Effects of C-Terminal Carboxylation on α-Conotoxin LsIA Interactions with Human α7 Nicotinic Acetylcholine Receptor: Molecular Simulation Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Homology Modeling of Amidated and Carboxylated LsIAs Anchoring to Human α7 nAChR
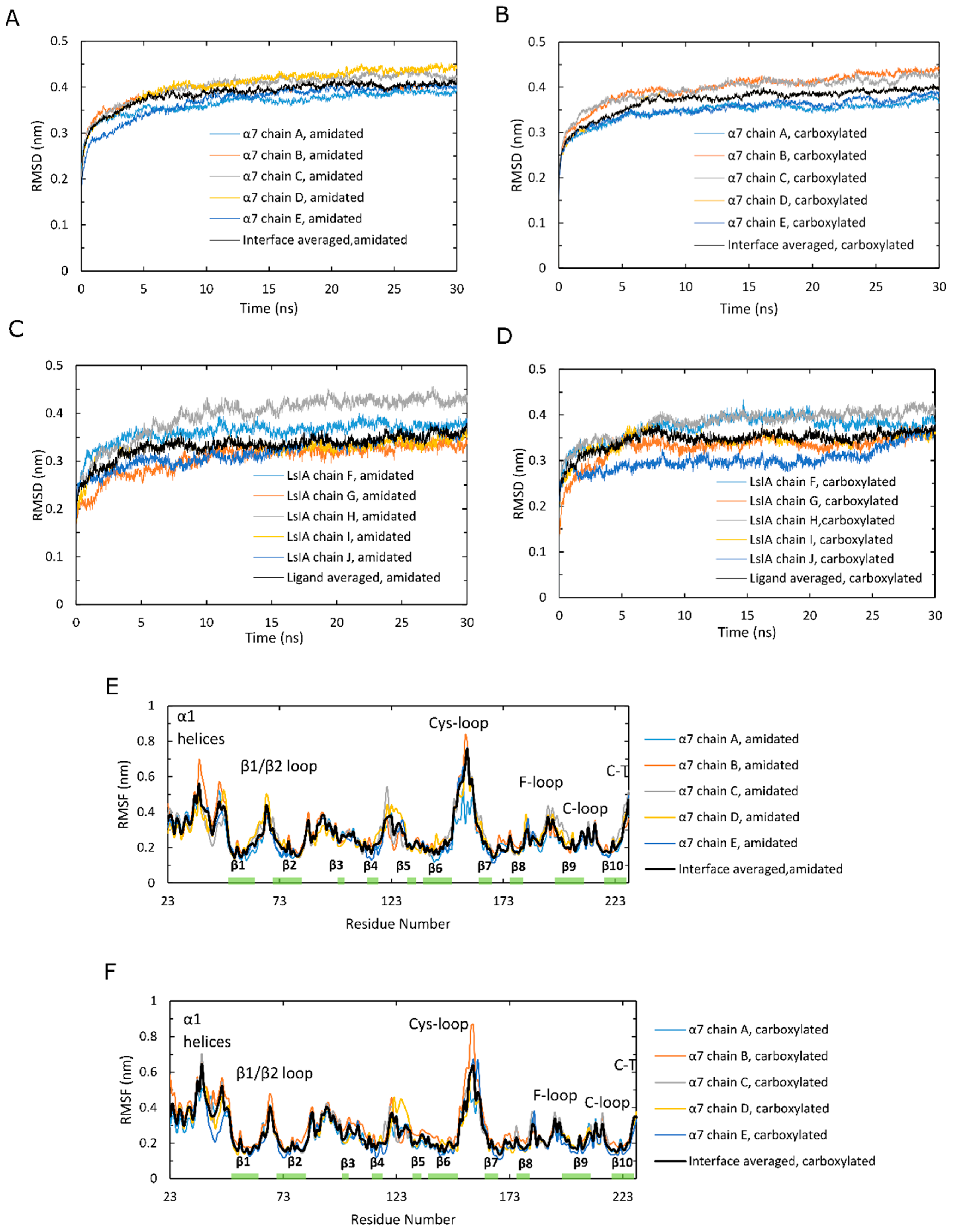
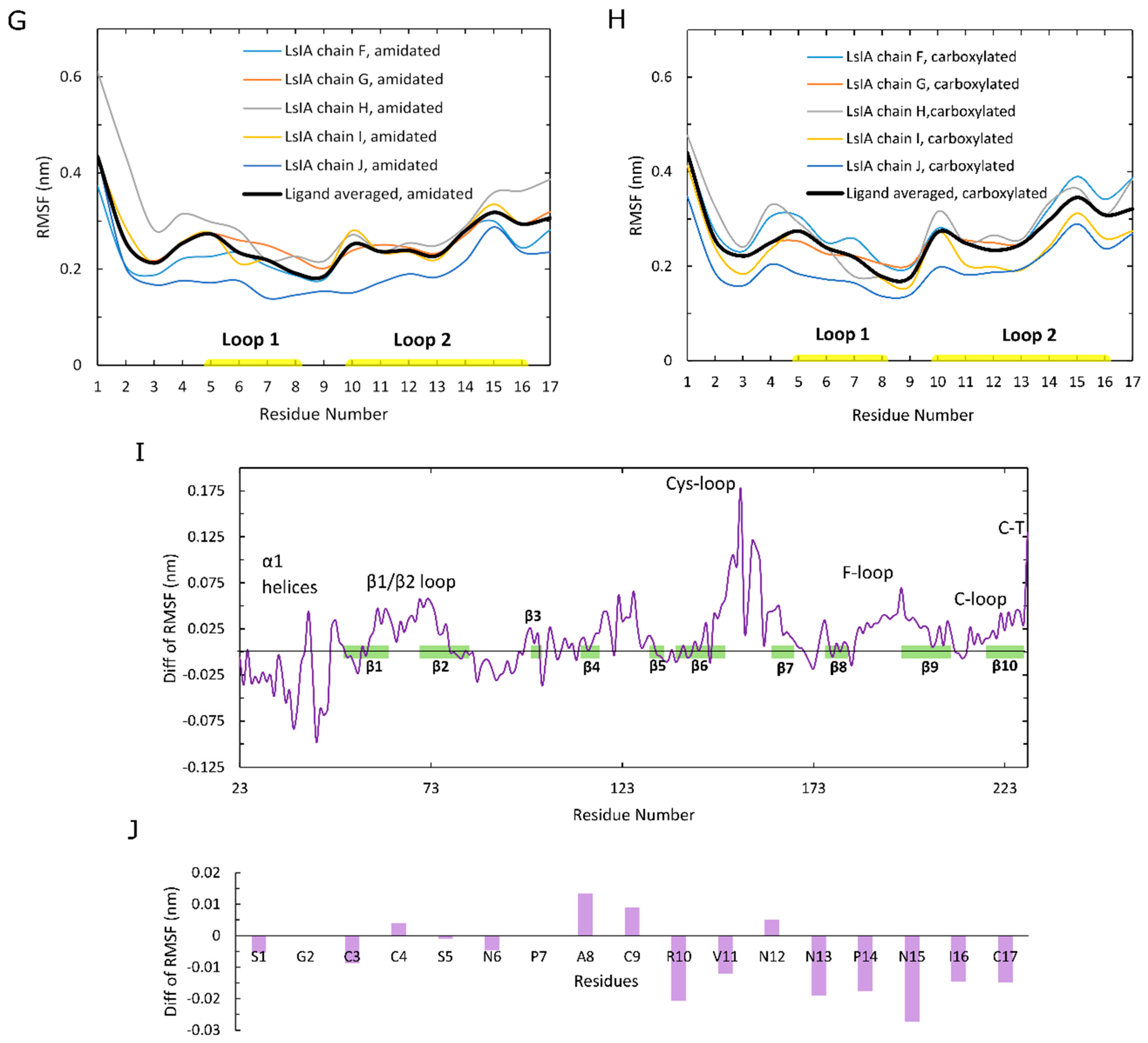
2.2. Amidated LsIA Binding Causes Higher Fluctuations in Loop C and the Juxtamembrane Cys-Loop Regions
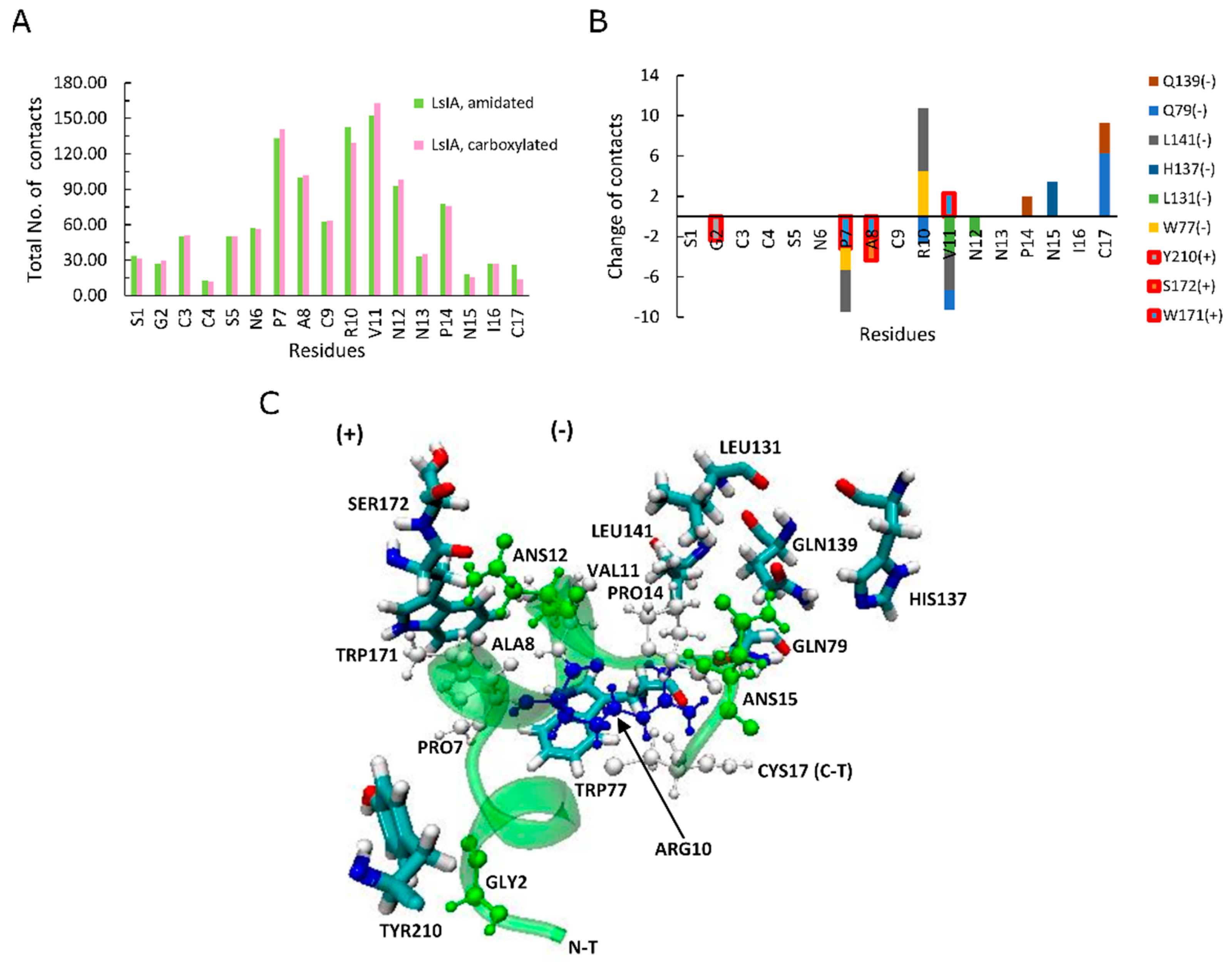
2.3. Carboxylation at LsIA C-Terminus Causes Indirect Loss of Contacts between Key Residue ARG10 and the α7 Complementary Face
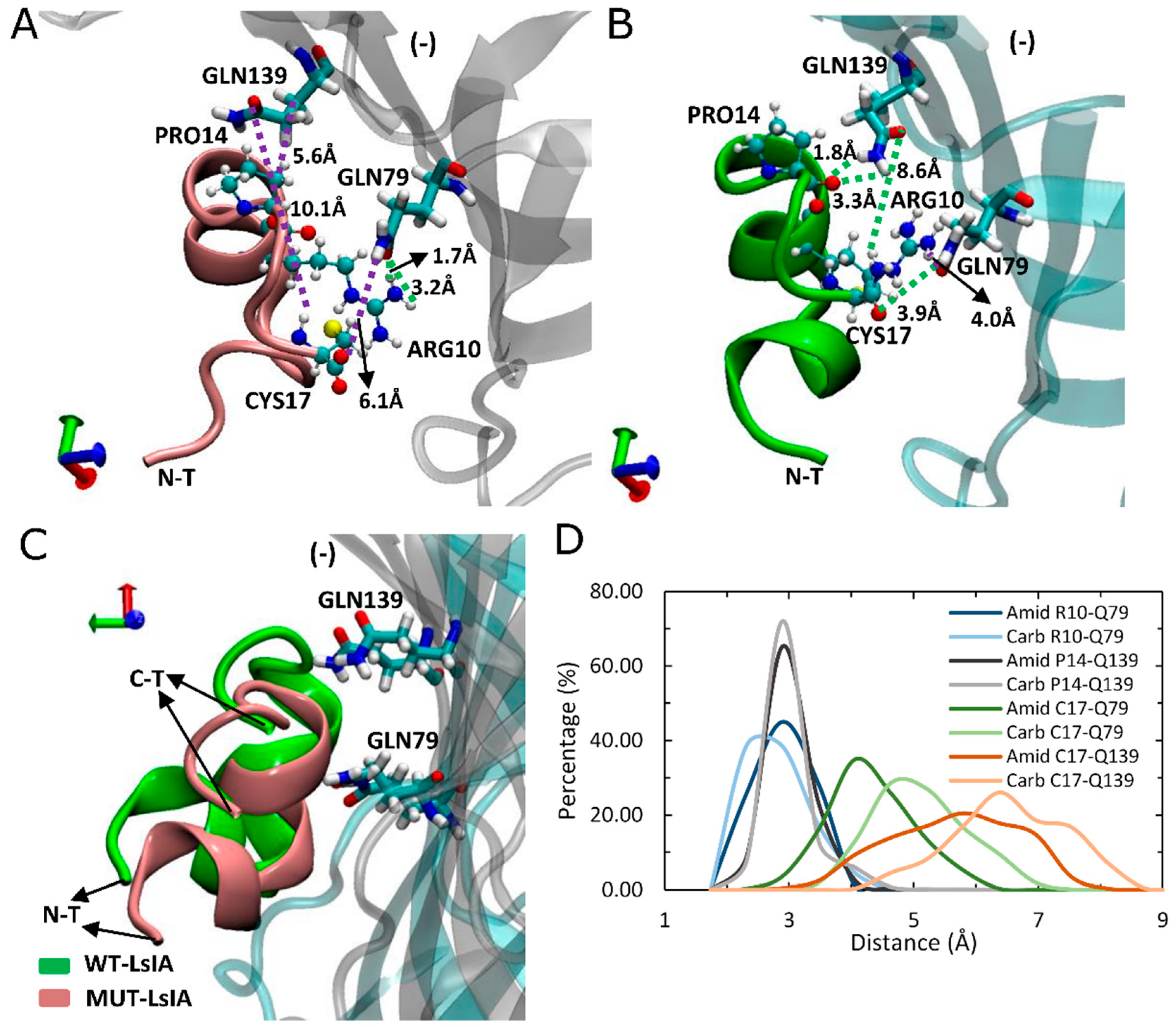
2.4. Amidated LsIA Favors Hydrogen Bonding and π-π Interactions between ARG10 and the α7(−) Subunit
2.5. Carboxylated LsIA Favors Hydrophobic Interactions with α7 Subunit Residues via PRO7 and VAL11
2.6. α7-TYR217 Forms Close Contacts with Both Amidated and Carboxylated LsIAs
2.7. An Intramolecular Salt-Bridge in Carboxylated LsIA Is Retained Upon Binding to α7, and Plays a Role in the Loss of Key Toxin-Receptor Contacts
2.8. Changes in Inter-Subunit Contacts on the Five Interfaces of Adjacent α7 Subunits
3. Conclusions
4. Methods
4.1. Homology Modeling
4.2. Molecular Dynamics Simulations
4.3. Structural Visualization and Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lebbe, E.K.M.; Peigneur, S.; Maiti, M.; Devi, P.; Ravichandran, S.; Lescrinier, E.; Ulens, C.; Waelkens, E.; D’Souza, L.; Herdewijn, P.; et al. Structure-Function Elucidation of a New α-Conotoxin, Lo1a, from Conus longurionis. J. Biol. Chem. 2014, 289, 9573–9583. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Corry, B. Role of acetylcholine receptor domains in ion selectivity. Biochim. Biophys. Acta (Bba) Biomembr. 2009, 1788, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef]
- Hogg, R.C.; Hopping, G.; Alewood, P.F.; Adams, D.J.; Bertrand, D. α-Conotoxins PnIA and [A10L]PnIA Stabilize Different States of the α7-L247T Nicotinic Acetylcholine Receptor. J. Biol. Chem. 2003, 278, 26908–26914. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Alpha7 Nicotinic Acetylcholine Receptor Is a Target in Pharmacology and Toxicology. Int. J. Mol. Sci. 2012, 13, 2219–2238. [Google Scholar] [CrossRef]
- Celie, P.H.N.; Kasheverov, I.E.; Mordvintsev, D.Y.; Hogg, R.C.; van Nierop, P.; van Elk, R. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, H.O.; Feuerbach, D. Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell. Mol. Life Sci. 2016, 73, 2511–2530. [Google Scholar] [CrossRef]
- D’Andrea, M.R.; Nagele, R.G. Targeting the Alpha 7 Nicotinic Acetylcholine Receptor to Reduce Amyloid Accumulation in Alzheimer’s Disease Pyramidal Neurons. Curr. Pharm. Des. 2006, 12, 677–684. [Google Scholar] [CrossRef]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Söderman, A.; Mikkelsen, J.D.; West, M.J.; Christensen, D.Z.; Jensen, M.S. Activation of nicotinic α7 acetylcholine receptor enhances long term potentation in wild type mice but not in APPswe/PS1ΔE9 mice. Neurosci. Lett. 2011, 487, 325–329. [Google Scholar] [CrossRef]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Talley, T.T.; Hansen, S.B.; Palmer, T.; Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake [alpha]-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Brejc, K.; van Dijk, W.J.; Klaassen, R.V.; Schuurmans, M.; Oost, J.V.D.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar] [CrossRef]
- Yi, M.; Tjong, H.; Zhou, H.-X. Spontaneous conformational change and toxin binding in α7 acetylcholine receptor: Insight into channel activation and inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 8280–8285. [Google Scholar] [CrossRef]
- Maricq, A.V.; Peterson, A.S.; Brake, A.J.; Myers, R.M.; Julius, D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 1991, 254, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Kasher, R.; Nicolas, A.; Guss, J.M.; Balass, M.; Fridkin, M.; Smit, A.B.; Brejc, K.; Sixma, T.K.; Katchalski-Katzir, E.; et al. The Binding Site of Acetylcholine Receptor as Visualized in the X-ray Structure of a Complex between α-Bungarotoxin and a Mimotope Peptide. Neuron 2001, 32, 265–275. [Google Scholar] [CrossRef]
- Dutertre, S.; Nicke, A.; Tyndall, J.D.A.; Lewis, R.J. Determination of α-conotoxin binding modes on neuronal nicotinic acetylcholine receptors. J. Mol. Recognit. 2004, 17, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef]
- Hansen, S.B.; Talley, T.T.; Radić, Z.; Taylor, P. Structural and Ligand Recognition Characteristics of an Acetylcholine-binding Protein from Aplysia californica. J. Biol. Chem. 2004, 279, 24197–24202. [Google Scholar] [CrossRef]
- Azam, L.; McIntosh, J.M. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 2009, 30, 771–783. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Zhmak, M.N.; Khruschov, A.Y.; Tsetlin, V.I. Design of New α-Conotoxins: From Computer Modeling to Synthesis of Potent Cholinergic Compounds. Mar. Drugs 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Rivier, J.; Clark, C.; Ramilo, C.A.; Corpuz, G.P.; Abogadie, F.C.; Mena, E.E.; Woodward, S.R.; Hillyard, D.R.; Cruz, L.J. Diversity of Conus neuropeptides. Science 1990, 249, 257–263. [Google Scholar] [CrossRef]
- Quiram, P.A.; McIntosh, J.M.; Sine, S.M. Pairwise Interactions between Neuronal α7Acetylcholine Receptors and α-Conotoxin PnIB. J. Biol. Chem. 2000, 275, 4889–4896. [Google Scholar] [CrossRef]
- Hoggard, M.F.; Rodriguez, A.M.; Cano, H.; Clark, E.; Tae, H.-S.; Adams, D.J.; Godenschwege, T.A.; Marí, F. In vivo and in vitro testing of native α-conotoxins from the injected venom of Conus purpurascens. Neuropharmacology 2017, 127, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Mcintosh, J.M.; Santos, A.D.; Olivera, B.M. Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu. Rev. Biochem. 1999, 68, 59–88. [Google Scholar] [CrossRef] [PubMed]
- Suresh, A.; Hung, A. Molecular simulation study of the unbinding of α-conotoxin [ϒ4E]GID at the α7 and α4β2 neuronal nicotinic acetylcholine receptors. J. Mol. Graph. Model. 2016, 70, 109–121. [Google Scholar] [CrossRef]
- Armishaw, C.J. Synthetic α-conotoxin mutants as probes for studying nicotinic acetylcholine receptors and in the development of novel drug leads. Toxins 2010, 2, 1471–1499. [Google Scholar] [CrossRef]
- Abraham, N.; Healy, M.; Ragnarsson, L.; Brust, A.; Alewood, P.F.; Lewis, R.J. Structural mechanisms for [alpha]-conotoxin activity at the human [alpha]3[beta]4 nicotinic acetylcholine receptor. Sci. Rep. 2017, 7, 45466. [Google Scholar] [CrossRef]
- Inserra, M.C.; Kompella, S.N.; Vetter, I.; Brust, A.; Daly, N.L.; Cuny, H.; Craik, D.J.; Alewood, P.F.; Adams, D.J.; Lewis, R.J. Isolation and characterization of α-conotoxin LsIA with potent activity at nicotinic acetylcholine receptors. Biochem. Pharmacol. 2013, 86, 791–799. [Google Scholar] [CrossRef]
- Chi, S.-W.; Kim, D.-H.; Olivera, B.M.; McIntosh, J.M.; Han, K.-H. Solution conformation of alpha-conotoxin GIC, a novel potent antagonist of alpha3beta2 nicotinic acetylcholine receptors. Biochem. J. 2004, 380, 347–352. [Google Scholar] [CrossRef]
- Dutertre, S.; Lewis, R.J. Toxin insights into nicotinic acetylcholine receptors. Biochem. Pharmacol. 2006, 72, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, Structure, and Activity of GID, a Novel α4/7-Conotoxin with an Extended N-terminal Sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar] [CrossRef]
- Quiram, P.A.; Sine, S.M. Structural Elements in α-Conotoxin ImI Essential for Binding to Neuronal α7 Receptors. J. Biol. Chem. 1998, 273, 11007–11011. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Kohda, D.; Hatanaka, H.; Lancelin, J.M.; Ishida, Y.; Oya, M.; Nakamura, H.; Inagaki, F.; Sato, K. Structure-activity relationships of. mu.-conotoxin GIIIA: Structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; et al. Structure/Function Characterization of μ-Conotoxin KIIIA, an Analgesic, Nearly Irreversible Blocker of Mammalian Neuronal Sodium Channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Smith, A.B.; Schroeder, C.I.; Yasuda, T.; Lewis, R.J. ω-Conotoxin CVID Inhibits a Pharmacologically Distinct Voltage-sensitive Calcium Channel Associated with Transmitter Release from Preganglionic Nerve Terminals. J. Biol. Chem. 2003, 278, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Kim, J.I.; Kobayashi, K.; Kodera, Y.; Maeda, T.; Sato, K. Three-Dimensional Structure in Solution of the Calcium Channel Blocker. omega.-Conotoxin MVIIA. Biochemistry 1995, 34, 10256–10265. [Google Scholar] [CrossRef]
- Rajendra, W.; Armugam, A.; Jeyaseelan, K. Toxins in anti-nociception and anti-inflammation. Toxicon 2004, 44, 1–17. [Google Scholar] [CrossRef]
- Livett, B.G.; Sandall, D.W.; Keays, D.; Down, J.; Gayler, K.R.; Satkunanathan, N.; Khalil, Z. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 2006, 48, 810–829. [Google Scholar] [CrossRef]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone Snails: A Big Store of Conotoxins for Novel Drug Discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Lee, C.-H.; Ho, Y.-S. Nicotinic Acetylcholine Receptor-Based Blockade: Applications of Molecular Targets for Cancer Therapy. Clin. Cancer Res. 2011, 17, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Quiram, P.A.; Jones, J.J.; Sine, S.M. Pairwise Interactions between Neuronal α7Acetylcholine Receptors and α-Conotoxin ImI. J. Biol. Chem. 1999, 274, 19517–19524. [Google Scholar] [CrossRef]
- Eddins, D.; Sproul, A.D.; Lyford, L.K.; McLaughlin, J.T.; Rosenberg, R.L. Glutamate 172, essential for modulation of L247T α7 ACh receptors by Ca2+, lines the extracellular vestibule. Am. J. Physiol. Cell Physiol. 2002, 283, C1454–C1460. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Chugunov, A.O.; Kudryavtsev, D.S.; Ivanov, I.A.; Zhmak, M.N.; Shelukhina, I.V.; Spirova, E.N.; Tabakmakher, V.M.; Zelepuga, E.A.; Efremov, R.G.; et al. High-Affinity α-Conotoxin PnIA Analogs Designed on the Basis of the Protein Surface Topography Method. Sci. Rep. 2016, 6, 36848. Available online: https://www.nature.com/articles/srep36848#supplementary-information (accessed on 11 October 2018). [CrossRef]
- Barrantes, F.J. The Nicotinic Acetylcholine Receptor Current Views and Future Trends; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Lebbe, E.K.M.; Peigneur, S.; Wijesekara, I.; Tytgat, J. Conotoxins targeting nicotinic acetylcholine receptors: An overview. Mar. Drugs 2014, 12, 2970–3004. [Google Scholar] [CrossRef] [PubMed]
- Boffi, J.C.; Marcovich, I.; Gill-Thind, J.K.; Corradi, J.; Collins, T.; Lipovsek, M.M.; Moglie, M.; Plazas, P.V.; Craig, P.O.; Millar, N.S.; et al. Differential Contribution of Subunit Interfaces to α9α10 Nicotinic Acetylcholine Receptor Function. Mol. Pharmacol. 2017, 91, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Lester, H.A.; Dibas, M.I.; Dahan, D.S.; Leite, J.F.; Dougherty, D.A. Cys-loop receptors: New twists and turns. Trends Neurosci. 2004, 27, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.; Sugiyama, N.; Sine, S.M.; Taylor, P. A model of the nicotinic receptor extracellular domain based on sequence identity and residue location. Biophys. J. 1997, 73, 52–66. [Google Scholar] [CrossRef]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef]
- Sahu, B.S.; Obbineni, J.M.; Sahu, G.; Singh, P.K.; Sonawane, P.J.; Sasi, B.K.; Allu, P.K.R.; Maji, S.K.; Bera, A.K.; Senapati, S.; et al. Molecular interactions of the physiological anti-hypertensive peptide catestatin with the neuronal nicotinic acetylcholine receptor. J. Cell Sci. 2012, 125, 2323–2337. [Google Scholar] [CrossRef]
- Peng, W.; Ding, F. Biomolecular recognition of antagonists by α7 nicotinic acetylcholine receptor: Antagonistic mechanism and structure–activity relationships studies. Eur. J. Pharm. Sci. 2015, 76, 119–132. [Google Scholar] [CrossRef]
- Hilf, R.J.C.; Dutzler, R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 2009, 457, 115–118. [Google Scholar] [CrossRef]
- Lee, W.Y.; Sine, S.M. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature 2005, 438, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N.; Fujiyoshi, Y. Gating movement of acetylcholine receptor caught by plunge-freezing. J. Mol. Biol. 2012, 422, 617–634. [Google Scholar] [CrossRef]
- Hibbs, R.E.; Gouaux, E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 2011, 474, 54. Available online: https://www.nature.com/articles/nature10139#supplementary-information (accessed on 15 January 2019). [CrossRef]
- Lummis, S.C.R.; Beene, D.L.; Lee, L.W.; Lester, H.A.; Broadhurst, R.W.; Dougherty, D.A. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005, 438, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, J.E.; Hénault, C.M.; Therien, J.P.D.; Sun, J. Nicotinic acetylcholine receptor–lipid interactions: Mechanistic insight and biological function. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 1806–1817. [Google Scholar] [CrossRef]
- Noridomi, K.; Watanabe, G.; Hansen, M.N.; Han, G.W.; Chen, L. Structural insights into the molecular mechanisms of myasthenia gravis and their therapeutic implications. eLife 2017, 6, e23043. [Google Scholar] [CrossRef]
- Armishaw, C.; Jensen, A.A.; Balle, T.; Clark, R.J.; Harpsøe, K.; Skonberg, C.; Liljefors, T.; Strømgaard, K. Rational Design of α-Conotoxin Analogues Targeting α7 Nicotinic Acetylcholine Receptors: Improved antagonistic activity by incorporation of proline derivatives. J. Biol. Chem. 2009, 284, 9498–9512. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.A.; Cuny, H.; Hung, A.; Clark, R.J.; Brust, A.; Akondi, K.; Alewood, P.F.; Craik, D.J.; Adams, D.J. Identifying Key Amino Acid Residues That Affect α-Conotoxin AuIB Inhibition of α3β4 Nicotinic Acetylcholine Receptors. J. Biol. Chem. 2013, 288, 34428–34442. [Google Scholar] [CrossRef]
- Ellison, M.; Gao, F.; Wang, H.-L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII Target Distinct Regions of the Human α7 Nicotinic Acetylcholine Receptor and Distinguish Human Nicotinic Receptor Subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef]
- Gay, E.A.; Giniatullin, R.; Skorinkin, A.; Yakel, J.L. Aromatic residues at position 55 of rat α7 nicotinic acetylcholine receptors are critical for maintaining rapid desensitization. J. Physiol. 2008, 586, 1105–1115. [Google Scholar] [CrossRef]
- Kang, T.S.; Vivekanandan, S.; Jois, S.D.S.; Kini, R.M. Effect of C-Terminal Amidation on Folding and Disulfide-Pairing of α-Conotoxin ImI. Angew. Chem. Int. Ed. 2005, 44, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Gao, F.; Bren, N.; Burghardt, T.P.; Hansen, S.; Henchman, R.H.; Taylor, P.; McCammon, J.A.; Sine, S.M. Agonist-mediated Conformational Changes in Acetylcholine-binding Protein Revealed by Simulation and Intrinsic Tryptophan Fluorescence. J. Biol. Chem. 2005, 280, 8443–8451. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. A Publ. Protein Soc. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Quigley, D.; Probert, M.I.J. Langevin dynamics in constant pressure extended systems. J. Chem. Phys. 2004, 120, 11432–11441. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Hung, A. Effects of C-Terminal Carboxylation on α-Conotoxin LsIA Interactions with Human α7 Nicotinic Acetylcholine Receptor: Molecular Simulation Studies. Mar. Drugs 2019, 17, 206. https://doi.org/10.3390/md17040206
Wen J, Hung A. Effects of C-Terminal Carboxylation on α-Conotoxin LsIA Interactions with Human α7 Nicotinic Acetylcholine Receptor: Molecular Simulation Studies. Marine Drugs. 2019; 17(4):206. https://doi.org/10.3390/md17040206
Chicago/Turabian StyleWen, Jierong, and Andrew Hung. 2019. "Effects of C-Terminal Carboxylation on α-Conotoxin LsIA Interactions with Human α7 Nicotinic Acetylcholine Receptor: Molecular Simulation Studies" Marine Drugs 17, no. 4: 206. https://doi.org/10.3390/md17040206
APA StyleWen, J., & Hung, A. (2019). Effects of C-Terminal Carboxylation on α-Conotoxin LsIA Interactions with Human α7 Nicotinic Acetylcholine Receptor: Molecular Simulation Studies. Marine Drugs, 17(4), 206. https://doi.org/10.3390/md17040206