Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations


Abstract
:
1. Introduction
1.1. Function of NaV
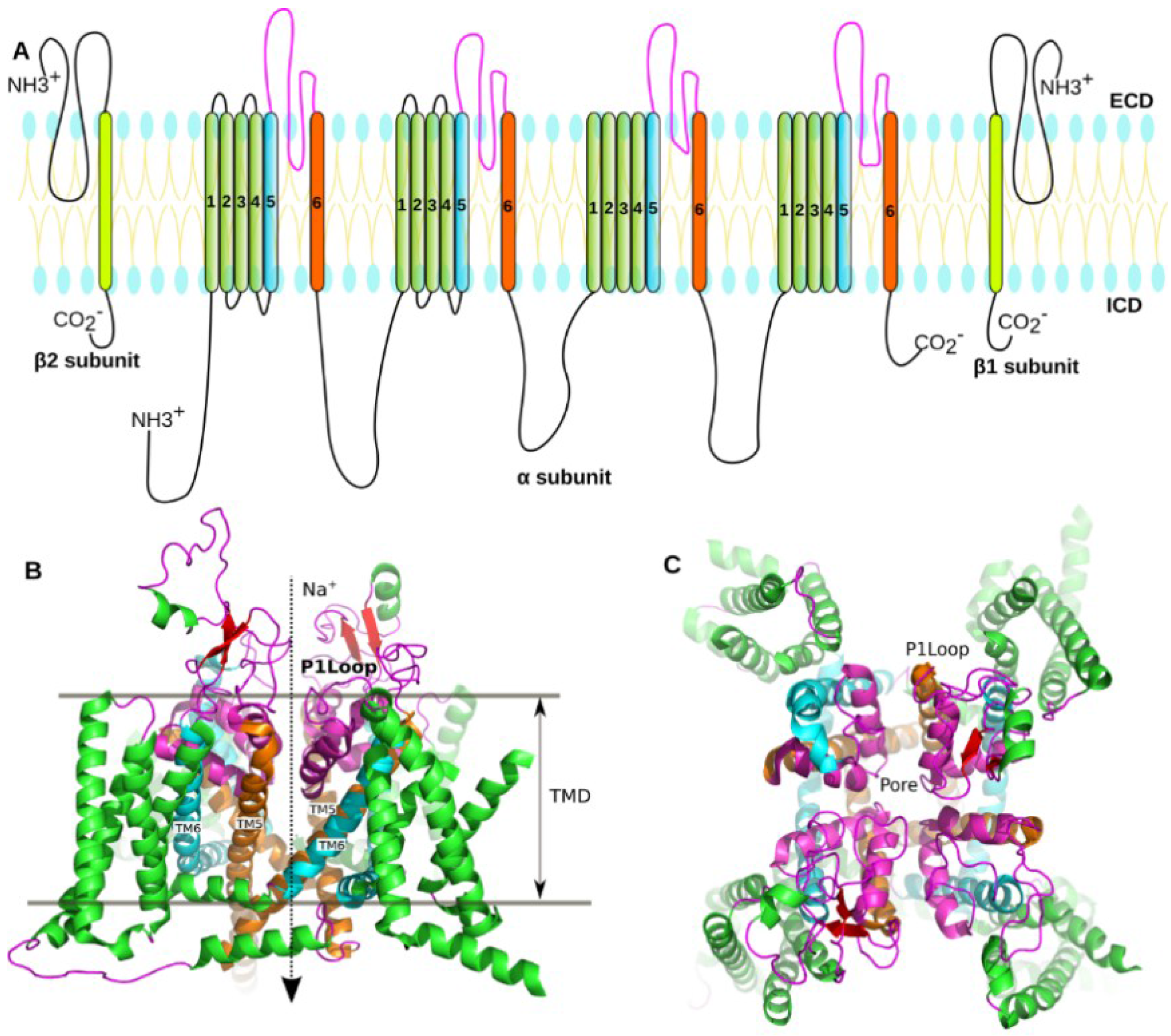
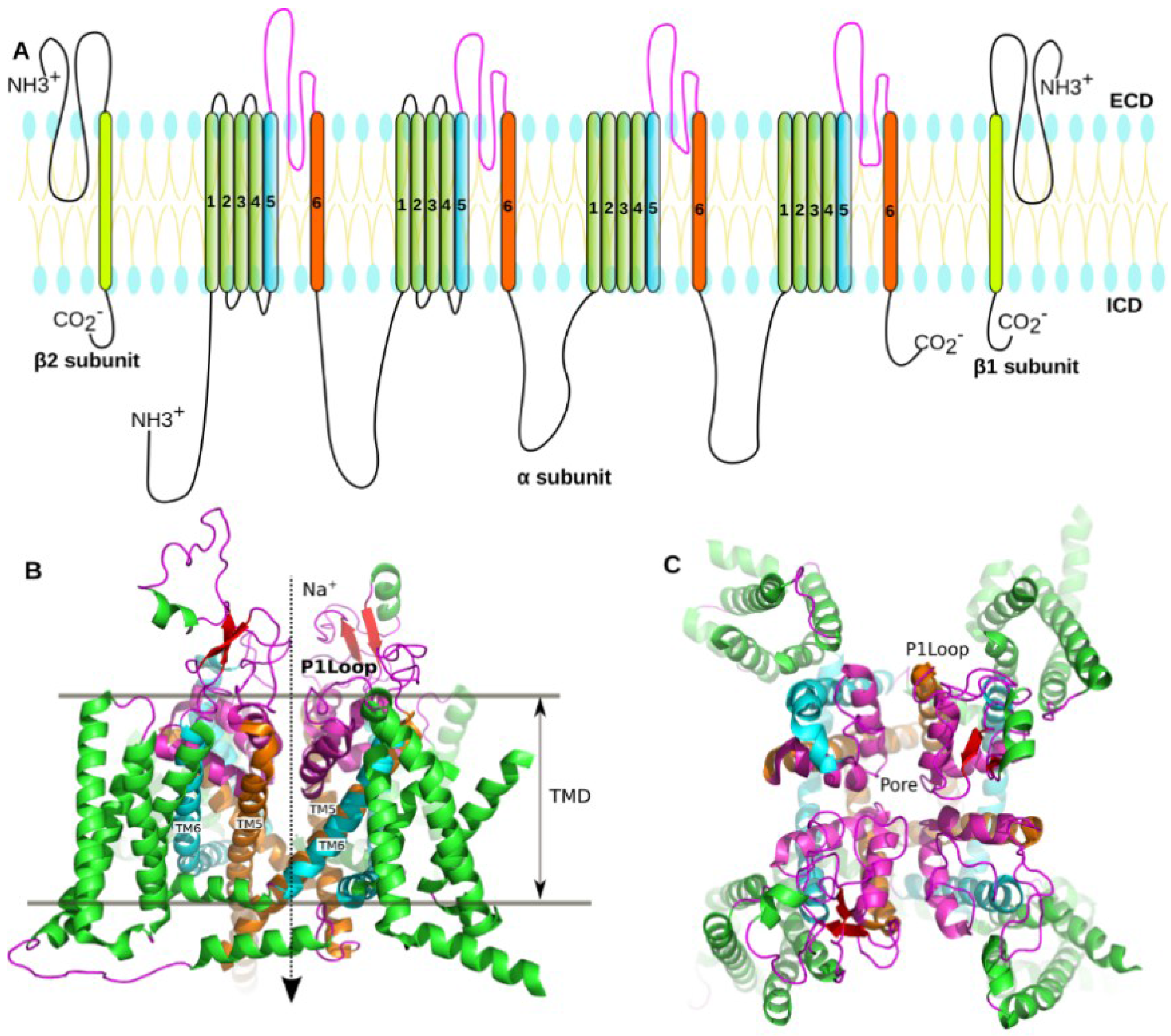
1.2. Structure of the NaV
1.3. Conotoxins
2. Methods
2.1. Flowchart for Specificity Prediction
2.2. Homology Modeling of the NaV1.4
2.3. PIIIA Docking
2.4. Restraint Molecular Dynamics Simulations
2.5. Molecular Dynamics Simulation of the NaV1.4 in Membrane
2.6. Binding Energy Calculations Using MMGB/SA
2.7. Decomposition of the Binding Energies
2.8. Binding Energies Calculations Using Umbrella Sampling
3. Results and Discussion
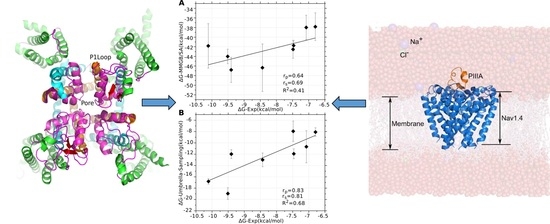
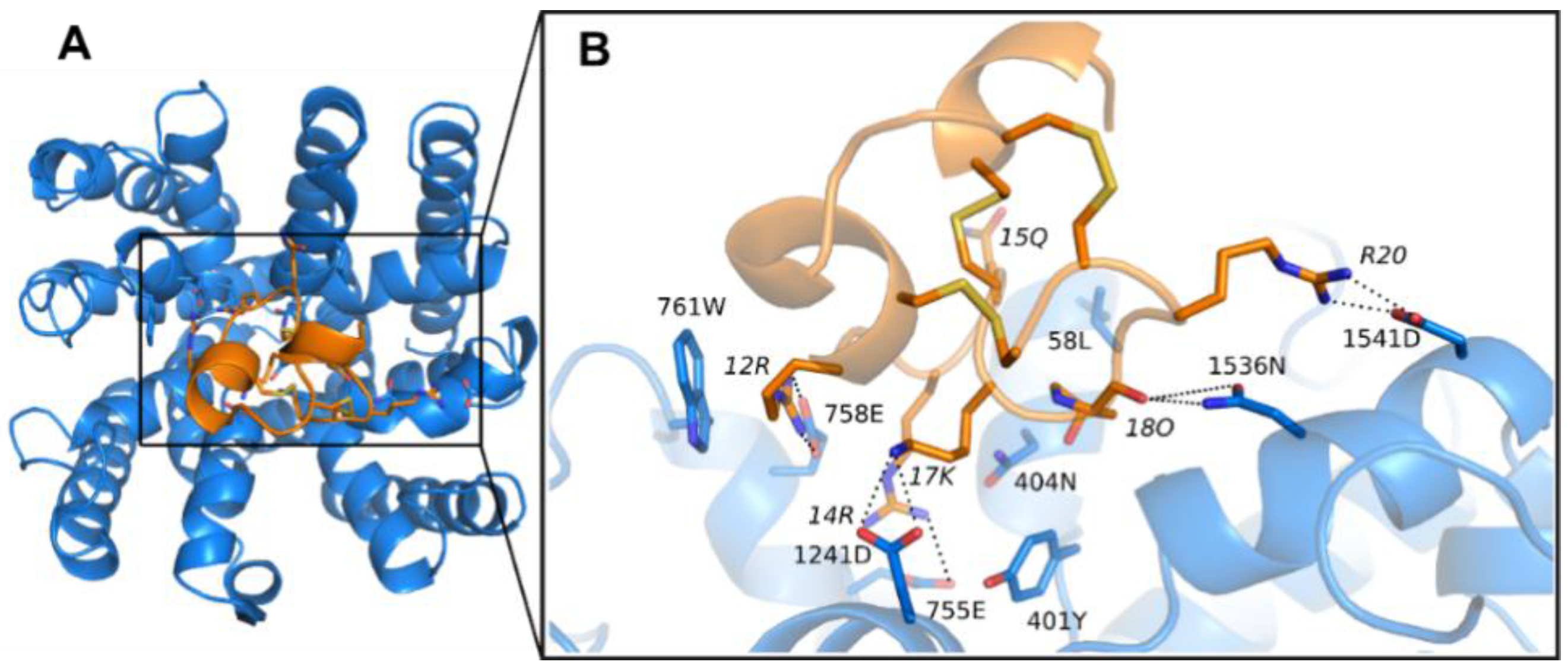
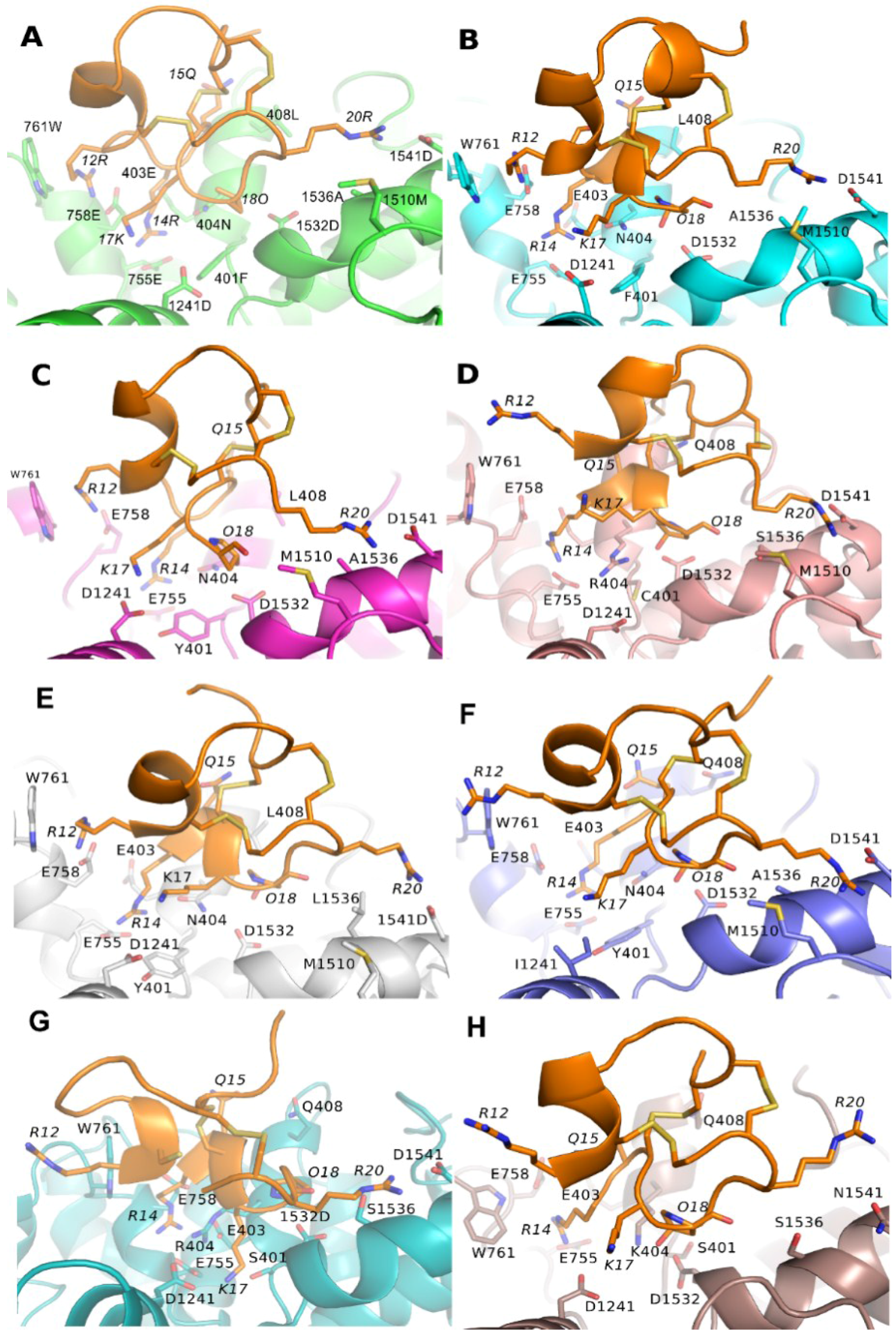
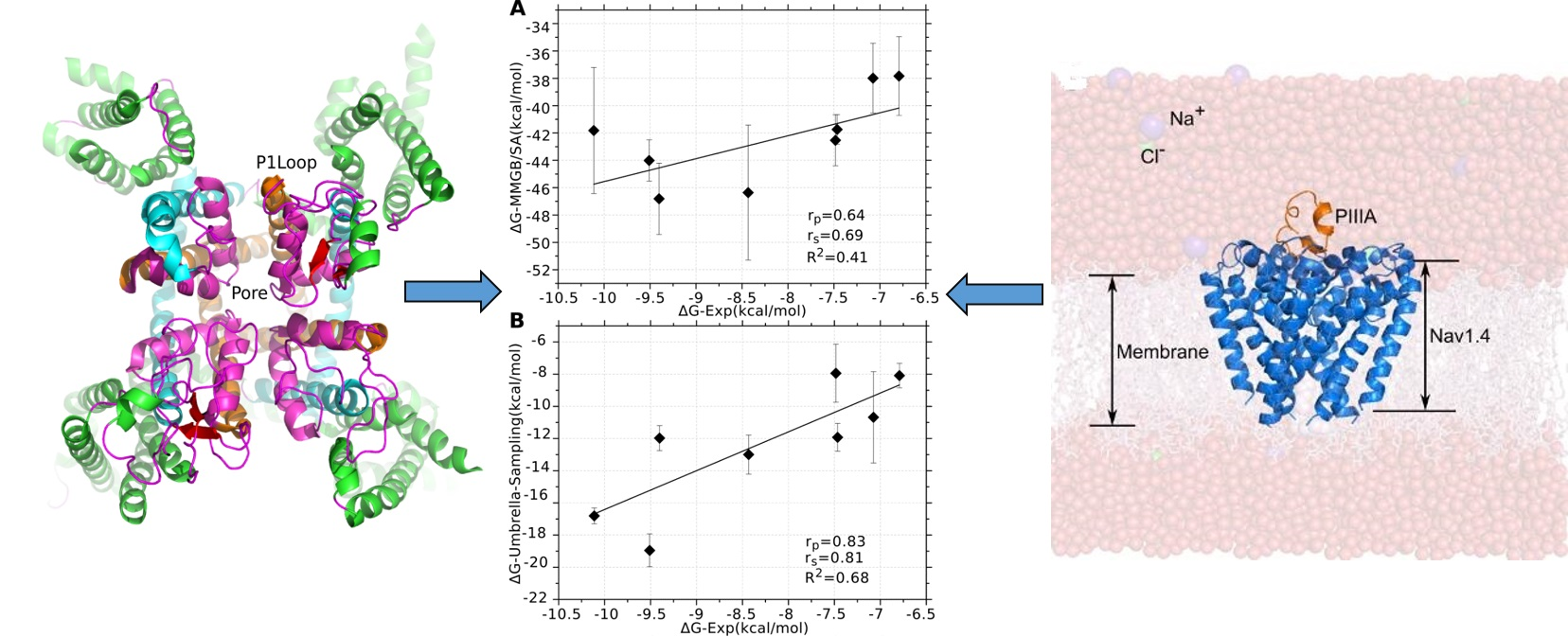
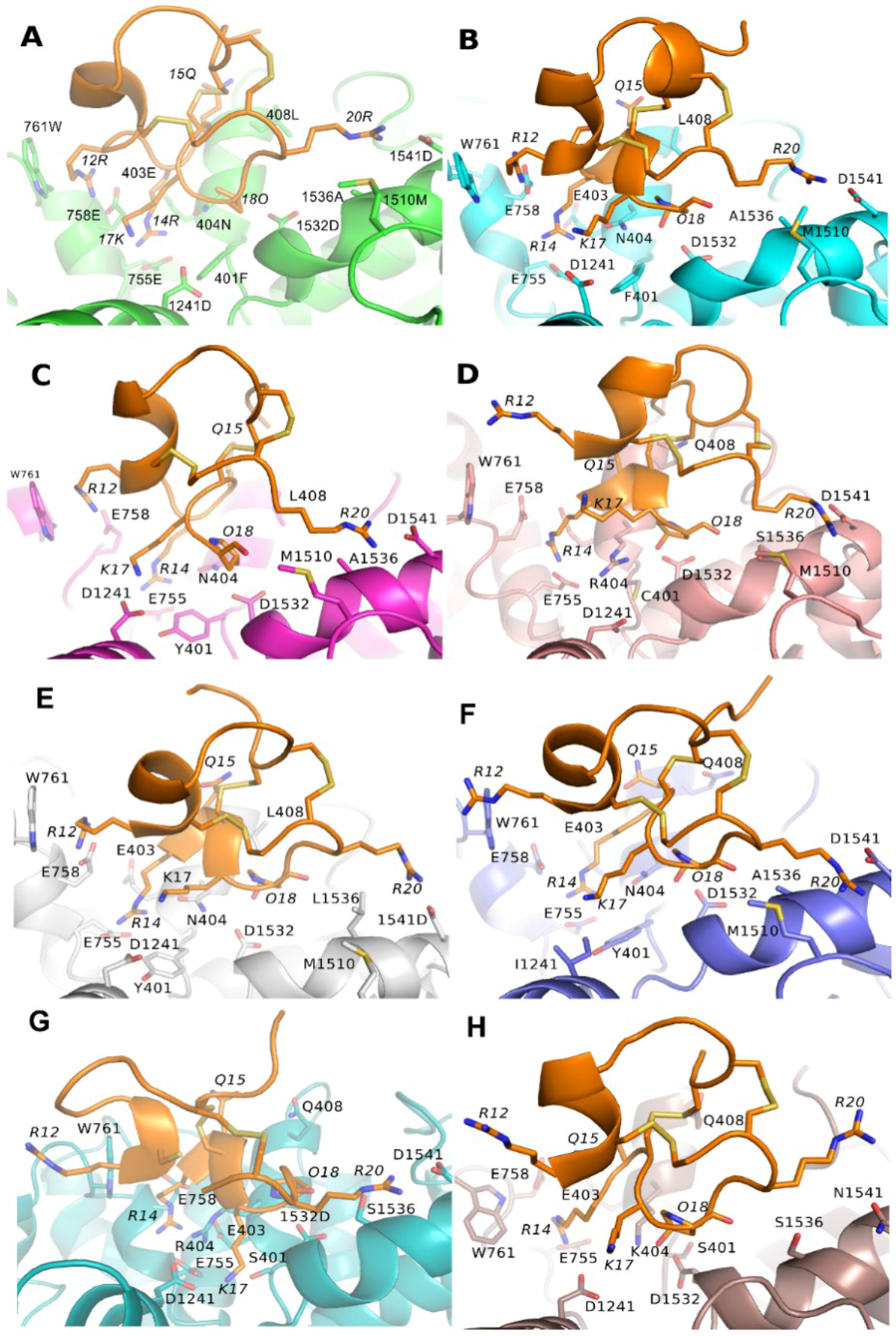
3.1. Prediction of the Binding Mode of the PIIIA at NaV1.4
3.2. Influences of the Membrane to the Conformation of the NaV1.4 in apo and NaV1.4 in Complex with PIIIA
3.3. Binding Modes of PIIIA at Other Subtypes of NaV
3.4. Prediction of the Specificity of PIIIA to Varied NaV Subtypes
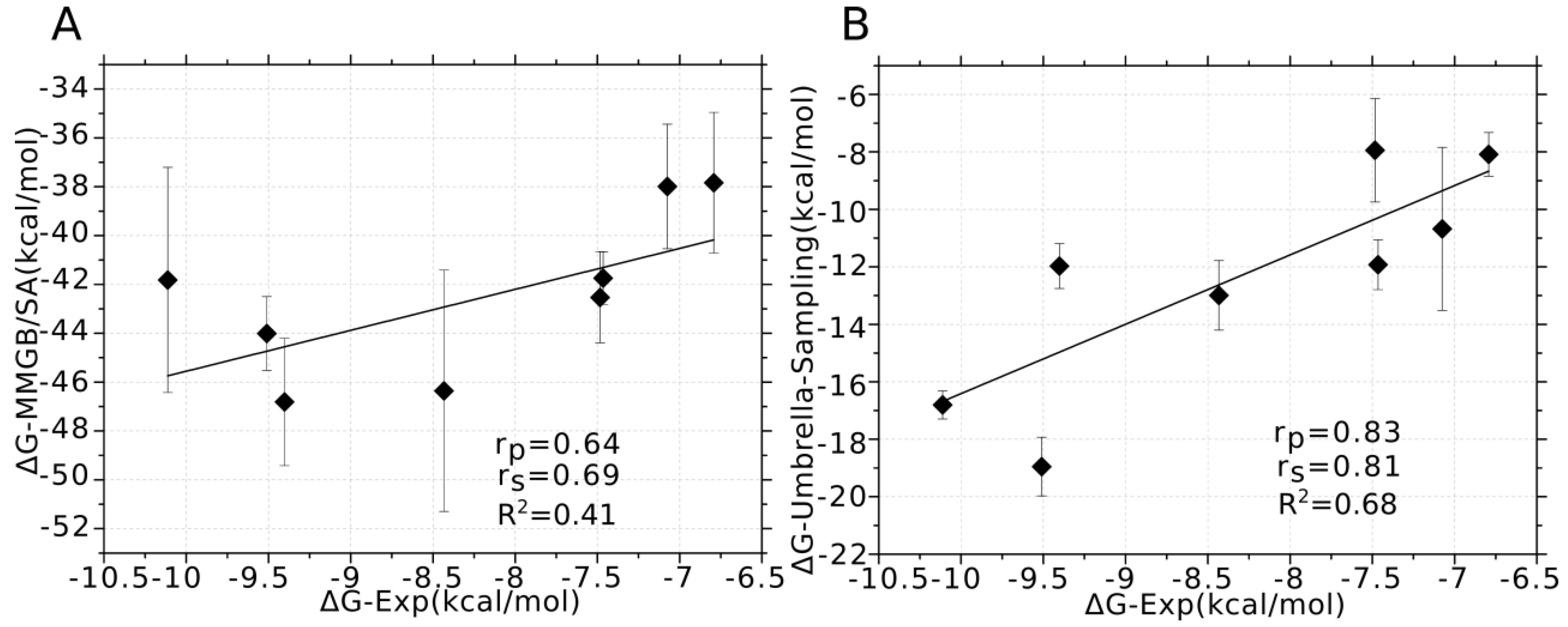
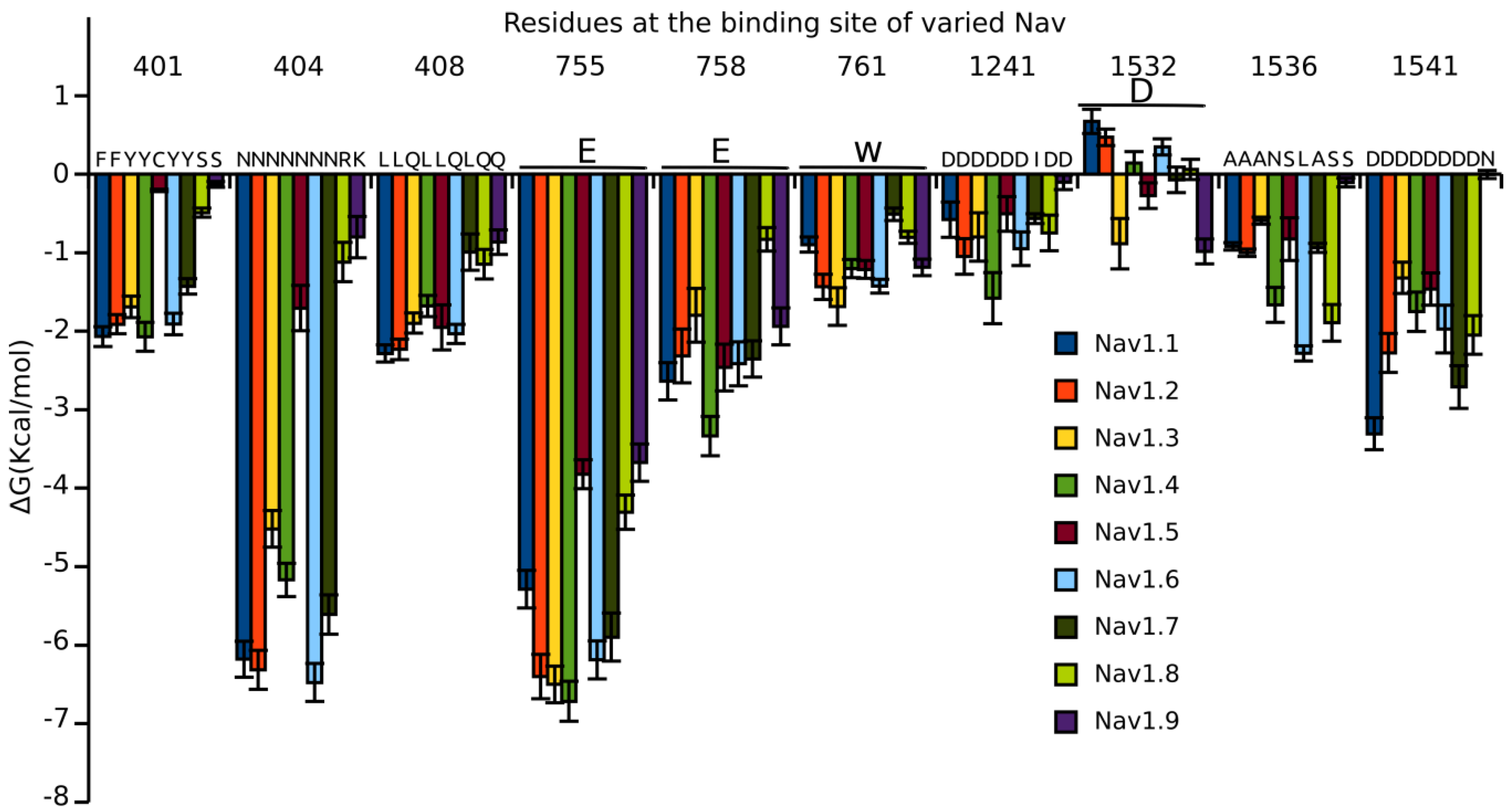
3.5. Binding Energy Decomposition Using MMGB/SA
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Ikeda, T.; Suzuki, H.; Takeshima, H.; Takahashi, T.; Kuno, M.; Numa, S. Expression of functional sodium channels from cloned cDNA. Nature 1986, 322, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Rudy, B. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J. Physiol. 1978, 283, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.P.; Waxman, S.G. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Investig. 2007, 117, 3603–3609. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L. Diversity of mammalian voltage-gated sodium channels. Ann. N. Y. Acad. Sci. 1999, 868, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhou, Q.; Pan, X.; Li, Z.; Wu, J.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhou, Q.; Wang, L.; Wu, J.; Zhao, Y.; Huang, G.; Peng, W.; Shen, H.; Lei, J.; Yan, N. Structure of the NaV 1.4-beta1 Complex from Electric Eel. Cell 2017, 170, 470.e11–482.e11. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Shon, K.J.; Olivera, B.M.; Watkins, M.; Jacobsen, R.B.; Gray, W.R.; Floresca, C.Z.; Cruz, L.J.; Hillyard, D.R.; Brink, A.; Terlau, H.; et al. mu-Conotoxin PIIIA, a new peptide for discriminating among tetrodotoxin-sensitive Na channel subtypes. J. Neurosci. 1998, 18, 4473–4481. [Google Scholar] [CrossRef] [PubMed]
- Safo, P.; Rosenbaum, T.; Shcherbatko, A.; Choi, D.Y.; Han, E.; Toledo-Aral, J.J.; Olivera, B.M.; Brehm, P.; Mandel, G. Distinction among neuronal subtypes of voltage-activated sodium channels by mu-conotoxin PIIIA. J. Neurosci. 2000, 20, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Yoshikami, D.; Azam, L.; Gajewiak, J.; Olivera, B.M.; Bulaj, G.; Zhang, M.M. mu-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 10302–10307. [Google Scholar] [CrossRef] [PubMed]
- Munasinghe, N.R.; Christie, M.J. Conotoxins That Could Provide Analgesia through Voltage Gated Sodium Channel Inhibition. Toxins 2015, 7, 5386–5407. [Google Scholar] [CrossRef] [PubMed]
- Korkosh, V.S.; Zhorov, B.S.; Tikhonov, D.B. Folding similarity of the outer pore region in prokaryotic and eukaryotic sodium channels revealed by docking of conotoxins GIIIA, PIIIA, and KIIIA in a NaVAb-based model of NaV1.4. J. Gen. Physiol. 2014, 144, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Tikhonov, D.B. Computational Structural Pharmacology and Toxicology of Voltage-Gated Sodium Channels. Curr. Top. Membr. 2016, 78, 117–144. [Google Scholar] [PubMed]
- Bagneris, C.; DeCaen, P.G.; Naylor, C.E.; Pryde, D.C.; Nobeli, I.; Clapham, D.E.; Wallace, B.A. Prokaryotic NaVMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc. Natl. Acad. Sci. USA 2014, 111, 8428–8433. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Watson, M.; Adams, D.J.; Hammarstrom, A.K.; Gage, P.W.; Hill, J.M.; Craik, D.J.; Thomas, L.; Adams, D.; Alewood, P.F.; et al. Solution structure of mu-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J. Biol. Chem. 2002, 277, 27247–27255. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal alpha7-nAChR by alpha-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M.; UniProt, C. UniProt Knowledgebase: A hub of integrated protein data. Database (Oxford) 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.; Giese, T.J.; Gohlke, H.; Götz, A.; Greene, D.; Homeyer, N.; et al. AMBER 2017; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- Frias Nunez, M. Plans for the establishment of central vaccination committees in the institutionalization of Medicine in Colombia. Cuad. Complut. Hist. Med. Cienc. 1993, 1, 89–102. [Google Scholar] [PubMed]
- Lavery, R.; Zakrzewska, K.; Beveridge, D.; Bishop, T.C.; Case, D.A.; Cheatham, T., 3rd; Dixit, S.; Jayaram, B.; Lankas, F.; Laughton, C.; et al. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res. 2010, 38, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Grossfield, A.; Pitman, M.C.; Feller, S.E.; Soubias, O.; Gawrisch, K. Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J. Mol. Biol. 2008, 381, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J. Correlations in liquid water for the TIP3P-Ewald, TIP4P-2005, TIP5P-Ewald, and SWM4-NDP models. J. Chem. Phys. 2012, 136, 064518. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, A.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N·Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Wittayanarakul, K.; Hannongbua, S.; Feig, M. Accurate prediction of protonation state as a prerequisite for reliable MM-PB(GB)SA binding free energy calculations of HIV-1 protease inhibitors. J. Comput. Chem. 2008, 29, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Northrup, S.H.; Pear, M.R.; Lee, C.Y.; McCammon, J.A.; Karplus, M. Dynamical theory of activated processes in globular proteins. Proc. Natl. Acad. Sci. USA 1982, 79, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. 1. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- McArthur, J.R.; Singh, G.; O’Mara, M.L.; McMaster, D.; Ostroumov, V.; Tieleman, D.P.; French, R.J. Orientation of mu-conotoxin PIIIA in a sodium channel vestibule, based on voltage dependence of its binding. Mol. Pharmacol. 2011, 80, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Tietze, A.A.; Tietze, D.; Ohlenschlager, O.; Leipold, E.; Ullrich, F.; Kuhl, T.; Mischo, A.; Buntkowsky, G.; Gorlach, M.; Heinemann, S.H.; et al. Structurally Diverse mu-Conotoxin PIIIA Isomers Block Sodium Channel NaV1.4. Angew. Chem. Int. Ed. 2012, 51, 4058–4061. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Zhang, M.M.; Gajewiak, J.; Azam, L.; Rivier, J.E.; Olivera, B.M.; Yoshikami, D. alpha- And beta-subunit composition of voltage-gated sodium channels investigated with mu-conotoxins and the recently discovered mu O-conotoxin GVIIJ. J. Neurophysiol. 2015, 113, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Wilson, M.J.; Azam, L.; Gajewiak, J.; Rivier, J.E.; Bulaj, G.; Olivera, B.M.; Yoshikami, D. Co-expression of Na(V)beta subunits alters the kinetics of inhibition of voltage-gated sodium channels by pore-blocking mu-conotoxins. Br. J. Pharmacol. 2013, 168, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Complex Structures between the N-Type Calcium Channel (Ca(V)2.2) and omega-Conotoxin GVIA Predicted via Molecular Dynamics. Biochemistry 2013, 52, 3765–3772. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Why the Drosophila Shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry 2013, 52, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Mahdavi, S.; Kuyucak, S. Computational Study of Binding of mu-Conotoxin GIIIA to Bacterial Sodium Channels Na(V)Ab and NaVRh. Biochemistry 2016, 55, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational Studies of Marine Toxins Targeting Ion Channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Suresh, A.; Hung, A. Molecular simulation study of the unbinding of alpha-conotoxin [Upsilon4E]GID at the alpha7 and alpha4beta2 neuronal nicotinic acetylcholine receptors. J. Mol. Graph. Model. 2016, 70, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.L.; Tabassum, N.; Jiang, T. Investigation of alpha-conotoxin unbinding using umbrella sampling. Bioorg. Med. Chem. Lett. 2016, 26, 1296–1300. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NaV | R12 a | R14 a | Q15 a | K17 a | O18 a | R20 a |
|---|---|---|---|---|---|---|
| NaV1.1 | E758, W761 | F401, N404, E755 | L408 | - | N404, D1533 | A1536, D1541 |
| NaV1.2 | E758, W761 | F401, N404, E758 | L408 | D1241 | N404, D1533 | A1536, D1541 |
| NaV1.3 | E758, W761 | E755 | L408 | D1241 | N404, D1533 | A1536, D1541 |
| NaV1.4 | E758, W761 | F401, N404, E755, E758 | L408 | D1241 | N404, D1533, N1536 | D1541 |
| NaV1.5 | W761 | E755, E758 | Q408 | - | R404 | D1541 |
| NaV1.6 | E758, W761 | Y401, N404, E755 | L408 | - | R1533 | L1536, D1541 |
| NaV1.7 | W761 | F401, N404, E755, E758 | Q408 | - | N404, D1533 | A1536, D1541 |
| NaV1.8 | W761 | N404, E755 | Q408 | D1241, G1536 | D1532, D1536 | S1541 |
| NaV1.9 | W761 | E755, E758 | Q408 | - | D1532, S1536 | N1541 |
| NaV | IC50 | MMGB/SA a | Umbrella Sampling b | Experimental (kcal/mol) c |
|---|---|---|---|---|
| NaV1.1 | 120 nm | −46.81 ± 2.61 | −11.97 ± 0.78 | −9.40 |
| NaV1.2 | 620 nm | −46.36 ± 4.94 | −12.99 ± 1.21 | −8.43 |
| NaV1.3 | 3.2 μm | −41.74 ± 1.08 | −11.93 ± 0.87 | −7.46 |
| NaV1.4 | 36 nm | −41.82 ± 4.61 | −16.81 ± 0.49 | −10.11 |
| NaV1.5 | 10 μm | −37.83 ± 2.88 | −8.09 ± 1.44 | −6.79 |
| NaV1.6 | 100 nm | −44.01 ± 1.51 | −18.95 ± 1.02 | −9.51 |
| NaV1.7 | >10 μm | −42.53 ± 1.86 | −7.94 ± 1.80 | −7.48 |
| NaV1.8 | >10 μm | −38.00 ± 2.55 | −10.68 ± 2.84 | −7.07 |
| NaV1.9 | - | −29.24 ± 5.03 | −9.41 ± 2.01 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Huang, W.; Jiang, T.; Yu, R. Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations. Mar. Drugs 2018, 16, 153. https://doi.org/10.3390/md16050153
Chen F, Huang W, Jiang T, Yu R. Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations. Marine Drugs. 2018; 16(5):153. https://doi.org/10.3390/md16050153
Chicago/Turabian StyleChen, Fangling, Wenxin Huang, Tao Jiang, and Rilei Yu. 2018. "Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations" Marine Drugs 16, no. 5: 153. https://doi.org/10.3390/md16050153
APA StyleChen, F., Huang, W., Jiang, T., & Yu, R. (2018). Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations. Marine Drugs, 16(5), 153. https://doi.org/10.3390/md16050153