Thuwalallenes A–E and Thuwalenynes A–C: New C15 Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp.

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
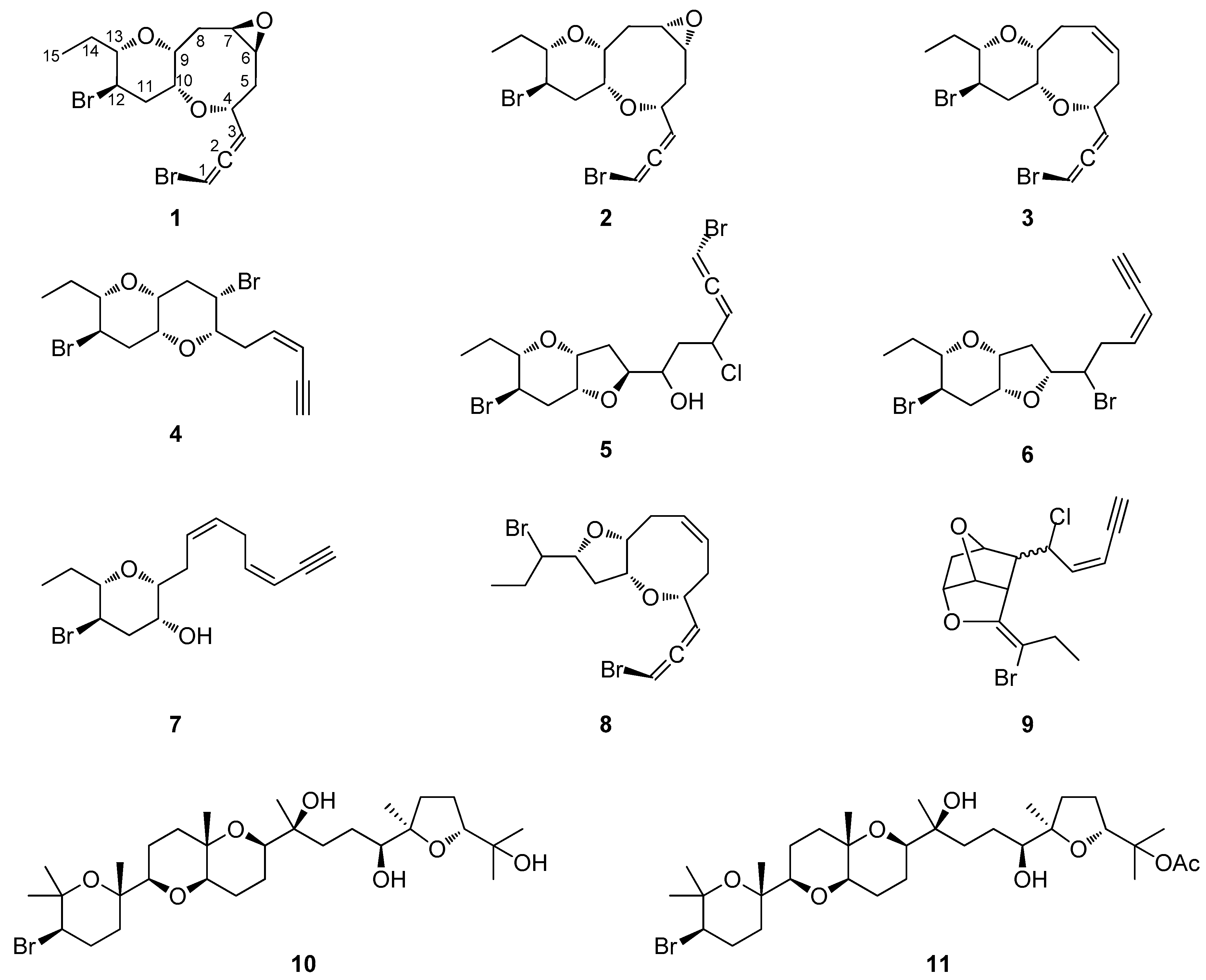
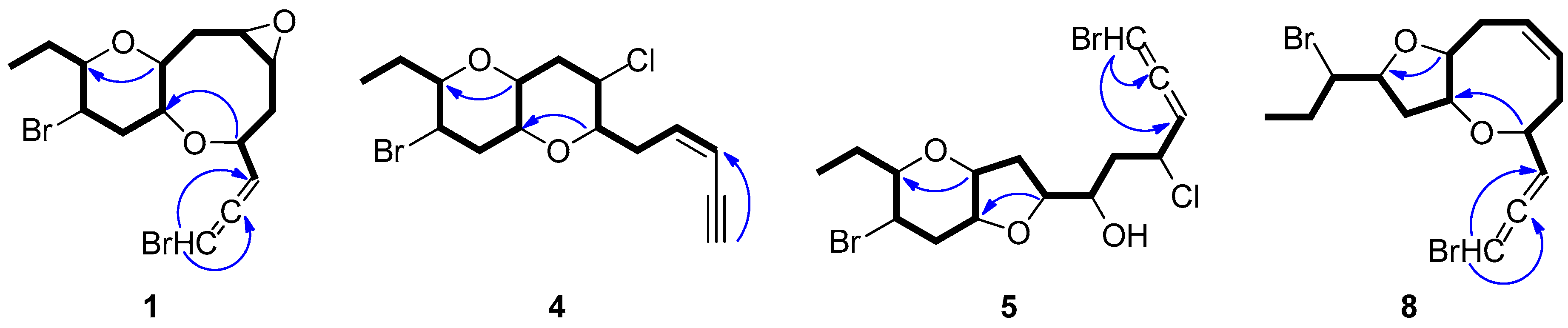
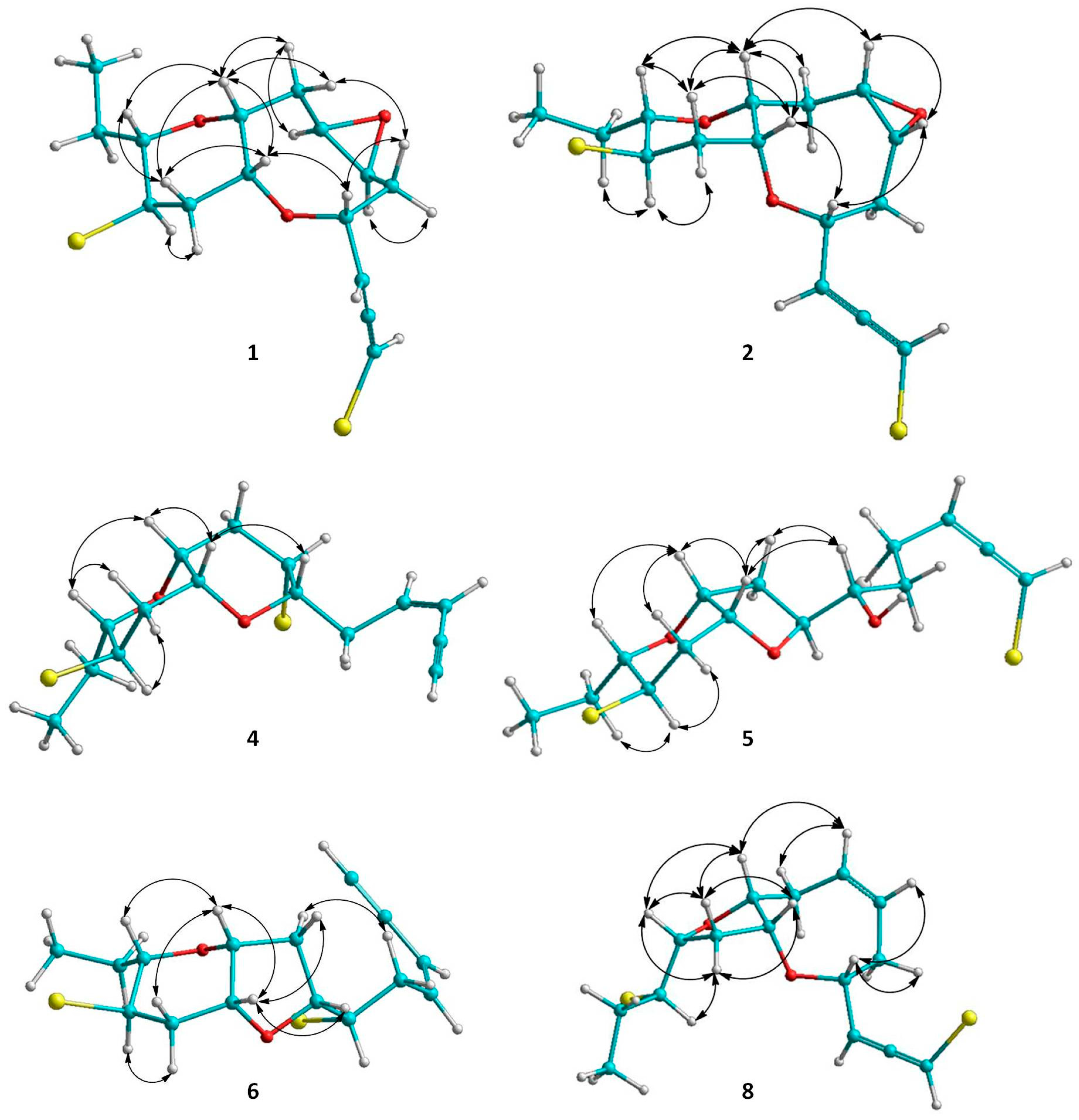
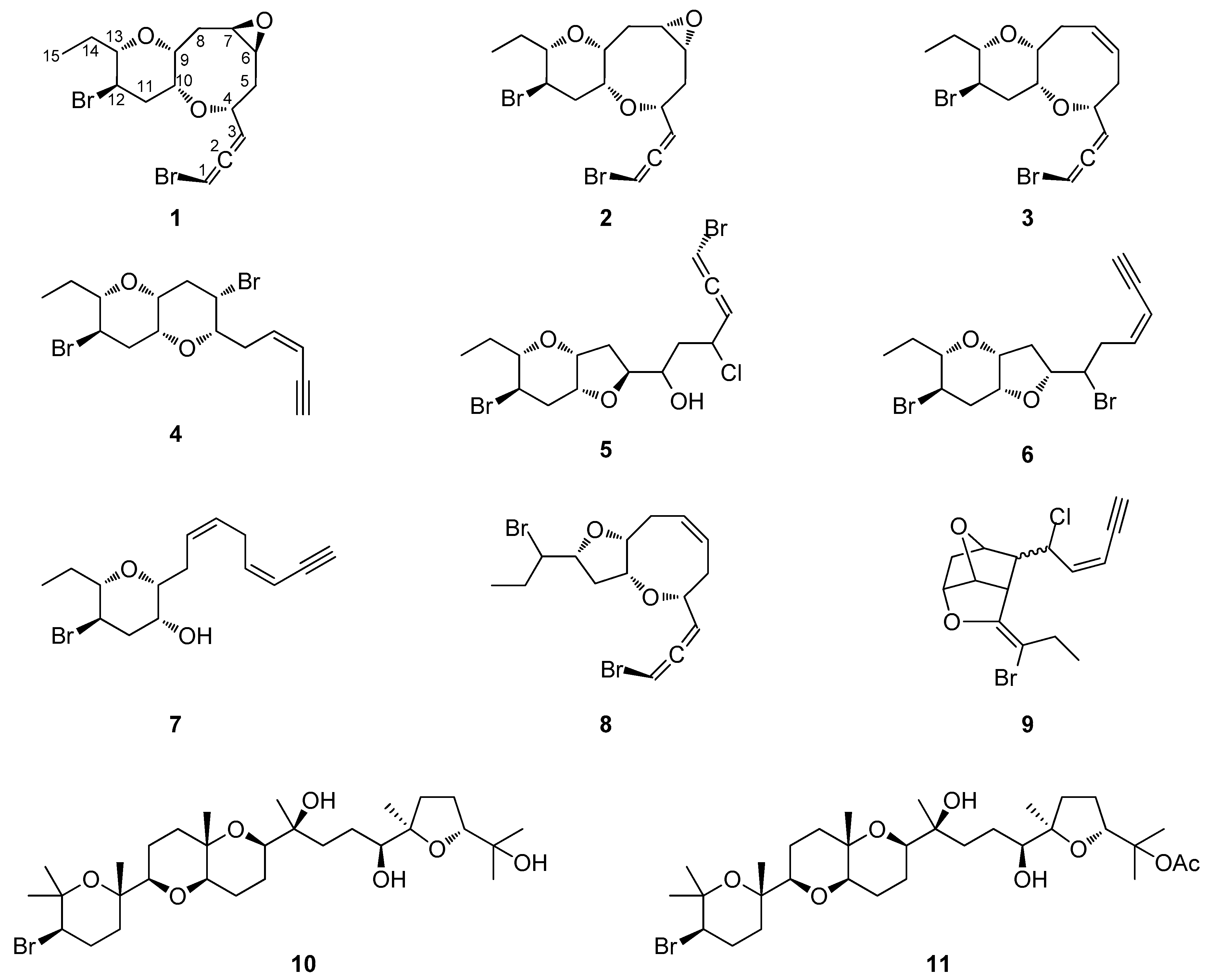
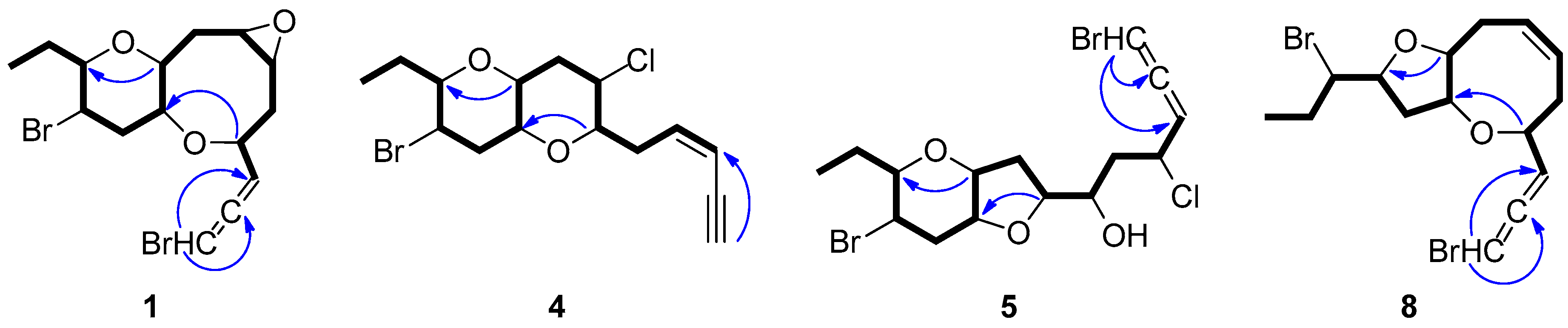
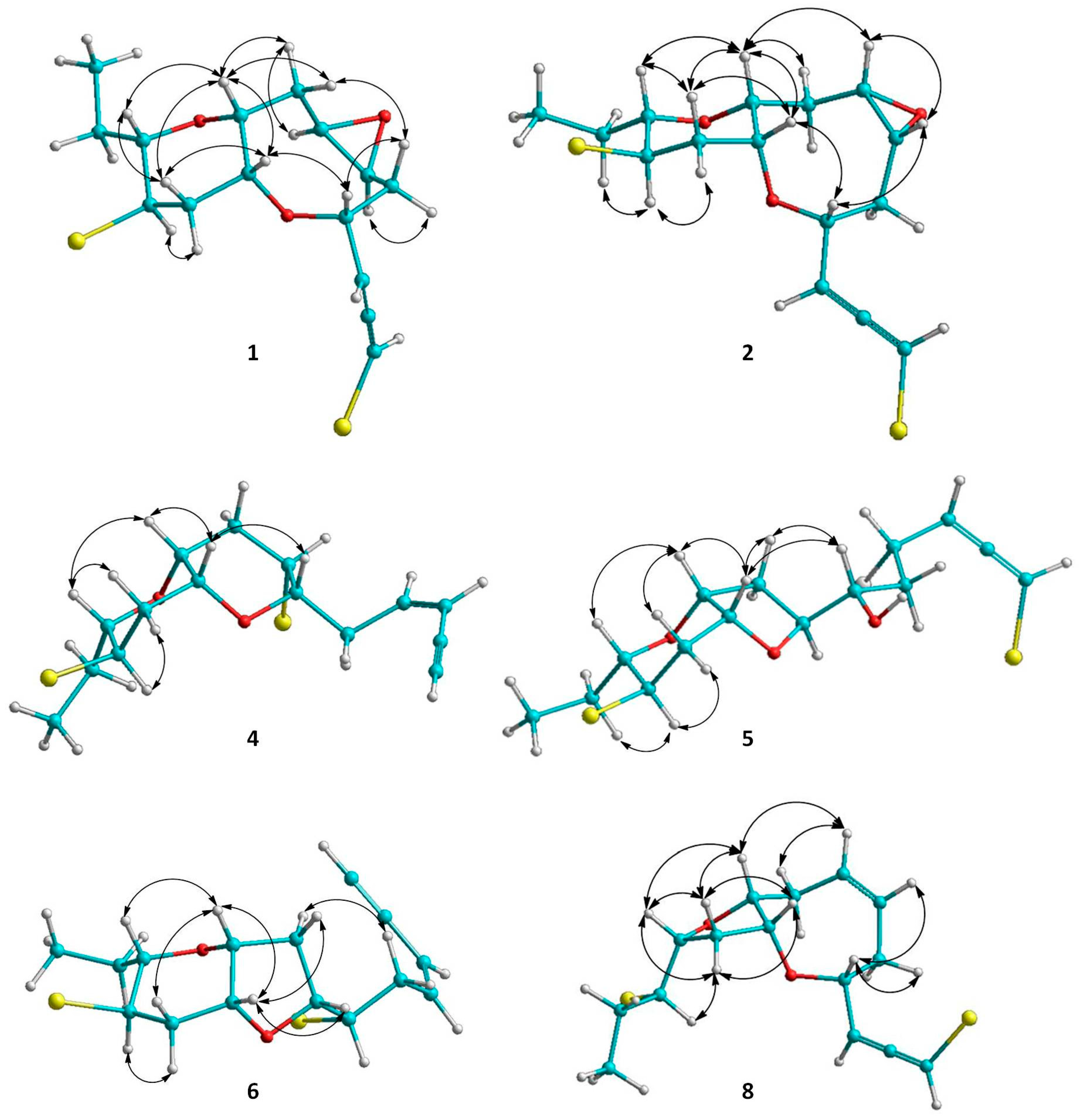
2.1. Structure Elucidation of the Isolated Metabolites
2.2. Evaluation of the Anti-inflammatory Activity of the Isolated Metabolites
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Cell Culture
3.5. Nitric Oxide Measurement
3.6. MTT Measurement
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guiry, M.D. AlgaeBase; Guiry, M.D., Guiry, G.M., Eds.; National University of Ireland: Galway, Ireland, 2019; Available online: http://www.algaebase.org/ (accessed on 5 October 2019).
- Harizani, M.; Ioannou, E.; Roussis, V. The Laurencia Paradox: An Endless Source of Chemodiversity. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 102, pp. 91–252. [Google Scholar]
- Paul, V.J.; Ritson-Williams, R.; Sharp, K. Marine chemical ecology in benthic environments. Nat. Prod. Rep. 2011, 28, 345–387. [Google Scholar] [CrossRef] [PubMed]
- MarinLit. A Database of the Marine Natural Products Literature. Available online: http://pubs.rsc.org/marinlit/ (accessed on 5 October 2019).
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Phan, C.-S.; Vairappan, C.S. New anti-bacterial halogenated tricyclic sesquiterpenes from Bornean Laurencia majuscula (Harvey) Lucas. Nat. Prod. Res. 2019, 33, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Perdikaris, S.; Mangoni, A.; Grauso, L.; Papazafiri, P.; Roussis, V.; Ioannou, E. Vagiallene, a rearranged C15 acetogenin from Laurencia obtusa. Org. Lett. 2019, 21, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, M.G.; Vyrla, D.; Harizani, M.; Doxaki, C.; Eliopoulos, A.G.; Roussis, V.; Ioannou, E.; Tsatsanis, C.; Kampranis, S.C. Neorogioltriol and related diterpenes from the red alga Laurencia inhibit inflammatory bowel disease in mice by suppressing M1 and promoting M2-like macrophage responses. Mar. Drugs 2019, 17, 97. [Google Scholar] [CrossRef] [PubMed]
- Kokkotou, K.; Ioannou, E.; Nomikou, M.; Pitterl, F.; Vonaparti, A.; Siapi, E.; Zervou, M.; Roussis, V. An integrated approach using UHPLC–PDA–HRMS and 2D HSQC NMR for the metabolic profiling of the red alga Laurencia: Dereplication and tracing of natural products. Phytochemistry 2014, 108, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Lhullier, C.; Falkenberg, M.; Ioannou, E.; Quesada, E.; Papazafiri, P.; Horta, P.A.; Schenkel, E.P.; Vagias, C.; Roussis, V. Cytotoxic halogenated metabolites from the Brazilian red alga Laurencia catarinensis. J. Nat. Prod. 2010, 73, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Ayyad, S.-E.; Al-Footy, K.O.; Alarif, W.M.; Sobahi, T.R.; Bassaif, S.A.; Makki, M.S.; Asiri, A.M.; Al Halwani, A.Y.; Badria, A.F.; Badria, F.A. Bioactive C15 acetogenins from the red alga Laurencia obtusa. Chem. Pharm. Bull. 2011, 59, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Hartshorn, M.P.; McLennan, T.J.; Munro, M.H.G.; Robinson, W.T.; Yorke, S.C. Thyrsiferol: A squalene-derived metabolite of Laurencia thyrsifera. Tetrahedron Lett. 1978, 1, 69–72. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, M.; Furusaki, A.; Matsumoto, T.; Kato, A.; Imanaka, Y.; Kurosawa, E. Teurilene and thyrsiferyl 23-acetate, meso and remarkably cytotoxic compounds from the marine red alga Laurencia obtusa (Hudson) Lamouroux. Tetrahedron Lett. 1985, 26, 1329–1332. [Google Scholar] [CrossRef]
- Lowe, G. The absolute configuration of allenes. J. Chem. Soc. Chem. Commun. 1965, 411–413. [Google Scholar]
- Elsevier, C.J.; Vermeer, P.; Gedanken, A.; Runge, W. Synthesis and absolute configurations of halogenoallenes. J. Org. Chem. 1985, 50, 364–367. [Google Scholar] [CrossRef]
- De Silva, E.D.; Schwartz, R.E.; Scheuer, P.J. Srilankenyne, a new metabolite from the sea hare Aplysia oculifera. J. Org. Chem. 1983, 48, 395–396. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Position | 1 1,2 | 2 1,3 | 3 4 | 4 1,3 | 5 1,3 | 6 1,5 | 7 1,5 | 8 4 |
|---|---|---|---|---|---|---|---|---|
| 1 | 75.7 | 73.7 | 73.8 | 82.1 | 75.1 | 80.0 | 80.7 | 74.2 |
| 2 | 201.5 | 201.9 | 201.0 | 79.9 | 200.5 | 81.8 | 78.6 | 200.9 |
| 3 | 102.7 | 102.0 | 103.2 | 111.1 | 102.1 | 110.5 | 108.0 | 103.8 |
| 4 | 73.6 | 75.6 | 78.0 | 140.2 | 54.5 | 141.6 | 142.9 | 79.9 |
| 5 | 31.2 | 36.3 | 34.2 | 35.6 | 40.9 | 34.2 | 28.0 | 34.5 |
| 6 | 52.3 | 52.4 | 129.6 | 78.3 | 70.6 | 55.6 | 127.9 | 128.9 |
| 7 | 52.7 | 51.8 | 129.4 | 46.4 | 81.5 | 80.9 | 126.0 | 130.1 |
| 8 | 32.6 | 29.9 | 30.1 | 36.5 | 35.8 | 36.1 | 29.1 | 28.8 |
| 9 | 76.3 | 76.2 | 79.7 | 70.7 | 77.7 | 76.1 | 79.7 | 85.9 |
| 10 | 78.6 | 79.5 | 77.7 | 75.6 | 78.3 | 79.4 | 69.1 | 83.1 |
| 11 | 43.5 | 42.1 | 42.7 | 40.5 | 38.4 | 37.7 | 43.0 | 39.7 |
| 12 | 48.6 | 48.6 | 49.3 | 47.1 | 47.4 | 47.4 | 47.6 | 81.7 |
| 13 | 83.7 | 82.7 | 83.2 | 82.1 | 80.2 | 80.6 | 83.5 | 61.9 |
| 14 | 26.0 | 26.0 | 26.4 | 25.7 | 26.1 | 26.0 | 25.5 | 28.1 |
| 15 | 9.2 | 9.3 | 9.6 | 7.9 | 8.8 | 8.8 | 9.1 | 11.6 |
| Position | 1 1 | 2 2 | 3 3 | 4 2 | 5 2 | 6 4 | 7 4 | 8 3 |
|---|---|---|---|---|---|---|---|---|
| 1 | 6.11 dd (5.6, 2.6) | 6.06 dd (5.5, 2.1) | 6.02 dd (5.6, 1.8) | 3.11 brs | 6.13 dd (5.7, 1.7) | 3.11 d (1.6) | 3.10 m | 6.06 dd (5.7, 2.5) |
| 3 | 5.56 dd (5.6, 4.5) | 5.52 dd (5.5, 5.5) | 5.52 dd (5.6, 5.6) | 5.55 br d (10.7) | 5.58 dd (6.9, 5.7) | 5.60 br dd (10.7, 1.6) | 5.46 m | 5.45 dd (5.7, 4.5) |
| 4 | 4.36 m | 4.18 ddd (11.0, 5.5, 2.1) | 4.01 m | 6.07 ddd (10.7, 7.3, 7.3) | 4.82 m | 6.15 ddd (10.7, 7.0, 7.0) | 5.93 ddd (10.9, 7.4, 7.4) | 3.90 m |
| 5 | α 2.25 dd (14.3, 4.5) | α 1.67 ddd (14.6, 11.0, 9.5) | α 2.56 m | a 2.61 m | a 1.99 m | a 3.02 dddd (16.0, 7.0, 2.9, 1.6) | 3.10 m | α 2.52 ddd (15.1, 10.4, 6.0) |
| β 1.65 m | β 2.36 m | β 2.16 brdd (14.2, 8.1) | b 2.73 ddd (14.4, 7.3, 7.3) | b 1.74 ddd (13.4, 11.0, 1.7) | b 2.80 ddd (16.0, 7.0, 7.0) | β 2.16 m | ||
| 6 | 3.09 ddd (10.8, 4.5, 4.5) | 3.00 m | 5.82 m | 3.30 m | 3.73 ddd (11.0, 6.1, 2.1) | 4.13 m | 5.48 m | 5.76 m |
| 7 | 3.19 ddd (9.3, 4.5, 4.5) | 2.87 ddd (11.4, 3.7, 3.7) | 5.71 m | 4.02 brs | 4.11 m | 4.16 m | 5.47 m | 5.79 m |
| 8 | α 2.59 ddd (14.6, 5.2, 4.5) | α 1.55 m | α 2.59 m | α 2.53 brd (15.7) | α 2.05 brdd (13.3, 6.2) | α 1.99 dd (14.4, 4.3) | 2.35 m | α 2.63 m |
| β 1.33 ddd (14.6, 9.3, 1.6) | β 2.39 m | β 2.23 m | β 2.31 ddd (15.7, 4.3, 4.3) | β 1.82 ddd (13.3, 9.6, 3.9) | β 2.23 ddd (14.4, 9.1, 5.2) | β 2.24 m | ||
| 9 | 3.58 brd (5.2) | 3.70 ddd (11.0, 5.3, 1.1) | 3.53 brdd (11.0, 5.3) | 3.59 brs | 4.12 m | 4.06 dd (5.2, 1.7) | 3.45 ddd (7.3, 7.3, 0.6) | 3.84 ddd (11.3, 7.7, 3.8) |
| 10 | 3.36 m | 3.55 brs | 3.63 brs | 3.48 brs | 3.91 brs | 3.77 m | 3.68 brs | 3.89 m |
| 11 | α 2.47 ddd (12.7, 4.2, 4.2) | α 2.57 ddd (12.9, 3.6, 3.6) | α 2.52 ddd (13.3, 3.5, 3.5) | α 2.62 m | α 2.70 ddd (14.5, 4.3, 2.4) | α 2.75 ddd (14.6, 4.5, 2.3) | α 2.57 ddd (13.7, 4.5, 3.6) | α 2.10 m |
| β 2.04 ddd (12.7, 12.7, 2.8) | β 2.08 ddd (12.9, 12.9, 3.6) | β 1.99 ddd (13.3, 13.3, 3.2) | β 2.08 m | β 2.13 ddd (14.5, 11.8, 3.5) | β 2.16 ddd (14.6,11.8, 3.7) | β 2.06 m | β 2.35 ddd (14.8, 8.9, 6.4) | |
| 12 | 4.11 ddd (12.7, 10.3, 4.2) | 4.01 ddd (12.9, 10.0, 3.6) | 4.02 m | 4.16 ddd (12.1, 10.0, 4.4) | 3.96 ddd (11.8, 10.2, 4.3) | 4.04 ddd (11.8, 10.2, 4.5) | 3.98 ddd (12.3, 9.9, 4.5) | 4.00 ddd (8.9, 8.9, 4.5) |
| 13 | 3.32 ddd (10.3, 7.9, 2.5) | 3.28 ddd (10.0, 10.0, 2.3) | 3.25 ddd (9.3, 9.3, 2.0) | 3.33 ddd (10.0, 6.0, 2.8) | 3.28 ddd (10.2, 7.8, 2.4) | 3.29 ddd (10.2, 7.4, 2.6) | 3.33 ddd (9.9, 9.9, 2.1) | 4.05 ddd (8.9, 8.9, 2.6) |
| 14 | a 1.98 dqd (14.8, 7.3, 2.5) | a 2.01 dqd (15.2, 7.4, 2.3) | a 2.01 dqd (14.9, 7.3, 2.0) | a 1.87 dqd (14.3, 7.3, 2.8) | a 1.97 m | a 1.94 dqd (14.7, 7.4, 2.6) | a 2.04 m | a 2.16 m |
| b 1.58 m | b 1.49 m | b 1.50 m | b 1.76 m | b 1.52 m | b 1.57 dqd (14.7, 7.4, 7.4) | b 1.49 m | b 1.73 m | |
| 15 | 0.95 t (7.3) | 0.93 t (7.4) | 0.96 t (7.3) | 0.98 t (7.3) | 0.93 t (7.4) | 0.92 t (7.4) | 0.96 t (7.4) | 1.06 t (7.3) |
| Compound | Inhibition of NO Production | Cytotoxicity (at 72 h) |
|---|---|---|
| 1 | 26.03 ± 3.73 | >15.62 |
| 2 | 13.23 ± 0.57 | >31.25 |
| 3 | 4.18 ± 0.48 | >15.62 |
| 4 | >62.5 | >62.50 |
| 5 | 12.41 ± 1.04 | >15.62 |
| 6 | 37.39 ± 2.51 | >15.62 |
| 8 | 3.98 ± 0.60 | >7.81 |
| 10 | 4.387 × 10−3 ± 0.851 × 10−3 | >0.10 |
| 11 | 2.633 × 10−3 ± 0.238 × 10−3 | >0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koutsaviti, A.; Daskalaki, M.G.; Agusti, S.; Kampranis, S.C.; Tsatsanis, C.; Duarte, C.M.; Roussis, V.; Ioannou, E. Thuwalallenes A–E and Thuwalenynes A–C: New C15 Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp. Mar. Drugs 2019, 17, 644. https://doi.org/10.3390/md17110644
Koutsaviti A, Daskalaki MG, Agusti S, Kampranis SC, Tsatsanis C, Duarte CM, Roussis V, Ioannou E. Thuwalallenes A–E and Thuwalenynes A–C: New C15 Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp. Marine Drugs. 2019; 17(11):644. https://doi.org/10.3390/md17110644
Chicago/Turabian StyleKoutsaviti, Aikaterini, Maria G. Daskalaki, Susana Agusti, Sotirios C. Kampranis, Christos Tsatsanis, Carlos M. Duarte, Vassilios Roussis, and Efstathia Ioannou. 2019. "Thuwalallenes A–E and Thuwalenynes A–C: New C15 Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp." Marine Drugs 17, no. 11: 644. https://doi.org/10.3390/md17110644
APA StyleKoutsaviti, A., Daskalaki, M. G., Agusti, S., Kampranis, S. C., Tsatsanis, C., Duarte, C. M., Roussis, V., & Ioannou, E. (2019). Thuwalallenes A–E and Thuwalenynes A–C: New C15 Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp. Marine Drugs, 17(11), 644. https://doi.org/10.3390/md17110644