Abstract
Glycolipids represent a broad class of natural products structurally featured by a glycosidic fragment linked to a lipidic molecule. Despite the large structural variety of these glycoconjugates, they can be classified into three main groups, i.e., glycosphingolipids, glycoglycerolipids, and atypical glycolipids. In the particular case of glycolipids derived from marine sources, an impressive variety in their structural features and biological properties is observed, thus making them prime targets for chemical synthesis. In the present review, we explore the chemistry and biology of this class of compounds.
1. Introduction
Glycolipids represent a broad class of biologically active natural products with a wide variety of molecular structures and biological functions, many of which are essential for life [1,2]. Despite their extensive structural diversity, glycolipids can be classified by the nature of the lipidic fragment. According to this classification, these glycoconjugates are divided into three main groups, i.e., glycoglycerolipids, glycosphingolipids, and those comprising the rest of the glycolipids possessing atypical lipidic moieties. Whereas glycoglycerolipids are mainly distributed in the realm of micro-organisms and plants, glycosphingolipids are extensively found in all living beings. In fact, these molecules are found on the surface of cell membranes and, together with glycoproteins and glycosaminoglycans, which are known as glycocalyx, play critical roles for cell growth, cellular recognition, adhesion, neuronal repair, and signal transduction, which are essential for health and involved in a number of diseases such as cancer and inflammatory processes and infections [3,4,5]. Given their molecular complexity and diversity, glycosphingolipids are classified into the following subgroups: (a) neutral, which can be subdivided into the cerebrosides that contain only one uncharged sugar, the diosylceramides with two sugar units, and the neutral glycosphingolipids with more than two (up to 30) uncharged sugars; and (b) acidic, which can be subdivided into the gangliosides, characterized by the presence of one or more neuraminic acid residues, and, finally the sulfatides, which contain at least one sugar residue with a sulfate group. In any case, all of the glycosphingolipids share a common ceramide lipid unit, consisting of a long-chain amino-alcohol fragment, which can be a sphingosine or a phytosphingosine unit (sphingoid base) linked to a fatty acid via an amide bond. In the case of the glycoglycerolipids, their core structure is comprised of a 1,2-diacyl glycerol attached to a mono- or an oligosaccharide molecule, although some variations with respect to this general structure can be found, as will be described later. In contrast to the glycosphingolipids, the glycoglycerolipids are present in nature in much lower abundance, which, combined with the difficulty of their isolation from natural sources, have significantly hampered extensive and detailed biological studies. The third group consists of the atypical glycolipids that include any glycoconjugate that contains a lipidic chain not present in the previous groups (Figure 1). The stunning and limitless wealth of secondary metabolites that marine organisms provide is extensive for this class of compounds, with a myriad of glycolipid-type natural products with impressive molecular diversity and a variety of biological activities, including antitumor, antiviral, and anti-inflammatory properties. Given the biological relevance and structural complexity of these classes of compounds, a large number of reviews [6,7,8,9,10,11,12], books, and book chapters [13,14,15] have been devoted to all aspects related to their chemistry and biology. More specifically, in the field of glycolipids of marine origin, several excellent reviews have been published, especially by Barnathan et al. [16], which represents an excellent description of all the glycolipids found in marine invertebrates, as well as by Li et al. [17], which focused on the chemistry and biology of glycoglycerolipids from marine organisms. In addition, numerous reviews have been reported on very specific compounds, such as KRN7000 [18] and related glycosphingolipids [19] due to their outstanding biological activities and their potential pharmacological activity. In light of this publication landscape, this review intends to give a chemical and biological perspective of these fascinating natural products with a particular emphasis on recent contributions that were not covered in the aforementioned reviews and highlighting the importance of their synthesis given the particularly intricate requirements to obtain sufficient amounts from their natural sources. This review also provides an updated state of the art of this field that can attract the interest of chemists and biologists, revealing the potential and prospects that these compounds may provide in biology, chemistry, and biomedicine for the future.
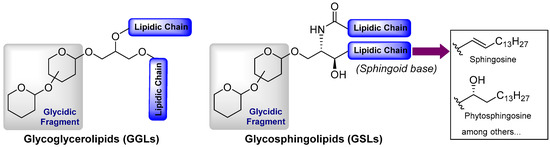
Figure 1.
General structural feature of glycolipids.
2. Chemistry and Biology of Glycosphingolipids
2.1. Neutral Glycosphingolipids
2.1.1. Cerebrosides
Acanthacerebrosides, Astrocerebrosides, and Asteriacerebrosides
Among the wide and important family of marine glycosphingolipids containing a phytosphingosine unit, which confers the glycolipids outstanding immunostimulant properties, as will be described later, the acanthacerebrosides and astrocerebrosides represent interesting members isolated from different starfish species. It was in 1988 when Komori et al. [20] discovered the first members of the acanthacerebrosides (A–F, 1–6) from the starfish Acanthaster planci. These compounds were isolated as a mixture, which hindered a complete structural elucidation and biological evaluations. However, subsequent purification efforts by the same group led to the isolation of one of their members, acanthacerebroside B (2), which could be isolated as a pure compound from the starfish Asterina pectinifera and allowed full structural characterization by a complete NMR spectroscopic analysis. In the same year, these authors described the first total synthesis of acanthacerebroside A (1), establishing unambiguously the structure and absolute configuration of this new family of cerebrosides [21]. In addition, the investigations of Komori et al. [22] with the starfish Astropecten latespinosus led to the discovery of three new related cerebrosides, which were named astrocerebrosides A (7), B (8), and C (9). Later, in 1991, the study of the starfish Asterias amurensis versicolor allowed Komori´s group to isolate new cerebrosides: asteriacerebrosides AߝF (10–15) [23]. Ten years later, Ishii et al. [24] discovered a new member of this class, asteriacerebroside G (16), from the starfish Asterias amurensis (Figure 2).
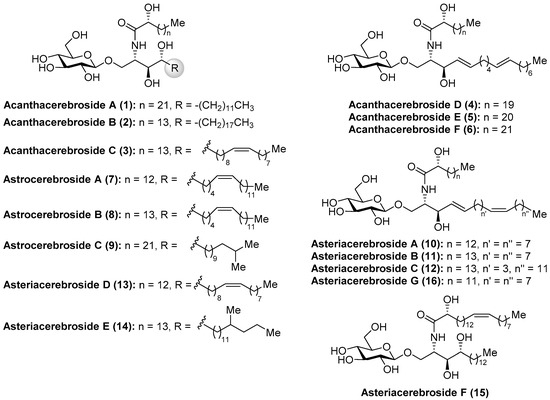
Figure 2.
Structures of the acanthacerebrosides (1–6), astrocerebrosides (7–9), and asteriacerebrosides (10–16).
More recently, from the same starfish (A. amurensis), Kim et al. identified new members of related asteriacerebrosides, whose structures were established by mass spectrometric techniques [25]. After the synthesis of acanthacerebroside A (1) by Komori in 1998, the Chida group [26] reported a new synthesis for acanthacerebroside A (1) and astrocerebroside A (7), in which the preparation of the phytosphingosine moiety was achieved using the commercially available L-quebrachitol (17) (Scheme 1). Accordingly, L-quebrachitol (17) was transformed into the azide 18 in seven steps, which was directed towards the phytosphingosine fragment 20, contained in acanthacerebroside A (1). The coupling of 20 with the donor trichloroacetimidate 21 via a glycosylation reaction mediated by BF3.OEt2 provided the corresponding acanthacerebroside A derivative, which was derivatized to the peracetylated 22 to facilitate purification. After removing the acetate protecting groups, natural acanthacerebroside A (1) was obtained. In a similar manner, astrocerebroside A (7) was efficiently prepared from the phytosphingosine 23, synthesized from the common epoxide 19, and the same glycosyl donor (21). From a biological standpoint, surprisingly only the asteriacerebrosides were evaluated, showing that asteriacerebrosides A (10), B (11), and G (16) displayed growth-promoting activity against the plant Brassica campestris. This was the first report of a promotive plant-growth activity of this class of compounds.
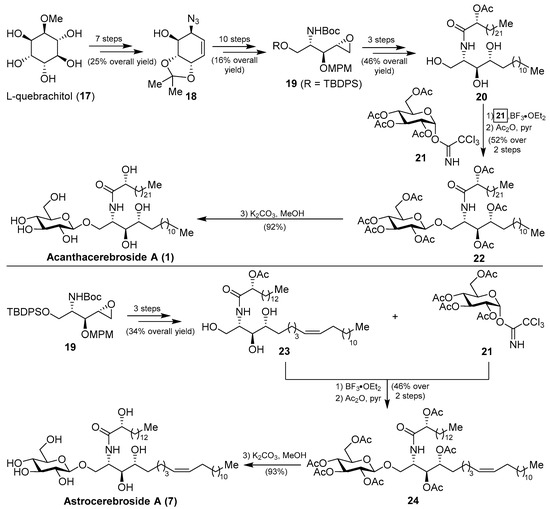
Scheme 1.
Total syntheses of acanthacerebroside A (1) and astrocerebroside A (7).
Agelasphins
Within the family of phytosphingosine-containing cerebrosides of marine origin, the agelasphins (25–31) (Figure 3) occupy a privileged position by virtue of their striking and promising biological properties. The presence of a galactosyl fragment instead of a glucosyl moiety confers upon them unexpected and intriguing biological properties. After the isolation of the first members of this family by Natori et al. in 1993 [27] from the marine sponge Agelas mauritianus, the agelasphins were rapidly recognized as antitumor compounds with weak toxicity, which elicited a great interest in chemical and biological circles. Despite these antitumoral properties, the agelasphins did not exhibit cytotoxicity against B16 melanoma cells at 20 µg/mL, which led Natori, Koezuka, et al. [28] to undertake further biological studies for a rational explanation of these intriguing properties. These studies revealed that these compounds, for example agelasphin-11 (28), were capable of stimulating the immune system via activation of NK cells, which explained not only their potent antitumor activity but also their immunostimulatory property. These outstanding biological findings encouraged the Koezuka’s group to achieve an extensive structure–activity relationship (SAR) study by preparation of a library of their analogues and subsequent antitumor activity evaluation from which the well-known analogue KRN7000 (32) was discovered [29]. At a molecular level, the activation of the immune system exerted by the agelasphins occurs because these compounds are potent ligands of the MHC class I-like CD1d protein, present on the surface of the antigen presenting cells (APCs), and, as a consequence of this potent interaction, an overwhelming response by the immune system is triggered. Thus, the invariant natural killer T cells (iNKT cells) are initially activated by producing high levels of cytokines, which, in turn, activate other antitumor effector cells, resulting in a strong immunological response by the organism against tumor cells. These important findings rapidly propelled KRN7000 (32) as a novel and promising anticancer agent, which recently entered into phase I clinical trials against various types of cancers [30]. As a consequence of its intriguing biological properties, KRN7000 has elicited widespread interest and excitement in both the biological and chemical fields. Indeed, an indication of this great interest is a flurry of activity directed toward the synthesis of a plethora of analogues for SAR studies [31,32]. Related to the agelasphins, Mangoni’s group [33] has recently identified a disaccharide derivative, named damicoside (33), from the marine sponge Axinella damicornis. This glycolipid represents the first disaccharide, structurally related to that of agelasphins, and with an immunostimulatory activity similar to the agelasphins, allowing the completion of the structure–activity relationship study of this fascinating class of compounds.
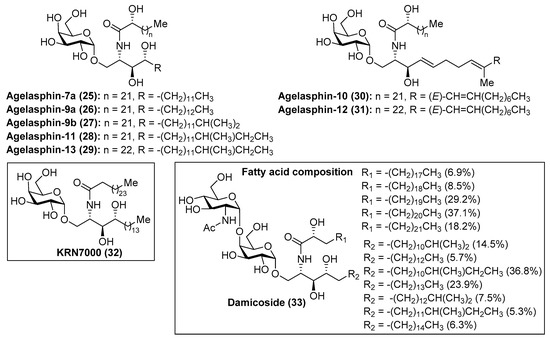
Figure 3.
Structures of the agelasphins (25–31), KRN7000 (32), and damicoside (33).
In order to confirm the structures of the agelasphins, Natori, together with Akimoto et al. [34], achieved the first total synthesis of an agelasphin member, agelasphin-9b (27). Accordingly, the construction of the phytosphingosine fragment commenced with the Wittig reaction of aldehyde 34 with phosphonium salt 35 to give a mixture of isomeric alcohols 36 in 68% yield. The alcohol 36 was then converted into the azide 37 in six steps in 45% overall yield. Subsequent reduction of the azide and acetylation of the corresponding amine with p-nitrophenyl ester 38 afforded a protected ceramide 39 in 40% overall yield. Reaction of 39 with galactosyl fluoride 40, under Mukaiyama’s glycosylation conditions, gave the corresponding α-galactoside in 36% yield, which was finally transformed into agelasphin-9b (27) after a deprotection of the protecting groups (Scheme 2). This synthesis was important not only because the authors were able to confirm the structures of these natural products, but also because the strategy for the future construction of these molecules was established and extended to the synthesis of other agelasphins and their analogues, such as KRN7000 (32) [18,19].
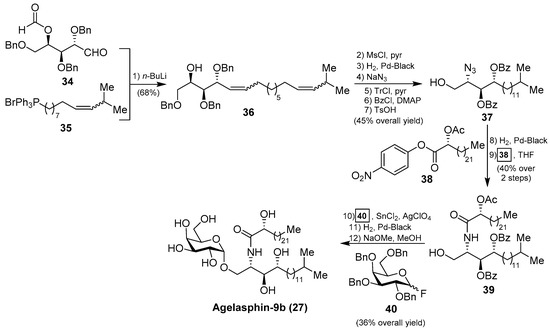
Scheme 2.
Total synthesis of agelasphin-9b (27).
Axidjiferosides
Barnathan et al. [35] described the isolation of axidjiferosides A–C (41–43) (Figure 4) from the marine sponge Axinyssa djiferi, collected from mangrove tree roots in Senegal. Interestingly, the structure of these compounds contained the unusual Δ6-phytosphingosine and also the unusual β-configuration of the galactopyranosyl unit. These compounds showed significant antimalarial activity, with an IC50 of 0.53 ± 0.2 µM against a chloroquine-resistant strain of Plasmodium falciparum. In addition, the axidjiferosides also showed antiplasmodial activity, with low cytotoxicity against various human cancer cell lines, and no significant antitrypanosomal and antileishmanial activities. In contrast to the α-galactosylceramide derivatives, such as the agelasphins, which displayed immunostimulating properties, the antimalarial activity exhibited by the axidjiferosides was ascribed by the authors to the β anomeric configuration present in these natural products.
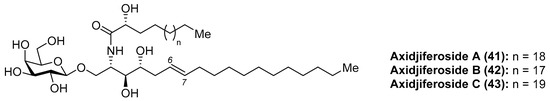
Figure 4.
Structures of the axidjiferosides (41–43).
Cerebrosides CE
Cerebrosides CE (44–48) (Figure 5) were isolated, together with ganglioside CG-1 (See Section Ganglioside CG-1), from the sea cucumber Cucumaria echinata [36]. These cerebrosides showed toxicity in a brine shrimp lethality assay with the rates of 11–27%. However, no further chemical and biological studies about these cerebrosides have been carried out so far.
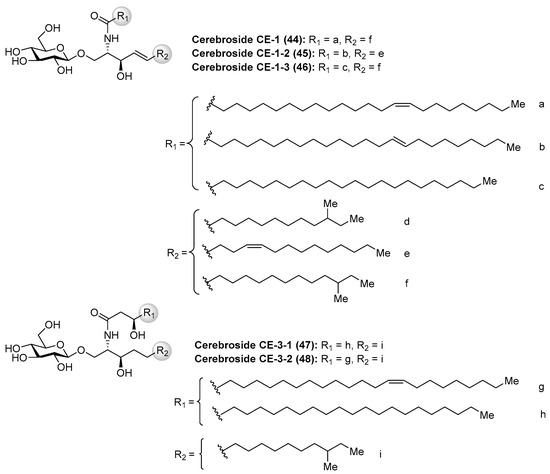
Figure 5.
Structures of the cerebrosides CE (44–48).
Halicylindrosides
Within the family of cerebrosides of marine origin, the halicylindrosides (49–58) (Figure 6) were isolated and characterized by Fusetani´s group in 1995 from the marine sponge Halichondria cylindrata [37]. These structural studies revealed that the halicylindrosides were a new family of phytosphingosine-containing cerebrosides, which promoted a great deal of expectation by virtue of their antifungal activity against Mortierella remanniana at 250 µg/disk and citotoxity against P388 murine leukemia cells at 6.8 µg/mL, respectively.
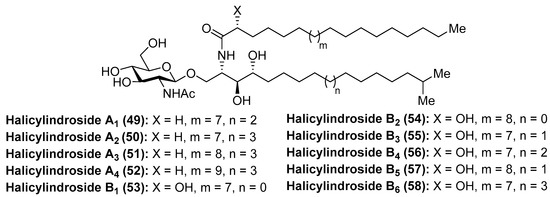
Figure 6.
Structures of the halicylindrosides (49–58).
The synthesis of the halicylindroside A homologue 59 was carried out by Murakami et al. [38], paving the way for the synthesis of the natural congeners. Thus, 61, which was prepared from N-benzoyl-d-glucosamine 60 in 87% over two steps, was transformed into the oxazoline derivative 63 in seven steps in 41% overall yield, involving regioselective O-methanesulfonation and diastereoselective Grignard addition as key steps. The coupling between 63 and N-chloroacetyl glucosyl chloride 64, which was chosen as a more efficient donor for the glycosylation reaction, was achieved by using silver trifluoromethanesulfonate as the promoter at 70 °C to obtain 65 in 75% yield. Reduction of the chloroacetyl 65 with n-Bu3SnH afforded acetamide 66 in 90% yield. Subsequent acid-catalyzed ring opening of the oxazoline 66, followed by reaction with palmitoyl chloride of the resulting amino alcohol provided 67, which was finally transformed into halicylindroside A homologue (59) after a basic treatment (Scheme 3). In a similar manner, a homologue of halicyndroside B (53) was obtained by using (2R)-acetoxypalmitoyl imide.
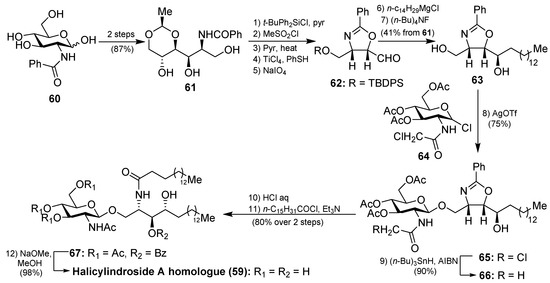
Scheme 3.
Total synthesis of halicylindroside A homologue 59.
Phallusides
Phallusides (68–71) (Figure 7) were isolated from the Mediterranean ascidian Phallusia fumigata and from the starfishes Allostichaster inaequalis and Cosmasterias lurida [39] and their structures were elucidated by a combination of spectroscopic and chemical degradation studies. These studies revealed the presence of an unusual sphingoid base, which corresponded to the 2-amino-9-methyl-d-erythro-(4E,8E,10E)-octadeca-4,8,10-triene-1,3-diol. Prior to these findings, Karlsson et al. [40] had described isolation and structure elucidation of a closely related glycosphingolipid from the sea anemone Metridium senite, which was assigned as Sch II (72). Interestingly, this compound was also isolated from the basidiomycete Schizophyllum commune some years later [41].
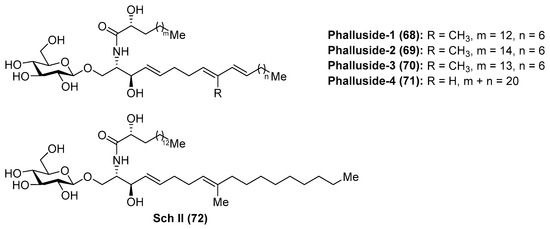
Figure 7.
Structures of the phallusides (68–71) and Sch II (72).
The biological activity evaluation of phallusides 1–3 (68–70) revealed that these compounds possessed significant activity as antifungal agents against several phytopathogenic fungi (Fusarium oxysporum f. sp. Niveum, F. solani f. sp. Cucurbitae, Pythium ultimum, and Alternaria solani) [42].
The first synthetic explorations of these compounds were attempted by Kocienski et al. [43] leading to the total syntheses of phalluside-1 (68) and Sch II (72) via a Cu (I)-mediated 1,2-metallate rearrangement of a lithiated glycal as a key step for the stereoselective synthesis of the sphingoid base. In this direction, the authors envisioned the synthesis of the azidosphingatrienine 79 from iodoalkene 73 through two sequentially 1,2-metallate rearrangements involving cyanocuprate intermediates, derived from lithium derivatives 74 and 77, to obtain the triene triol 78. Conversion of 78 into the azide 79 proceeded in eight steps in 17% overall yield. The key Schmidt glycosylation reaction of the alcohol 79 with the donor 80, using BF3.OEt2 as a promoter, afforded the glycoside 81 in a modest 34% yield. To complete the synthesis, a Staudinger reaction of the azide 81, followed by an N-acetylation of the resulting amine with the acid 82 generated protected phalluside 83 in 63% yield over two steps. Final debenzoylation under basic conditions gave phalluside 68 in a 72% yield. For the synthesis of Sch II (72), the authors employed the same synthetic strategy using the same lithium derivatives 74 and 77 but starting from a different iodoalkene (Scheme 4).
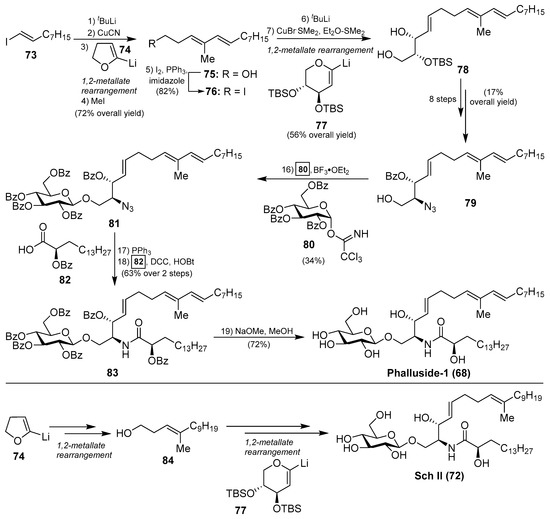
Scheme 4.
Total synthesis of phalluside-1 (68) and Sch II (72).
2.1.2. Diosylceramides
Amphiceramides
Amphiceramides A (85) and B (86) were isolated from the Caribbean sponge Amphimedon compressa by Mangoni et al. [44] (Figure 8). The structures of both glycolipids contain an unusual Δ6-phytosphingosine unit, which can be found in other secondary metabolites such as the aforementioned axidjiferosides A–C (41–43). In addition, amphiceramide A (85) contains an uncommon N-acetyl-β-glucosamine, which has never been found in any natural product. Amphiceramide B (86) was the first glycosphingolipid that possesses an allolactose [Gal(1 β→6)Glc] residue β-linked to the ceramide moiety.
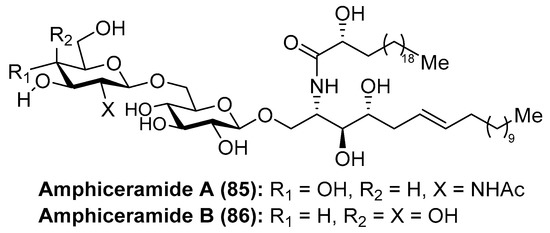
Figure 8.
Structures of the amphiceramides A (85) and B (86).
Plakosides
Fattorusso et al. [45] described the isolation of plakosides (87–90) (Figure 9) from the marine sponge Plakortis simplex, which featured the presence of a cyclopropane ring in the lipidic chain and a 2-O-prenylation of the carbohydrate moiety. Interestingly, in contrast to the immunostimulating property of related glycosphingolipids, such as agelasphins, the plakosides exhibited potent immunosuppressive activity, with plakosides A (87) and B (88) as inhibitors of the proliferative response of lymph-node cells when T cells were stimulated with concavaline A in all doses tested (0.01–10 µg/mL). In addition, these natural products did not display cytotoxic property. This intriguing immunosuppressive activity was ascribed to the presence of the susbstituent on C-2 of the inner monosaccharide. Some years later, the same authors isolated two new related glycosphingolipids, plakosides C (89) and D (90), from another marine-sponge species, Ectyoplasia ferox [46]. Further investigations led to the conclusion that the plakosides are in fact biosynthesized by sponge-associated bacterial symbionts and not from the sponge itself [46].
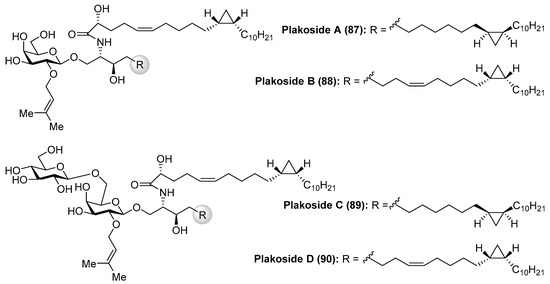
Figure 9.
Structures of plakosides 87–90.
The interesting molecular structures of the plakosides, combined with their intriguing immunosuppressive activity, have prompted their total synthesis to further investigate their biological properties. Thus, Nicolaou et al. [47] reported an efficient and stereoselective total synthesis of these glycosphingolipids, which provided not only sufficient amounts of the compounds for further biological studies, but also some analogues that allowed the determination of key structural factors for the immunosuppressive activity through a structure–activity-relationship study. Having prepared the cyclopropane-containing derivatives 91 and 92 in stereoselective manner, a first assembly of 91 with the corresponding galactosyl fluoride 93 was achieved in an excellent 93% yield in a stereoselective glycosylation reaction mediated by SnCl2/AgOTf. The introduction of the prenyl group was then carried out in two steps to obtain the key intermediate 94, which was prepared for the final steps (Scheme 5). These steps consisted of an amide coupling between 94 and the fatty acid 92, followed by a global protecting group deprotection in two additional steps.
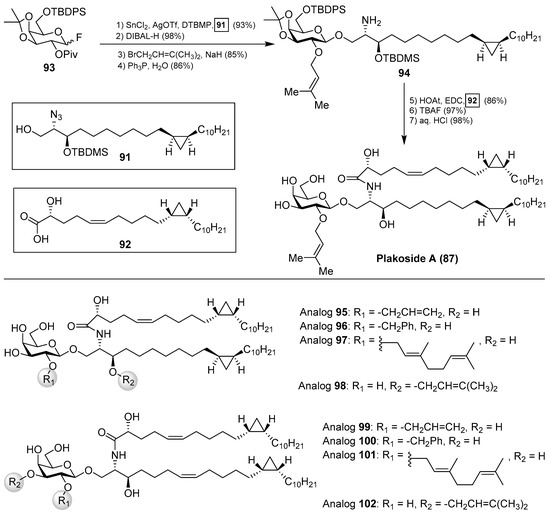
Scheme 5.
Total syntheses of plakoside A (87) and its analogues 95–102.
In a similar manner, the synthesis of plakoside B (88) was efficiently accomplished by using the suitable sphingosine derivative. This synthetic strategy was extended to the preparation of a small collection of plakoside analogues 95–102, in which the effect of the prenyl group upon the biological activities was analysed by modification of this group. In fact, with all these plakosides in hand, their biological activities were evaluated in three in vitro assays (the mixed-lymphocyte-reaction proliferation (MLR) assay, the concanavalin A response assay, and the murine bone marrow cell proliferation assay). Surprisingly, the authors found that all these compounds (95–102), and the synthetic plakosides A (87) and B (88), displayed modest immunosuppressive activity compared to the reported activities for the natural compounds. Among them, analogue 95 was the most active in the series, albeit in a modest range, with an IC50 value of 7.1 µM in the MLR proliferation assay. The discrepancy between the immunosuppressive activities of the synthetic plakosides and of the natural products reported in the literature could be explained by the work of Mori et al., who corrected the absolute configuration initially assigned for the plakosides [48,49]. These authors demonstrated, through their total synthesis described in Scheme 6, that the absolute configurations of the stereogenic carbons of the cyclopropane rings present in both lipidic chains, through compounds 103 and 104, were opposite to those initially proposed by the Fattorusso’s group [50,51]. The incorrect configurations assigned for plakoside A (108) could explain the poor immunosuppressive activity exhibited by the synthetic plakosides A and B prepared by Nicolaou et al. which corresponded to the stereoisomers of the naturally ocurring counterparts. It is interesting to point out that the structures of the synthetic plakosides obtained by Nicolaou’s group possessed the same spectroscopic data and optical rotations as those from naturally occurring plakosides. This fact led them to conclude that the synthetic plakosides were the same as the naturally occurring counterparts.
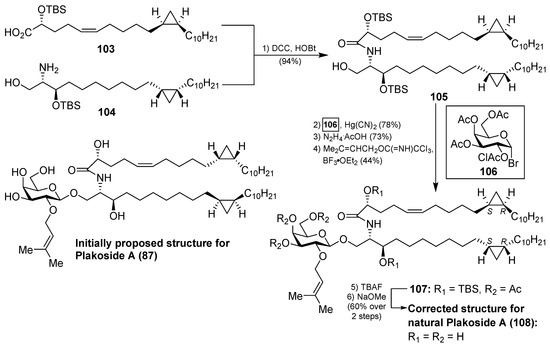
Scheme 6.
Total synthesis of the corrected structure of plakoside A (108).
Terpiosides
Terpioside A (109) and B (110) (Figure 10) were isolated from the marine sponge Terpios sp. by Costantino et al. [52] and represent the first glycosphingolipids reported from sponges of the genus Terpios. Later, Cutignano et al. [53] reported the isolation of the terpiosides from the Antarctic sponge Lyssodendoryx flabellata. The structure of terpioside A (109) was secured by a combination of extensive spectroscopic analysis, as well as chemical degradation studies, revealing the presence of a unique sugar moiety, comprised of an α-fucofuranoside, linked to the 3-position of a β-glucopyranoside, which is the sugar residue linked to the ceramide. Thus, terpioside A (109) represents the first natural glycosphingolipid that contains an l-fucose in a furanose form. On the other hand, terpioside B (110) contains a pentasaccharide chain, which possesses two terminal α-l-fucofuranose units. The biological activity evaluation of terpioside B (110) led to the discovery that this compound was capable of inhibiting LPS-induced NO release, displaying higher activity than simpler glycosphingolipids, such as terpioside A (109) and monoglucosylceramide [54].
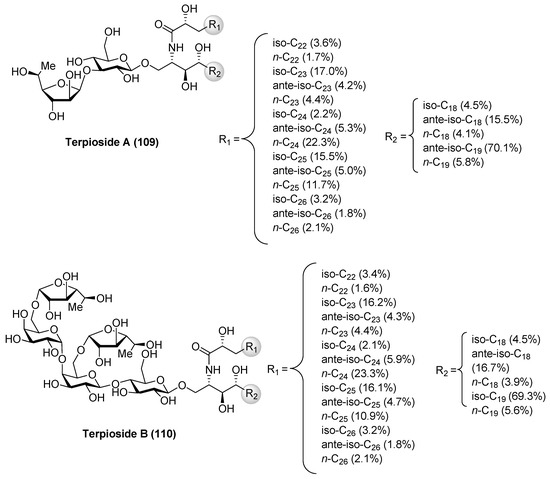
Figure 10.
Structures of terpiosides A (109) and B (110).
2.1.3. Neutral Glycosphingolipids with Oligosaccharide Chains
Agelagalastatin
Agelagalastatin (111) was isolated from the Western-Pacific marine sponge Agelas sp. as a mixture of two isomers (111a and 111b) through a human cancer cell line bioassay carried out by Pettit et al. [55]. Agelagalastatin showed significant in vitro antitumoral activity against various human cancer cell lines, including lung NCI-H460, brain SF-295, renal A498, colon KM20L2, melanoma SK-MEL-5, and the ovarian OVCAR-3, with a range of GI50 values from 0.77 µg/mL to 2.8 µg/mL. Structurally, this natural product contains an unprecedented digalactofuranosyl unit, which has not been found before in a natural product.
This intriguing structure, in conjuction with its antitumor activity, drew the attention of many synthetic chemists and, as a consequence, a total synthesis of agelagalastatin (111) was reported by Kim et al. [56] based on an α-selective glycosylation of the ceramide 115a or 115b with the trisaccharide fluoride 114, prepared from monosaccharide derivatives 112 and 113 after several steps. This key glycosylation reaction, performed in the presence of SnCl2, AgClO4, and DTBMP, furnished 116a and 116b in 72 and 73% yields, respectively. Completion of the synthesis involved the final deprotection of the benzyl, O-acetyl, and O-benzoyl groups in 70 and 74% overall yields to obtain agelagalastatins 111a and 111b, respectively (Scheme 7).
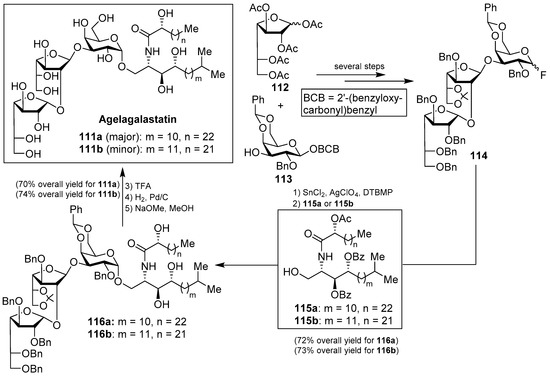
Scheme 7.
Structures of agelagalastatins (111a and 111b) and their total synthesis.
Clarhammosides
The clarhamnosides 117axby (Figure 11) were isolated as an inseparable mixture of different members from the marine sponge Agelas clathrodes by Costantino et al. [57]. The combination of 2D NMR and CD spectroscopic techniques allowed the structural determination of this complex glycosphingolipid mixture. Clarhamnosides are the first α-galactoglycosphingolipids with a l-rhamnose unit in the sugar head. In addition, the sequential two 1,2-cis-α-d-galactopyranosidic linkages are also rare in nature.
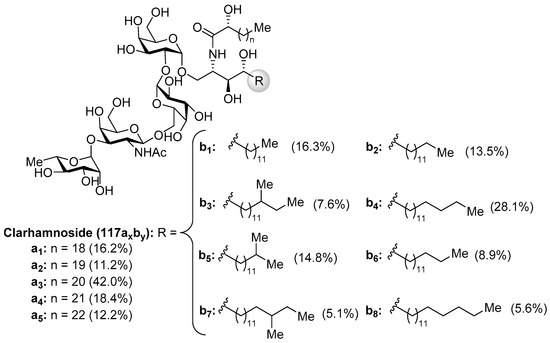
Figure 11.
Structures of the clarhamnosides (117axby).
The first total synthesis of a clarhamnoside member (117a2b6) was carried out by Li et al. [58]. According to this work, the glycosylation reaction between 118 and 119, promoted by the NIS/AgOTf system, afforded a 3:1 α:β separable mixture of the disaccharide 120 in 76% yield. Removal of the benzoyl group in 120 gave the disaccharide 121 in 86% yield, which was used as an acceptor for the next glycosylation reaction. In this reaction, 121 was reacted with donor trifluoroacetimidate 122 employing TMSOTf and 4 Å MS at −20 °C to provide the β-glycoside 123 in 76% yield. Conversion of the N-phthalimido group into an acetamido group by sequential treatment with ethylenediamine and Ac2O afforded 124 in 72% yield in two steps. The reduction of 124, employing H2S in pyridine/water, was prior to the corresponding coupling of the resulting amine with α-hydroxy acid 125 to provide 126 in 66% yield in two steps. Final hydrogenolysis afforded the corresponding tetrasaccharide intermediate, which was acetylated to obtain clarhamnoside peracetate 127. Removal of the acetyl groups furnished clarhamnoside 117a2b6 in a moderately good yield (Scheme 8).
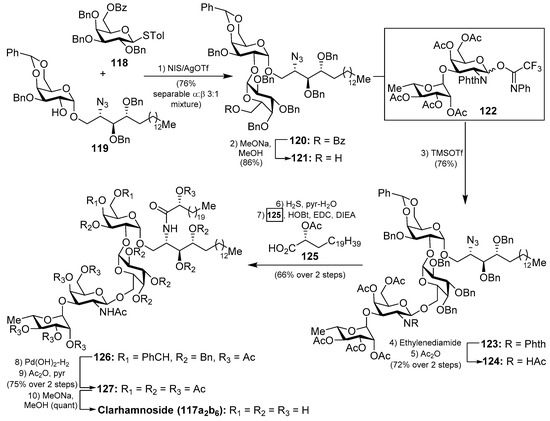
Scheme 8.
Total synthesis of clarhamnoside 117a2b6.
Vespariosides
After the isolation of vesparioside A (128) by Mangoni et al. [59] from Spheciospongia vesparia in 2005, the same authors in 2008 reported the isolation and structural elucidation of a unique furanose-rich glycosphingolipid that was named vesparioside B (129) (Figure 12) [60]. Through an exhaustive spectroscopic analysis, supported by theoretical and degradation studies, the authors were able to establish the absolute configuration of this stunning molecule, which was unambiguously confirmed through the total synthesis carried out by Yang et al. in 2016 [61]. In this total synthesis, after an extensive exploration of various strategies, the authors decided to construct initially the glycosphingolipid derivative 133, via a glycosylation reaction of the donor trichloroacetimidate 131, prepared from the disaccharide 130, and the phytosphingosine acceptor 132.
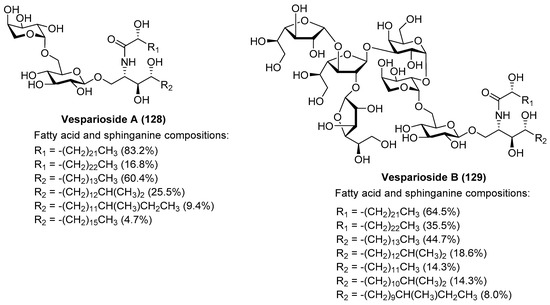
Figure 12.
Structures of vespariosides A (128) and B (129).
The resulting glycosphingolipid 133 was then prepared for assembly with the tetrasaccharide fragment in the form of thioglycoside 135. To this end, the removal of the PMB group of 133 by treatment with DDQ provided the alcohol 134, which was reacted with the donor 135 under the action of the promoter NIS/TfOH. The resulting complex glycosphingolipid 136, obtained in excellent yield and stereoselectively, was then transformed into natural vesparioside B (129) after removal of all the protecting groups in a 60% overall yield in three steps (Scheme 9).
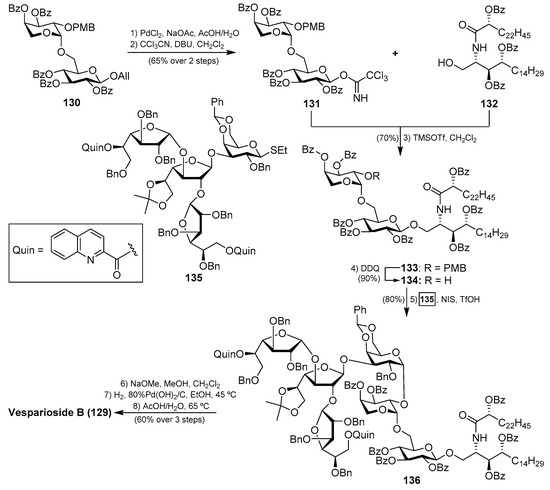
Scheme 9.
Total synthesis of vesparioside B (129).
Interestingly, the preparation of the tetrasaccharide 135 required the stereoselective synthesis of 1,2-cis-α-galactofuranoside units, for which some methods have been recently developed. In this case, Yang’s group has developed an elegant methodology based on a hydrogen-bond-mediated aglycone delivery (HAD) strategy for which they employed a 2-quinolinecarbonyl (Quin) functionality. This group, acting as an H-bond acceptor and incorporated in the α-face of the galactofuranoside ring, displays a strong α-stereocontrol effect in glycosylation reactions when furanosyl thioglycoside donors are employed in reactions with various acceptors. The application of this strategy in the synthesis of tetrasaccharide 135 commenced with the glycosylation reaction of the readily available galactofuranosyl acceptor 137 and the donor 138, which was armed with the Quin group at the 5-position. This reaction delivered exclusively the α-disaccharide 139 in an excellent 95% yield, proving the reliability and validity of this strategy. The assembly of the third galactofuranosyl derivative proved more challenging, as the use of the 5-O-Quin derivative 138 as a donor did not work, resulting in recovered starting materials. Fortunately, when the 6-O-Quin derivative 141 was used instead, the reaction afforded the expected trisaccharide 142 as a 6:1 α/β separable mixture in an 85% combined yield. Finally, the fourth glycoside was incorporated through a glycosylation reaction with the trichloroacetamidate derivative of 142 and the thioglycoside acceptor 143 to deliver tetrasaccharide 135 in a 55% yield as a single β-anomer (Scheme 10). This total synthesis not only confirmed the molecular structure of this fascinating glycosphingolipid, but also, more importantly, provided the opportunity for further biological exploration of this class of compounds, which was not achievable due to the scarcity of material from natural sources. These biological studies with synthetic vespariosides are currently in progress.
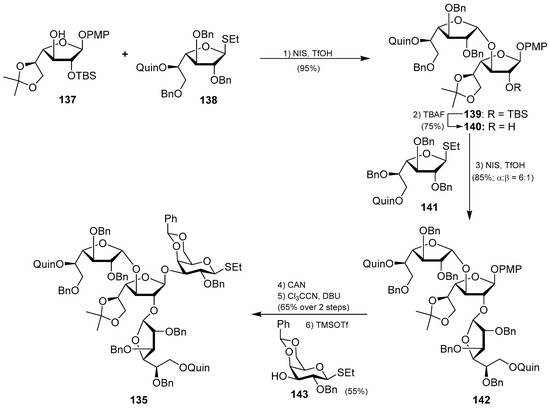
Scheme 10.
Synthesis of the tetrasaccharide unit 135 of vespariosides.
2.2. Acidic Glycosphingolipids
2.2.1. Gangliosides
Structurally characterized by the presence of one or more sialic acid units, the gangliosides of a marine origin are almost exclusively found in echinoderms, mainly from urchins. In other marine invertebrates, it has been demonstrated that the sialic acid unit is replaced by a glucuronic acid residue. Despite the limited distribution of this class of glycosphingolipids in the marine realm, a huge number of natural products belonging to this category has been discovered. As a consequence, we have selected for this section the most biologically relevant gangliosides.
Ganglioside Hp-s1
Ganglioside Hp-s1 (144) was isolated from the ovary of the sea urchin Diadema setosum or from the sperm of the sea urchin Hemicentrotus pulcherrimus [62]. This compound showed an interesting neuritogenic activity towards the rat pheochromocytoma PC-12 cell line in the presence of nerve growth factor (NGF) and, when compared to ganglioside GM-1 (25.4%), its effect was better (34.0%). Together with ganglioside Hp-s1, ganglioside DSG-A (145) (Figure 13), isolated also from D. setosum, displayed similar neuritogenic activity, which can be useful for the treatment of neurodegenerative diseases [63].
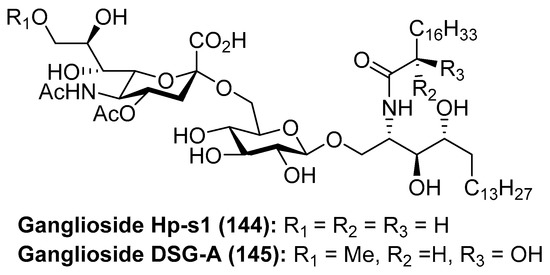
Figure 13.
Structures of ganglioside Hp-s1 (144) and DSG-A (145).
Several syntheses of these natural products have been reported [64]. Initial synthetic efforts were carried out by Tsai et al. [65], who prepared the ganglioside Hp-s1 analogue 151. The synthesis was accomplished by a sequence of chemoselective glycosylation reactions as key steps. Thus, the disaccharide 148 was obtained by the sialylation of the acceptor 147 with the sialyl donor 146a, mediated by NIS/TfOH as a promoter at −30 °C in the presence of 4 Å molecular sieves, in 63% yield as an inseparable α:β 3.2:1 mixture. The next glycosylation reaction involved the resulting disaccharide 148 with the azidosphingosine derivative 149 in the presence of AgOTf and 3 Å molecular sieves at room temperature to obtain the resulting disaccharide derivative as a separable mixture of αβ and ββ anomers in 62% combined yield. From the corresponding αβ anomer, a Staudinger reaction was followed by an amidation process to obtain 150 in 54% yield in two steps. Final debenzylation and deacetylation reactions afforded the ganglioside Hp-s1 analogue 151 in 78% overall yield (Scheme 11). This analogue exhibited neurogenetic activity towards the human neuroblastoma cell line SH-SY5Y without the presence of NGF.
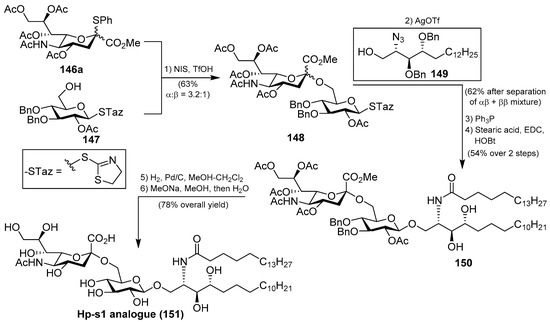
Scheme 11.
Total synthesis of Hp-s1 analogue 151.
A different synthetic strategy was used in the first total synthesis of ganglioside Hp-s1 (144), achieved by Luo et al. [64], based on two key glycosylation reactions. According to this synthetic strategy, after an extensive optimization of the glycosylation reactions, the authors found that the first reaction of phytosphingosine 152 with benzyl protected imidate 153, using TMSOTf as a promoter at −30 °C to room temperature, followed by a deacetylation step, afforded 154 in 82% (β:α 3.2:1) yield. The second glycosylation reaction involved the glycosyl acceptor 154 and the sialyl donor 146b, mediated by NIS/TfOH as a promoter, to obtain 155 in 84% yield (αβ:ββ 3.9:1). From 155, the conversion of the azide group into the corresponding amine, through a Staudinger reaction, followed by amide formation, and final deprotection of acetyl, benzyl, and acetonide groups delivered ganglioside Hp-s1 (144) in 43% overall yield from 155 (Scheme 12).
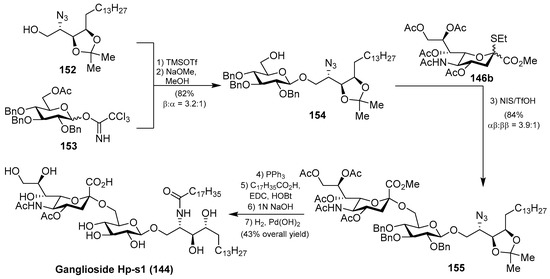
Scheme 12.
Total synthesis of ganglioside Hp-s1 (144).
The same authors carried out an SAR study involving the synthesis of six Hp-s1 analogues (156–161) by replacing the glucosyl unit of Hp-s1 with α-glucose (157), α- and β-galactose (158 and 159), and α- and β-mannose (160 and 161), including the simple cerebroside 156 [66]. After the biological evaluation, the authors found that the C-2 hydroxyl group of the glucosyl unit played a crucial role in the stimulation of neurite outgrowth of SH-SY5Y cells. In addition, it was found that analogue 158 activated NKT cells, although it was inactive on neurite outgrowth of SH-SY5Y cells (Figure 14).
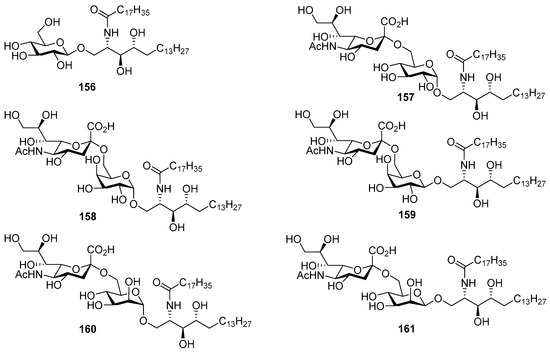
Figure 14.
Gangliosides Hp-s1 analogues 156–161.
Based on these synthetic studies, the same authors developed an efficient method for α-selective sialylation based on a pre-activated 5-N,4-O-carbamate thiosialoside donor using p-TolSCl/AgOTf as promoters which was extended to the synthesis of gangliosides Hp-s1 and DSG-A [67]. Tsai et al. also achieved the total synthesis of ganglioside DSG-A (145) through a chemoselective glycosylation reaction in a [1 +1 + 2] synthetic strategy for the assembly of the four fragments of this molecule [68].
Gangliosides HLG
Gangliosides HLG-1 (162), HLG-2 (163) and HLG-3 (164) (Figure 15) were isolated from the sea cucumber Holothuria leucospilota by Higuchi et al. [69]. These compounds showed similar neuritogenic activity toward the rat pheochromocytoma cell line, PC-12 cell, in the presence of NGF as other marine-derived gangliosides. Structurally, these gangliosides contain a unique α(2,4) linkage between sialic acids, which captured the attention of many synthetic chemists.
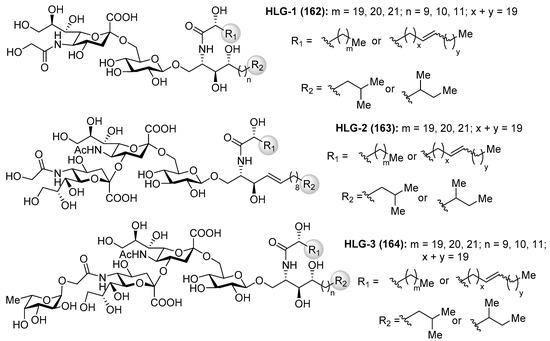
Figure 15.
Structures of gangliosides HLG-1, -2, and -3 (162–164).
The first total synthesis of ganglioside HLG-2 (163) was carried out by Kiso et al. [70]. To construct the glycan unit, sialylation at C-4 hydroxyl group of the sialic acid residue was achieved using the 1,5-lactam-sialyl unit 166 as a novel glycosyl acceptor and readily prepared from 165 by a basic treatment (refluxing NaOMe in MeOH) in 95% yield. The glycosylation reaction of 166 with Troc-sialyl donor 167 in the presence of NIS/TfOH took place exclusively at C-4 hydroxyl group in a stereoselective manner to give the trisaccharide 168 in 69% yield (α:β 60:9). To complete the synthesis, the next glycosylation reaction involved the donor 169, obtained in eight steps from the trisaccharide 168, and the ceramide acceptor 170 in the presence of TMSOTf as a promoter, to obtain the corresponding ganglioside in 49% yield, which was finally transformed into ganglioside HLG-2 (163) in a quantitative yield after a basic treatment (Scheme 13).
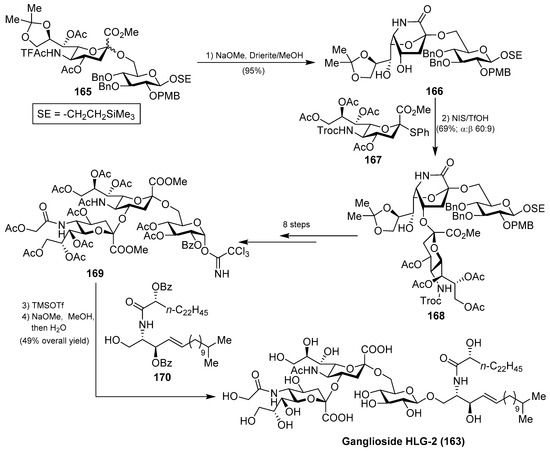
Scheme 13.
Total synthesis of ganglioside HLG-2 (163).
Interesting was the synthetic approach by Ye et al., who achieved the synthesis of the glycan portion of ganglioside HLG-2 in a highly efficient and stereoselective manner, involving two key glycosylation reactions: first, for the construction of the disaccharide 173 using the phosphate donor 172, in the presence of TMSOTf, in 95% yield; and, second, for the generation of the trisaccharide 176 by the use of NIS/TfOH in 96% yield, both with excellent α-stereoselectivity. From 176, final conversion of functional groups and deprotection reactions afforded the trisaccharide core of ganglioside HLG-2 (177) [71] (Scheme 14).
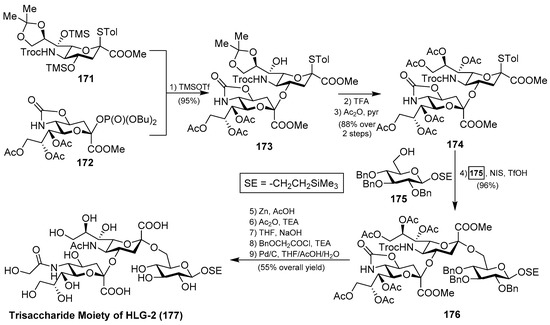
Scheme 14.
Total synthesis of the trisaccharide core 177 of ganglioside HLG-2 (163).
Ganglioside GP-3
Ganglioside GP-3 (178) was isolated from the starfish Asterina pectinifera by Higuchi et al. [72]. As previous gangliosides, GP-3 also exhibited neuritogenic activity toward the rat pheochromocytoma PC-12 cells, in the presence of the NGF, displaying, in this case, a lower effect when compared with ganglioside GM-1. Structurally, the glycan part contains nine monosaccharide units in a unique saccharide sequence with two internal sialic acid residues and three furanose residues.
The total synthesis of ganglioside GP-3 (178) was achieved by Kiso et al. [73]. Briefly, the final glycosylation reaction of the octasaccharide donor 179 with the glucosphingolipic derivative 180 was promoted by TMSOTf to furnish the protected ganglioside GP-3 181 in 77% yield. A final deprotection sequence, which included aqueous TFA and basic treatments, afforded ganglioside GP-3 (178) (Scheme 15).
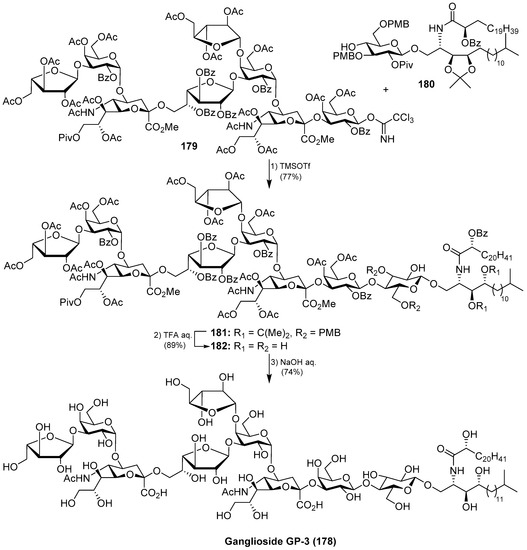
Scheme 15.
Total synthesis of ganglioside GP-3 (178).
Ganglioside CG-1
Ganglioside CG-1 (183), isolated from the sea cucumber Cucumaria echinata by Higuchi et al. [36], showed neuritogenic activity toward the rat pheochromocytoma PC-12 cells. The same authors carried out a partial synthesis of a related CG-1 ganglioside, which lacked the sulfate group in the natural product, by a coupling of 2-thioglycoside 185 with 184, which was prepared from the natural acanthacerebroside (1), in the presence of NIS/TfOH, to obtain the corresponding α-sialoside in 31% yield. This advanced precursor of CG-1 was then subjected to stepwise deprotections to obtain the related CG-1 ganglioside 186 in 93% yield [74] (Scheme 16). This synthetic study can delineate the path towards the total synthesis of this natural product.
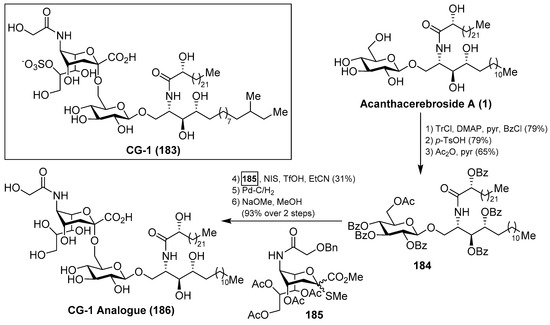
Scheme 16.
Total synthesis of ganglioside CG-1 analogue 186.
Gangliosides SJG
Gangliosides SJG-1 (187) and SJG-2 (188) were isolated from the sea cucumber Stichopus japonicus by Higuchi et al. [75,76] (Figure 16). As the gangliosides described in previous sections, the SJG gangliosides also showed neuritogenic activity toward the rat pheochromocytoma PC12 cells in the presence of NGF. The effect of SJG-2 was higher than that of mammalian ganglioside (proportion of neurite bearing cells SJG-2 64.8%, SJG-1 35.4%, and GM-1 47.0%).
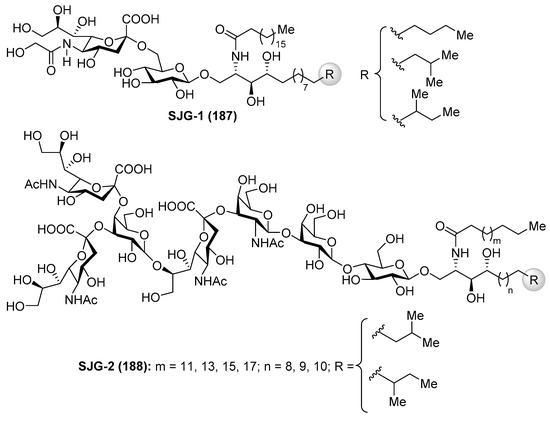
Figure 16.
Structures of gangliosides SJG (187 and 188).
The same authors carried out an SAR study of a few gangliosides from Echinoderms involving trisialo-gangliosides (SJG-2 and LLG-5), disialo-gangliosides (LLG-3 [77], GAA-7 [78], HLG-2, LMG-4, HLG-3, and GP-3), and monosialo-gangliosides (AG-2, AG-3, HLG-1, SJG-1, LMG-2). Evaluation of the neuritogenic activity on a rat pheochromocytoma cell line (PC-12 cells) indicated that (a) the presence of sialic acid is essential; (b) the presence of two terminal sialic acids is key for strong activity; (c) gangliosides having an 8-O-Me sialic acid showed stronger activity; (d) gangliosides possessing sialic acid inside of the oligosaccharide unit showed better activity; (e) the different activity showed by HLG-1 and SJG-1 (44.7 and 35.4%, respectively), which have the same sugar unit, is due to the difference in the structure of their ceramide units; (f) SJG-2, LLG-5, LLG-3, and GAA-7 have better effect than mammalian ganglioside GM-1, which has positive effects in neuritogenical diseases; and (g) these gangliosides showed no activity without the presence of NGF [79].
Gangliosides CEG
Gangliosides CEG-3 (189), CEG-4 (190), CEG-5 (191), CEG-6 (192), CEG-8 (193), and CEG-9 (194) were isolated from the sea cucumber Cucumaria echinata by Higuchi et al. [80,81] (Figure 17). These compounds showed neuritogenic activity toward the rat pheochromocytoma cell line PC-12 in the presence of a nerve growth factor.
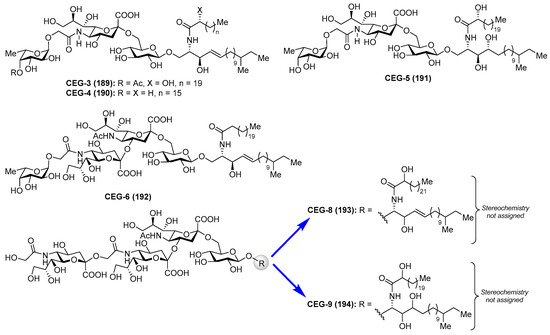
Figure 17.
Structures of gangliosides CEG (189–194).
2.2.2. Sulfatides
Axinelloside A
Axinelloside A (195), a highly sulfated liposaccharide, was isolated from the lipophilic extract of the Japanease marine sponge Axinella infundibula by Fusetani et al. [82]. The structure of axinelloside A (195) was elucidated after an excellent spectroscopic work based on 2D NMR and MS techniques, determining the presence of twelve sugars including scyllo-inositol, d-arabinose, five d-galactoses, and five l-fucose units, to which one (R)-3-hydroxy-octadecanoic acid, three molecules of (E)-2-hexadecenoic acids and 19 sulfates groups were attached. This compound shows similar structure to sulfated polysaccharides from the marine sponge Chondrilla nucula and Dysidea fragilis (Figure 18).
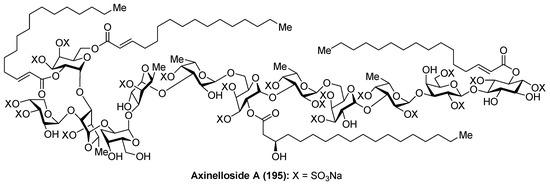
Figure 18.
Structure of axinelloside A (195).
From a biological standpoint, axinelloside A (195) strongly inhibited the activity of human telomerase with an IC50 value of 0.4 µM (2 µg/mL). In this biological activity, it is possible that the sulfate groups play a key role, since it was proven that the dictyodendrins [83], a family of sulfated pyrrolocarbazoles isolated from the sponge Dictyodendrilla verongiformis, lost all telomerase inhibitory activity when their sulfate groups were removed. The mechanism of action of the telomerase inhibitory activity of axinelloside A (195) remains unknown.
Encouraged by the telomerase inhibitory activity of axinelloside A (195), Walczak et al. [84] achieved the synthesis of the scyllo-inositol fragment from d-glucose through a Ferrier rearrangement of a vinyl-acetate derivative and stereoslective reduction of the resulting ketone. The same authors established a synthetic sequence for the preparation of a series of simple sulfated d-galactosyl liposaccharides, inspired by axinelloside A (195), in order to evaluate their potential as new telomerase inhibitors [85]. However, the biological activities of these galactose derivatives have not been reported so far.
3. Chemistry and Biology of Glycoglycerolipids
3.1. Aminoglycoglycerolipids and Related Glycoglycerolipids
Within this class of glycoglycerolipids, it is noteworthy to mention avrainvilloside (196), isolated from the Dominican green alga Avrainvillea nigricans by Taglialatela-Scafati et al. [86]. Related to avrainvilloside (196), the aminoglycoglycerolipid 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol (197a) was isolated from a marine alga, designated as UM2972M, by Kingston et al. This compound displayed potent inhibitory activity against the Myt1-kinase (IC50 = 0.12 mg/mL), an enzyme involved in the regulation of the cdc2/cyclin B kinase activity, which is essential for the growth of tumor cells [87]. Schmidt et al. [88] achieved the total synthesis of aminoglycoglycerolipid 197a by glycosylation reaction of the imidate 198 and the alcohol 199, promoted by TMSOTf, to obtain 200 in a 46% combined yield, as a separable α:β 78:22 mixture of anomers. Deprotection of the major α-anomer 200, followed by an acylation step to introduce a palmitic-acid unit, afforded 201 in 55% yield in two steps. The final Staudinger reaction, followed by an amide coupling with the last palmitic acid unit and global deprotection, gave aminoglycoglycerolipid 197a in 79% overall yield (Scheme 17).
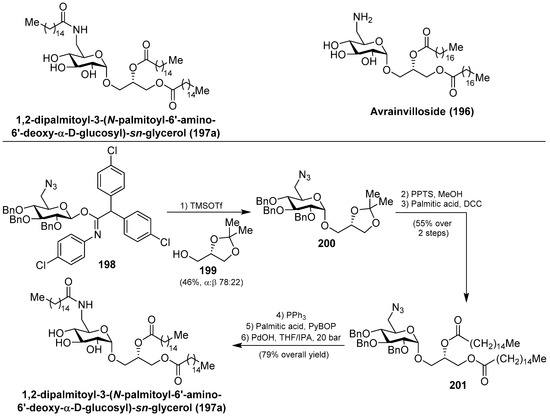
Scheme 17.
Synthesis of 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol 197a.
Later, Li et al. [89] completed the synthesis of the aminoglycoglyceropilpid 197a as well as its acyl analogues (197b–h) in an efficient method employing the trichloroacetimidate donor 202 to obtain 200, by reaction with the glycerol derivative 199, in 88% yield and high α-selectivity (α:β 33:1) as the key step. The same authors synthesized different mannosyl (203a–h) and galactosyl analogues related to 197 in a similar manner. Given the antiviral activity displayed by many glycoglycerolipids isolated from algae, the authors evaluated the inhibitory activity of these amino derivatives against the influenza A virus (IAV) by the cytophatic effects inhibition assay. These analogues displayed inhibition of the viral replication in MDCK cells and it was found that the type of the linkages and the length of the acids could influence the activity. Among them, 203g showed the best activity with an IC50 of 69.9 µM [90,91]. Later, new analogues 204–207 (Figure 19) were prepared and tested for anti-IAV activity, and the authors concluded that acylamino and glycerol groups of the glycolipids were essential for the inhibitory activity on IAV multiplication [92]. From this new series, 204d was the most potent, with an IC50 of 60.8 µM, and may provide a point of exploration of unique aminoglycoglycerolipids in drug discovery for pneumonia caused by viruses. In fact, this compound was selected for preliminary inhibitory studies on IAV infection in vivo. The results of this study showed that 204d exhibited a significant reduction of viral titers in the lungs of IAV-infected mice at a dose of 5 mg/Kg/d, with a striking increase of the survival rate (90%) compared to the infected group treated with oseltamivir.
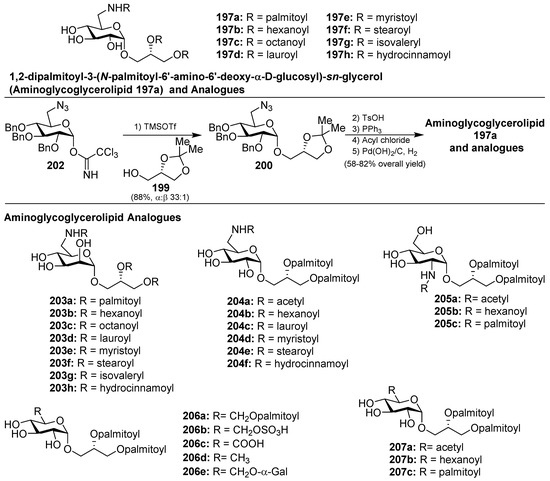
Figure 19.
Structures of aminoglycoglycerolipids based on 197a.
3.2. Crasserides and Isocrasserides
Crasserides (208a–m) (Figure 20), initially isolated by Fattorusso et al. [93] from the Caribbean sponge Pseudoceratina crassa (family Dysideidae), represent unique glycolipids that resemble common glycoglycerolipids by the replacement of an usual sugar by a five-membered cyclitol and the 1-O-acyl group present in the glycerol unit by an 1-O-alkyl group. Since their first isolation, the crasserides have been found in several families of sponges, including Verongula gigantea (family Dysideidae), Aplysina fulva (family Aplysinidae), A. cauliformis (family Aplysinidae), Neofibularia nolitangere (family Mycalidae), Agelas clathrodes (family Agelasidae), A. dispar (family Agelasidae), A. conifera, A. longissima, Plakortis simplex (family Plakinidae), Ectyoplasia ferox (family Raspailiidae), and Siphonodictyon coralliphagum (family Niphatidae) [94]. The structures of these interesting natural products were established and their stereochemistry was determined by exhaustive NMR spectroscopic analyses. Preliminary biological evaluations of these compounds revealed that the crasserides are potent feeding deterrents, with a concentration as low as 30 µg/cm2 of food pellets according to the antifeedant assay on the fish Carassius auratus. Almost ten years later, the same authors discovered from the same sponges a new family of related compounds, which are termed isocrasserides (209a–m) [94], which accompanied the crasserides in minor amounts, demonstrating that these compounds are genuine natural metabolites and not artifacts, as initially suspected.
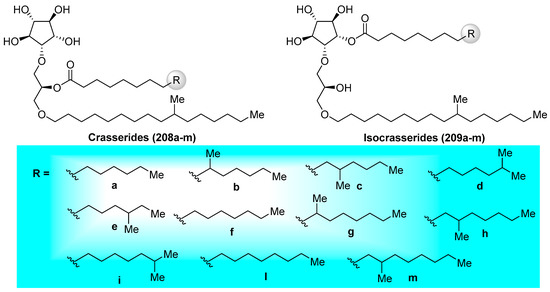
Figure 20.
Structures of the crasserides (208a–m) and the isocrasserides (209a–m).
3.3. Myrmekiosides
The myrmekiosides (210–212) belong to the family of the glycoglycerolipids, isolated from the marine sponges Myrmekioderma sp. and Trikentrion loeve by Barnathan’s group and displayed potent antitumor activity [95]. In fact, their preliminary biological evaluations revealed that myrmekiosides altered the tumor cell morphology of H-ras transformed NIH3T3 fibroblasts to normal at concentrations of 5 µg/mL. Later, Barnathan et al. isolated a new and related glycoglycerolipid from M. dendyi, which was named myrmekioside E (213), and found that its peracetylated derivative (214) exerted significant cytotocixity against NSCLC-N6 and lung tumor A549 cells with IC50 values of 7.3 and 9.7 µM, respectively [96]. Like metabolites isolated from a marine origin, the scarcity of a bioactive material demanded its chemical synthesis as the only way to provide access in sufficient quantities for further biological evaluations. Prompted by this requirement, together with the unique mono-O-alkyl-diglycosylglycerol structure, Li’s group published in 2015 in what represents the first and, so far, only total synthesis of myrmekioside A (210) [97]. Accordingly, careful inspection of this unprecedented molecular framework led the authors to prepare the xylosylglycerol 215 as the starting material to incorporate in subsequent steps other sugar units. Thus, the glycerol moiety was elaborated for the next glycosylation reaction with the trichloroacetimidate 216 to obtain, in very high yield, diglycosylglycerol 217, after removal of the acetate group under basic conditions. Compound 217 was then reacted with thioglycoside 218, using NIS/TfOH as a promoter, to give the corresponding glycoglycerol in an excellent 90% yield. At this point, the authors had to replace the acetyl protecting groups by benzyl ethers, and then the resulting derivative 219 was driven to the completion of the synthesis of myrmekioside A (210), for which the etherification reaction to introduce the pending lipidic chain was carried out by reaction of the alcohol resulting from the desilylation process of 219 with the mesylate 220 (Scheme 18). This synthesis demonstrated the proposed structure for this natural product and, in addition, that the modular character of the synthetic strategy could be useful for the preparation of other myrmekioside derivatives and analogues for further biological studies.
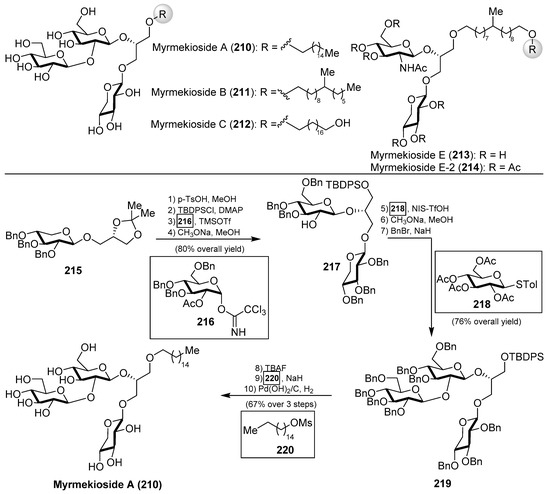
Scheme 18.
Structures of the myrmekiosides (210–214) and total synthesis of myrmekioside A (210).
3.4. Nigricanosides
Isolated as methyl esters (223 and 224) from the green alga Avrainvillea nigricans in Dominica [98], the nigricanosides (221 and 222) represent a unique and unprecedented class of glycolipids with outstanding antitumoral activity in the low nM range with an IC50 value of 3 nM for 221 against human breast cancer MCF-7 and colon cancer HCT-116 cell lines (Figure 21). More intriguing was the recognition of the ability of the nigricanosides to promote a polimerization of tubulins as the mechanism of action of their antiproliferative property. However, the tremendous scarcity of these natural products from the natural sources (Only 800 µg and 400 µg of the methyl esters of nigricanosides A and B, 223 and 224, respectively, were obtained from 28 Kg of wet material!) represents a significant hurdle to gain further insight into the biological properties and to establish the absolute configuration of the compounds. Consequently, the total synthesis represents the only means to determine unambiguously the stereochemistry of the chiral centers and to have enough material for further biological screenings.
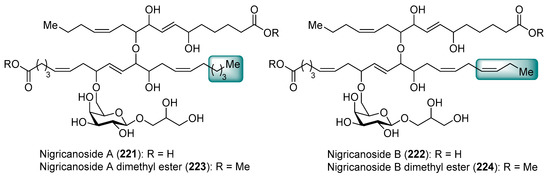
Figure 21.
Structures of nigricanosides A (221), B (222), and their dimethyl esters, 223 and 224.
Thus, several synthetic approaches have been initiated for the lipidic fragments [99,100,101], but many of them have not solved the stereochemical assignment problem. In fact, this stereochemical assignment represents a formidable synthetic challenge because of the presence of seven chiral centers in the lipidic unit, which could provide up to 128 possible stereoisomers, assuming that the configuration for the sugar unit and the geometry of the double bonds were correctly established (Scheme 19). This synthetic challenge was taken on by the McMillan [102] and Ready [103] groups, which used a flexible synthetic strategy capable of delivering all the possible stereoisomers. In addition, the knowledge of the stereochemistry of a related 20-C fatty acid, trioxilin A3 (225), isolated and identified as a hydrolysis product of the natural epoxide hepoxilin A3, led the authors to propose this stereochemistry for the 20-C unit of the nigricanosides. Thus, according to Scheme 19, they prepared the ester 226 in a convergent and efficient way from (R)-glycidol as the chiral source, which was prepared for the subsequent assembly of the 16-C fatty acid unit via an ether linkage. To this aim, the free alcohol of 226 was treated with 2-bromo acetic acid and, after the incorporation of the chiral auxiliary 227, the alkylation with (Z)-1-iodo-2-hexene afforded, with a complete stereoselectivity, the corresponding alkylated product 228. The use of the enantiomer ent-227 provided the corresponding stereoisomer at this position. By continuation of the product 228, the elongation of the second fatty acid was achieved in four additional steps, in which the chiral ester (R)-229 was incorporated via a hydrozirconation reaction with the Schwartz reagent with a subsequent transmetalation with dimethylzinc. In a similar manner, the authors used the enantiomer (S)-229 to generate all the possible stereochemical combinations of the lipidic fragment. The completion of the lipidic moiety was undertaken in five additional steps, in which the resulting product 230 was prepared for the introduction of the sugar moiety. In this event, the alcohol derived from the deprotection of the SEM group was treated with the galactosyl triflate 231 to obtain glycolipid 232, which was finally directed towards the nigricanoside A methyl ester via a glycosylation reaction of the trichloroacetimidate derivative of 232 with the alcohol 199. Unfortunately, the resulting synthetic nigricanoside 223a did not match with the reported natural product and, furthermore, was inactive against HCT-116 and MCF7 tumoral cell lines. A detailed comparative analysis of the 1H NMR spectra of synthetic diester 223a and natural nigricanoside A dimethyl ester 223 revealed that the most important differences were located at the C7-C8 trans olefin region. These spectroscopic differences led the authors to propose a C6/C9 syn relationship for the natural product instead of the initially proposed anti relative configuration.
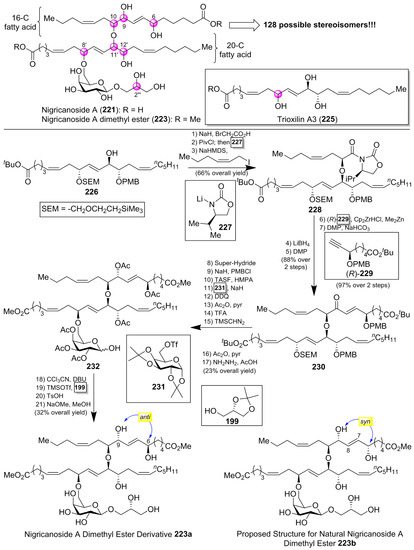
Scheme 19.
Synthesis of proposed natural nigricanoside A dimethyl ester (223a).
Consequently, the nigricanoside derivative 223b was prepared in the same way as for 223a, and all its spectroscopic and physical properties matched with those reported for the natural product dimethyl ester. Despite this structural correspondence, it was surprising to find that neither 223b nor its epimer at the glycerol subunit displayed cytotoxicity against HCT116 or MCF7 cells up to 10 µM. Due to the lack of cytotoxicity for the synthetic material compared to the reported activity for the natural product an explanation is warranted. One such explanation may be the presence of an unidentified product in the fractions corresponding to the natural nigrecanosides as the molecule responsible for the antitumoral activity exhibited by these natural products. The brilliant and outstanding synthetic work by McMillan and Ready’s groups allowed the stereochemical assignment of these fascinating natural products. However, the biological properties of the synthetic nigricanosides have opened an important uncertainty about the identity of the compound responsible for the antitumoral activity detected for the isolated compounds, which must be resolved in future investigations.
4. Chemistry and Biology of Atypical Glycolipids
4.1. Agminosides
Agminosides A–E (233–237) were isolated from the New Zealand sponge Raspailia agminata by Northcote et al. [104] (Figure 22). Structurally, these compounds possess only one type of aglycone, and contain up to six partially acetylated glucose residues that differ only in the level of acetylation and the number of sugars. Their structural similarity made their separation challenging, which was achieved only after repetitive normal-phase chromatography, which was more difficult due to the lack of a chromophore. Mass spectrometry-guided isolation and extensive NMR analysis, together with chemical derivatization, were used for the identification of their structures.
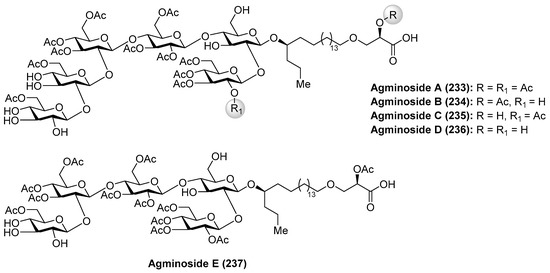
Figure 22.
Structures of agminosides A–E (233–237).
4.2. Ancorinosides
Ancorinoside A (238) was isolated from the marine sponge Ancorina sp. by Ohta et al. [105] through bioassay-guided purification of the crude extract. This natural product is the first metabolite that possesses a tetramic acid ring derived from a d-amino acid, as well as a unique 21-methylene side chain. This compound was able to inhibit the blastulation of the starfish (Asterina pectinifera) embryo. Some years later, ancorinosides B–D (239–241) were isolated from the marine sponge Penares sollasi by Fusetani et al. [106] (Figure 23). Their structures were elucidated by spectroscopic and chemical methods, consisting of a tetramic acid glycoside related to ancorinoside A (238). In contrast to ancorinoside A (238), ancorinosides B–D (239–241) inhibited membrane type I matrix metalloproteinase (MT1-MMP) with IC50 values in the range of 180–500 µg/mL. An SAR study carried out by the same authors, wherein the aglycone of ancorinoside B (242a), its methyl ester 242b, and tenuazonic acid (243) were tested, showed that 243 exhibited the best results, while 242a and 242b were slightly more potent than ancorinoside B (239). These results indicated that the two carboxylic acid groups were irrelevant for the biological profile and pointed out the importance of the tetramic acid group for this biological activity. In the same year, Ikegami et al. [107] isolated the magnesium salt of ancorinoside A (238) from the marine sponge Ancorina sp., which exhibited an inhibitory activity of the blastulation of the starfish Asterina pectinifera embryo similar to ancorinoside A (238). This result indicated that the presence of Mg2+ ions, which act as transmembrane transport and influence the fluidity and permeability of the membrane, did not affect the inhibitory activity of ancorinoside A (238).
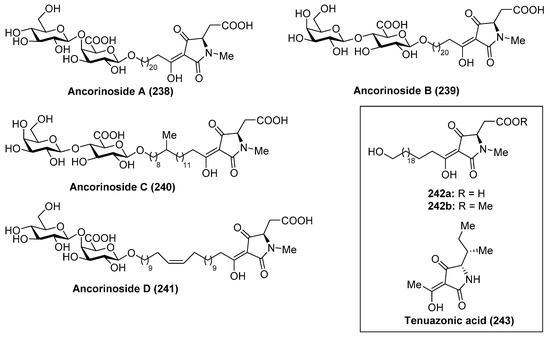
Figure 23.
Structures of ancorinosides A–D (238–241).
A total synthesis of ancorinoside A (238) was achieved in 1.6% yield in 18 steps by Schobert et al. [108], as described in Scheme 20. The key steps were the Schmidt glycosylation for the introduction of the lipidic spacer, a TEMPO oxidation to the uronic acid, functionalisation of the spacer terminus, and final Dieckmann cyclisation for the construction of the tetramic residue. Thus, the authors started the synthesis from peracetylated β-d-galactose, which was rapidly transformed into the disaccharide 244 in six steps in 49% overall yield. Oxidative deprotection, employing CAN, followed by generation of the corresponding imidate, afforded a separable α/β mixture of 245 in 56% yield over two steps. From 245, the glycosylation reaction with a spacer precursor 246 was achieved by the use of TMSOTf as an activator to obtain a mixture of the desired disaccharide 248 together with deacetyl 247, which was reacetylated to generate 248 in a 73% yield in two steps. Subsequent debenzylation reaction, TEMPO oxidation of the alcohol to the corresponding uronic acid, esterification of the acid, removal of the silyl group, followed by Dess–Martin periodinane oxidation to the corresponding aldehyde, and a final E-olefination with Ley’s β-ketophosphonate 249 afforded 250 in 29% overall yield. The coupling between 250 and 251 was achieved by the use of Ley’s protocol mediated by a silver salt to obtain the β-ketoamide 252 in 63% yield. Debenzylation of 252, followed by a final base-induced Dieckmann cyclisation with concomitant deacetylation, afforded ancorinoside A (238) (Scheme 20).
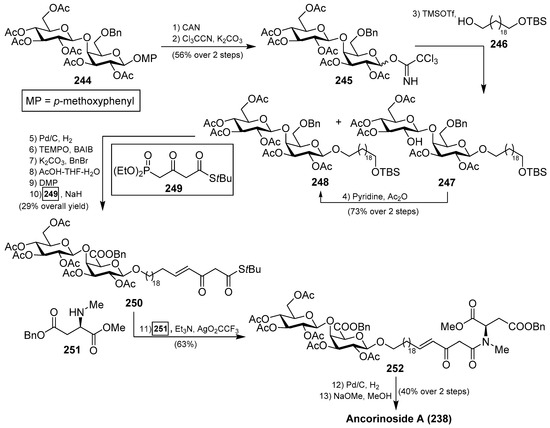
Scheme 20.
Total synthesis of ancorinoside A (238).
4.3. Bartolosides
Bartoloside A (253) was isolated from the cyanobacterium Nodosilinea sp. LEGE 06102 [109] and bartolosides B–D (254–256) were isolated from the cyanobacterium Synechocystis salina LEGE 06155 through a bioassay-guided fractionation of the crude extract by Balskus et al. (Figure 24). A year later, Leao et al. [110] isolated bartolosides E–K (257–263) from the strain LEGE 06099 of S. salina.
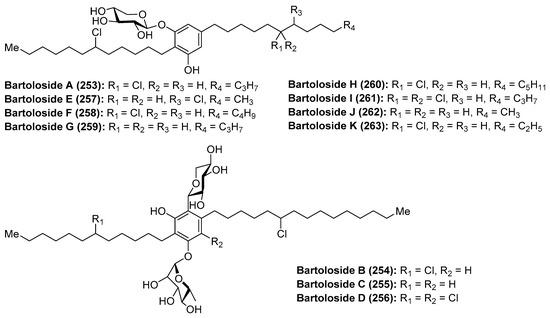
Figure 24.
Structures of bartolosides A–K (253–263).
Structurally, these glycolipids from marine cyanobacteria possess unique structures featured by the presence of a dialkyl-resorcinol core, decorated by one or two xylose units with chlorine groups in the aliphatic chains and a C-glycosyl moiety in the case of bartolosides B (254), C (255), and D (256). In the case of bartolosides E–K (257–263), these can be considered as analogues of bartoloside A (253) that differ in the alkyl chain lengths or halogenation patterns.
The chlorinated dialkylresorcinol core of the bartolosides conferred a challenge for their structural elucidation, which was finally established by combined biosynthetic and bioinformatic analysis. With regards to their biological features, the bartolosides are not known to possess strong biological activities, but their high abundance inside the cells suggests that they may have an important biological role that has to be investigated.
4.4. Caminosides
The caminosides (264–270) (Figure 25) are glycolipids with interesting antimicrobial activity isolated from the marine sponge Caminus sphareoconia by Andersen et al. [111]. Firstly, caminoside A (264) was discovered after a biological screening of crude extracts of the marine sponge for its ability to inhibit the secretion of Escherichia coli-secreted proteins (Esps) by enteropathogenic E. coli (EPEC), a major cause of infantile diarrhea that actually represents the main cause of mortality of children in developing countries. Interestingly, pathogenic E. coli, and not nonpathogenic E. coli, employed the named type III secretory apparatus to deliver Esps. Thus, compounds capable of inhibiting the type III secretion system would affect the pathogenic E. coli and not the comensal E. coli flora. Actually, the inhibition of the type III secretion system produced the attenuation of the pathogenicity without killing the bacteria. Thus, caminoside A (264) displayed an IC50 of 20 µM, not inhibiting the growth of Gram-negative bacteria such as E. coli (MIC > 100 µg/mL). In addition, caminoside A (264) showed significant antibacterial activity against methicillin-resistant Staphylococcus aureus (MIC 12 µg/mL) and vancomycin-resistant Enterococcus (MIC 12 µg/mL). Some years later, the same authors isolated and identified new members of the caminosides (B–D, 265–267) with similar biological activities [112]. In all the cases, the authors prepared the peracetylated derivatives 268–270 to facilitate their separation and structural determination. On the other hand, the unusual structure of these glycolipids is featured by the presence of a nonglycerol lipidic chain, whose stereochemistry at C-10 position was recently determined by circular dichroism [113].
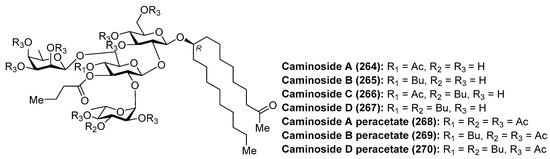
Figure 25.
Structures of caminosides A–D (264–267) and their peracetylated derivatives (268–270).
The first synthesis of caminoside A (264) was reported by Yu et al. [114], wherein they succeeded in sorting out the different issues concerning the stereochemistry of the glycosidic bonds, particularly the 1,2-cis-β-mannopyranoside-type linkage of the 6-deoxy-talose unit. This issue was solved by the use of a 2-O-Lev (levulinyl) fucosyl derivative to obtain stereoselectively the β-glycosidic linkage with a glucopyranosyl derivative, and then an inversion of the C-2 configuration via oxidation/reduction of the alcohol. Having constructed the disaccharide 274, its assembly with the glucosyl derivative 273, prepared via glycosylation of the trichloroacetimidate 271 and alcohol 272, was undertaken after removal of the acetate and Wacker oxidation of 273. The resulting trisaccharide 275 was then prepared for the introduction of the 6-deoxy-l-glucosyl unit, for which 275 was benzylated at the 2-OH group of the deoxy-talose fragment, followed by the selective removal of the 2-(azidomethyl)benzoyl group (Azmb) that was possible in the presence of the acetate and butyrate groups by treatment with tributylphosphine. With the resulting acceptor 277 in hand, the reaction with the donor 278, under the action of catalytic TMSOTf, provided the corresponding caminoside derivative in 53% yield, exclusively as the α-anomer, which was finally subjected to a global deprotection step and acetylation to obtain caminoside A peracetate (268) (Scheme 21). More recently, Li et al. have reported the total synthesis of caminoside B (265) in a closely related strategy, as summarized in Scheme 22, through key intermediates 282–285 and by efficient and stereoselective glycosylation reactions [115].
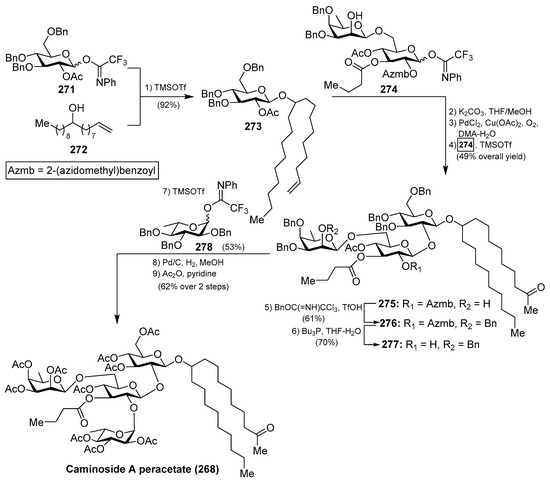
Scheme 21.
Total synthesis of caminoside A peracetate (268).
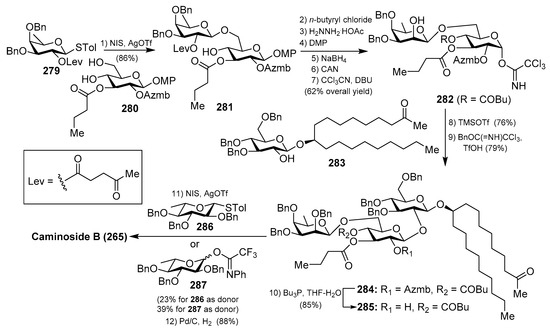
Scheme 22.
Total synthesis of caminoside B (265).
4.5. Clathrosides and Isoclathrosides
Clathrosides A–C (288–290) and isoclathrosides A–C (291–293) (Figure 26), isolated from the Caribbean sponge Agelas clathrodes by Mangoni et al. [116], are glycosides containing a very long chain alcohol derived from fatty acids. The structural differences between the clathrosides and the isoclathrosides can be found in the configuration and in the branching of the alkyl chains. Stereostructures of the clathrosides were elucidated by NMR and CD spectroscopy, mass spectrometry, and chemical degradation studies. Clathroside A (288) and isoclathroside A (291) did not show significant activity on the immune system of mammals despite the structural similarity between clathrosides and the immunoactive simplexides, which will be described in the following section.
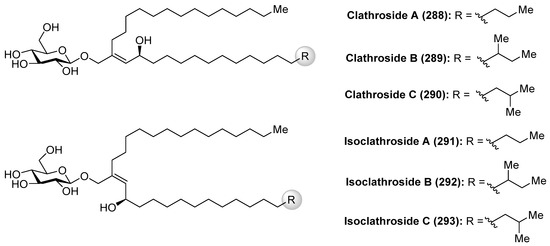
Figure 26.
Structures of clathrosides A–C (288–290) and isoclathrosides A–C (291–293).
4.6. Discoside
Discoside (294) (Figure 27) was isolated as a mixture of homologues from the marine sponge Discodermia dissoluta by Fattorusso et al. [117]. The complete stereostructure of discoside was determined by mass spectrometric and NMR spectroscopic techniques, together with CD analysis of degradation products. Interestingly, discoside (294) represents the first reported glycolipid with a 4,6-O-diacylated α-linked to the 2-hydroxyl group of a myo-inositol unit [2-O-(4,6-di-O-acyl-α-d-mannopyranosyl)-myo-inositol]. The myo-inositol mannoside is a well-known building block of phosphatidylinositol mannosides and of their multiglycosylated form, lipomannans, which exhibit immunoregulatory effects. However, such types of compounds have never been reported from marine sponges, 1-O-pentadecanoyl-2-O-(6-O-heptadecanoyl-α-d-mannopyranosyl)-myo-inositol being the only closely discoside-type compound, which was isolated from various strains of Propionibacterium. This finding suggests that discoside is synthesized by symbiotic cyanobacteria associated with the marine sponge.
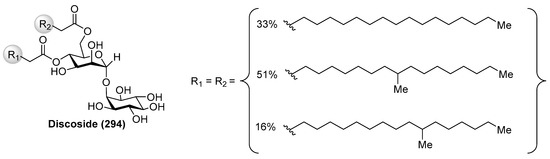
Figure 27.
Structure of discoside (294).
The synthesis of 4,6-di-O-octadecanoyl discoside (300) was recently accomplished by Florence et al., starting with the preparation of the thiomannoside donor 296. From α-d-phenylthiomannoside 295, sequential protection of the 4- and 6-OH, benzylation of the remaining two hydroxyl groups, acidic methanolysis of the anisylidene acetal, followed by esterification under Steglich conditions, provided the donor 296 in 34% overall yield. The key glycosylation reaction between the donor 296 and the acceptor 297, which was synthesized in six steps from a readily available orthoformate derivative of myo-inositol, was achieved with the activation of 296 using NIS and TESOTf, followed by the reaction with 297 to give a mixture of separable anomers 298 and 299 in 43% yield (α:β ratio 1:1.5). The total synthesis was completed by final debenzylation of the minor α isomer 299 to provide 4,6-di-O-octadecanoyl discoside (300) in a 98% yield (Scheme 23) [118].
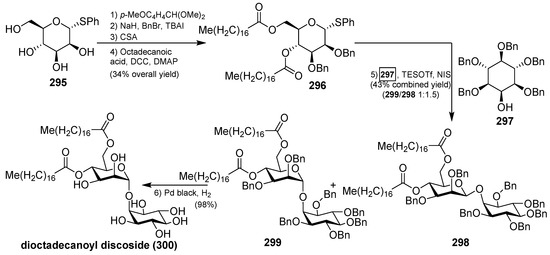
Scheme 23.
Total synthesis of dioctadecanoyl discoside 300.
4.7. Erylusamides
Erylusamides A–D (301–304) were isolated from the marine sponge Erylus cf. deficiens by Santos et al. through a bioassay-guided fractionation in order to find inhibitors of indoleamine 2,3-dioxygenase (IDO) [119]. The structures of erylusamides were established by NMR spectroscopy, HREIMS and chemical derivatization. These compounds possess a pentasaccharide moiety with unusual highly acetylated d-glucose, together with d-xylose and d-galactose units, and unprecedented aglycones, which are structurally featured by long chains of dihydroxyketo amides.
On the other hand, the related erylusamines A–E (305–309) were isolated from the marine sponge Erylus placenta by Fusetani et al. [120,121]. These compounds are interleukin-6 (IL-6) receptor antagonists. Erylusamine TA (310), erylusine (311), and erylusidine (312) (Figure 28) were isolated from Erylus cf. lendenfeldi by Kashman et al. [122]. These compounds were not tested for any biological activities due to the fact that they were not isolated as pure compounds but together with a small amount of other homologues.
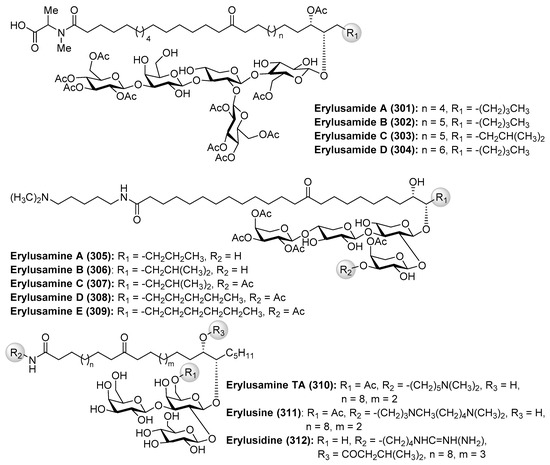
Figure 28.
Structures of erylusamides A–D (301–304) and related glycolipids (305–312).
4.8. Ieodoglucomides and Ieodoglycolipid
Ieodoglucomides A (313) and B (314) (Figure 29) were isolated from a marine-derived bacterium, Bacillus licheniformis, in 2012 by Shin et al. [123] and their structures were elucidated by a combination of NMR spectroscopic analysis of the natural products with the spectroscopic and physical analyses of the products derived from their acid hydrolysis. Iedoglucomides are unique glycolipopeptides that contain an amino acid (l-ala for ieodoglucomide A and glycine for iedoglucomide B), an unprecedented fatty acid (14-hydroxy-15-methylhexadecanoic acid), succinic acid, and a sugar (β-d-glucose). Iedoglucomide B (314) showed moderate antimicrobial activity against Gram-positive and Gram-negative bacteria, antifungal as well as growth inhibition against lung cancer (NCI-H23) and stomach cancer (NUGC-3) cell lines with GI50 values of 25.18 and 17.78 µg/mL, respectively.
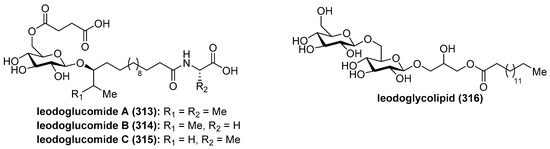
Figure 29.
Structures of ieodoglucomides A–C (313–314) and ieodoglycolipid (316).
Ieodoglucomide C (315) and ieodoglycolipid (316) were isolated from the fermentation broth of the marine-derived bacterium Bacillus licheniformis [124]. In contrast to iedoglucomide B (314), these compounds showed good antibiotic properties against Staphylococcus aureus, Bacillus subtilis, B. cereus, Salmonella typhi, Escherichia coli, and Pseudomonas aeruginosa with MIC values in the 0.01–0.05 µM range. In addition, these compounds inhibit the mycelial growth of plant pathogenic fungi Aspergillus niger, Rhizoctonia solani, Botrytis cinerea, and Colletotrichum acutatum as well as the human pathogen Candida albicans, with MICs values of 0.03–0.05 µM. The antimicrobial profiles of ieodoglucomide C (315) and ieodoglycolipid (316) suggest that they could be potential bioprobes for the development of useful antibiotics and fungicides.
Total syntheses of ieodoglucomide A (313) and B (314) were carried out by Reddy et al. [125] starting from d-glucose and using β-glycosylation and olefin cross-methatesis reactions as the key steps. Thus, the synthesis started with the conversion of 317, prepared from d-glucose in two steps, into the corresponding α-trichloroacetimidate, which was employed in the key glycosylation reaction, promoted by TMSOTf, with the alcohol 318 to obtain the β-glycoside 319 in 67% yield in two steps. The introduction of the succinyl group onto the sugar to generate 320 was accomplished in six steps in 41% overall yield from 319. Next, the key olefin cross-metathesis between 320 and 321 or 322, under the action of the Grubbs 2nd generation catalyst, gave the protected glycolipids 323 and 324 in excellent 86% and 85% yields, respectively. Final hydrogenation provided ieodoglucomides A (313) and B (314) in 92 and 90% yields, respectively (Scheme 24).
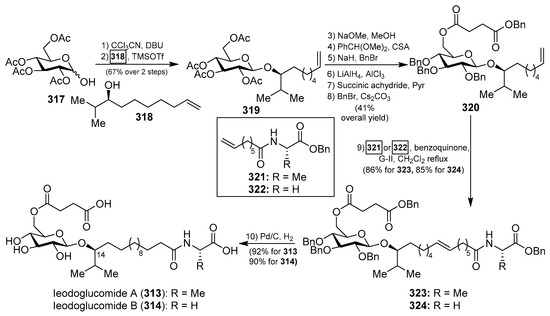
Scheme 24.
Total synthesis of ieodoglucomides A (313) and B (314).
In order to identify analogs of ieodoglucomides with improved biological properties, the same authors prepared the α-isomers of ieodoglucomides A and B (328 and 329) and their C-14-epimers (331 and 332) (Scheme 25) [126]. As described in Scheme 25, the syntheses of these analogues were accomplished according to the same synthetic strategy employed for the synthesis of the natural products, starting from the α-anomer 325 for the synthesis of the analogues 328 and 329, and from the epimer 330 for the syntheses of the analogues 331 and 332. These stereochemical analogues, together with the natural ieodoglucomides, were tested for cytotoxicity against various cancer cell lines and found that ieodoglucomide A (313) and B (314), as well as their α-isomers (328 and 329), did not exhibit cytotoxicity in any cell lines tested. However, their α-C14 epimers (331 and 332) showed an inhibition of proliferation of DU145 and HeLa cells with IC50 values in the range of 15.2–35.5 µM. In addition, this biological study demonstrated that the activity of caspases-3 and -9 increased when these cell lines were treated with these compounds 331 and 332, suggesting that the α-epi isomers of the ieodoglucomides led to the activation of pathway-initiating apoptosis in DU145 and HeLa cells.

Scheme 25.
Total synthesis of α-isomers of ieodoglucomide A (328) and B (329) and their α-14-epi analogues (331 and 332).
4.9. Pachymoside A
Pachymoside A (333) (Figure 30) was isolated from the North Sea marine sponge Pachymatisma johnstonia through a bioassay-guided fractionation in order to find inhibitors of the bacterial type III secretion system (TTSS), carried out by Andersen et al. [127]. Its structure was elucidated by NMR spectral analysis and chemical degradation. This structural study revealed that pachymoside A (333) possesses complete acetylation of the galactose residues (the eight galactose hydroxyls are acetylated) and only partial acetylation of the glucose residues (the C-6 hydroxyls of β-glucose and d-glucose are acetylated). Despite its promising activity as a TTSS inhibitor, pachymoside A (333) is not a true TTSS inhibitor. It seems that the biological activity exhibited is due to its ability to activate extracelular bacterial proteases that rapidly degrade the excreted Esps, resulting in a false indication of inhibition.
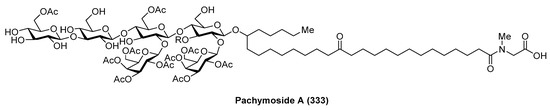
Figure 30.
Structure of pachymoside A (333).
4.10. Plaxyloside
Plaxyloside (334) (Figure 31) was isolated as its peracetate from the Caribbean sponge Plakortis simplex by Fattorusso et al. [128]. Its structure was established by NMR spectral analysis and chemical methods. Plaxyloside (334) is a glycolipid with a long linear polyisoprenoid alcohol aglycone and a linear carbohydrate chain of β-xylopyranose units and represents the first natural oligosaccharide that contains more than three (1→3) linked xylopyranosides. The biological activity of this compound was not evaluated.
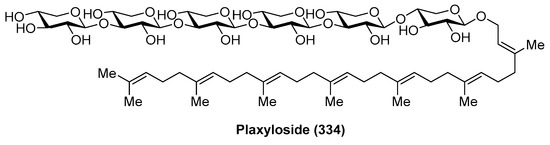
Figure 31.
Structure of plaxyloside (334).
4.11. Roselipins
Roselipins (335–338) (Figure 32) were isolated from the culture broth of fungus Gliocladium roseum KF-1040 by Omura et al. [129,130,131]. Later, a mixture of five new roselipins derivatives, separated in two fractions, named roselipins mixture 1–2 (339a–b) and mixture 3–5 (340a–c), were isolated from an extract of the fungus Clonostachys candelabrum by Singh et al. [132]. These compounds differ by the position of esterification of the arabinitol residue. Overall, the roselipins have a unique structure featured by a highly methylated C-20 fatty acid skeleton modified with d-mannose and d-arabinitol residues. Extensive biological evaluations of the roselipins revealed that these compounds displayed a broad range of biological activities. In particular, these compounds inhibit diacylglycerol acyltransferase (DGAT) with IC50 values ranging from 15 to 22 µM in an enzyme assay system using rat-liver microsomes, showing that are selective inhibitors of DGAT2 (IC50 = 30–50 µM) [133]. An SAR study carried out by Omura et al. [134] revealed that demannosyl roselipins 3A (341) and 3B (342), prepared from the natural products via enzymatic degradation, conserved DGAT inhibitory activity, in contrast to the dearabinitoyl derivatives (343 and 344), which completely lost their activity, suggesting that the arabinitoyl fatty acid core is essential for eliciting DGAT inhibitory activity. In addition, the roselipin derivatives 3A (341) and 3B (342) were more potent in the cell assay than the roselipins 335–338, indicating that these derivatives are more membrane-permeable than natural roselipins. These compounds also exhibited antimicrobial activity against Saccharomyces cervisiae and Aspergillus niger and showed a cytotoxic effect on Raji cells at 39 µM. Furthermore, the mixture of roselipins 2A and 2B (336 and 338) inhibited HIV-1 integrase with IC50 = 8.5 µM [135]. On the other hand, Ondeyka et al. [136] reported that roselipins 2A (336), 2B (338), and 1A (335) blocked the CXCR3 receptor interaction of IP-10 ligand with IC50 = 14.6, 23.5, and 41 µM. The roselipins were also identified as anthelmintic compounds [133].
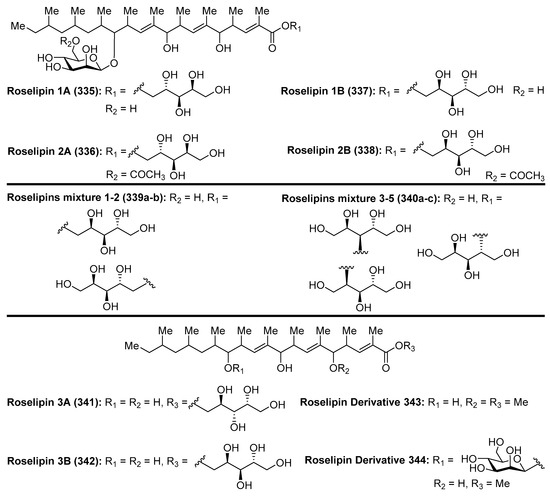
Figure 32.
Structures of the roselipins (335–340) and derivatives 341–344.
4.12. Simplexides
The same marine sponge that produces the plakosides and plaxyloside (Sections Plakosides and Plaxyloside), Plakortis simplex, has provided other types of glycolipids with immunosuppressive properties, the simplexides (345a–e), which were isolated as a mixture of related disaccharides [137]. Even though these compounds could not be separated by HPLC techniques, the authors were able to establish their structures via degradation and subsequent CG-MS analyses of the resulting methyl esters of the lipidic chains. Despite the remarkable structural differences with the plakosides, the simplexides share a very similar biological profile, showing potent inhibitory activity of the proliferarion of T cells in the murine immune system when stimulated with concanavalin A through a noncytotoxic mechanism. Further biological studies demonstrated that the immunosuppressive effects of the simplexides are due to the induction that they exert on the expression and release of cytokines and chemokines from human monocytes by direct interaction with the CD1d receptors, expressed in these cells [138]. In addition, the simplexides induce the expansion of iNKT (natural killer T cells with an invariant T cell receptor alpha chain). An interesting synthesis for the simplexides was recently developed by Yingxia et al. [139], in which thioglycosides were employed to construct the complete simplexide scaffold in a one-pot glycosylation method. Thus, the armed donor 346 reacted with the disarmed acceptor 347 by the action of the promoter system NIS/AgOTf to obtain, with a high α-selectivity, the disaccharide 348. Without isolation of this product, the corresponding alcohol 349 was added to the crude mixture with additional promoters (NIS/AgOTf) to proceed with a second glycosylation reaction to provide 350a–c, which were finally isolated in excellent yields and stereoselectivity. Final deprotection of 350a–c gave simplexides 345a–c (Scheme 26).
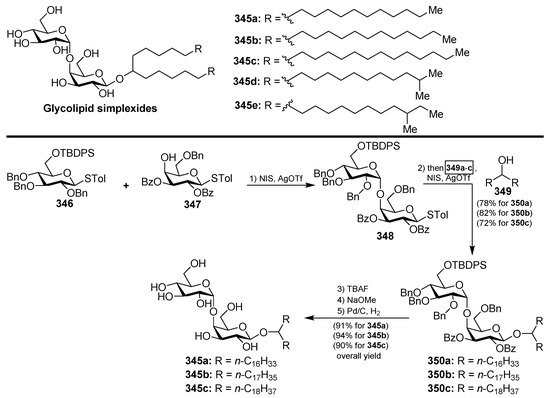
Scheme 26.
Structures and synthesis of the simplexides (345a–c).
More recently, a synthesis of this class of glycolipids by Yu et al. [140] exploited the cross-metathesis reaction to build the lipidic chain from the allyl disaccharide 356, efficiently prepared from glycosyl derivatives 351 and 352 via trichloroacetimidates. The crucial cross-metathesis reaction was achieved by the action of the Grubbs 2nd generation catalyst to obtain simplexides 357a–h in very good yields, which were transformed into the final simplexides 345b and 358a–g in two additional steps (Scheme 27). This new efficient and concise synthesis of the simplexides and analogues will allow the determination of the structural factors that govern their interesting biological activities, a study that has yet to be completed by the authors.
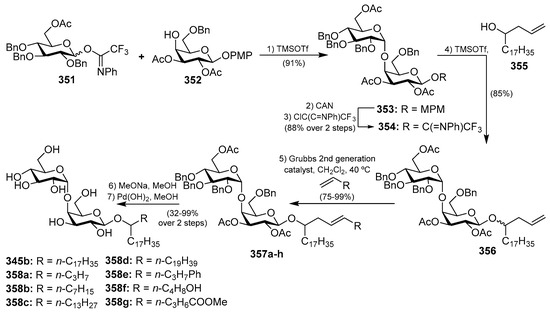
Scheme 27.
Synthesis of the natural simplexide 345b and analogues 358a–g.
5. Conclusions
Carbohydrates are often found as primary metabolites in the form of monomers, oligomers, or polymers, and play essential functions for life. However, in the realm of natural products, carbohydrates can be found mainly as components of glycoconjugates. In this case, they play important roles in conferring certain physical, chemical, and biological properties to the carrier molecules, being essential in cellular-recognition processes. A relevant example of the glycoconjugates are glycolipids and, notably, the glycolipids from a marine origin, which represent an important class of natural products with wide structural diversity and a broad range of biological activities, including antitumoral, antibiotic, antiviral, antimalarial, immunostimulatory, and neuritogenic activities. However, their difficult accessibility from natural sources, coupled with their extreme scarcity, has made their chemical synthesis essential to provide their access for additional studies. In addition, further understanding of their mechanisms of biological action allows for the rational design and synthesis of new analogues. Through the present review, we have presented the molecular and biological diversity of the glycoclipids derived from marine sources, giving special emphasis on their syntheses as an important tool to confirm their molecular structures, gain insight into their biological activities, and the design of analogues for the development of new drugs. In conclusion, the readers have hopefully realized a greater interest in this class of natural products and the awesome power of chemical synthesis for the development of valuable bioactive compounds based on these natural products.
Funding
This research received no external funding.
Acknowledgments
This work was financially supported by the Ministerio de Economía y Competitividad (CTQ2014-60223-R). I.C.-S. thanks Ministerio de Educación, Cultura y Deporte for a fellowship (Programme FPU). We thank J. I. Trujillo from Pfizer (Groton, CT) for assistance in the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sasaki, D. Glycolipids: New Research; Nova Biomedical: New York, NY, USA, 2007; ISBN 978-1-60456-216-3. [Google Scholar]
- Sweely, C.C. Biochemistry of Lipids, Lipoproteins and Membranes; Benjamin/Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Hakomori, S.-I. Structure and Function of Sphingolipids in Transmembrane Signalling and Cell-Cell Interactions. Biochem. Soc. Trans. 1993, 21, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological Roles of Oligosaccharides: All of the Theories are Correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T. A View on Sphingolipids and Disease. Chem. Phys. Lipids 2011, 164, 590–606. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, T.; van den Berg, R.J.B.H.N.; Boot, R.G.; van der Marel, G.A.; Overkleeft, H.S.; Aerts, J.M.F.G. Glycosphingolipids—Nature, Function, and Pharmacological Modulation. Angew. Chem. Int. Ed. 2009, 48, 8848–8869. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Tashiro, T. Sphingolipids and Glycosphingolipids—Their Synthesis and Bioactivities. Heterocycles 2011, 83, 951–1003. [Google Scholar] [CrossRef]
- Vankar, Y.D.; Schmidt, R.R. Chemistry of Glycosphingolipids—Carbohydrate Molecules of Biological Significance. Chem. Soc. Rev. 2000, 29, 201–216. [Google Scholar] [CrossRef]
- Farwanah, H.; Kolter, T. Lipidomics of Glycosphingolipids. Metabolites 2012, 2, 134–164. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.X.; Chen, J.H. The Cerebrosides. Nat. Prod. Rep. 2003, 20, 509–534. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Bergter, E.; Sassaki, G.L.; de Souza, L.M. Structural Analysis of Fungal Cerebrosides. Front. Microbiol. 2011, 2, 239. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T. Ganglioside Biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef] [PubMed]
- Kates, M. Glycolipids, Phosphoglycolipids and Sulfoglycolipis. Handbook of Lipid Research; Springer: Boston, MA, USA, 1990; Volume 6. [Google Scholar]
- Schnaar, R.L.; Suzuki, A.; Stanley, P. Glycosphingolipids. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; pp. 129–141. [Google Scholar]
- Kulkarni, S.S. Synthesis of Glycosphingolipids. In Glycochemical Synthesis: Strategies and Applications; Hung, S.-C., Zulueta, M.M.L., Eds.; Wiley: Hoboken, NJ, USA, 2016; pp. 293–326. [Google Scholar]
- Barnathan, G.; Couzinet-Mossion, A.; Wielgosz-Collin, G. Glycolipids from Marine Invertebrates. In Outstanding Marine Molecules: Chemistry, Biology, Analysis, 1st ed.; La Barre, S., Kornprobst, J.-M., Eds.; Wiley-VCH Verlag GmbH and Co., KGaA: Weinheim, Germany, 2014; pp. 99–162. ISBN 9783527681501. [Google Scholar]
- Zhang, J.; Li, C.; Yu, G.; Guan, H. Total Synthesis and Structure–Activity Relationship of Glycoglycerolipids from Marine Organisms. Mar. Drugs 2014, 12, 3634–3659. [Google Scholar] [CrossRef] [PubMed]
- Banchet-Cadeddu, A.; Hénon, E.; Dauchez, M.; Renault, J.-H.; Monneaux, F.; Haudrechy, A. The Stimulating Adventure of KRN7000. Org. Biomol. Chem. 2011, 9, 3080–3104. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.L.; Teyton, L.; Bendelac, A.; Savage, P.B. Stimulation of Natural Killer T Cells by Glycolipids. Molecules 2013, 18, 15662–15688. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Higuchi, R.; Isobe, R.; Komori, T. Isolation and Structure of Six New Cerebrosides. Liebigs Ann. Chem. 1988, 1988, 19–24. [Google Scholar] [CrossRef]
- Sugiyama, S.; Honda, M.; Komori, T. Synthesis of Acanthacerebroside A. Liebigs Ann. Chem. 1990, 1990, 1063–1068. [Google Scholar] [CrossRef]
- Higuchi, R.; Kagoshima, M.; Komori, T. Structures of Three New Cerebrosides, Astrocerebroside A, B, and C and of Related Nearly Homogeneous Cerebrosides. Liebigs Ann. Chem. 1990, 1990, 659–663. [Google Scholar] [CrossRef]
- Higuchi, R.; Jhou, J.X.; Inukai, K.; Komori, T. Isolation and Structure of Six New Cerebrosides, Asteriacerebrosides A–F, and Two Known Cerebrosides, Astrocerebroside A and Acanthacerebroside C. Liebigs Ann. Chem. 1991, 1991, 745–752. [Google Scholar] [CrossRef]
- Ishii, T.; Okino, T.; Mino, Y. A Ceramide and Cerebroside from the Starfish Asterias amurensis Lütken and Their Plant-Growth Promotion Activities. J. Nat. Prod. 2006, 69, 1080–1082. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Park, Y.S.; Rho, J.-R.; Kim, Y.H. Structural Determination of Cerebrosides Isolated from Asterias amurensis Starfish Eggs using High-Energy Collision-Induced Dissociation of Sodium-Adducted Molecules. Rapid Commun. Mass Spectrom. 2011, 25, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Chida, N.; Sakata, N.; Murai, K.; Tobe, T.; Nagase, T.; Ogawa, S. Total Synthesis of Acanthacerebroside A and Astrocerebroside A via a Chiral Epoxide Intermediate Derived from l-Quebrachitol. Bull. Chem. Soc. Jpn. 1998, 71, 259–272. [Google Scholar] [CrossRef]
- Natori, T.; Koezuka, Y.; Higa, T. Agelasphins, Novel α-Galactosylceramides from the Marine Sponge Agelas mauritianus. Tetrahedron Lett. 1993, 34, 5591–5592. [Google Scholar] [CrossRef]
- Natori, T.; Morita, M.; Akimoto, K.; Koezuka, Y. Agelasphins, Novel Antitumor and Immunostimulatory Cerebrosides from the Marine Sponge Agelas mauritianus. Tetrahedron 1994, 50, 2771–2784. [Google Scholar] [CrossRef]
- Morita, M.; Motoki, K.; Akimoto, K.; Natori, T.; Sakai, T.; Sawa, E.; Yamaji, K.; Koezuka, Y.; Kobayashi, E.; Fukushima, H. Structure-Activity Relationship of α-Galactosylceramides against B16-Bearing Mice. J. Med. Chem. 1995, 38, 2176–2187. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Punt, C.J.A.; Ando, Y.; Ruijter, R.; Nishi, N.; Peters, M.; von Blomberg, B.M.E.; Scheper, R.J.; van der Vliet, H.J.J.; van den Eertwegh, A.J.M.; et al. A Phase I Study of the Natural Killer T-Cell Ligand α-Galactosylceramide (KRN7000) in Patients with Solid Tumors. Clin. Cancer Res. 2002, 8, 3702–3709. [Google Scholar] [PubMed]
- Motoki, K.; Kobayashi, E.; Uchida, T.; Fukushima, H.; Koezuka, Y. Antitumor Activities of α-, β-Monogalactosylceramides and Four Diastereomers of an α-Galactosylceramide. Bioorg. Med. Chem. Lett. 1995, 5, 705–710. [Google Scholar] [CrossRef]
- Reddy, B.G.; Silk, J.D.; Salio, M.; Balamurugan, R.; Shepherd, D.; Ritter, G.; Cerundolo, V.; Schmidt, R.R. Nonglycosidic Agonists of Invariant NKT Cells for Use as Vaccine Adjuvants. ChemMedChem 2009, 4, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; D’Esposito, M.; Fattorusso, E.; Mangoni, A.; Basilico, N.; Parapini, S.; Taramelli, D. Damicoside from Axinella damicornis: The Influence of a Glycosylated Galactose 4-OH Group on the Immunostimulatory Activity of α-Galactoglycosphingolipids. J. Med. Chem. 2005, 48, 7411–7417. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, K.; Natori, T.; Morita, M. Synthesis and Stereochemistry of Agelasphin-9b. Tetrahedron Lett. 1993, 34, 5593–5596. [Google Scholar] [CrossRef]
- Farokhi, F.; Grellier, P.; Clément, M.; Roussakis, C.; Loiseau, P.; Genin-Seward, E.; Kornprobst, J.-M.; Barnathan, G.; Wielgosz-Collin, G. Antimalarial Activity of Axidjiferosides, New β-Galactosylceramides from the African Sponge Axinyssa djiferi. Mar. Drugs 2013, 11, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Hara, E.; Miyamoto, T.; Higuchi, R.; Isobe, R.; Honda, S. Isolation and Structure of Biologically Active Glycosphingolipids from the Sea Cucumber Cucumaria echinata. Eur. J. Org. Chem. 1998, 1998, 371–378. [Google Scholar] [CrossRef]
- Li, H.; Matsunaga, S.; Fusetani, N. Halicylindrosides, Antifungal and Cytotoxic Cerebrosides from the Marine Sponge Halichondria cylindrata. Tetrahedron 1995, 51, 2273–2280. [Google Scholar] [CrossRef]
- Murakami, T.; Taguchi, K. Stereocontrolled Synthesis of Novel Phytosphingosine-type Glucosaminocerebrosides. Tetrahedron 1999, 55, 989–1004. [Google Scholar] [CrossRef]
- Durin, R.; Zubia, E.; Ortega, M.J.; Naranjo, S.; Salv, J. Phallusides, New Glucosphingolipids from the Ascidian Phallusia fumigata. Tetrahedron 1998, 54, 14597–14602. [Google Scholar] [CrossRef]
- Karlsson, K.-A.; Leffler, H.; Samuelsson, B.E. Characterization of Cerebroside (Monoglycosylceramide) from the Sea Anemone, Metridium senile. Biochim. Biophys. Acta 1979, 574, 79–93. [Google Scholar] [CrossRef]
- Kawai, G.; Ikeda, Y. Fruiting-Inducing Activity of Cerebrosides Observed with Schizophyllum commune. Biochim. Biophys. Acta 1982, 719, 612–618. [Google Scholar] [CrossRef]
- Hammami, S.; Bergaoui, A.; Boughalleb, N.; Romdhane, A.; Khoja, I.; Ben Halima Kamel, M.; Mighri, Z. Antifungal Effects of Secondary Metabolites Isolated from Marine Organisms Collected from the Tunisian Coast. C. R. Chim. 2010, 13, 1397–1400. [Google Scholar] [CrossRef]
- Black, F.J.; Kocienski, P.J. Synthesis of Phalluside-1 and Sch II using 1,2-Metallate Rearrangements. Org. Biomol. Chem. 2010, 8, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A.; Teta, R. Amphiceramide A and B, Novel Glycosphingolipids from the Marine Sponge Amphimedon compressa. Eur. J. Org. Chem. 2009, 2009, 2112–2119. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Mangoni, A.; Di Rosa, M.; Ianaro, A. Glycolipids from Sponges. 6. Plakoside A and B, Two Unique Prenylated Glycosphingolipids with Immunosuppressive Activity from the Marine Sponge Plakortis simplex. J. Am. Chem. Soc. 1997, 119, 12465–12470. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Mangoni, A. Glycolipids from Sponges. Part 9: Plakoside C and D, Two Further Prenylated Glycosphingolipids from the Marine Sponge Ectyoplasia ferox. Tetrahedron 2000, 56, 5953–5957. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Li, J.; Zenke, G. Total Synthesis and Biological Evaluation of Glycolipids Plakosides A, B and Their Analogs. Helv. Chim. Acta 2000, 83, 1977–2006. [Google Scholar] [CrossRef]
- Seki, M.; Kayo, A.; Mori, K. Synthesis of (2S,3R,11S,12R,2′′′R,11′′′S,12′′′R)-Plakoside A, a Prenylated and Immunosuppressive Marine Galactosphingolipid with Cyclopropane-Containing Alkyl Chains. Tetrahedron Lett. 2001, 42, 2357–2360. [Google Scholar] [CrossRef]
- Seki, M.; Mori, K. Synthesis of a Prenylated and Immunosuppressive Marine Galactosphingolipid with Cyclopropane-Containing Alkyl Chains: (2S,3R,11S,12R,2′′′R, 5′′′Z,11′′′S,12′′′R)-Plakoside A and Its (2S,3R,11R,12S,2′′′R,5′′′Z,11′′′R,12′′′S) Isomer. Eur. J. Org. Chem. 2001, 2001, 3797–3809. [Google Scholar] [CrossRef]
- Mori, K.; Tashiro, T.; Akasaka, K.; Ohrui, H.; Fattorusso, E. Determination of the Absolute Configuration at the Two Cyclopropane Moieties of Plakoside A, an Immunosuppressive Marine Galactosphingolipid. Tetrahedron Lett. 2002, 43, 3719–3722. [Google Scholar] [CrossRef]
- Tashiro, T.; Akasaka, K.; Ohrui, H.; Fattorusso, E.; Mori, K. Determination of the Absolute Configuration at the Two Cyclopropane Moieties of Plakoside A, an Immunosuppressive Marine Galactosphingolipid. Eur. J. Org. Chem. 2002, 2002, 3659–3665. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A.; Teta, R. Terpioside from the Marine Sponge Terpios sp., the First Glycosphingolipid Having an l-Fucofuranose Unit. Eur. J. Org. Chem. 2008, 2008, 2130–2134. [Google Scholar] [CrossRef]
- Cutignano, A.; De Palma, R.; Fontana, A. A Chemical Investigation of the Antarctic Sponge Lyssodendoryx flabellata. Nat. Prod. Res. 2012, 26, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Mangoni, A.; Teta, R.; Panza, E.; Ianaro, A. Terpioside B, a Difucosyl GSL from the Marine Sponge Terpios sp. is a Potent Inhibitor of NO Release. Bioorg. Med. Chem. 2010, 18, 5310–5315. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Xu, J.; Gingrich, D.E.; Williams, M.D.; Doubek, D.L.; Chapuis, J.-C.; Schmidt, J.M. Antineoplastic agents. Part 395. Isolation and Structure of Agelagalastatin from the Papua New Guinea Marine Sponge Agelas sp. Chem. Commun. 1999, 915–916. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, B.-Y.; Jeon, H.B.; Kim, K.S. Total Synthesis of Agelagalastatin. Org. Lett. 2006, 8, 3971–3974. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fatturusso, E.; Imperatore, C.; Mangoni, A. Glycolipids from Sponges. 13. Clarhamnoside, the First Rhamnosylated α-Galactosylceramide from Agelas clathrodes. Improving Spectral Strategies for Glycoconjugate Structure Determination. J. Org. Chem. 2004, 69, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Li, C.; Liu, Y.; Zhang, Z.; Li, Y. Concise Synthesis of Clarhamnoside, a Novel Glycosphingolipid Isolated from the Marine Sponge Agela clathrodes. Carbohydr. Res. 2007, 342, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Vesparioside from the Marine Sponge Spheciospongia vesparia, the First Diglycosylceramide with a Pentose Sugar Residue. Eur. J. Org. Chem. 2005, 2005, 368–373. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Glycolipids from Sponges. 20. J-Coupling Analysis for Stereochemical Assignments in Furanosides: Structure Elucidation of Vesparioside B, a Glycosphingolipid from the Marine Sponge Spheciospongia vesparia. J. Org. Chem. 2008, 73, 6158–6165. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.-C.; Zhu, S.-Y.; Cao, H.; Yang, J.-S. Total Synthesis of Marine Glycosphingolipid Vesparioside B. J. Am. Chem. Soc. 2016, 138, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Ijuin, T.; Kitajima, K.; Song, Y.; Kitazume, S.; Inoue, S.; Haslam, S.M.; Morris, H.R.; Dell, A.; Inoue, Y. Isolation and identification of novel sulfated and nonsulfated oligosialyl glycosphingolipids from sea urchin sperm. Glycoconj. J. 1996, 13, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Tanabe, K.; Miyamoto, T.; Kusumoto, T.; Inagaki, M.; Higuchi, R. Isolation and Structure of a Monomethylated Ganglioside Possessing Neuritogenic Activity from the Ovary of the Sea Urchin Diadema setosum. Chem. Pharm. Bull. 2008, 56, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-S.; Sawant, R.C.; Yang, S.-A.; Liao, Y.-J.; Liao, J.-W.; Badsara, S.S.; Luo, S.-Y. Synthesis of ganglioside Hp-s1. RSC Adv. 2014, 4, 47752–47761. [Google Scholar] [CrossRef]
- Tsai, Y.-F.; Shih, C.-H.; Su, Y.-T.; Yao, C.-H.; Lian, J.-F.; Liao, C.-C.; Hsia, C.-W.; Shui, H.-A.; Rani, R. The Total Synthesis of a Ganglioside Hp-s1 Analogue Possessing Neuritogenic Activity by Chemoselective Activation Glycosylation. Org. Biomol. Chem. 2012, 10, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.-T.; Yeh, C.-H.; Yang, S.-A.; Lin, C.-Y.; Tai, H.-J.; Shelke, G.B.; Reddy, D.M.; Yu, A.L.; Luo, S.-Y. Design, Synthesis, and Biological Evaluation of Ganglioside Hp-s1 Analogues Varying at Glucosyl Moiety. ACS Chem. Neurosci. 2016, 7, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.B.; Chen, B.-R.; Yang, S.-A.; Kuo, T.-M.; Syu, Y.-L.; Ko, Y.-C.; Luo, S.-Y. Mild and Highly α-Selective O-Sialylation Method Based on Pre-Activation: Access to Gangliosides Hp-s1, DSG-A, and Their Analogues. Asian J. Org. Chem. 2017, 6, 1556–1560. [Google Scholar] [CrossRef]
- Wu, Y.-F.; Tsai, Y.-F.; Guo, J.-R.; Yu, C.-P.; Yu, H.-M.; Liao, C.-C. First Total Synthesis of Ganglioside DSG-A Possessing Neuritogenic Activity. Org. Biomol. Chem. 2014, 12, 9345–9349. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Matsubara, R.; Kaneko, M.; Miyamoto, T.; Higuchi, R. Isolation and Structure of a Biologically Active Ganglioside Molecular Species from the Sea Cucumber Holothuria leucospilota. Chem. Pharm. Bull. 2001, 49, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Iwayama, Y.; Ando, H.; Ishida, H.; Kiso, M. A First Total Synthesis of Ganglioside HLG-2. Chem. Eur. J. 2009, 15, 4637–4648. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.-F.; Wang, Y.; Xiong, D.-C.; Ye, X.-S. Stereoselective Synthesis of the Trisaccharide Moiety of Ganglioside HLG-2. J. Org. Chem. 2014, 79, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Inoue, S.; Inagaki, K.; Sakai, M.; Miyamoto, T.; Komori, T.; Inagaki, M.; Isobe, R. Isolation and Structure of a New Biologically Active Ganglioside Molecular Species from the Starfish Asterina pectinifera. Chem. Pharm. Bull. 2006, 54, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Sawa, M.; Tamai, H.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. The Total Synthesis of Starfish Ganglioside GP3 Bearing a Unique Sialyl Glycan Architecture. Chem. Eur. J. 2016, 22, 8323–8331. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Mori, T.; Sugata, T.; Yamada, K.; Miyamoto, T. Partial Synthesis of a Sea Cucumber Ganglioside Analogue from a Starfish Cerebroside. Eur. J. Org. Chem. 1999, 1999, 145–147. [Google Scholar] [CrossRef]
- Kaneko, M.; Kisa, F.; Yamada, K.; Miyamoto, T.; Higuchi, R. Structure of Neuritogenic Active Ganglioside from the Sea Cucumber Stichopus japonicus. Eur. J. Org. Chem. 1999, 1999, 3171–3174. [Google Scholar] [CrossRef]
- Kaneko, M.; Kisa, F.; Yamada, K.; Miyamoto, T.; Higuchi, R. Structure of a New Neuritogenic-Active Ganglioside from the Sea Cucumber Stichopus japonicus. Eur. J. Org. Chem. 2003, 2003, 1004–1008. [Google Scholar] [CrossRef]
- Tamai, H.; Ando, H.; Tanaka, H.-N.; Hosoda-Yabe, R.; Yabe, T.; Ishida, H.; Kiso, M. The Total Synthesis of the Neurogenic Ganglioside LLG-3 Isolated from the Starfish Linckia laevigata. Angew. Chem. Int. Ed. 2011, 50, 2330–2333. [Google Scholar] [CrossRef] [PubMed]
- Tamai, H.; Imamura, A.; Ogawa, J.; Ando, H.; Ishida, H.; Kiso, M. First Total Synthesis of Ganglioside GAA-7 from Starfish Asterias amurensis versicolor. Eur. J. Org. Chem. 2015, 2015, 5199–5211. [Google Scholar] [CrossRef]
- Kaneko, M.; Yamada, K.; Miyamoto, T.; Inagaki, M.; Higuchi, R. Neuritogenic Activity of Gangliosides from Echinoderms and Their Structure-Activity Relationship. Chem. Pharm. Bull. 2007, 55, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Kisa, F.; Yamada, K.; Miyamoto, T.; Inagaki, M.; Higuchi, R. Isolation and Structure of Biologically Active Monosialo-Gangliosides from the Sea Cucumber Cucumaria echinata. Chem. Pharm. Bull. 2006, 54, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Kisa, F.; Yamada, K.; Miyamoto, T.; Inagaki, M.; Higuchi, R. Isolation and Structure of Biologically Active Disialo- and Trisialo-Gangliosides from the Sea Cucumber Cucumaria echinata. Chem. Pharm. Bull. 2006, 54, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Warabi, K.; Hamada, T.; Nakao, Y.; Matsunaga, S.; Hirota, H.; van Soest, R.W.M.; Fusetani, N. Axinelloside A, an Unprecedented Highly Sulfated Lipopolysaccharide Inhibiting Telomerase, from the Marine Sponge, Axinella infundibula. J. Am. Chem. Soc. 2005, 127, 13262–13270. [Google Scholar] [CrossRef] [PubMed]
- Warabi, K.; Matsunaga, S.; van Soest, R.W.M.; Fusetani, N. Dictyodendrins A-E, the First Telomerase-Inhibitory Marine Natural Products from the Sponge Dictyodendrilla verongiformis. J. Org. Chem. 2003, 68, 2765–2770. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Walczak, M.A. Synthesis of Asymmetrically Substituted scyllo-Inositol. Tetrahedron Lett. 2016, 57, 3281–3283. [Google Scholar] [CrossRef]
- Guang, J.; Rumlow, Z.A.; Wiles, L.M.; O’Neill, S.; Walczak, M.A. Sulfated Liposaccharides Inspired by Telomerase Inhibitor Axinelloside A. Tetrahedron Lett. 2017, 58, 4867–4871. [Google Scholar] [CrossRef]
- Andersen, R.J.; Taglialatela-Scafati, O. Avrainvilloside, a 6-Deoxy-6-aminoglucoglycerolipid from the Green Alga Avrainvillea nigricans. J. Nat. Prod. 2005, 68, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-N.; Tang, S.; Johnson, R.K.; Mattern, M.P.; Lazo, J.S.; Sharlow, E.R.; Harich, K.; Kingston, D.G.I. New Glycolipid Inhibitors of Myt1 Kinase. Tetrahedron 2005, 61, 883–887. [Google Scholar] [CrossRef]
- Göllner, C.; Philipp, C.; Dobner, B.; Sippl, W.; Schmidt, M. First Total Synthesis of 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol—A Glycoglycerolipid of a Marine Alga with a High Inhibitor Activity against Human Myt1-Kinase. Carbohydr. Res. 2009, 344, 1628–1631. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Li, C.; Guan, H.; Yu, G. Synthesis of Glycoglycerolipid of 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol and its Analogues, Inhibitors of Human Myt1-Kinase. Carbohydr. Res. 2012, 355, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, Y.; Wang, W.; Zhang, X.; Li, C.; Guan, H. Synthesis and Antiviral Evaluation of 6′-acylamido-6′-deoxy-α-d-mannoglycerolipids. Carbohydr. Res. 2013, 381, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, Y.; Zhang, J.; Zhao, Z.; Yu, G.; Guan, H. Synthesis of 6′-acylamido-6′-deoxy-α-d-galactoglycerolipids. Carbohydr. Res. 2013, 376, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhang, J.; Ma, H.; Sun, L.; Zhang, X.; Yu, G.; Guan, H.; Wang, W.; Li, C. Synthesis and Anti-Influenza A Virus Activity of 6′-amino-6′-deoxy-glucoglycerolipids Analogs. Mar. Drugs 2016, 14, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Mangoni, A. Isolation of Five-Membered Cyclitol Glycolipids, Crasserides: Unique Glycerides from the Sponge Pseudoceratina crassa. J. Org. Chem. 1993, 58, 186–191. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Glycolipids from Sponge. 11. Isocrasserides, Novel Glycolipids with a Five-Membered Cyclitol Widely Distributed in Marine Sponges. J. Nat. Prod. 2002, 65, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Higuchi, K.; Kato, A.; Murakami, N.; Kobayashi, M. Myrmekiosides A and B, Novel Mono-O-alkyl-diglycosylglycerols Reversing Tumor Cell Morphology of ras-Transformed Cells from a Marine Sponge of Myrmekioderma sp. Tetrahedron 1999, 55, 14865–14870. [Google Scholar] [CrossRef]
- Farokhi, F.; Wielgosz-Collin, G.; Robic, A.; Debitus, C.; Malleter, M.; Roussakis, C.; Kornprobst, J.-M.; Barnathan, G. Antiproliferative Activity against Human non-Small Cell Lung Cancer of Two O-alkyl-diglycosylglycerols from the Marine Sponges Myrmekioderma dendyi and Trikentrion laeve. Eur. J. Med. Chem. 2012, 49, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, C.; Sun, L.; Yu, G.; Guan, H. Total Synthesis of Myrmekioside A, a Mono-O-alkyl-diglycosylglycerol from Marine Sponge Myrmekioderma sp.: Total Synthesis of Myrmekioside A. Eur. J. Org. Chem. 2015, 2015, 4246–4253. [Google Scholar] [CrossRef]
- Williams, D.E.; Sturgeon, C.M.; Roberge, M.; Andersen, R.J. Nigricanosides A and B, Antimitotic Glycolipids Isolated from the Green Alga Avrainvillea nigricans Collected in Dominica. J. Am. Chem. Soc. 2007, 129, 5822–5823. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, T.; Fujiwara, K.; Okamoto, S.; Kondo, Y.; Akiba, U.; Ishigaki, Y.; Katoono, R.; Suzuki, T. Double Bond Formation Based on Nitroaldol Reaction and Radical Elimination: A Prototype Segment Connection Method for the Total Synthesis of Nigricanoside A Dimethyl Ester. Tetrahedron Lett. 2018, 59, 1846–1850. [Google Scholar] [CrossRef]
- Kinashi, N.; Fujiwara, K.; Tsunoda, T.; Katoono, R.; Kawai, H.; Suzuki, T. A stereoselective Method for the Construction of the C8′–O–C6′′ Ether of Nigricanoside-A: Synthesis of Simple Models for the C20 Lipid Chain/Galactosyl Glycerol Segment. Tetrahedron Lett. 2013, 54, 4564–4567. [Google Scholar] [CrossRef]
- Kurashina, Y.; Kuwahara, S. Stereoselective Synthesis of a Protected Form of (6R,7E,9S,10R,12Z)-6,9,10-trihydroxy-7,12-hexadecadienoic Acid. Biosci. Biotechnol. Biochem. 2012, 76, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Espindola, A.P.D.M.; Crouch, R.; DeBergh, J.R.; Ready, J.M.; MacMillan, J.B. Deconvolution of Complex NMR Spectra in Small Molecules by Multi Frequency Homonuclear Decoupling (MDEC). J. Am. Chem. Soc. 2009, 131, 15994–15995. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Koswatta, P.; DeBergh, J.R.; Fu, P.; Pan, E.; MacMillan, J.B.; Ready, J.M. Structure Elucidation of Nigricanoside A through Enantioselective Total Synthesis. Chem. Sci. 2015, 6, 2932–2937. [Google Scholar] [CrossRef] [PubMed]
- Wojnar, J.M.; Northcote, P.T. The Agminosides: Naturally Acetylated Glycolipids from the New Zealand Marine Sponge Raspailia agminata. J. Nat. Prod. 2011, 74, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Ohta, E.; Ikegami, S. Ancorinoside A: A Novel Tetramic Acid Glycoside from the Marine Sponge, Ancorina sp. which Specifically Inhibits Blastulation of Starfish Embryos. J. Org. Chem. 1997, 62, 6452–6453. [Google Scholar] [CrossRef]
- Fujita, M.; Nakao, Y.; Matsunaga, S.; Seiki, M.; Itoh, Y.; van Soest, R.W.M.; Fusetani, N. Ancorinosides B–D, Inhibitors of Membrane Type 1 Matrix Metalloproteinase (MT1-MMP), from the Marine Sponge Penares sollasi Thiele. Tetrahedron 2001, 57, 1229–1234. [Google Scholar] [CrossRef]
- Ohta, E.; Ohta, S.; Ikegami, S. Ancorinoside A Mg Salt from the Marine Sponge, Ancorina sp., which Specifically Inhibits Blastulation of Starfish Embryos. Tetrahedron 2001, 57, 4699–4703. [Google Scholar] [CrossRef]
- Petermichl, M.; Schobert, R. Total Synthesis of the Diglycosidic Tetramic Acid Ancorinoside A. Chem. Eur. J. 2017, 23, 14743–14746. [Google Scholar] [CrossRef] [PubMed]
- Leão, P.N.; Nakamura, H.; Costa, M.; Pereira, A.R.; Martins, R.; Vasconcelos, V.; Gerwick, W.H.; Balskus, E.P. Biosynthesis-Assisted Structural Elucidation of the Bartolosides, Chlorinated Aromatic Glycolipids from Cyanobacteria. Angew. Chem. Int. Ed. 2015, 54, 11063–11067. [Google Scholar] [CrossRef] [PubMed]
- Afonso, T.B.; Costa, M.S.; Rezende de Castro, R.; Freitas, S.; Silva, A.; Schneider, M.P.C.; Martins, R.; Leão, P.N. Bartolosides E–K from a Marine Coccoid Cyanobacterium. J. Nat. Prod. 2016, 79, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Robertson, M.; Gauthier, A.; Finlay, B.B.; van Soest, R.; Andersen, R.J. Caminoside A, an Antimicrobial Glycolipid Isolated from the Marine Sponge Caminus sphaeroconia. Org. Lett. 2002, 4, 4089–4092. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Robertson, M.; Gauthier, A.; Finlay, B.B.; MacMillan, J.B.; Molinski, T.F.; van Soest, R.; Andersen, R.J. Caminosides B−D, Antimicrobial Glycolipids Isolated from the Marine Sponge Caminus sphaeroconia. J. Nat. Prod. 2006, 69, 173–177. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, J.B.; Linington, R.G.; Andersen, R.J.; Molinski, T.F. Stereochemical Assignment in Acyclic Lipids Across Long Distance by Circular Dichroism: Absolute Stereochemistry of the Aglycone of Caminoside A. Angew. Chem. Int. Ed. 2004, 43, 5946–5951. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Han, X.; Yu, B. First Total Synthesis of Caminoside A, an Antimicrobial Glycolipid from Sponge. Synlett 2005, 3, 437–440. [Google Scholar] [CrossRef]
- Zhang, Z.; Zong, C.; Song, G.; Lv, G.; Chun, Y.; Wang, P.; Ding, N.; Li, Y. Total Synthesis of Caminoside B, a Novel Antimicrobial Glycolipid Isolated from the Marine Sponge Caminus sphaeroconia. Carbohydr. Res. 2010, 345, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Glycolipids from Sponges. Part 17. Clathrosides and Isoclathrosides, Unique Glycolipids from the Caribbean Sponge Agelas clathrodes. J. Nat. Prod. 2006, 69, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, L.; Costantino, V.; Fattorusso, E.; Mangoni, A. Glycolipids from Sponges. Part 16. Discoside, a Rare myo-Inositol-Containing Glycolipid from the Caribbean Sponge Discodermia dissoluta. J. Nat. Prod. 2005, 68, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Florence, G.J.; Aslam, T.; Miller, G.J.; Milne, G.D.S.; Conway, S.J. Synthesis of the Marine Glycolipid Dioctadecanoyl Discoside. Synlett 2009, 19, 3099–3102. [Google Scholar] [CrossRef]
- Gaspar, H.; Cutignano, A.; Grauso, L.; Neng, N.; Cachatra, V.; Fontana, A.; Xavier, J.; Cerejo, M.; Vieira, H.; Santos, S. Erylusamides: Novel Atypical Glycolipids from Erylus cf. deficiens. Mar. Drugs 2016, 14, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Sata, N.; Asai, N.; Matsunaga, S.; Fusetani, N. Erylusamines, IL-6 Receptor Antagonists, from the Marine Sponge, Erylus placenta. Tetrahedron 1994, 50, 1105–1110. [Google Scholar] [CrossRef]
- Fusetani, N.; Sata, N.; Matsunaga, S. Isolation and Structure Elucidation of Erylusamine B, a New Class of Marine Natural Products, which Blocked an IL-6 Receptor, from the Marine Sponge Erylus placenta Thiele. Tetrahedron Lett. 1993, 34, 4067–4070. [Google Scholar] [CrossRef]
- Goobes, R.; Rudi, A.; Kashman, Y.; Ilan, M.; Loya, Y. Three New Glycolipids from a Red Sea Sponge of the Genus Erylus. Tetrahedron 1996, 52, 7921–7928. [Google Scholar] [CrossRef]
- Tareq, F.S.; Kim, J.H.; Lee, M.A.; Lee, H.-S.; Lee, Y.-J.; Lee, J.S.; Shin, H.J. Ieodoglucomides A and B from a Marine-Derived Bacterium Bacillus licheniformis. Org. Lett. 2012, 14, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Tareq, F.S.; Lee, H.-S.; Lee, Y.-J.; Lee, J.S.; Shin, H.J. Ieodoglucomide C and Ieodoglycolipid, New Glycolipids from a Marine-Derived Bacterium Bacillus licheniformis 09IDYM23. Lipids 2015, 50, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.R.; Jithender, E.; Prasad, K.R. Total Syntheses of the Proposed Structure for Ieodoglucomides A and B. J. Org. Chem. 2013, 78, 4251–4260. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.R.; Jithender, E.; Singh, A.; Ummanni, R. Stereoisomers of Ieodoglucomides A and B: Synthesis and Evaluation of Anticancer Activity. Synthesis 2014, 46, 822–827. [Google Scholar] [CrossRef]
- Warabi, K.; Zimmerman, W.T.; Shen, J.; Gauthier, A.; Robertson, M.; Finlay, B.B.; van Soest, R.; Andersen, R.J. Pachymoside A—A Novel Glycolipid Isolated from the Marine Sponge Pachymatisma johnstonia. Can. J. Chem. 2004, 82, 102–112. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Plaxyloside from the Marine Sponge Plakortis simplex: An Improved Strategy for NMR Structural Studies of Carbohydrate Chains. Eur. J. Org. Chem. 2001, 2001, 4457–4462. [Google Scholar] [CrossRef]
- Ōmura, S.; Tomoda, H.; Tabata, N.; Ohyama, Y.; Abe, T.; Namikoshi, M. Roselipins, Novel Fungal Metabolites Having a Highly Methylated Fatty Acid Modified with a Mannose and an Arabinitol. J. Antibiot. 1999, 52, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, H.; Ohyama, Y.; Abe, T.; Tabata, N.; Namikoshi, M.; Yamaguchi, Y.; Masuma, R.; Ōmura, S. Roselipins, Inhibitors of Diacylglycerol Acyltransferase, Produced by Gliocladium roseum KF-1040. J. Antibiot. 1999, 52, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Tabata, N.; Ohyama, Y.; Tomoda, H.; Abe, T.; Namikoshi, M.; Ōmura, S. Structure Elucidation of Roselipins, Inhibitors of Diacylglycerol Acyltransferase Produced by Gliocladium roseum KF-1040. J. Antibiot. 1999, 52, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Ayers, S.; Zink, D.L.; Mohn, K.; Powell, J.S.; Brown, C.M.; Bills, G.; Grund, A.; Thompson, D.; Singh, S.B. Anthelmintic Constituents of Clonostachys candelabrum. J. Antibiot. 2010, 63, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Inokoshi, J.; Kawamoto, K.; Takagi, Y.; Matsuhama, M.; Ōmura, S.; Tomoda, H. Expression of Two Human Acyl-CoA: Diacylglycerol Acyltransferase Isozymes in Yeast and Selectivity of Microbial Inhibitors toward the Isozymes. J. Antibiot. 2009, 62, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, H.; Tabata, N.; Ohyama, Y.; Ōmura, S. Core Structure in Roselipins Essential for Eliciting Inhibitory Activity against Diacylglycerol Acyltransferase. J. Antibiot. 2003, 56, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Collado, J.; Singh, S.B.; Jayasuriya, H.; Dewey, R.; Polishook, J.D.; Dombrowski, A.W.; Zink, D.L.; Platas, G.; Pelaez, F.; et al. Isolation, Structure, and HIV-1-Integrase Inhibitory Activity of Structurally Diverse Fungal Metabolites. J. Ind. Microbiol. Biotechnol. 2003, 30, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Ondeyka, J.G.; Herath, K.B.; Jayasuriya, H.; Polishook, J.D.; Bills, G.F.; Dombrowski, A.W.; Mojena, M.; Koch, G.; DiSalvo, J.; DeMartino, J.; et al. Discovery of Structurally Diverse Natural Product Antagonists of Chemokine Receptor CXCR3. Mol. Divers. 2005, 9, 123–129. [Google Scholar] [CrossRef]
- Costantino, V.; Fattorusso, E.; Mangoni, A.; Di Rosa, M.; Ianaro, A. Glycolipids from Sponges. VII. Simplexides, Novel Immunosuppressive Glycolipids from the Caribbean Sponge Plakortis simplex. Bioorg. Med. Chem. Lett. 1999, 9, 271–276. [Google Scholar] [CrossRef]
- Loffredo, S.; Staiano, R.I.; Granata, F.; Costantino, V.; Borriello, F.; Frattini, A.; Lepore, M.T.; Mangoni, A.; Marone, G.; Triggiani, M. Simplexide Induces CD1d-Dependent Cytokine and Chemokine Production from Human Monocytes. PLoS ONE 2014, 9, 111326. [Google Scholar] [CrossRef] [PubMed]
- Lü, G.; Wang, P.; Liu, Q.; Zhang, Z.; Zhang, W.; Li, Y. Reactivity-based One-pot Synthesis of Immunosuppressive Glycolipids from the Caribbean Sponge Plakortis simplex. Chin. J. Chem. 2009, 27, 2217–2222. [Google Scholar]
- Li, J.; Li, W.; Yu, B. A Divergent Approach to the Synthesis of Simplexides and Congeners via a Late-Stage Olefin Cross-Metathesis Reaction. Org. Biomol. Chem. 2013, 11, 4971–4974. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).