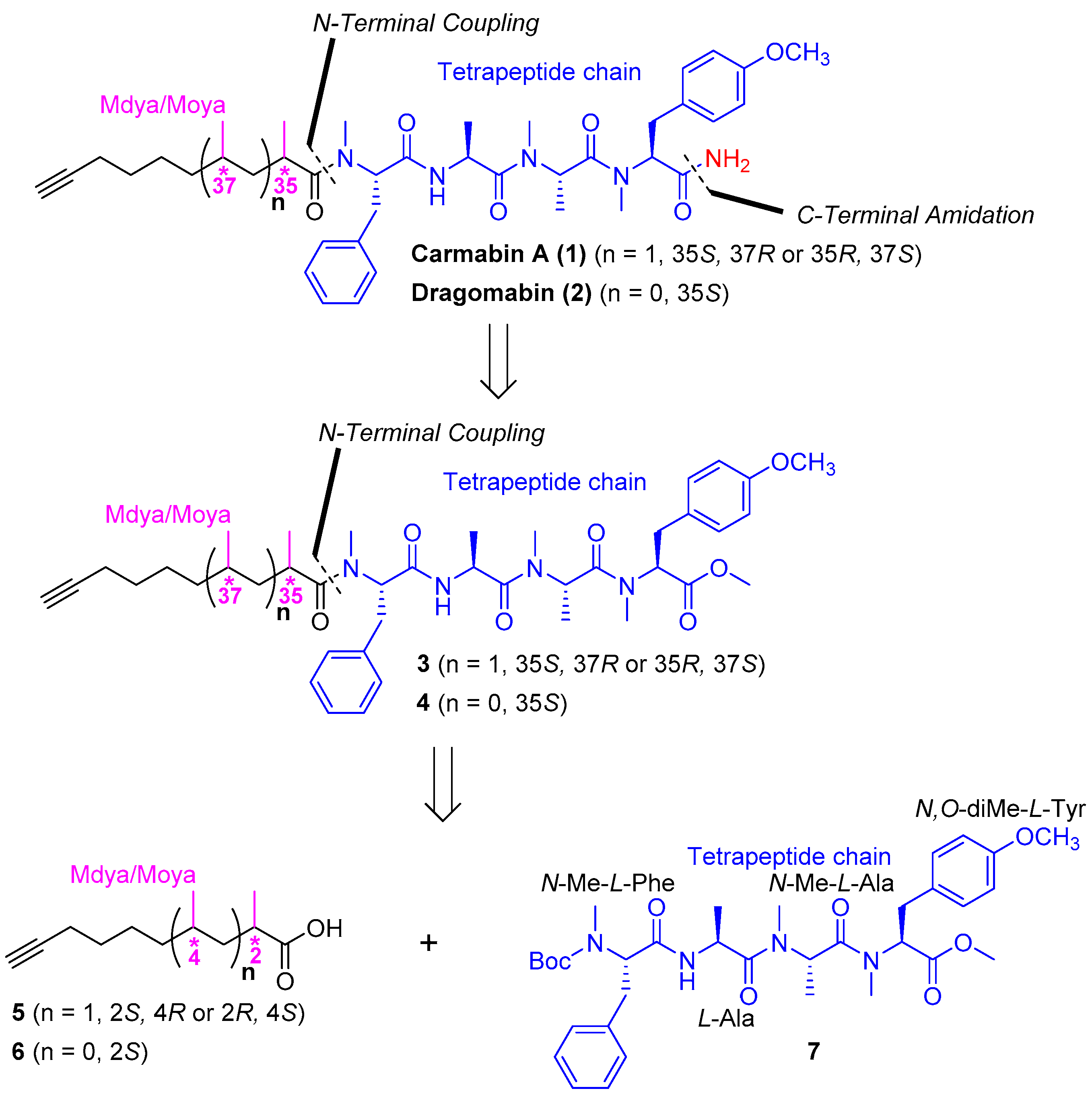
3.2. Chemistry
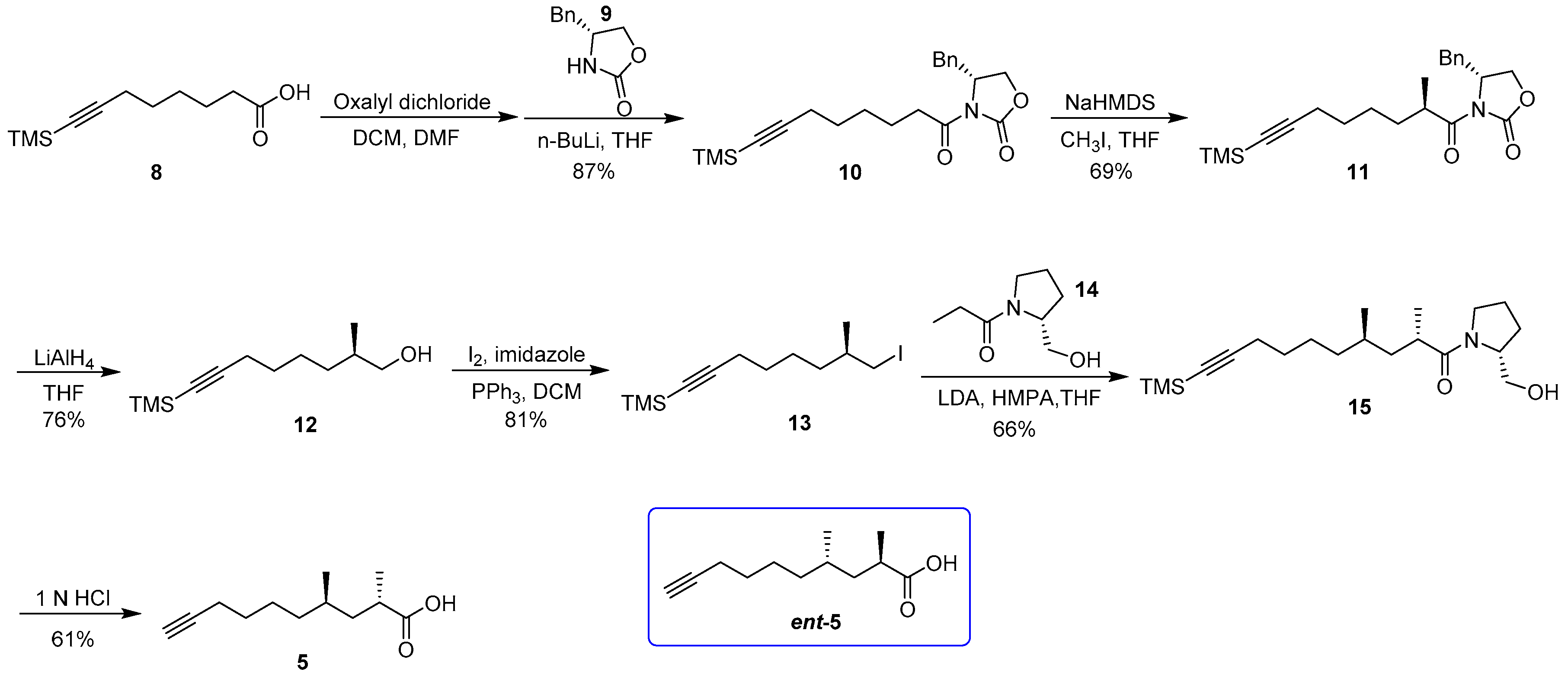
(2S,4R)-2,4-Dimethyldec-9-ynoic acid (
5) was obtained following the procedure for the preparation of compound (2
R,4
S)-2,4-dimethyldec-9-ynoic acid(
ent-5) [
14].
(R)-4-Benzyl-3-(8-(trimethylsilyl)oct-7-ynoyl)oxazolidin-2-one (10) Yield: 87%; −39.5 (c 1.0, CHCl3); IR (KBr) νmax 2959, 2932, 2172, 1796, 1701, 1244, 1056, 759, 644 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.35–7.20 (m, 5H), 4.70–4.64 (m, 1H), 4.22–4.16 (m, 2H), 3.30 (dd, J = 13.3, 3.1 Hz, 1H), 3.02–2.85 (m, 2H), 2.76 (dd, J = 13.3, 9.7 Hz, 1H), 2.25 (t, J = 7.0 Hz, 2H), 1.76–1.67 (m, 2H), 1.60–1.43 (m, 4H), 0.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 173.3, 153.6, 135.4, 129.5, 129.1, 127.5, 107.4, 84.7, 66.3, 55.3, 38.1, 35.5, 28.5, 28.4, 23.9, 19.8, 0.3; HRESIMS m/z 394.1815 [M + Na]+ (calcd. for C21H29NNaO3Si+, 394.1809).
(R)-4-Benzyl-3-((R)-2-methyl-8-(trimethylsilyl)oct-7-ynoyl)oxazolidin-2-one (11) Yield: 69%; −52.3 (c 1.0, CHCl3); IR (KBr) νmax 2939, 2862, 2174, 1783, 1699, 1211, 761, 640 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.35–7.20 (m, 5H), 4.67 (qd, J = 6.6, 2.8 Hz, 1H), 4.23–4.14 (m, 2H), 3.73–3.67 (m, 1H), 3.27 (dd, J = 13.3, 2.9 Hz, 1H), 2.77 (dd, J = 13.3, 9.6 Hz, 1H), 2.22 (t, J = 7.0Hz, 2H), 1.77–1.75 (m, 1H), 1.53–1.35 (m, 5H), 1.23 (d, J = 6.8 Hz, 3H), 0.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.3, 153.2, 135.5, 129.6, 129.1, 127.5, 107.4, 84.7, 66.2, 55.5, 38.1, 37.8, 32.9, 28.7, 26.5, 19.9, 17.4, 0.3; HRESIMS m/z 408.1969 [M + Na]+ (calcd. for C22H31NNaO3Si+, 408.1965).
(R)-2-Methyl-8-(trimethylsilyl)oct-7-yn-1-ol (12) Yield: 76%; +6.3 (c 1.0, CHCl3); IR (KBr) νmax 3372, 2958, 2937, 2175, 1250, 1044, 760, 640 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.54–3.40 (m, 2H), 2.23 (t, J = 6.9 Hz, 2H), 1.67–1.60 (m, 1H), 1.55–1.35 (m, 5H), 1.28 (t, J = 5.5 Hz, 1H), 1.19–1.06 (m, 1H), 0.92 (d, J = 6.7 Hz, 3H), 0.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 107.7, 84.6, 68.4, 35.8, 32.6, 28.9, 26.2, 19.9, 16.6, 0.3; compound 12 was not observed by HRESIMS analysis due to complete fragmentation.
(R)-(8-Iodo-7-methyloct-1-yn-1-yl)trimethylsilane (13) Yield: 81%; −2.8 (c 1.0, CHCl3); IR (KBr) νmax 2959, 2935, 2174, 1249, 1195, 760, 640 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.25–3.13 (m, 2H), 2.23 (t, J = 6.9 Hz, 2H), 1.53–1.32 (m, 6H), 1.28–1.18 (m, 1H), 0.98 (d, J = 6.4 Hz, 3H), 0.15 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 107.5, 84.8, 36.0, 34.7, 28.6, 26.1, 20.7, 19.9, 17.8, 0.3; compound 13 was not observed by HRESIMS analysis due to complete fragmentation.
(2S,4R)-1-((R)-2-(Hydroxymethyl)pyrrolidin-1-yl)-2,4-dimethyl-10-(trimethylsilyl)dec-9-yn-1-one (15) Yield: 66%; +30.2 (c 1.0, CHCl3); IR (KBr) νmax 3431, 2959, 2935, 2174, 1620, 1463, 1249, 1052, 759, 640 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.21 (dd, J = 7.7, 2.1 Hz, 1H), 4.23 (dd, J = 13.7, 5.8 Hz, 1H), 3.69–3.46 (m, 4H), 2.62 (dd, J = 13.4, 6.7 Hz, 1H), 2.20 (t, J = 6.9 Hz, 2H), 2.07–1.84 (m, 3H), 1.59–1.23 (m, 10H), 1.12 (d, J = 6.7 Hz, 3H), 0.86 (d, J = 5.8 Hz, 3H), 0.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 178.7, 107.6, 84.5, 68.0, 61.3, 48.0, 40.7, 36.9, 35.9, 30.5, 28.9, 28.5, 26.1, 24.7, 19.9, 19.5, 17.4, 0.3; HRESIMS m/z 374.2490 [M + Na]+ (calcd. for C20H37NNaO2Si+, 374.2486).
(2S,4R)-2,4-Dimethyldec-9-ynoic acid (5) Yield: 61%; +5.6 (c 1.0, CHCl3); IR (KBr) νmax 3309, 2937, 2862, 2118, 1706, 1292, 633 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.59–2.50 (m, 1H), 2.19 (td, J = 6.9, 2.4 Hz, 2H), 1.94 (t, J = 2.4 Hz, 1H), 1.59–1.31 (m, 9H), 1.16 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 182.7, 84.8, 68.3, 40.9, 37.1, 36.5, 30.5, 28.8, 26.1, 19.4, 18.5, 17.0; HRESIMS m/z 195.1388 [M − H ]− (calcd. for C12H19O−, 195.1391); the optical rotation of ent-5 ( −5.8 (c 1.0, CHCl3)).
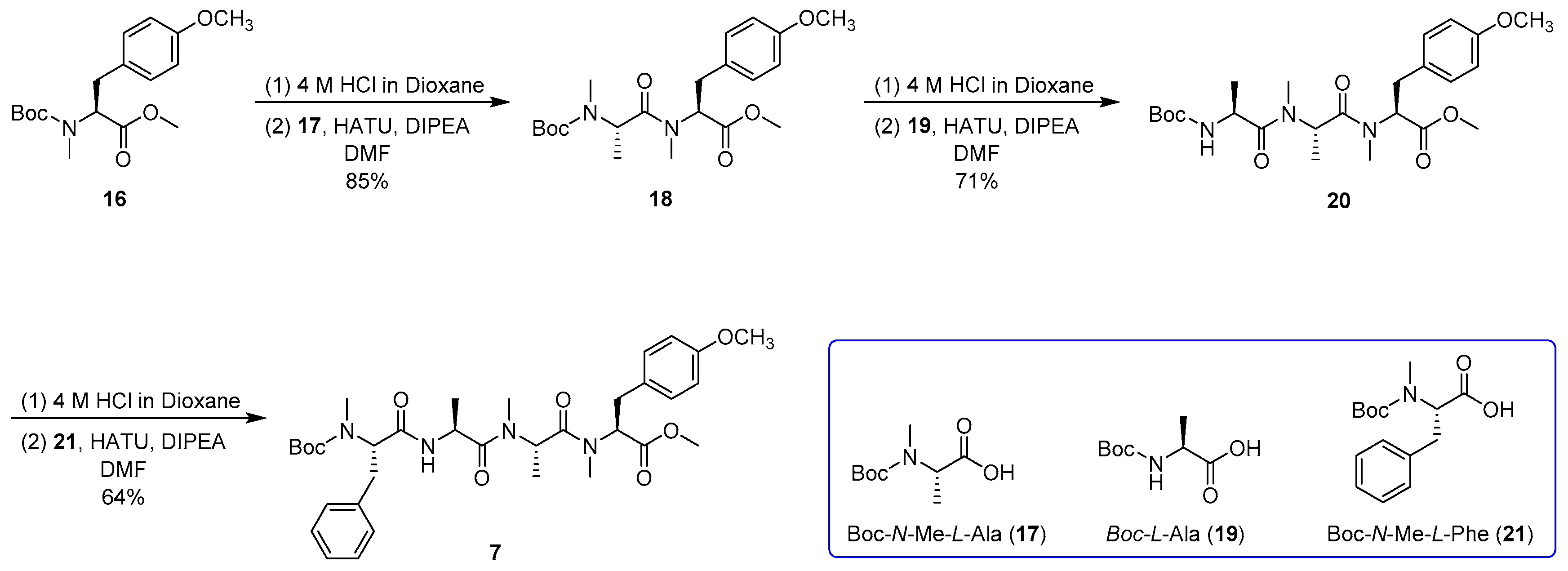
Boc-N-Me-l-Ala-N,O-diMe-l-Tyr (18) Compound 16 (30.0 g, 92.8 mmol) was dissolved in 4.0 M HCl in dioxane (150 mL). After the mixture had been stirred for 3 h at room temperature, the whole mixture was concentrated to afford an HCl salt as a white solid and used directly for the next step. To the solution of the HCl salt, compound 17 (37.7 g, 185 mmol), and HATU (70.3 g, 185 mmol) in DMF (150 mL) was added DIPEA (48.0 g, 371 mmol) at 0 °C under argon, and then the mixture was warmed to room temperature and stirred overnight. After being diluted with EtOAc (800 mL), the whole mixture was washed with 1% HCl (3 × 150 mL), 5% aqueous NaHCO3 (3 × 150 mL), and brine (3 × 150 mL), and the organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and purified by silica gel column chromatography (10/1 to 3/1 petroleum ether/EtOAc) to afford compound 18 (32.2 g, 85%) as a colorless oil: −121.2 (c 1.0, CHCl3); IR (KBr) νmax 2978, 1745, 1657, 1515, 1249, 833, 561 cm−1; 1H NMR (400 MHz, CDCl3) mixture of rotamers δ 7.14, 7.09 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 8.3 Hz, 1H), 6.84, 6.80 (d, J = 8.5 Hz, 2H), 5.37, 5.12, 5.05 (dd, J = 11.4, 4.8 Hz, 1H), 5.02, 4.80, 4.70, 4.63, 4.33 (q, J = 6.6 Hz, 1H), 3.76 (s, 3H), 3.73 (s, 3H), 3.35–3.28 (m, 1H), 3.03–2.93 (m, 1H), 2.85, 2.84, 2.80 (s, 3H), 2.58, 2.55, 2.23, 2.19 (s, 3H), 1.44, 1.43, 1.42 (s, 9H), 1.19, 1.09, 0.97, 0.90 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) mixture of rotamers δ 171.9, 171.7, 171.3, 171.1, 158.7, 158.6, 155.3, 155.2, 130.1, 130.0, 129.8, 129.2, 128.5, 114.3, 114.1, 80.1, 80.0, 61.0, 60.6, 59.5, 58.6, 55.4, 52.5, 52.4, 51.7, 50.8, 49.5, 34.6, 33.8, 32.9, 31.9, 30.0, 29.8, 28.9, 28.6, 28.4, 28.1, 14.4; HRESIMS m/z 431.2155 [M + Na]+ (calcd. for C21H32N2NaO6+, 431.2153).
Boc-l-Ala-N-Me-l-Ala-N,O-diMe-l-Tyr (20) Compound 18 (31.0 g, 75.9 mmol) was dissolved in 4.0 M HCl in dioxane (150 mL). After the mixture had been stirred for 3 h at room temperature, the whole mixture was concentrated to afford an HCl salt as a white solid and used directly for the next step. To the solution of the HCl salt, compound 19 (37.7 g, 152 mmol), and HATU (57.7 g, 152 mmol) in DMF (150 mL) was added DIPEA (39.2 g, 303 mmol) at 0 °C under argon, and then the mixture was warmed to room temperature and stirred overnight. After being diluted with EtOAc (800 mL), the whole mixture was washed with 1% HCl (3 × 150 mL), 5% aqueous NaHCO3 (3 × 150 mL), and brine (3 × 150 mL), and the organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and purified by silica gel column chromatography (5/1 to 1/1 petroleum ether/EtOAc) to afford compound 20 (25.8 g, 71%) as a colorless oil: −93.4 (c 1.0, CHCl3); IR (KBr) νmax 3445, 2981, 1743, 1637, 1516, 1249, 1172, 614, 556 cm–1; 1H NMR (400 MHz, CDCl3) mixture of rotamers δ 7.09 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 8.5 Hz, 1H), 5.47–5.33 (m, 2H), 4.85–4.37 (m, 1H), 3.77, 3.75 (s, 3H), 3.74, 3.71 (s, 3H), 3.34, 3.26 (dd, J = 14.8, 4.9 Hz, 1H), 2.97–2.88 (m, 1H), 2.92, 2.85, 2.71 (s, 3H), 2.83, 2.77, 2.28 (s, 3H), 1.41 (s, 9H), 1.27, 1.18, 1.11, 0.81 (d, J = 6.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) mixture of rotamers δ 172.3, 171.3, 171.1, 170.7, 158.8, 155.1, 130.3, 129.9, 129.7, 128.8, 128.4, 114.3, 114.2, 114.1, 79.8, 58.2, 58.1, 55.4, 55.36, 52.6, 52.5, 49.7, 44.0, 46.6, 34.5, 33.8, 31.9, 31.8, 30.3, 30.1, 29.7, 29.2, 28.5, 19.0, 18.8, 14.1; HRESIMS m/z 502.2528 [M + Na]+ (calcd. for C24H37N3NaO7+, 502.2524).
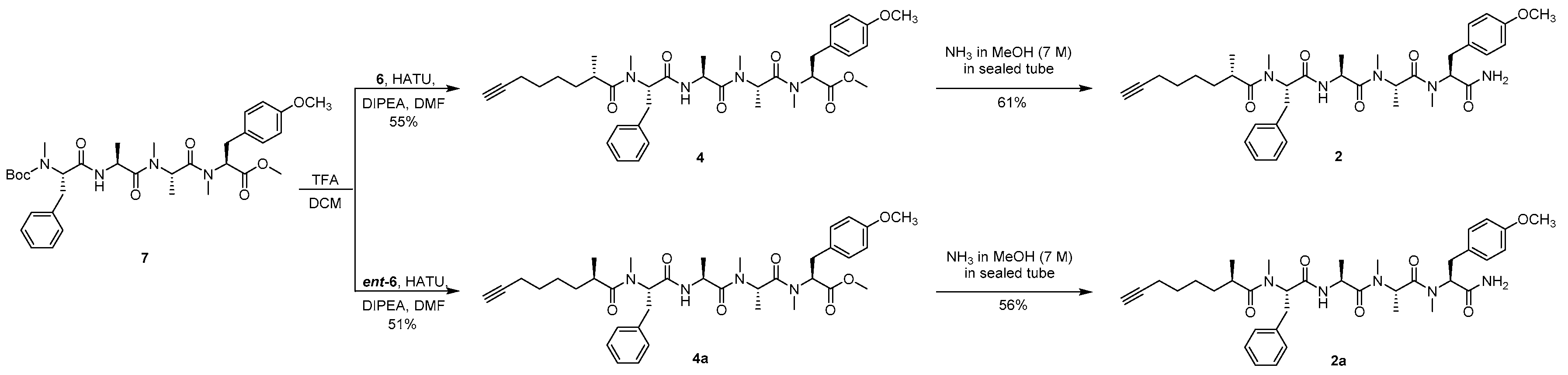
Boc-N-Me-l-Phe-l-Ala-N-Me-l-Ala-N,O-diMe-l-Tyr (7) Compound 20 (25.0 g, 52.1 mmol) was dissolved in 4.0 M HCl in dioxane (125 mL). After the mixture had been stirred for 3 h at room temperature, the whole mixture was concentrated to afford an HCl salt as a white solid and used directly for the next step. To the solution of the HCl salt, compound 21 (29.1 g, 104 mmol), and HATU (39.6 g, 104 mmol) in DMF (125 mL) was added DIPEA (26.9 g, 208 mmol) at 0 °C under argon, and then the mixture was warmed to room temperature and stirred overnight. After being diluted with EtOAc (600 mL), the whole mixture was washed with 1% HCl (3 × 120 mL), 5% aqueous NaHCO3 (3 × 120 mL), and brine (3 × 120 mL), and the organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and purified by silica gel column chromatography (2/1 to 1/2 petroleum ether/EtOAc) to afford compound 7 (21.3 g, 64%) as a colorless oil: −100.6 (c 1.0, CHCl3); IR (KBr) νmax 3402, 2924, 1744, 1644, 1515, 1248, 1174, 615, 556 cm−1; 1H NMR (400 MHz, CDCl3) mixture of rotamers δ 7.27–7.15 (m, 5H), 7.14, 7.12 (d, J = 8.8 Hz, 2H), 6.96 (br, 1H), 6.85, 6.81 (d, J = 8.6 Hz, 2H), 5.40, 4.82 (m, 1H), 5.39–5.31 (m, 1H), 4.81–4.64 (m, 2H), 3.77 (s, 3H), 3.75, 3.72 (s, 3H), 3.42–3.26 (m, 2H), 3.00–2.89 (m, 2H), 2.86, 2.75, 2.72 (s, 3H), 2.72, 2.32 (s, 3H), 1.46, 1.37, 1.32 (s, 9H), 1.25, 1.14 (d, J = 6.8 Hz, 3H), 1.19, 0.81 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) mixture of rotamers δ 171.7, 171.7, 171.6, 171.3, 171.2, 171.1, 158.9, 158.8, 130.3, 129.9, 129.2, 128.9, 128.6, 126.6, 114.4, 114.2, 58.4, 55.5, 52.5, 49.7, 48.1, 45.7, 34.3, 33.8, 32.0, 29.7, 29.2, 28.3, 18.4, 14.2, 14.1; HRESIMS m/z 663.3368 [M + Na]+ (calcd. for C34H48N4NaO8+, 663.3364).
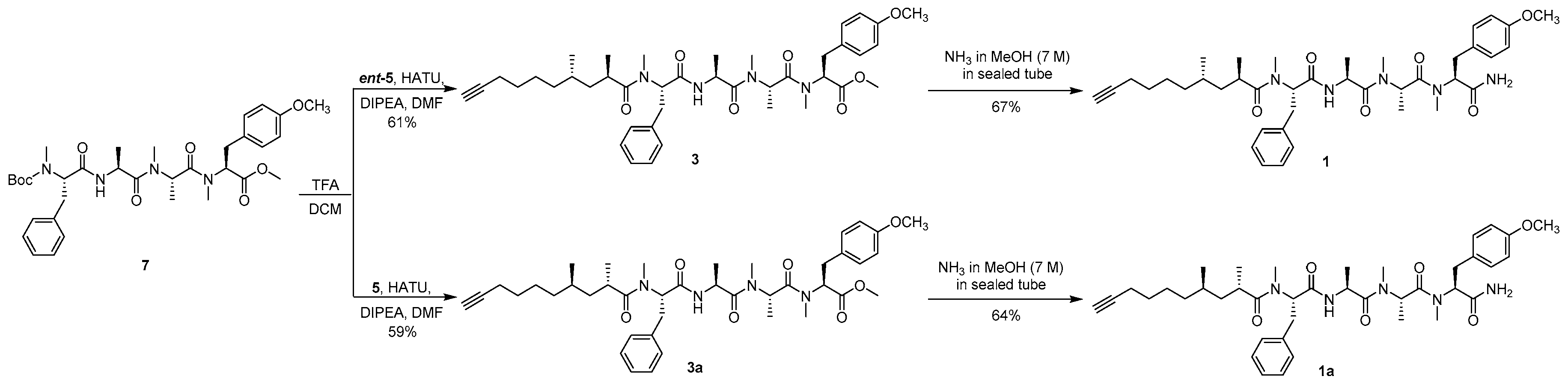
Methyl (2S,5S,8S,11S,14R,16S)-11-benzyl-2-(4-methoxybenzyl)-3,5,6,8,12,14,16-heptamethyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazadocos-21-ynoate (3) To the solution of compound 7 (200 mg, 0.312 mmol) in DCM (1.8 mL) was added TFA (0.2 mL). After the mixture had been stirred for 6 h at room temperature, toluene (2 mL) was added and then the whole mixture was concentrated under reduced pressure. The residue was dissolved with water (3 mL) and extracted with petroleum ether (3 × 1 mL). The aqueous phase was adjusted to pH 8–9 with Na2CO3 and extracted with DCM (3 × 10 mL), and the combined organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated to afford a free amine as a colorless oil and used directly for the next step. To the solution of the free amine, ent-5 (122 mg, 0.622 mmol), and HATU (237 mg, 0.624 mmol) in DMF (1 mL) was added DIPEA (84.7 mg, 0.655 mmol) at 0 °C under argon, and then the mixture was warmed to room temperature and stirred overnight. After being diluted with EtOAc (10 mL), the whole mixture was washed with 1% HCl (3 × 2 mL), 5% aqueous NaHCO3 (3 × 2 mL), and brine (3 × 2 mL), and the organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and purified by silica gel column chromatography (100/1 to 50/1 DCM/MeOH) to afford compound 3 (137 mg, 61%) as a colorless oil: −150.6 (c 0.10, CHCl3); IR (KBr) νmax 3446, 2934, 2115, 1743, 1636, 1515, 1248, 1179, 620, 560 cm−1; 1H NMR (400 MHz, CDCl3) mixture of rotamers δ 7.26–7.16 (m, 5H), 7.12, 7.09 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 7.3 Hz, 1H), 6.85, 6.80 (d, J = 8.6 Hz, 2H), 5.48 (dd, J = 10.5, 6.2 Hz, 1H), 5.40, 4.86 (q, J = 6.6 Hz, 1H), 5.34, 4.81 (dd, J = 12.0, 4.8 Hz, 1H), 4.73, 4.62 (p, J = 6.8 Hz, 1H), 3.77 (s, 3H), 3.75, 3.71 (s, 3H), 3.38–3.23 (m, 2H), 3.04–2.84 (m, 2H), 2.90, 2.89 (s, 3H), 2.85, 2.71 (s, 3H), 2.80, 2.31 (s, 3H), 2.63–2.58 (m, 1H), 2.19–2.12 (m, 2H), 1.94 (t, J = 2.5 Hz, 1H), 1.47–1.39 (m, 2H), 1.31–1.22 (m, 2H), 1.21–0.96 (m, 5H), 1.18, 0.80 (d, J = 6.7 Hz, 3H), 1.17, 1.11 (d, J = 6.8 Hz, 3H), 1.05, 1.03 (d, J = 6.7 Hz, 3H), 0.72, 0.70 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) mixture of rotamers δ 178.2, 178.2, 171.4, 171.4, 171.2, 171.1, 170.8, 170.7, 169.6, 169.6, 158.8, 158.7, 137.1, 137.0, 130.3, 129.8, 129.0, 128.9, 128.5, 128.3, 126.7, 114.3, 114.1, 84.7, 68.3, 61.0, 58.4, 57.0, 56.7, 55.4, 52.4, 49.6, 48.0, 45.6, 45.5, 40.9, 40.8, 36.5, 34.4, 33.9, 33.8, 33.7, 32.0, 31.2, 31.0, 30.2, 30.1, 29.6, 29.1, 28.8, 25.8, 19.6, 18.4, 18.3, 18.1, 17.0, 14.1, 14.0; HRESIMS m/z 741.4203 [M + Na]+ (calcd. for C41H58N4NaO7+, 741.4198).
(2R,4S)-N-((S)-1-(((S)-1-(((S)-1-(((S)-1-Amino-3-(4-methoxyphenyl)-1-oxopropan-2-yl)(methyl)amino)-1-oxopropan-2-yl)(methyl)amino)-1-oxopropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-N,2,4-trimethyldec-9-ynamide (carmabin A (
1)) Compound
3 (80.0 mg, 0.111 mmol) was added to a sealed tube followed by anhydrous NH
3 solution (7.0 M in MeOH, 1 mL). After the mixture had been stirred for 20 h at 60 °C, the whole mixture was concentrated and purified by silica gel column chromatography (50/1 to 20/1 DCM/MeOH) to afford a colorless oil; n-pentane (2.5 mL) was added, and then the mixture was stirred for 1 h at room temperature and filtered to afford compound
1 (52.3 mg, 67%) as a white powder:
−175.6 (c 0.44, CHCl
3); IR (KBr) ν
max 3448, 2935, 2115, 1634, 1513, 1249, 1179, 670, 619 cm
−1; HRESIMS
m/
z 726.4205 [M + Na]
+ (calcd. for C
40H
57N
5NaO
6+, 726.4201).
1H and
13C NMR data in the
Supporting Information.
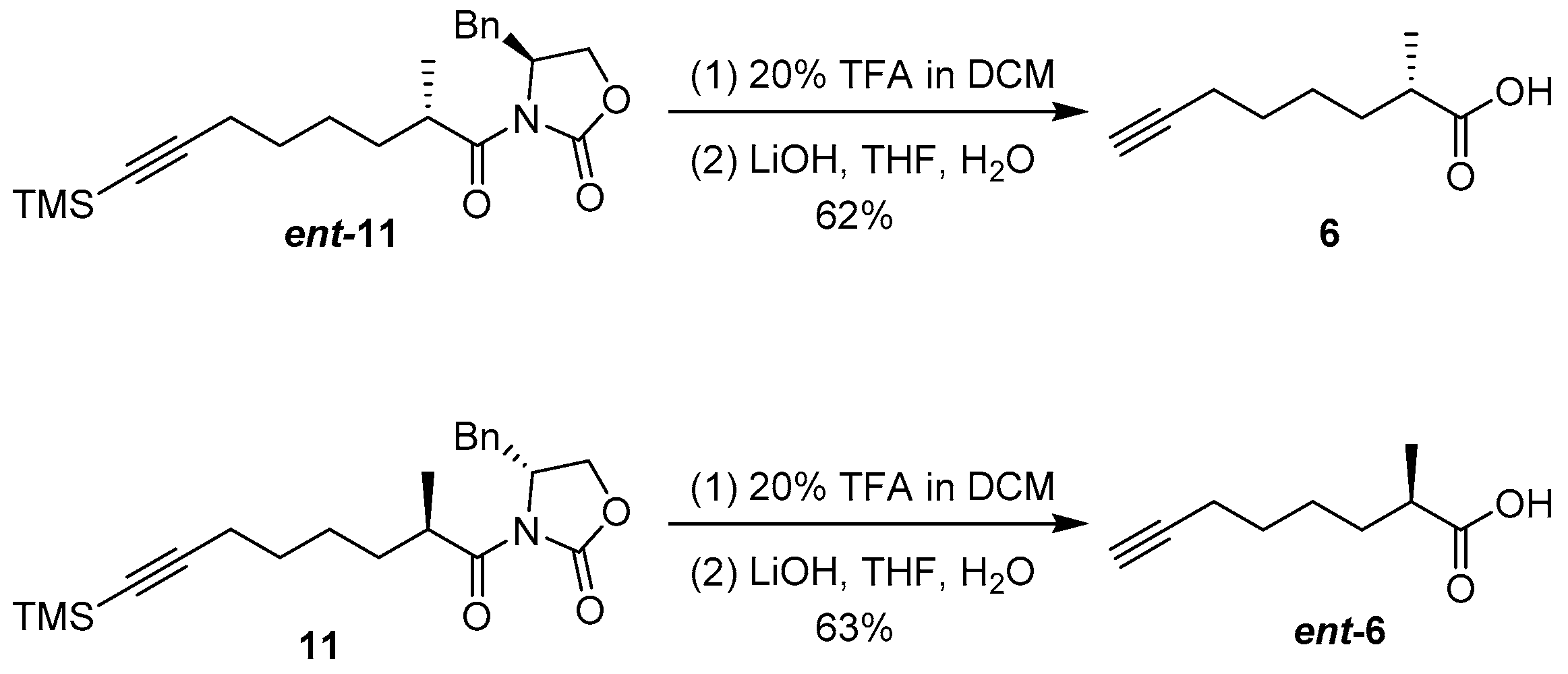
(S)-2-Methyloct-7-ynoic acid (6) To the solution of compound ent-11 (1.00 g, 2.59 mmol) in DCM (8 mL) was added TFA (2 mL). After the mixture had been stirred for 3 h at room temperature, toluene (10 mL) was added and then the whole mixture was concentrated under reduced pressure. The residue was dissolved with water (10 mL) and adjusted to pH 8–9 with Na2CO3 and extracted with DCM (3 × 20 mL), and the combined organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated to afford a crude oil and used directly for the next step. To the crude oil in THF (10 mL) and water (2.5 mL) was added LiOH·H2O (435 mg, 10.4 mmol) and H2O2 (2.90 mL, 20.7 mmol, 30% in water) at 0 °C. After the mixture had been stirred for 3 h at room temperature. The whole mixture was adjusted to pH 2–3 with 1 N HCl and extracted with EtOAc (3 × 30 mL), and the combined organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated and purified by silica gel column chromatography (petroleum ether/EtOAc = 50/1 − 30/1) to afford compound 6 (248 mg, 62%) as a colorless oil: +13.2 (c 0.90, CHCl3); IR (KBr) νmax3304, 2942, 2865, 2118, 1706, 1236, 638 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.52–2.42 (m, 1H), 2.20 (td, J = 6.9, 2.5 Hz, 2H), 1.94 (t, J = 2.5 Hz, 1H), 1.75–1.65 (m, 1H), 1.58–1.42 (m, 5H), 1.19 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 182.9, 84.4, 68.5, 39.3, 33.1, 28.4, 26.4, 18.4, 17.0; compound 6 was not observed by HRESIMS analysis due to complete fragmentation.
(R)-2-methyloct-7-ynoic acid (
ent-6) Compound
ent-6 was obtained from compound
11 (800 mg, 2.07 mmol) following the same procedure for the preparation of compound
6 (200 mg, 63%) colorless oil:
−13.0 (c 0.90, CHCl
3);
1H NMR (400 MHz, CDCl
3) δ 2.52–2.43 (m, 1H), 2.20 (td,
J = 6.9, 2.5 Hz, 2H), 1.94 (t,
J = 2.5 Hz, 1H), 1.74–1.65 (m, 1H), 1.58–1.42 (m, 5H), 1.19 (d,
J = 7.0 Hz, 3H). The spectroscopic data are in agreement with those reported in the literature [
9].
Methyl(2S,5S,8S,11S,14S)-11-benzyl-2-(4-methoxybenzyl)-3,5,6,8,12,14-hexamethyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazaicos-19-ynoate (4) Compound 4 was obtained from compound 7 (200 mg, 0.312 mmol) and compound 6 (95.9 mg, 0.622 mmol) following the procedure for the preparation of compound 3, (116 mg, 55%), colorless oil: −59.8 (c 0.10, CHCl3); IR (KBr) νmax 3444, 2935, 2115, 1744, 1642, 1515, 1248, 1179, 701, 618 cm−1; 1H NMR (400 MHz, CDCl3) mixture of rotamers δ 7.27–7.14 (m, 5H), 7.12, 7.09 (d, J = 8.9 Hz, 2H), 6.94 (d, J = 7.2 Hz, 1H), 6.86, 6.80 (d, J = 8.6 Hz, 2H), 5.47 (dd, J = 10.1, 6.2 Hz, 1H), 5.41, 4.84 (dd, J = 12.9, 6.2 Hz, 1H), 5.33, 4.76 (dd, J = 12.1, 7.3 Hz, 1H), 4.73, 4.62 (p, J = 6.9 Hz, 1H), 3.77 (s, 3H), 3.75, 3.72 (s, 3H), 3.39–3.24 (m, 2H), 3.02–2.91 (m, 2H), 2.90, 2.89 (s, 3H), 2.85, 2.78, 2.71 (s, 3H), 2.80, 2.34, 2.31 (s, 3H), 2.60–2.52 (m, 1H), 2.20–2.13 (m, 2H), 1.94–1.91 (m, 1H), 1.72–1.64 (m, 1H), 1.53–1.47 (m, 2H), 1.40–1.30 (m, 3H), 1.18, 1.10, 0.81 (d, J = 6.7 Hz, 3H), 1.18, 1.11, 1.05 (d, J = 6.8 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) mixture of rotamers δ 177.7, 177.6, 171.4, 171.3, 171.1, 170.9, 170.7, 170.5, 169.4, 169.4, 168.4, 158.7, 158.6, 137.4, 137.0, 136.9, 130.1, 129.7, 129.6, 129.1, 128.8, 128.7, 128.3, 128.1, 127.0, 126.5, 114.2, 114.0, 113.9, 84.4, 84.3, 68.5, 68.3, 68.2, 62.9, 60.9, 58.2, 56.9, 56.8, 55.3, 55.2, 52.4, 52.3, 49.5, 47.9, 45.8, 45.5, 45.3, 35.8, 35.4, 34.5, 34.3, 33.7, 33.6, 33.3, 33.2, 32.9, 31.9, 31.3, 31.2, 30.0, 29.6, 29.5, 29.0, 28.5, 28.4, 26.7, 26.6, 26.2, 18.2, 18.1, 18.0, 17.3, 14.0, 13.9; HRESIMS m/z 699.3732 [M + Na]+ (calcd. for C38H52N4NaO7+, 699.3728.
(S)-N-((S)-1-(((S)-1-(((S)-1-(((S)-1-Amino-3-(4-methoxyphenyl)-1-oxopropan-2-yl)(methyl)amino)-1-oxopropan-2-yl)(methyl)amino)-1-oxopropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-N,2-dimethyloct-7-ynamide (
2) Compound
2 was obtained from compound
4 (80.0 mg, 0.118 mmol) following the procedure for the preparation of compound
1, (47.7 mg, 61%), white powder:
−163.8 (
c 0.50, CHCl
3); IR (KBr) ν
max 3447, 2935, 2114, 1634, 1514, 1249, 1081, 700, 617; HRESIMS
m/
z 684.3736 [M + Na]
+ (calcd. for C
37H
51N
5NaO
6+, 684.3732).
1H and
13C NMR data are in the
Supplementary Materials.
Methyl (2S,5S,8S,11S,14R)-11-benzyl-2-(4-methoxybenzyl)-3,5,6,8,12,14-hexamethyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazaicos-19-ynoate (4a) Compound 4a was obtained from compound 7 (200 mg, 0.312 mmol) and compound ent-6 (95.9 mg, 0.622 mmol) following the procedure for the preparation of compound 3, (108 mg, 51%), colorless oil: −105.0 (c 0.10, CHCl3); IR (KBr) νmax 3447, 2931, 2115, 1636, 1385, 1271, 1179, 670, 615 cm−1; 1H NMR (400 MHz, CDCl3) mixture of rotamers δ 7.27–7.16 (m, 5H), 7.12, 7.09 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 7.1 Hz, 1H), 6.85, 6.80 (d, J = 8.7 Hz, 2H), 5.45 (dd, J = 10.1, 6.2 Hz, 1H), 5.40, 4.83 (dd, J = 12.9, 6.4 Hz, 1H), 5.34, 4.76 (dd, J = 11.9, 4.8 Hz, 1H), 4.73, 4.62 (p, J = 6.9 Hz, 1H), 3.77 (s, 3H), 3.75, 3.72 (s, 3H), 3.38–3.26 (m, 2H), 3.05–2.92 (m, 2H), 2.91, 2.89 (s, 3H), 2.85, 2.77, 2.71 (s, 3H), 2.81, 2.34, 2.31 (s, 3H), 2.55–2.50 (m, 1H), 2.08–2.03 (m, 2H), 1.94–1.91 (m, 1H), 1.49–1.41 (m, 1H), 1.39–1.31 (m, 2H), 1.21–1.06 (m, 3H), 1.19, 1.10, 0.81 (d, J = 6.8 Hz, 3H), 1.17, 1.11, (d, J = 6.8 Hz, 3H), 1.07, 0.44 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) mixture of rotamers δ 177.7, 177.6, 171.4, 171.2, 171.0, 170.8, 170.6, 169.5, 169.5, 158.8, 158.7, 137.5, 137.2, 137.1, 130.2, 129.8, 129.2, 128.9, 128.8, 128.5, 126.6, 114.3, 114.1, 84.5, 68.3, 63.2, 61.0, 58.4, 57.2, 56.8, 55.3, 52.4, 52.4, 49.6, 47.9, 45.5, 45.4, 36.0 34.4, 33.7, 33.7, 33.3, 32.0, 31.4, 31.1, 30.1, 29.6, 29.1, 28.5, 26.3, 18.3, 18.2, 18.1, 17.4, 17.4, 14.1, 14.0; HRESIMS m/z 699.3732 [M + Na]+ (calcd. for C38H52N4NaO7+, 699.3728).
(R)-N-((S)-1-(((S)-1-(((S)-1-(((S)-1-Amino-3-(4-methoxyphenyl)-1-oxopropan-2-yl)(methyl)amino)-1-oxopropan-2-yl)(methyl)amino)-1-oxopropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-N,2-dimethyloct-7-ynamide (dragomabin (
2a)) Compound
2a was obtained from compound
4a (80.0 mg, 0.118 mmol) following the procedure for the preparation of compound
1, (43.8 mg, 56%), white powder:
−118.3 (
c 0.50, CHCl
3); IR (KBr) ν
max 3448, 2936, 2114, 1636, 1515, 1249, 1179, 668, 616; HRESIMS
m/
z 684.3736 [M + Na]
+ (calcd. for C
37H
51N
5NaO
6+, 684.3732).
1H and
13C NMR data are in the
Supplementary Materials.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





