng, G.P.; Peng, Y.B.; Ren, M.T.; Cheng, X.L.; Li, P. Oleanane-type triterpene glucuronides from the roots of Glycyrrhiza uralensis. Fischer. Planta Med. 2010, 76, 1457–1463. [Google Scholar] [CrossRef]
- Doi, Y.; Ishibashi, M.; Yamaguchi, N.; Kobayashi, J. Isolation of apo-9′-fucoxanthinone from the cultured marine dinoflagellate Amphidinium sp. J. Nat. Prod. 1995, 58, 1097–1099. [Google Scholar] [CrossRef]
- Darr, A.; Calabro, A. Synthesis and characterization of tyramine-based hyaluronan hydrogels. J. Mater. Sci. Mater. Med. 2009, 20, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Konidaris, K.F.; Gasser, G.; Tonks, N.K. A potent, selective and orally bioavailable inhibitor of the protein tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 2018, 293, 1517–1525. [Google Scholar] [CrossRef]
- Maheswari, N.; Karthikeyan, C.; Trivedi, P.; Moorthy, N.S. Recent advances in protein tyrosine phosphatase 1B targeted drug discovery for type II diabetes and obesity. Curr. Drug Targets 2018, 19, 551–575. [Google Scholar] [CrossRef]
- Okano, M.; Fukamiya, N.; Fujita, T.; Matsumoto, H.; Aratani, T. Constituents of marine algae VIII. Non polar constituents of Hizikia fusiformis (Harvey) Okamura. Bull. Jpn. Soc. Sci. Fish. 1985, 51, 1305–1310. [Google Scholar] [CrossRef]
- Shibata, E.; Kanno, T.; Tsuchiya, A.; Kuribayashi, K.; Tabata, C.; Nakano, T.; Nishizaki, T. Free fatty acids inhibit protein tyrosine phosphatase 1B and activate Akt. Cell. Physiol. Biochem. 2013, 32, 871–879. [Google Scholar] [CrossRef]
- Hunnicutt, J.W.; Hardy, R.W.; Williford, J.; McDonald, J.M. Saturated fatty acid-induced insulin resistance in rat adipocytes. Diabetes 1994, 43, 540–545. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shin, K.H.; Kim, B.K.; Lee, S. Anti-diabetic activities of fucosterol from Pelvetia siliquosa. Arch. Pharm. Res. 2004, 27, 1120–1122. [Google Scholar] [CrossRef]
- Jung, H.A.; Bharkta, H.K.; Min, B.S.; Choi, J.S. Fucosterol activates the insulin signaling pathway in insulin resistant HepG2 cells via inhibiting PTP1B. Arch. Pharm. Res. 2016, 39, 1454–1464. [Google Scholar] [CrossRef]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; McDowell, R.S.; Hansen, S.K. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef]
- Na, M.K.; Cui, L.; Min, B.S.; Bae, K.H.; Yoo, J.K.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitory activity of triterpenes isolated from Astilbe koreana. Bioorg. Med. Chem. Lett. 2006, 16, 3273–3276. [Google Scholar] [CrossRef] [PubMed]
- Akao, T.; Hattori, M.; Kanaoka, M.; Yamamoto, K.; Namba, T.; Kobashi, K. Hydrolysis of glycyrrhizin to 18β-glycyrrhetyl monoglucuronide by lysosomal β-d-glucuronidase of animal livers. Biochem. Pharmacol. 1991, 41, 1025–1029. [Google Scholar] [CrossRef]
- Takeda, S.; Ishihara, K.; Wakui, Y.; Amagaya, S.; Maruno, M.; Akao, T.; Kobashi, K. Bioavailability study of glycyrrhetic acid after oral administration of glycyrrhizin in rats; relevance to the intestinal bacterial hydrolysis. J. Pharm. Pharmacol. 1996, 48, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Villorbina, G.; Roura, L.; Camps, F.; Joglar, J.; Fabriàs, G. Enzymatic desaturation of fatty acids: Δ11 desaturase activity on cyclopropane acid probes. J. Org. Chem. 2003, 68, 2820–2829. [Google Scholar] [CrossRef]
- Li, T.; Zhzng, X.D.; Song, Y.W.; Liu, W.A. A microplate based screening method for a-glucosidase inhibitors. Chin. J. Clin. Pharmacol. Ther. 2005, 10, 1128–1134. [Google Scholar]
- Cui, L.; Na, M.; Oh, H.; Bae, E.Y.; Jeong, D.G.; Ryu, S.E.; Kim, S.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitors from Morus root bark. Bioorg. Med. Chem. Lett. 2006, 16, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Seong, S.H.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Moracin derivatives from Morus Radix as dual BACE1 and cholinesterase inhibitors with antioxidant and anti-glycation capacities. Life Sci. 2018, 210, 20–28. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) a | Enzyme Kinetics | ||
|---|---|---|---|---|
| PTP1B | α-Glucosidase | Ki (μM) b | Inhibition Type c | |
| 1 | 6.59 ± 0.09 | 48.05 ± 3.37 | ‒ | ‒ |
| 2 | 49.39 ± 1.39 | 93.63 ± 3.68 | ‒ | ‒ |
| 3 | 13.65 ± 0.49 | 113.44 ± 2.47 | ‒ | ‒ |
| 4 | > 150 | > 150 | ‒ | ‒ |
| 5 | 50.58 ± 1.86 | > 150 | ‒ | ‒ |
| 6 | 16.70 ± 0.36 | > 150 | 24.43 | Non-competitive |
| 7 | 4.86 ± 1.36 | > 200 | ‒ | ‒ |
| 8 | 13.58 ± 0.10 | 111.51 ± 1.44 | ‒ | ‒ |
| 9 | 10.68 ± 0.17 | 34.85 ± 2.39 | ‒ | ‒ |
| 10 | 16.43 ± 0.07 | > 200 | ‒ | ‒ |
| 11 | NT | NT | ‒ | ‒ |
| 12 | 11.51 ± 0.52 | 43.90 ± 0.77 | ‒ | ‒ |
| 13 | 4.92 ± 0.01 | > 150 | 4.13 | Non-competitive |
| 14 | 174.19 ± 5.44 | 133.84 ± 3.86 | ‒ | ‒ |
| 15 | >400 | > 250 | ‒ | ‒ |
| 16 | >400 | > 250 | ‒ | ‒ |
| 17 | >400 | > 300 | ‒ | ‒ |
| 18 | 110.33 ± 0.39 | > 150 | ‒ | ‒ |
| 19 | 49.39 ± 1.39 | > 150 | ‒ | ‒ |
| 20 | 323.21 ± 0.84 | > 250 | ‒ | ‒ |
| 21 | 188.06 ± 3.21 | 273.23 ± 5.65 | ‒ | ‒ |
| 22 | 10.40 ± 0.75 | 113.30 ± 0.70 | 3.17 | Competitive |
| 23 | 26.07 ± 0.59 | 128.72 ± 3.88 | 16.23 | Competitive |
| Ursolic acid d | 7.31 ± 0.16 | ‒ | ‒ | ‒ |
| Acarbose d | ‒ | 158.41 ± 1.05 | ‒ | ‒ |
| Compounds | B-Score (kcal/mol) | H-Bonds Interacting Residues | Hydrophobic Interacting Residues |
|---|---|---|---|
| 5 | −8.10 | Glu200 | Leu192, Asn193, Glu276, Gly277, Lys279, Phe196, Phe280 |
| 6a (24R and 28R) | −7.90 | Glu200 | Pro188, Ala189, Leu192, Asn193, Glu276, Phe196, Phe280 |
| 6b (24S and 28R) | −8.66 | Glu200 | Pro188, Ala189, Leu192, Asn193, Phe196, Glu276, Gly277, Lys279, Phe280 |
| 13 | −5.03 | Lys150, Tyr153, Asn193, Lys197 | Lys150, Ser151, Tyr152, Tyr153, Ala189, Pro188, Leu272, Ser187, Glu276, Leu192, Asn193, Lys197, Phe280 |
| 22 | −9.09 | Lys116, Lys120 (Salt bridge) | Lys120, Phe182, Gly183, Asp265, Thr263, Glu115, Arg221 |
| 23 | −8.81 | Asp48, Lys116 (Salt bridge), Gly183 | Tyr46, Val49, Ala217, Gln262, Phe182 |
| Standard A a | −11.23 | Arg24, Tyr46, Asp48, Ser216, Ala217, Arg221, Arg254, Gln262 | Ser28, Val49, Lys116, Lys120, Cys215, Ile219, Gly220, Met258, Gly259 |
| Standard B a | −10.98 | Asn193, Glu276 | Ala189, Leu192, Phe196, Gly277, Lys279, Phe280, Ile281, Met282 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seong, S.H.; Nguyen, D.H.; Wagle, A.; Woo, M.H.; Jung, H.A.; Choi, J.S. Experimental and Computational Study to Reveal the Potential of Non-Polar Constituents from Hizikia fusiformis as Dual Protein Tyrosine Phosphatase 1B and α-Glucosidase Inhibitors. Mar. Drugs 2019, 17, 302. https://doi.org/10.3390/md17050302
Seong SH, Nguyen DH, Wagle A, Woo MH, Jung HA, Choi JS. Experimental and Computational Study to Reveal the Potential of Non-Polar Constituents from Hizikia fusiformis as Dual Protein Tyrosine Phosphatase 1B and α-Glucosidase Inhibitors. Marine Drugs. 2019; 17(5):302. https://doi.org/10.3390/md17050302
Chicago/Turabian StyleSeong, Su Hui, Duc Hung Nguyen, Aditi Wagle, Mi Hee Woo, Hyun Ah Jung, and Jae Sue Choi. 2019. "Experimental and Computational Study to Reveal the Potential of Non-Polar Constituents from Hizikia fusiformis as Dual Protein Tyrosine Phosphatase 1B and α-Glucosidase Inhibitors" Marine Drugs 17, no. 5: 302. https://doi.org/10.3390/md17050302
APA StyleSeong, S. H., Nguyen, D. H., Wagle, A., Woo, M. H., Jung, H. A., & Choi, J. S. (2019). Experimental and Computational Study to Reveal the Potential of Non-Polar Constituents from Hizikia fusiformis as Dual Protein Tyrosine Phosphatase 1B and α-Glucosidase Inhibitors. Marine Drugs, 17(5), 302. https://doi.org/10.3390/md17050302