Recent Synthesis and Discovery of Brefeldin A Analogs

Abstract
:1. Introduction
2. Results
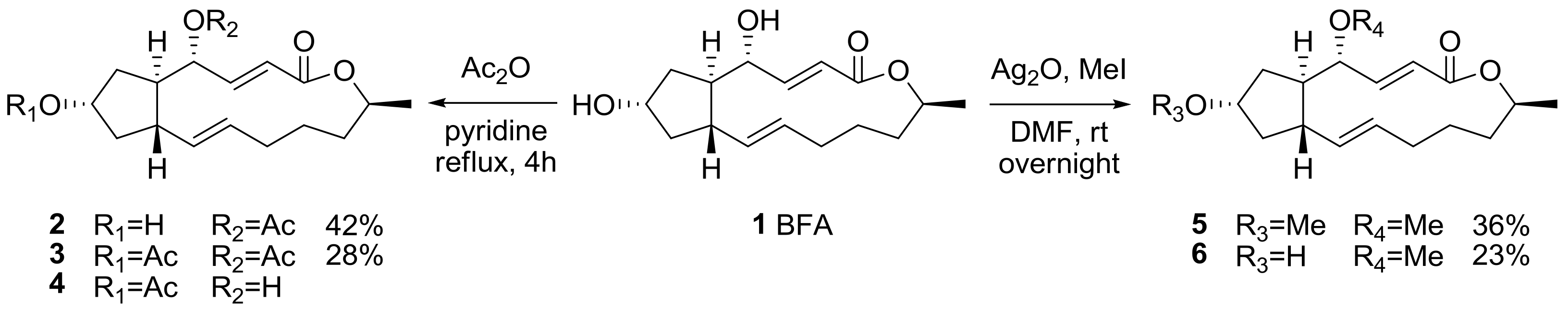
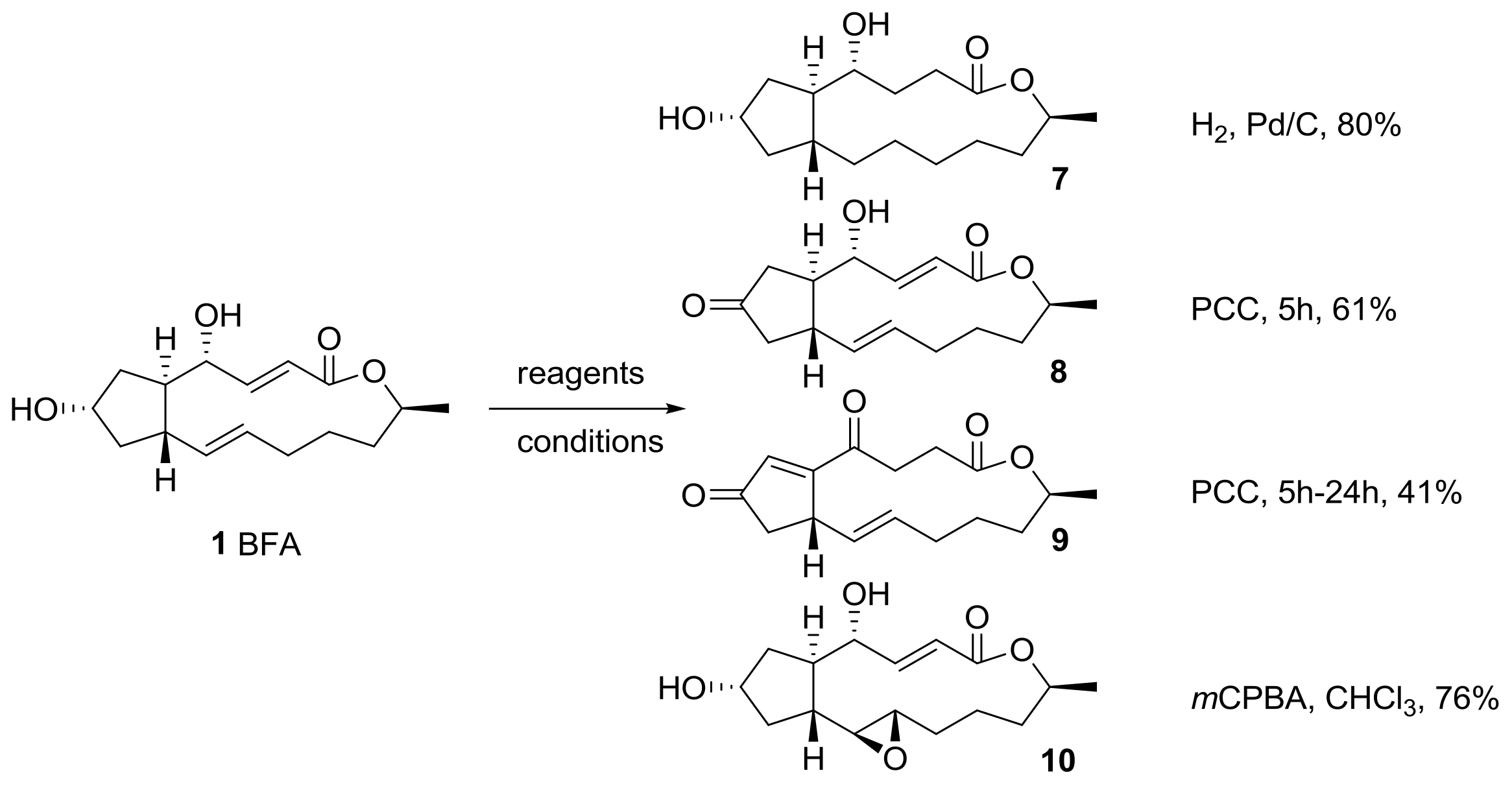
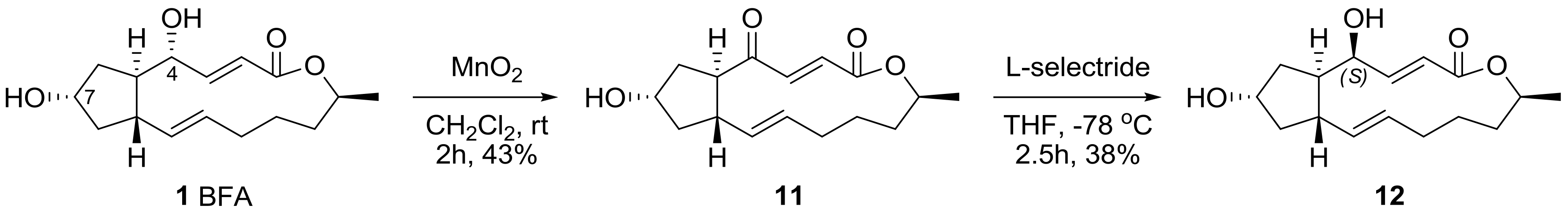
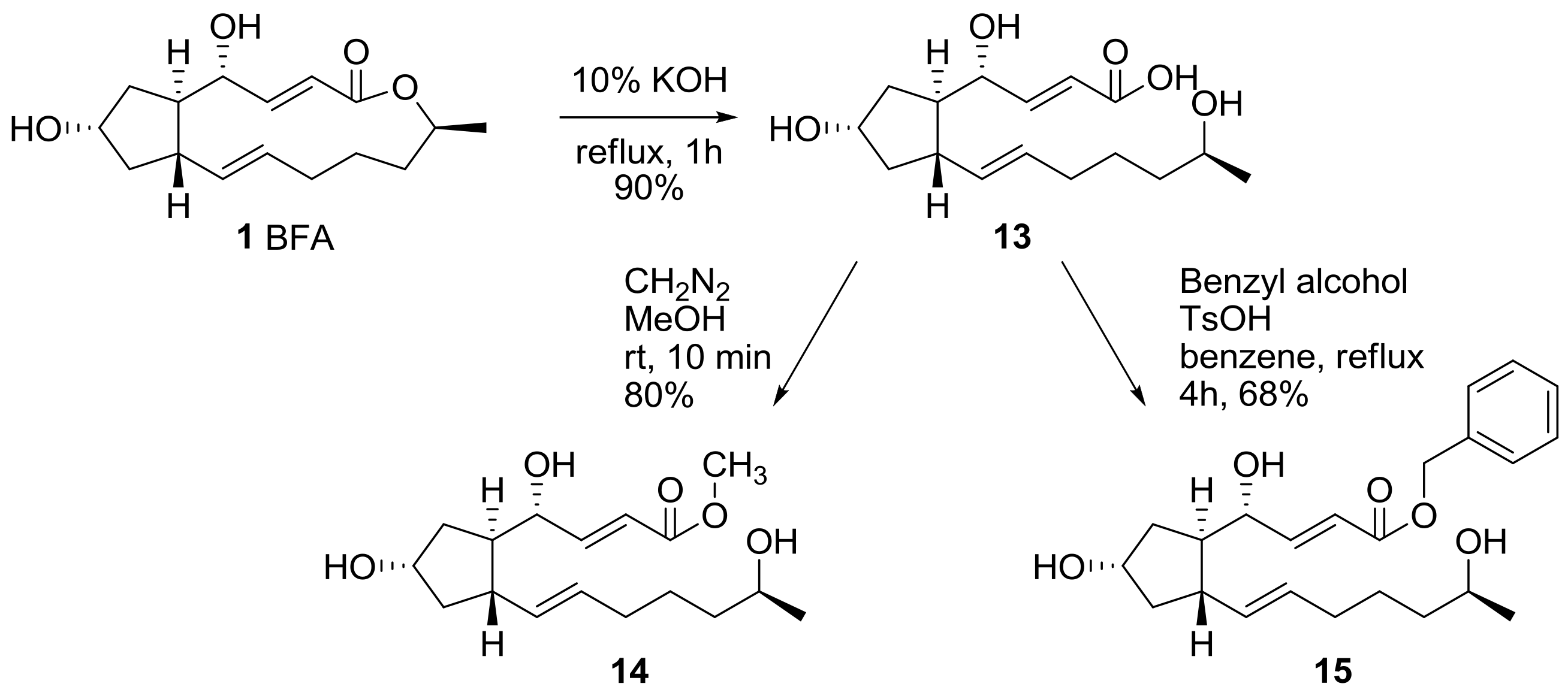
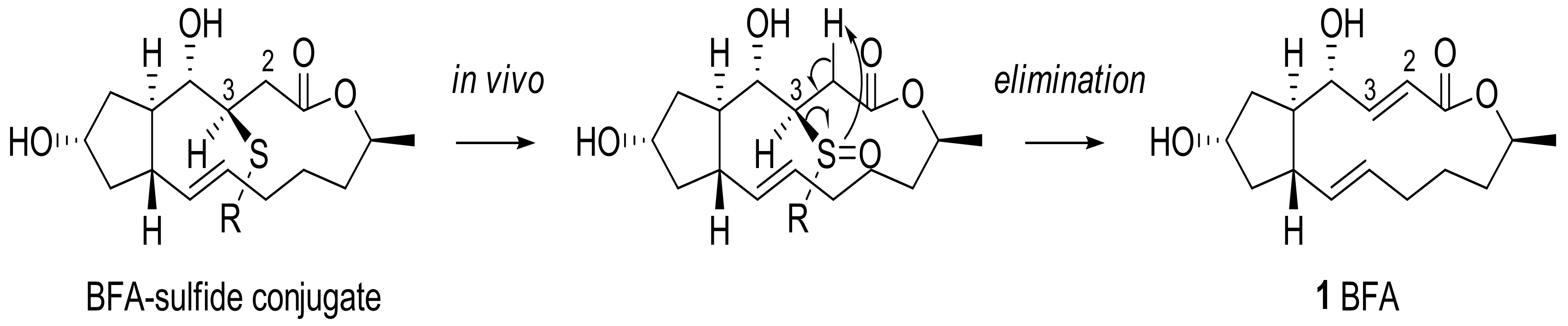
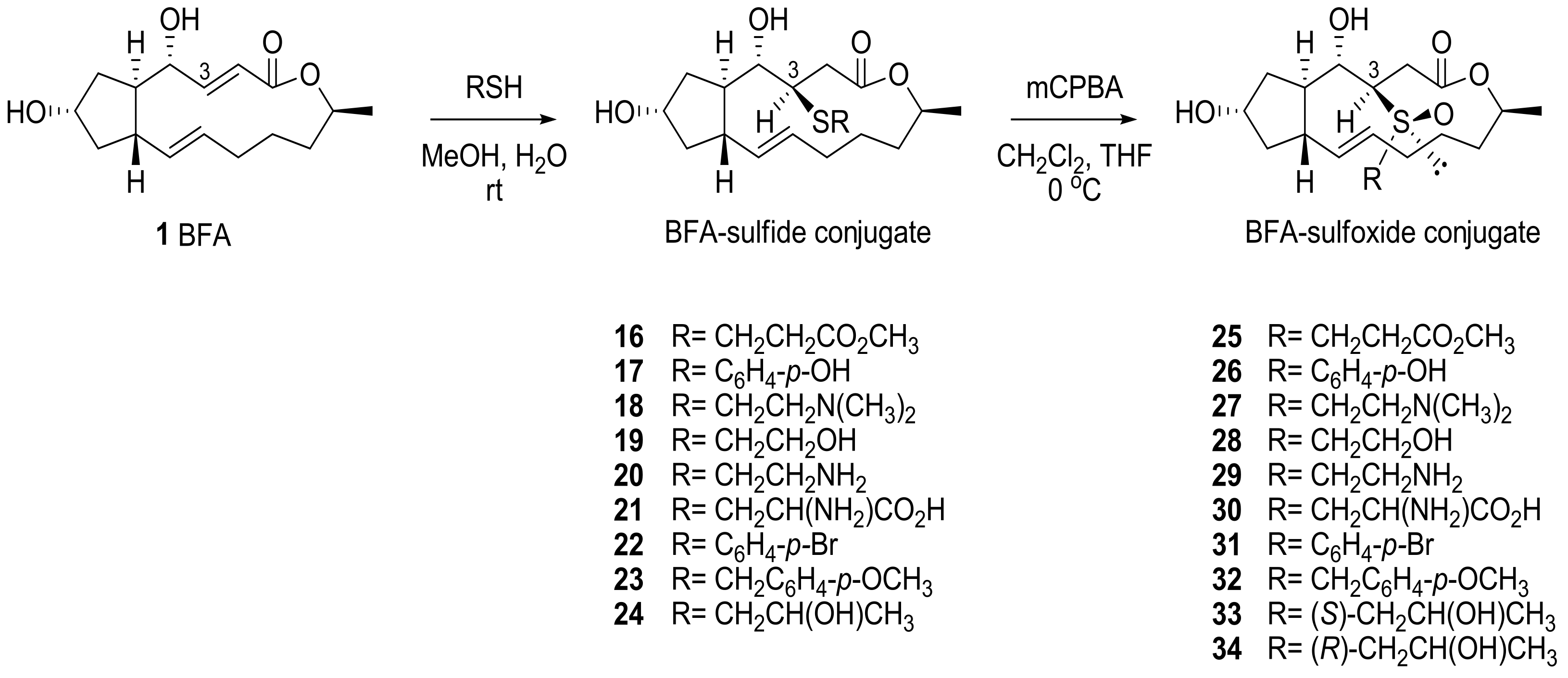
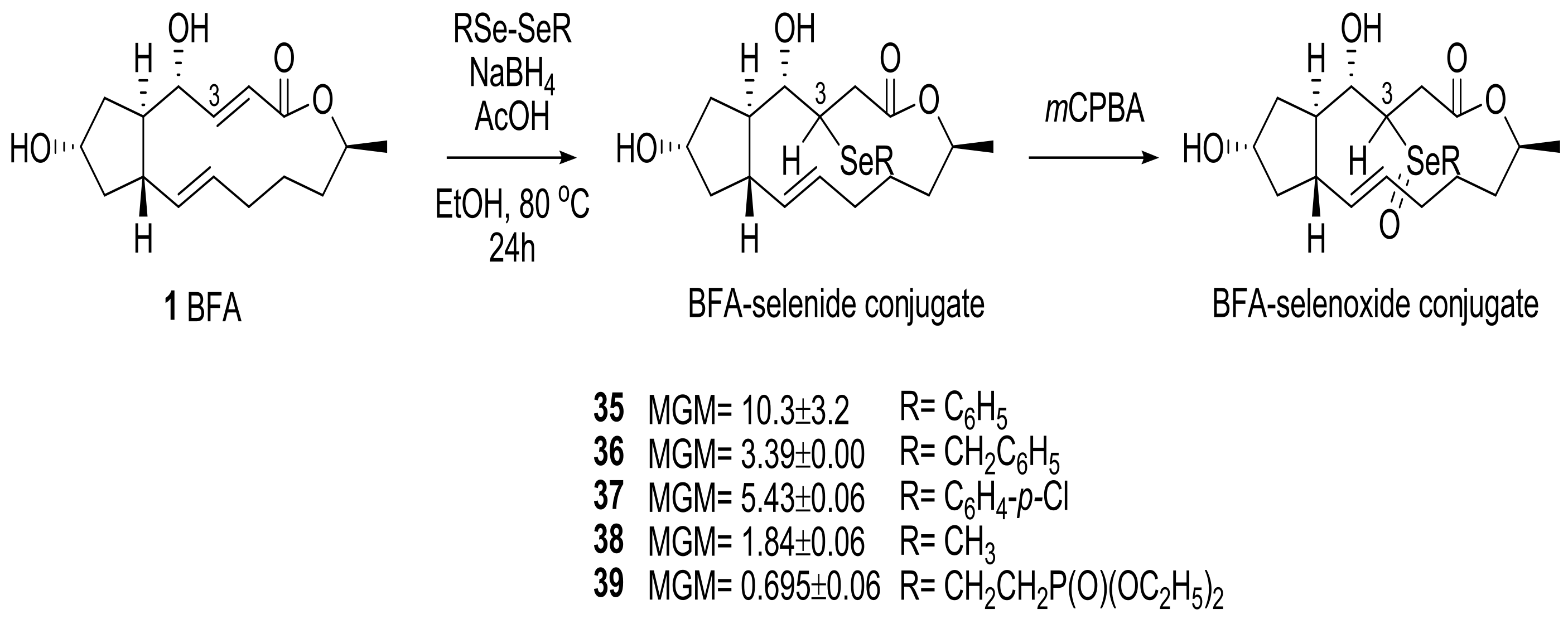
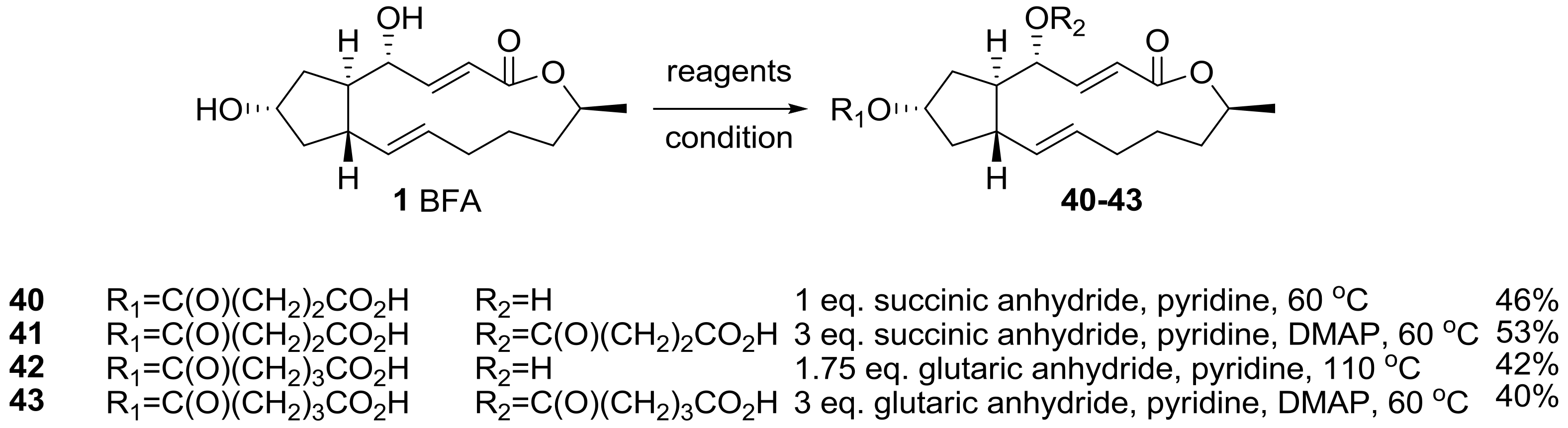
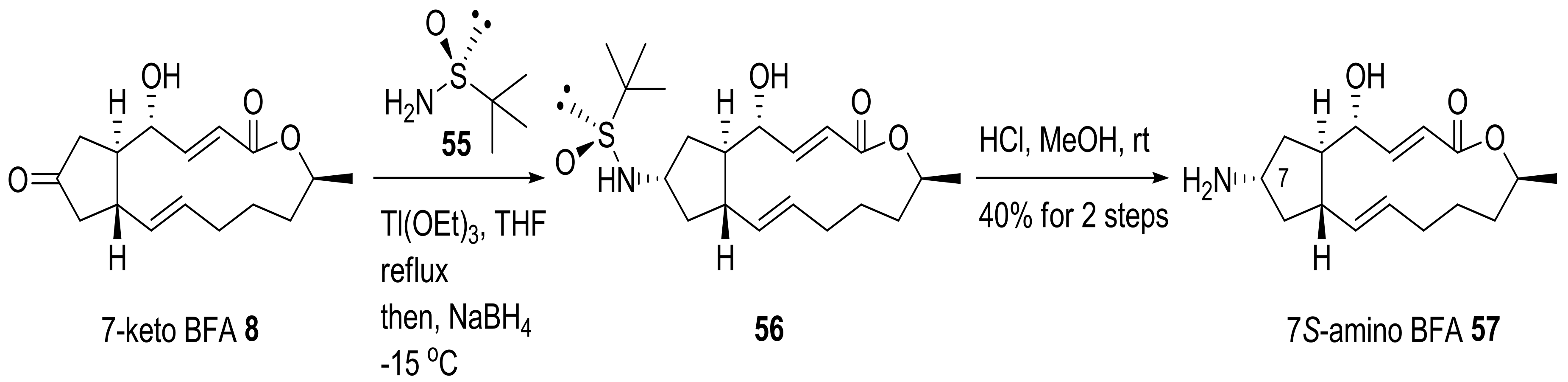
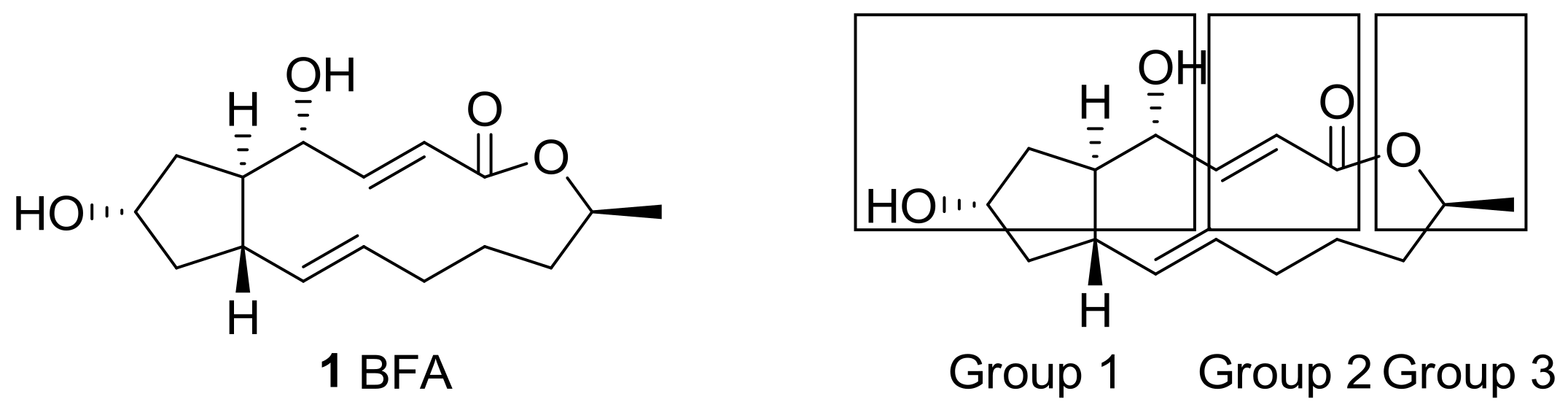
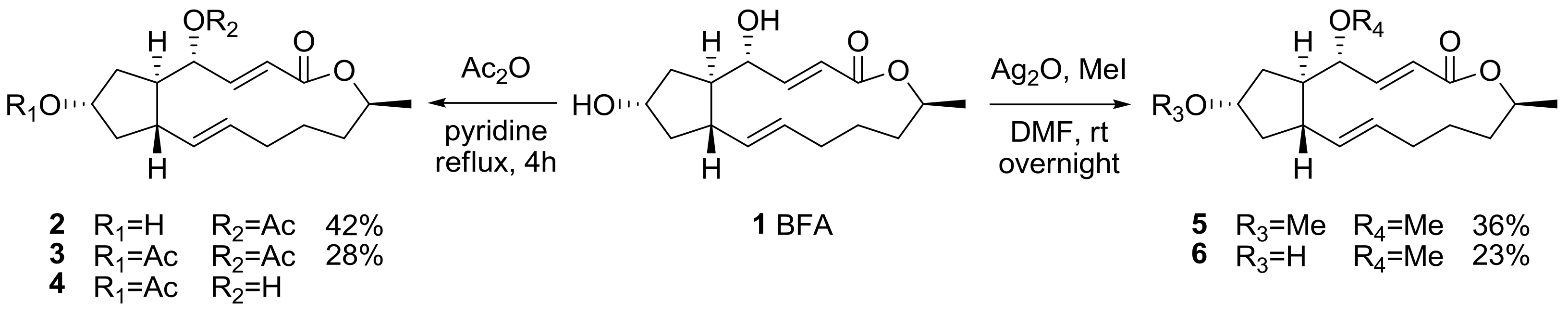
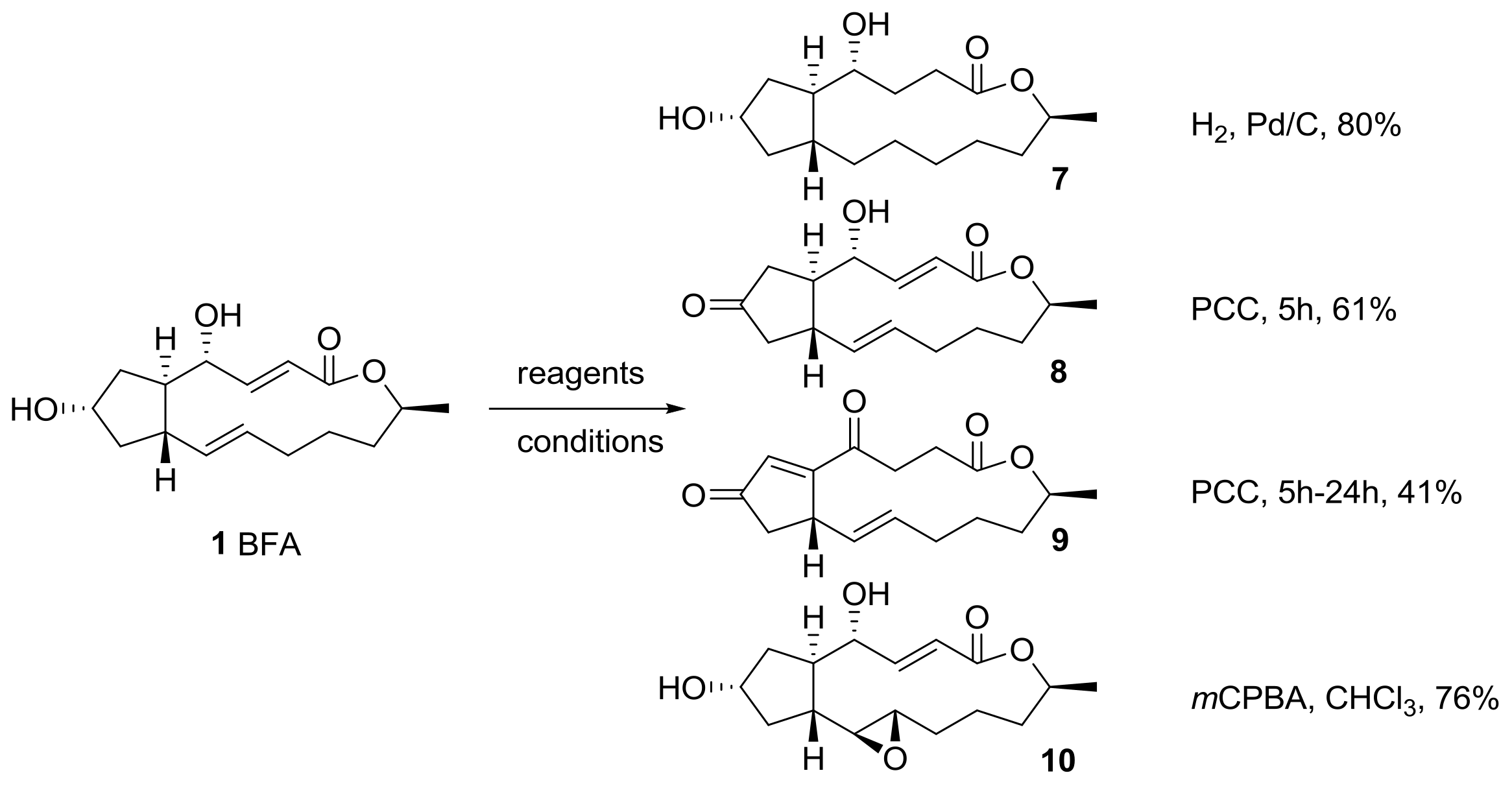
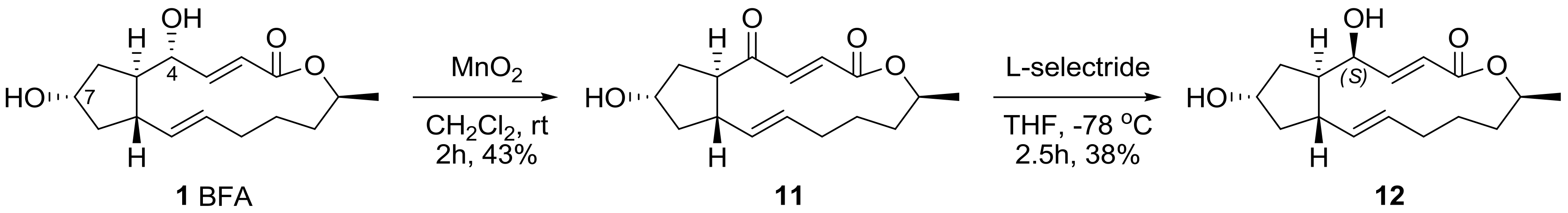
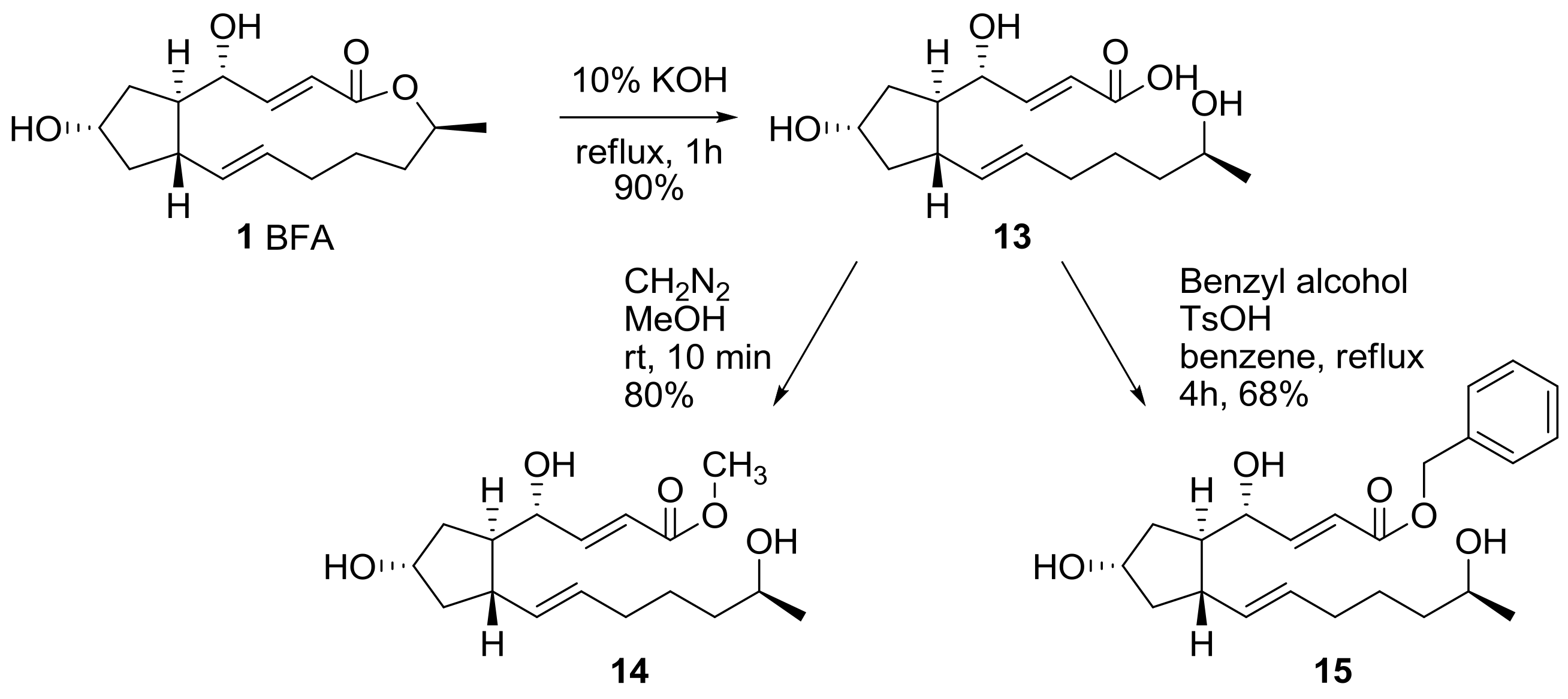
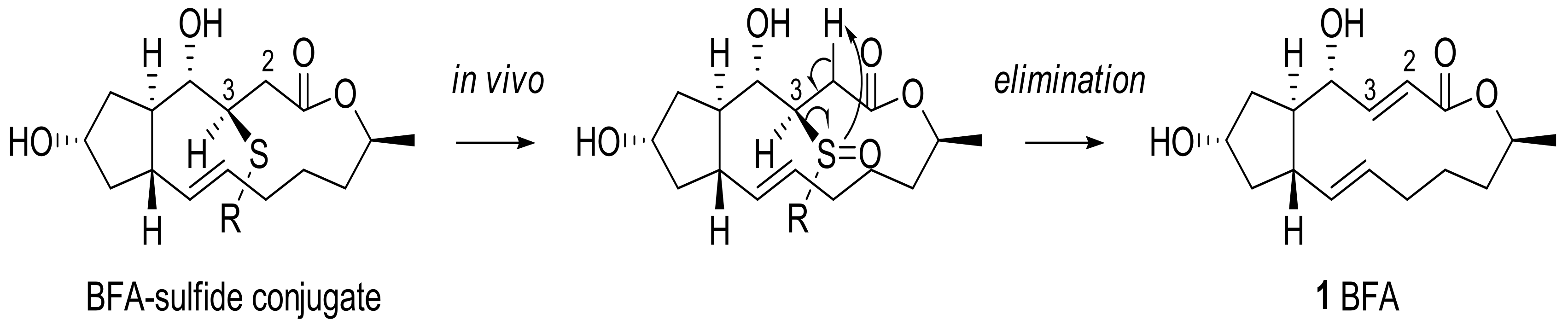
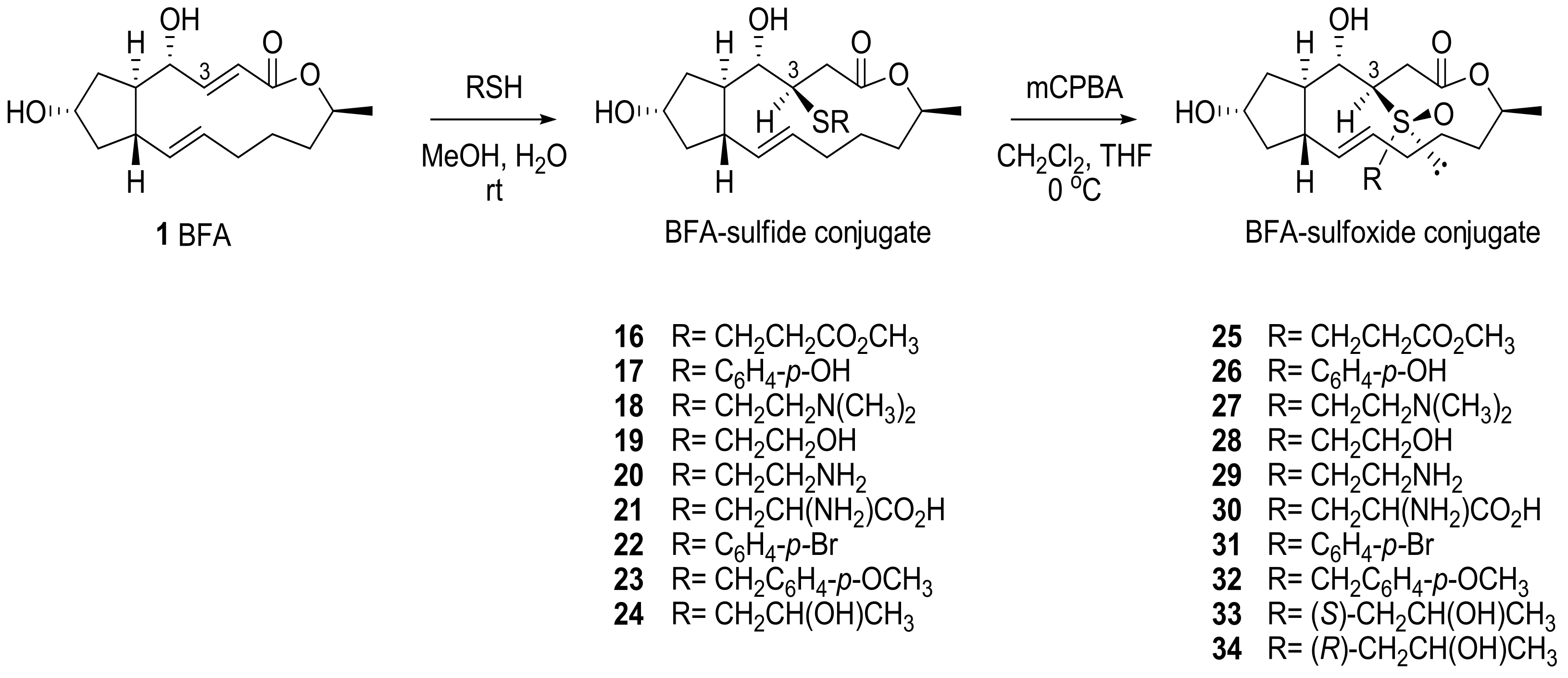
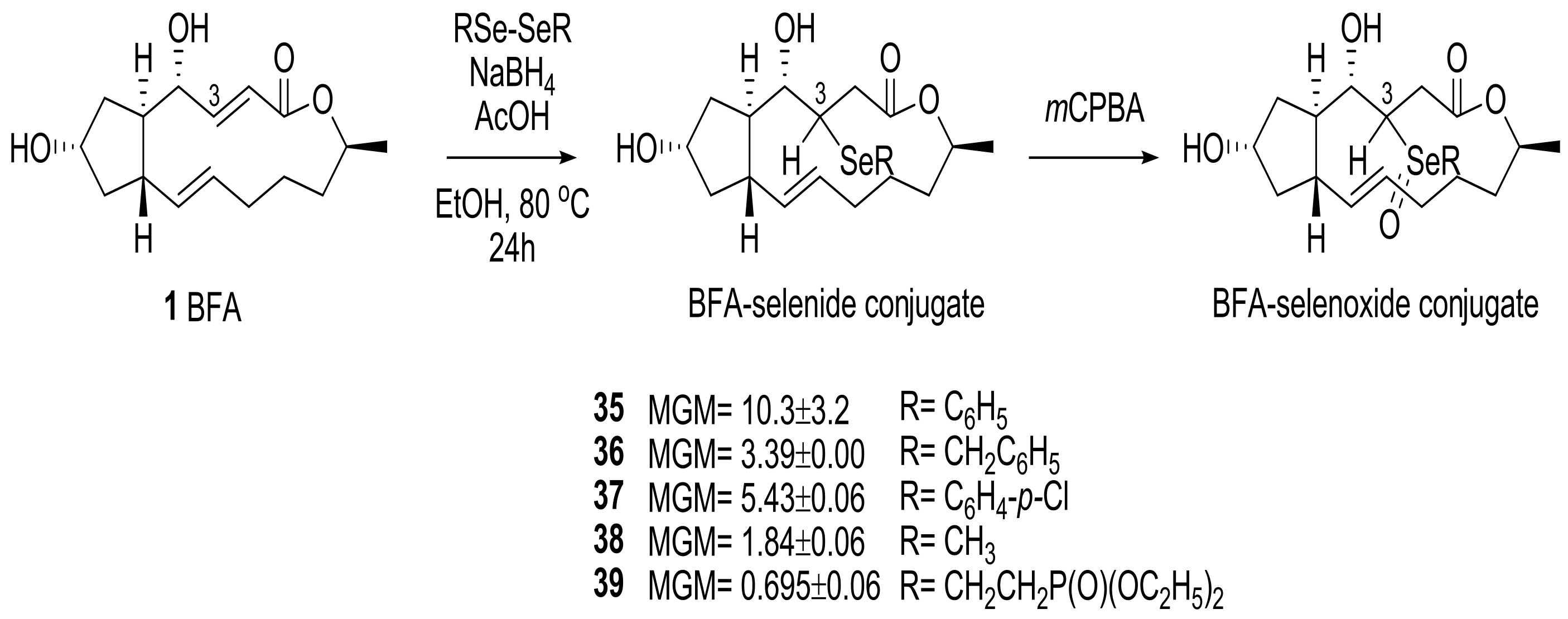
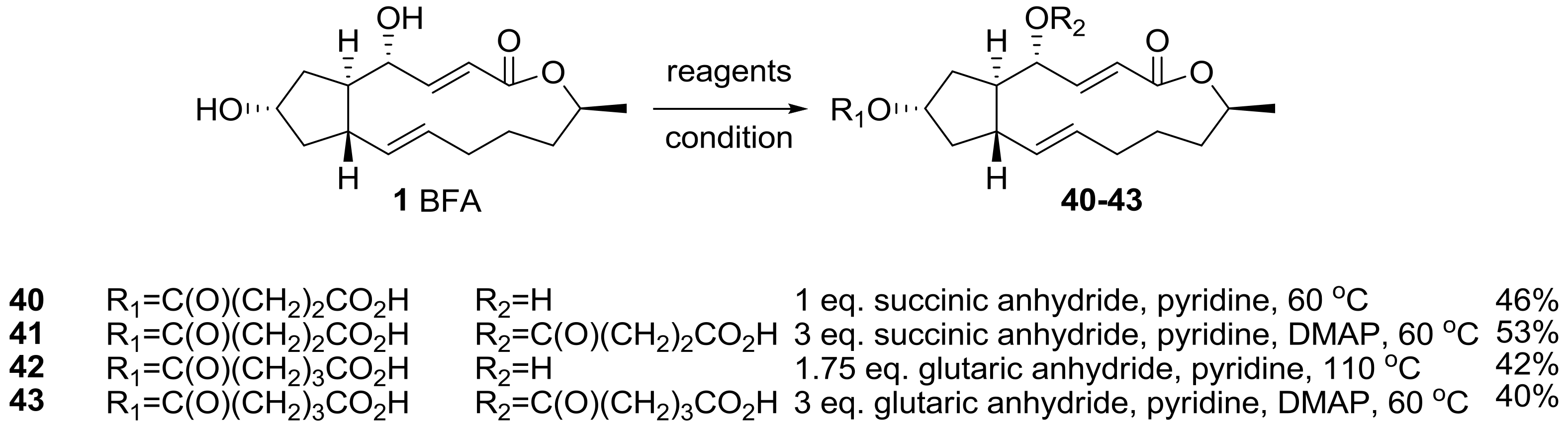
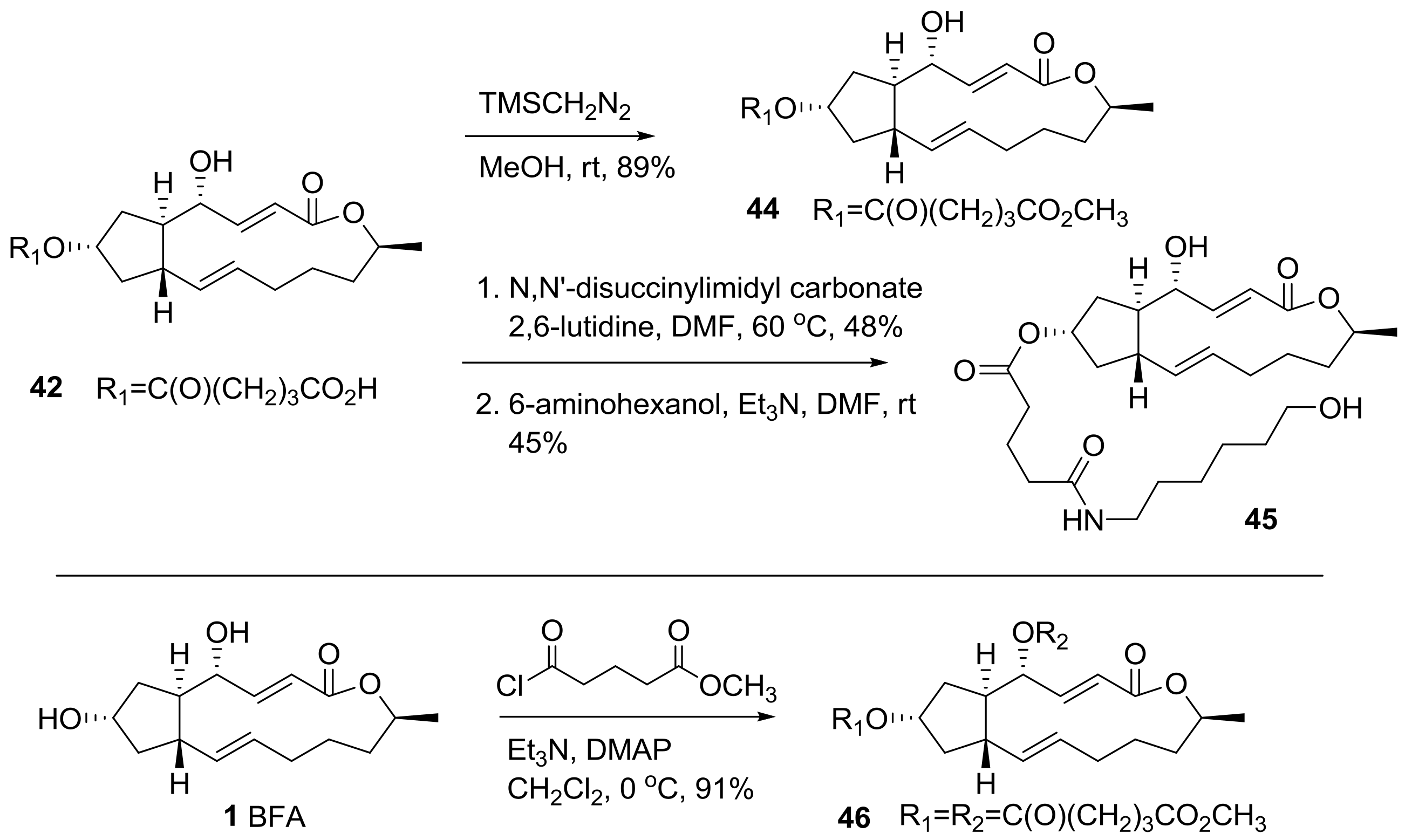
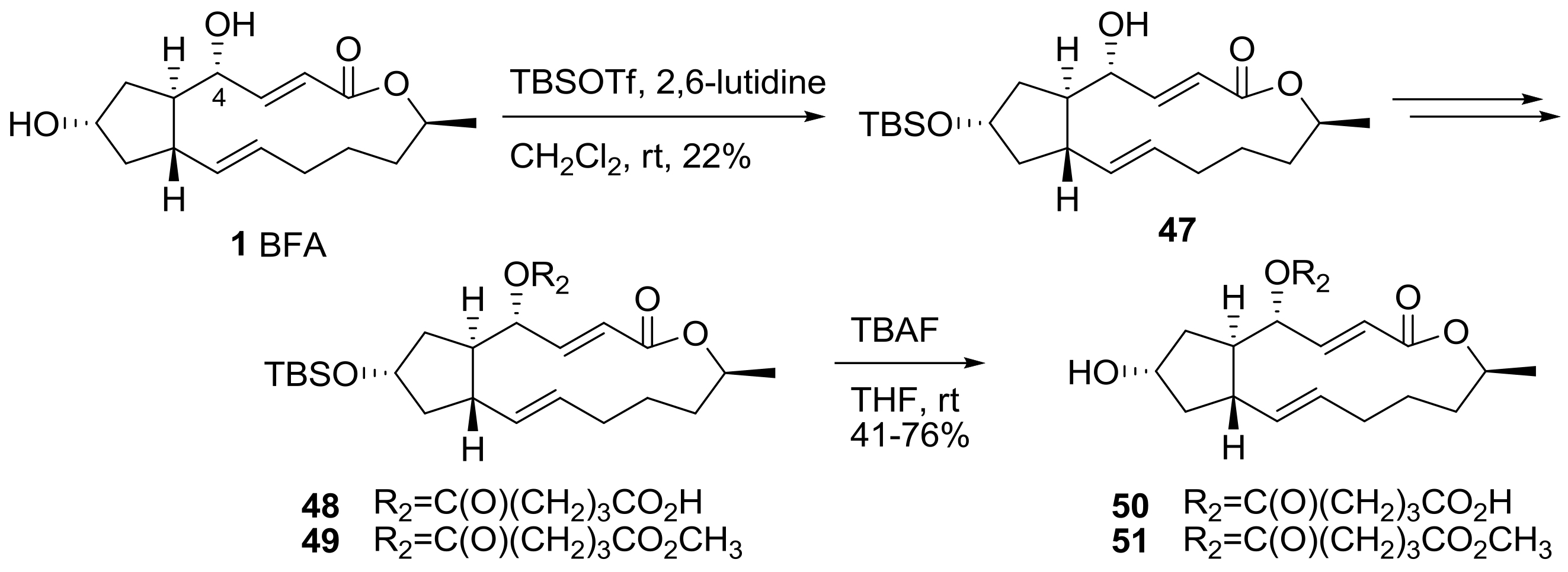
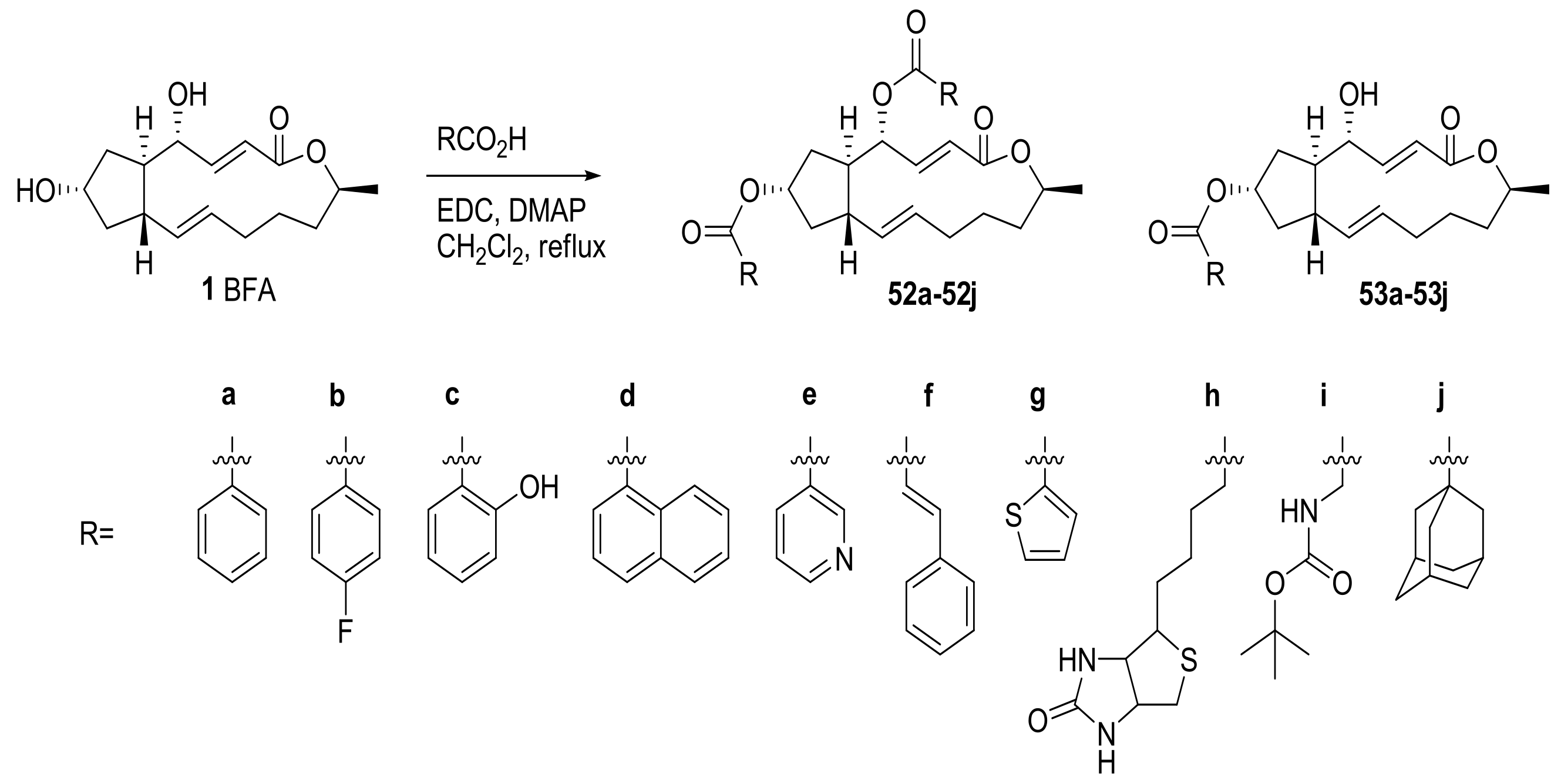
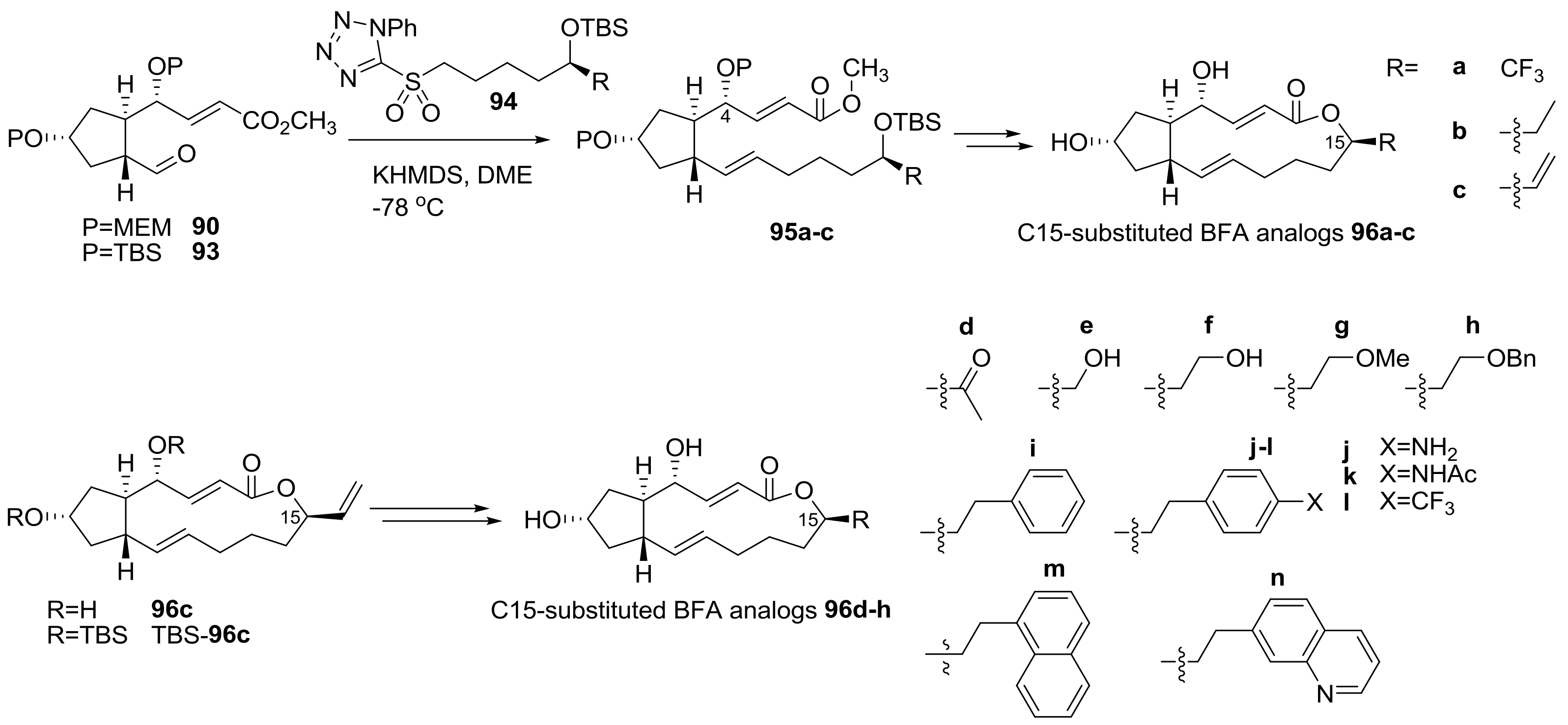
2.1. Modification of BFA
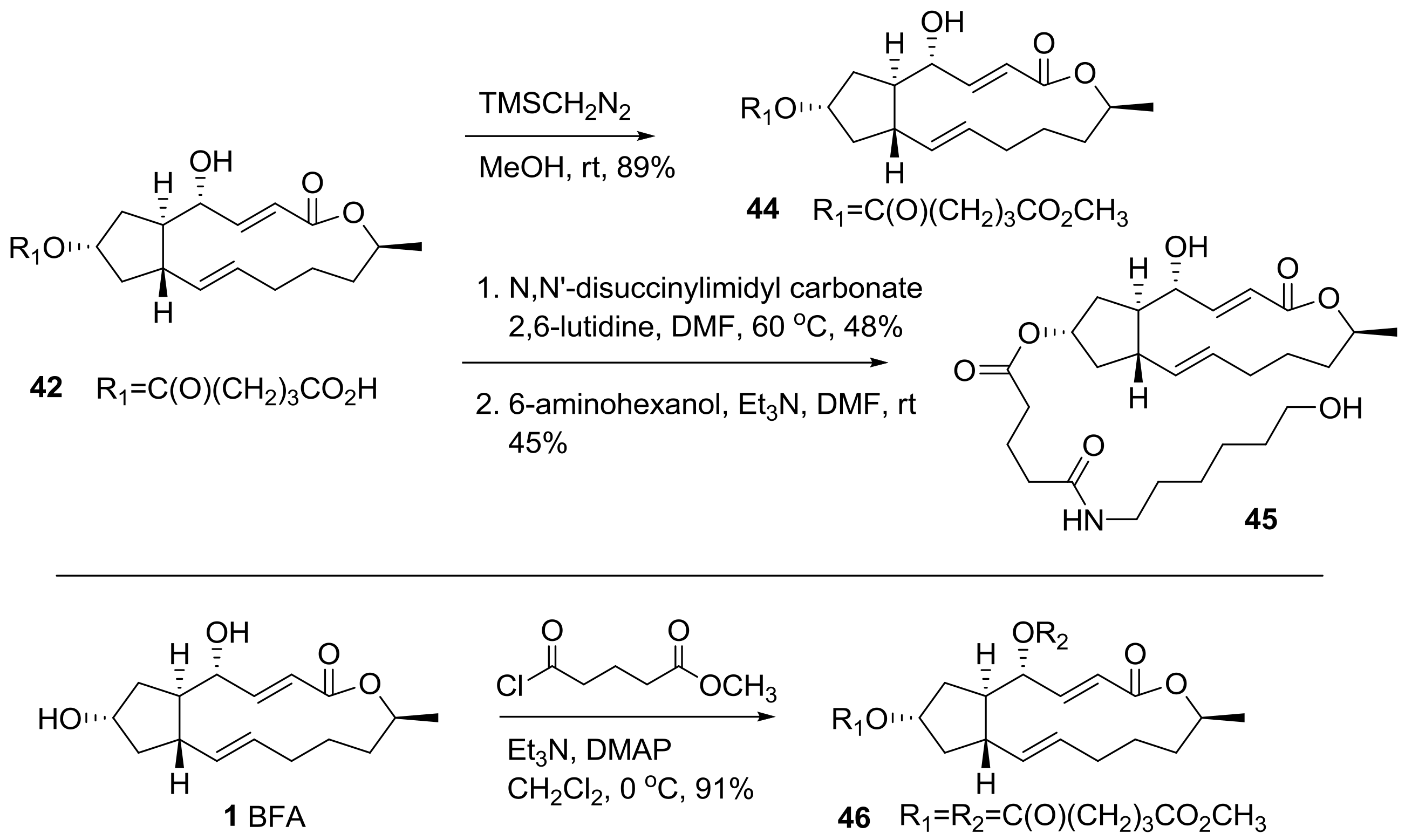
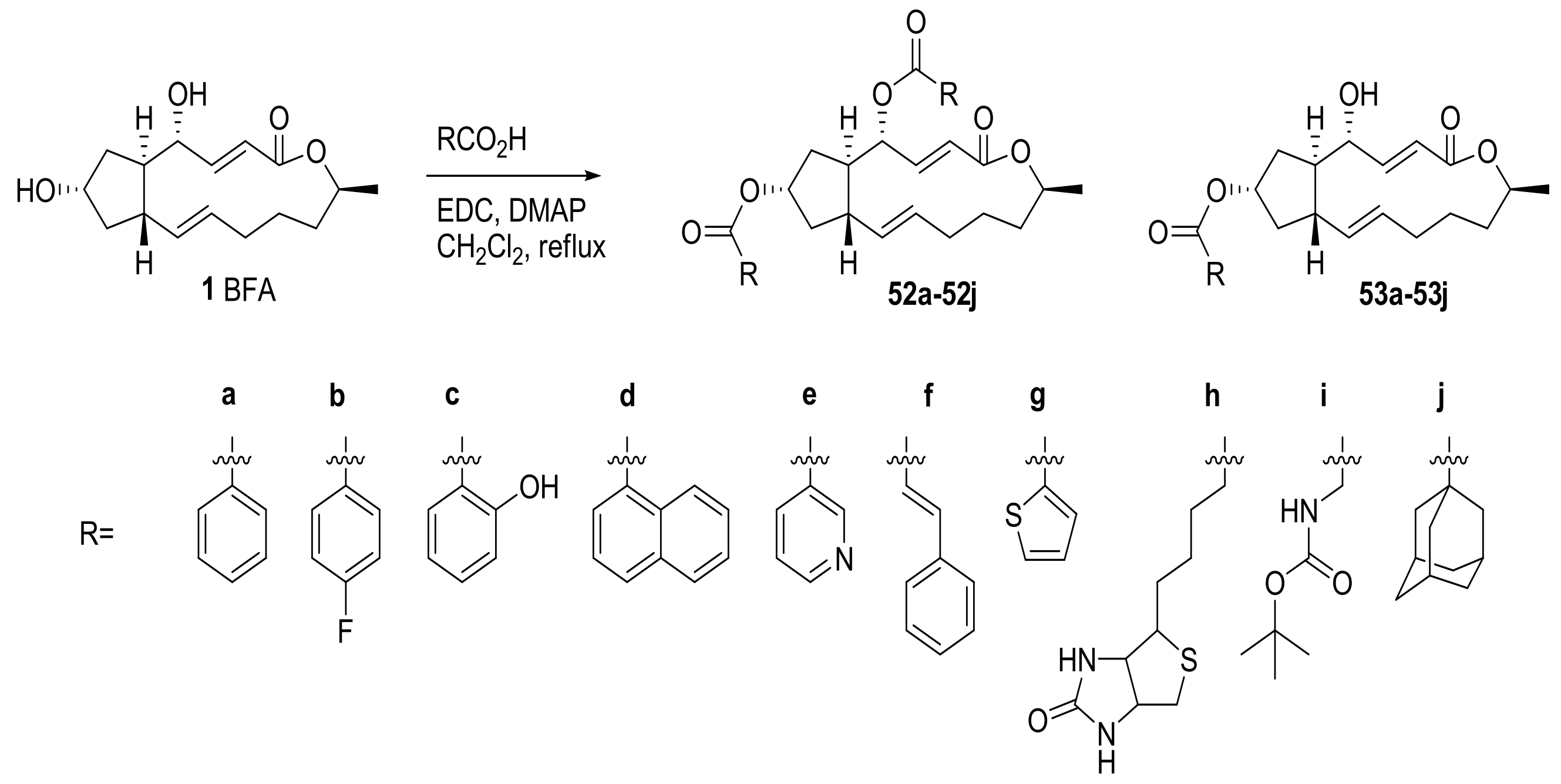
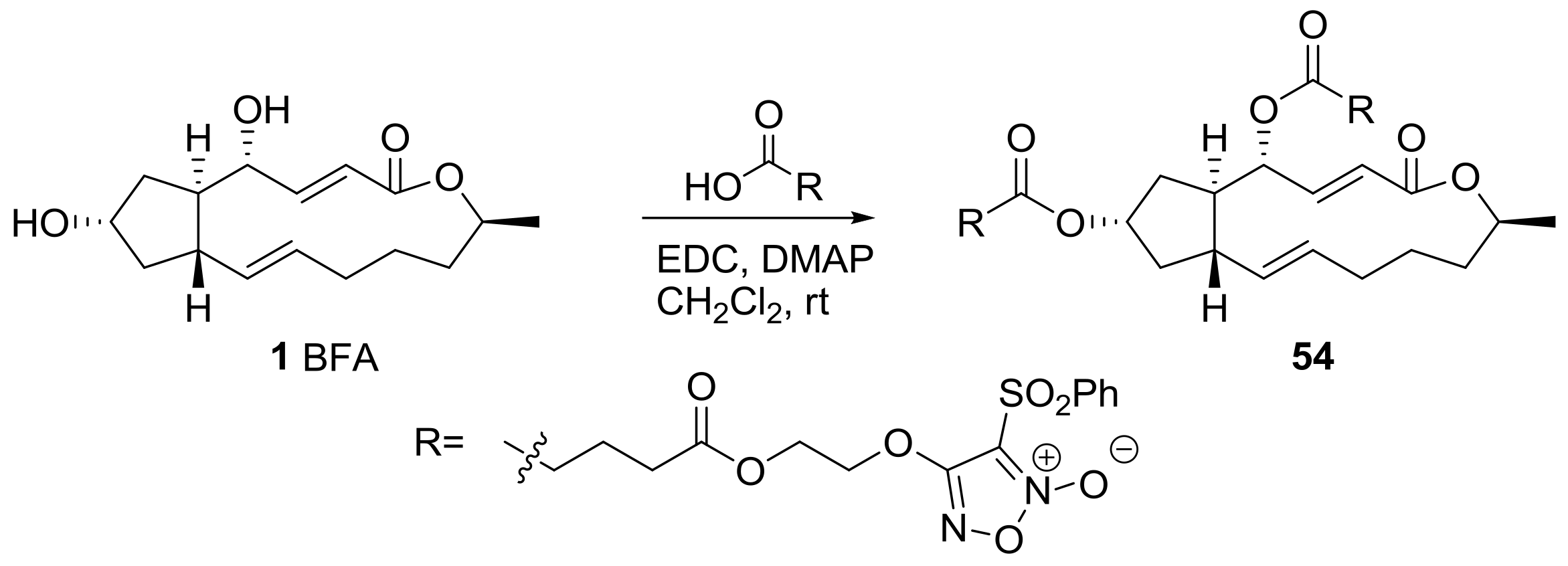
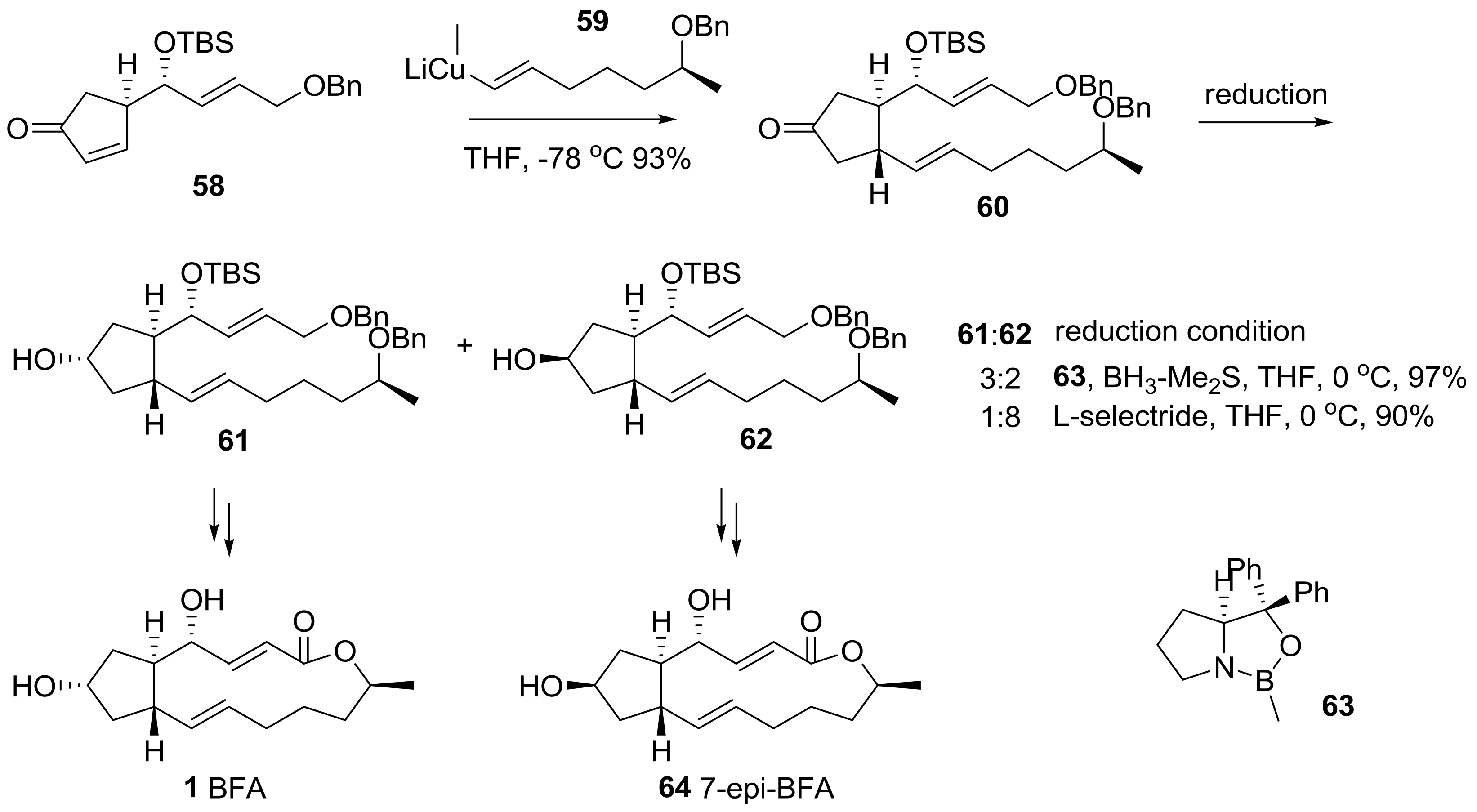
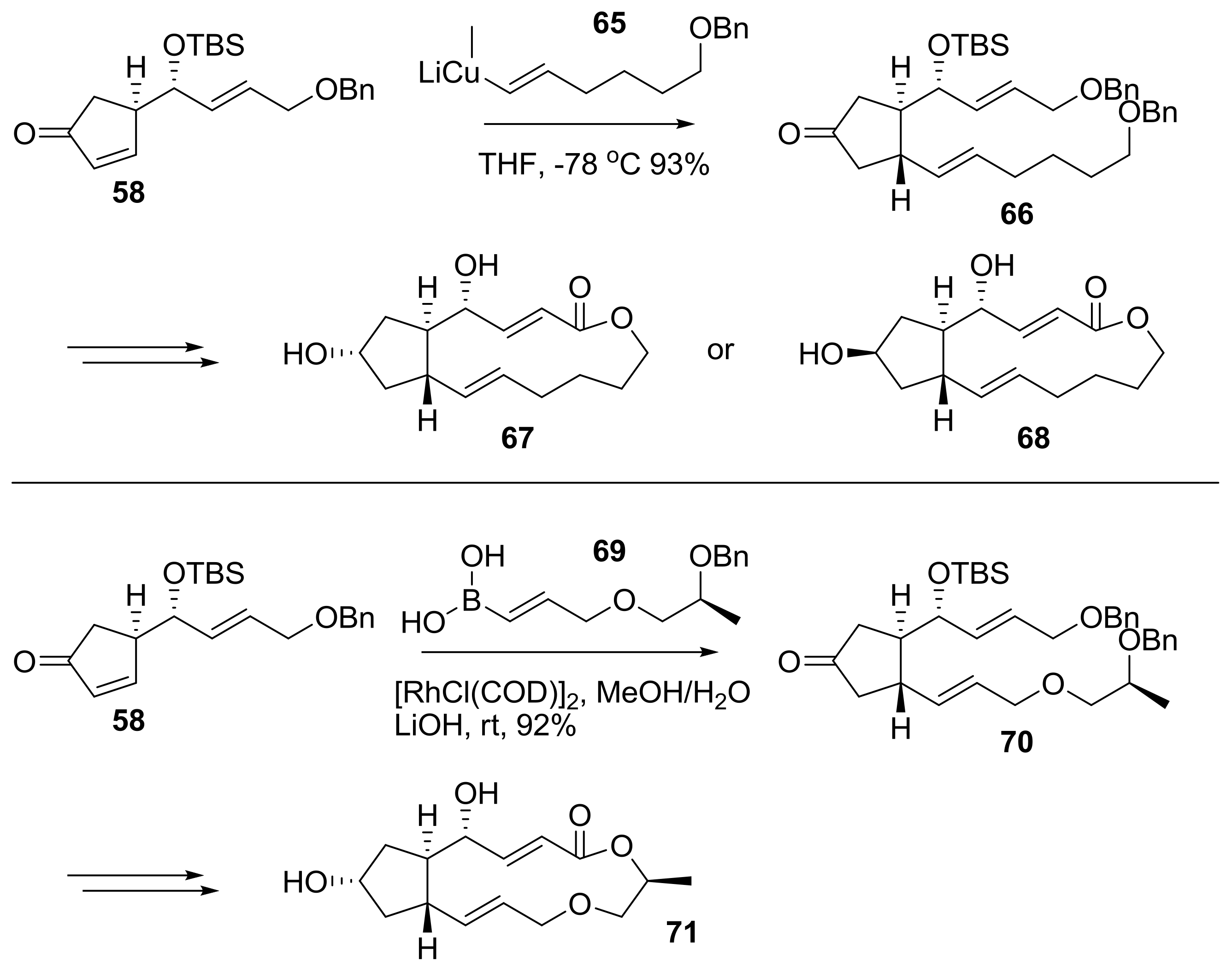
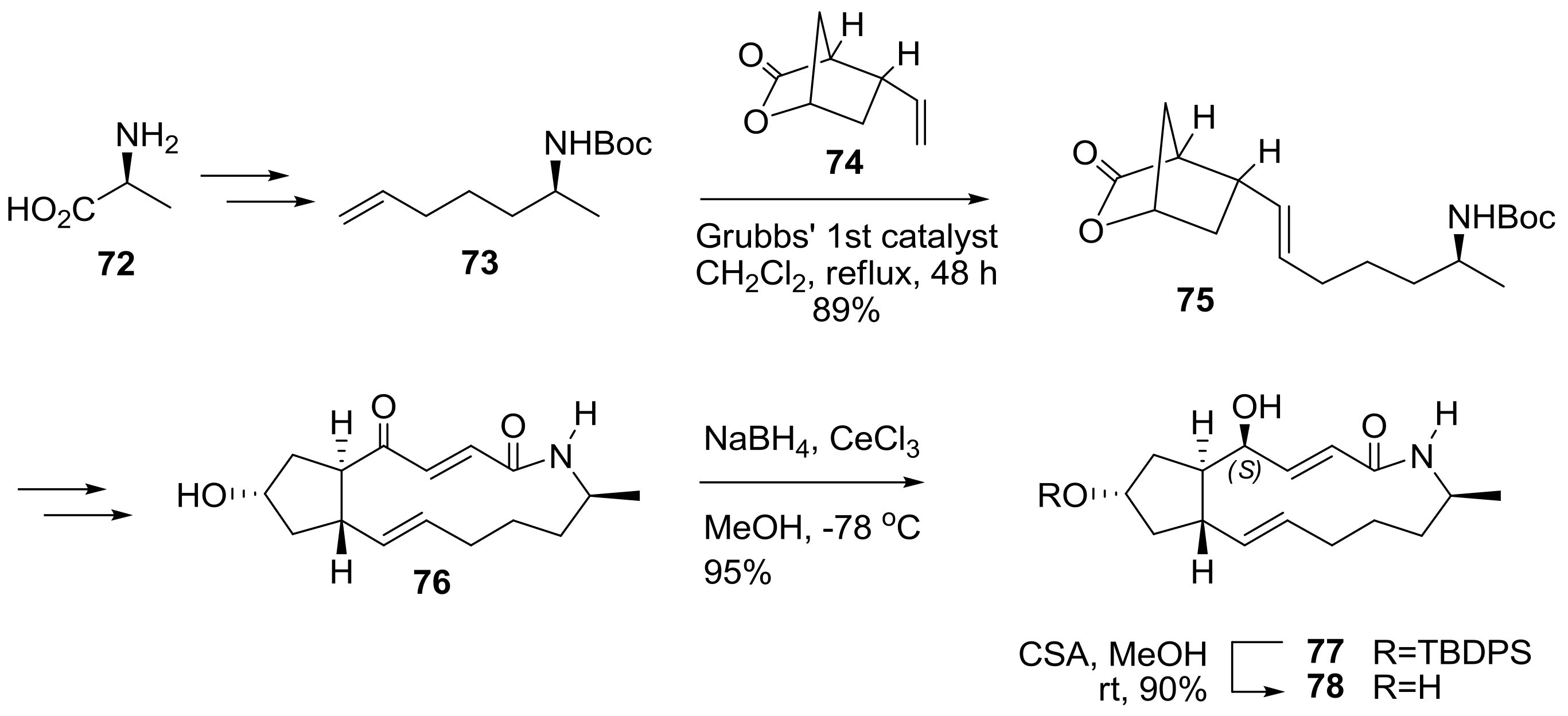
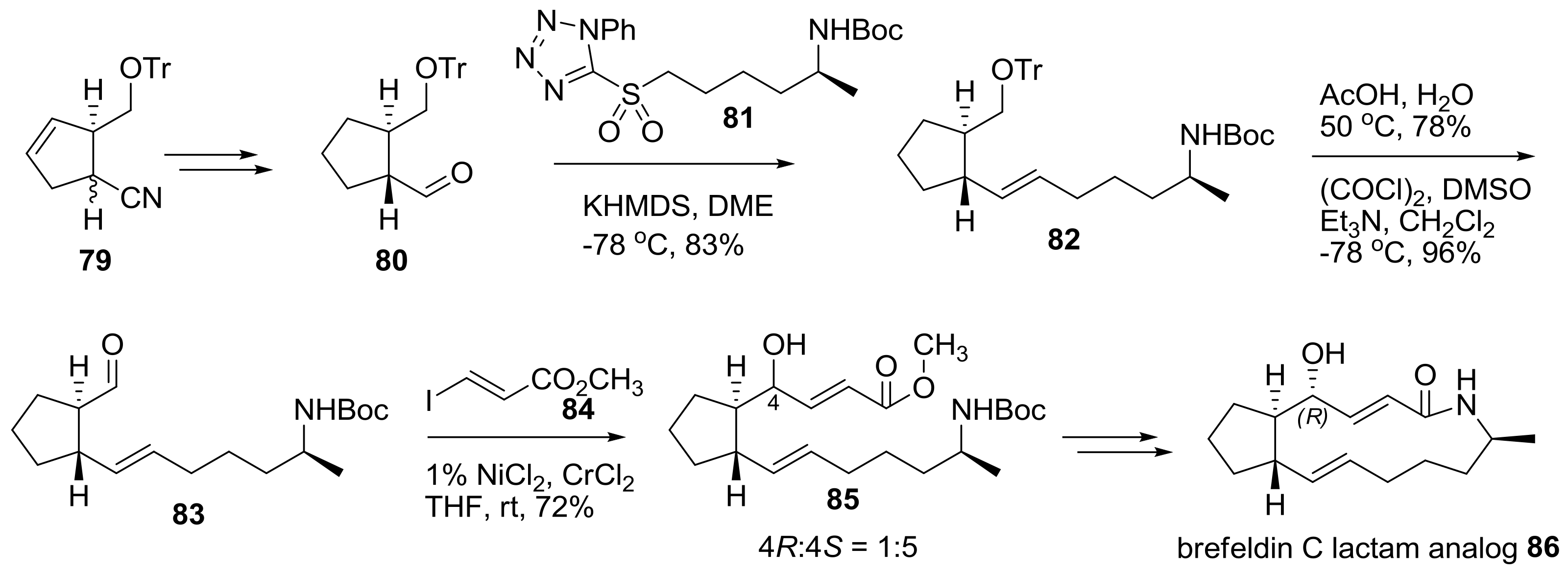
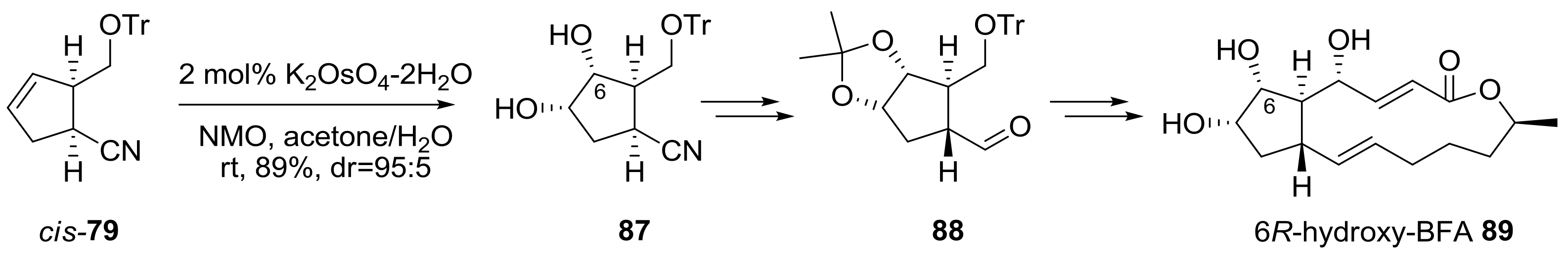
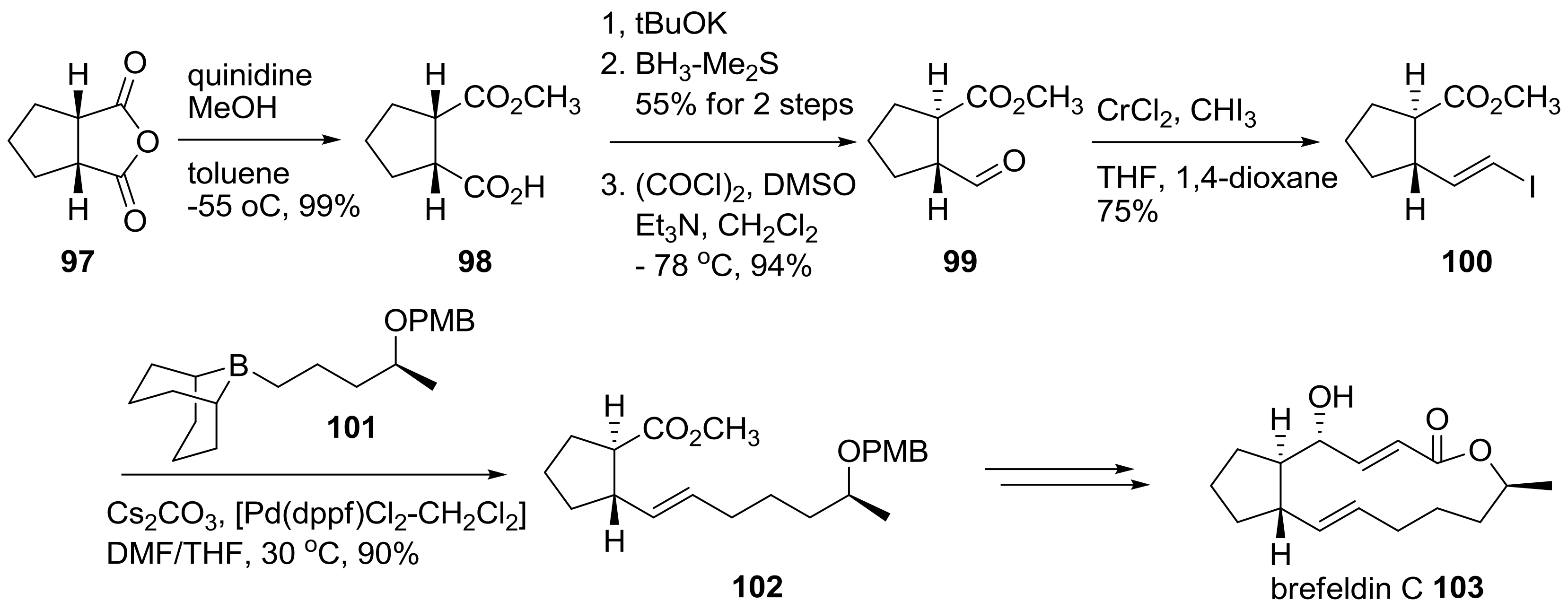
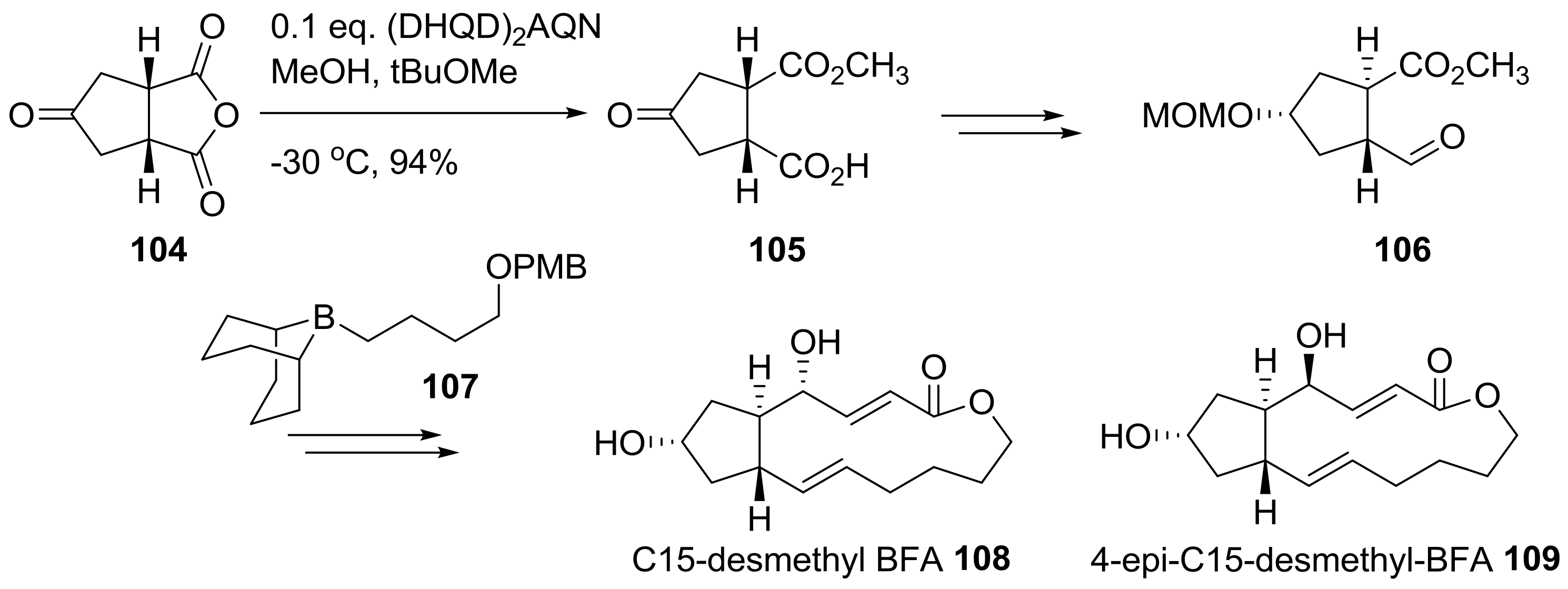
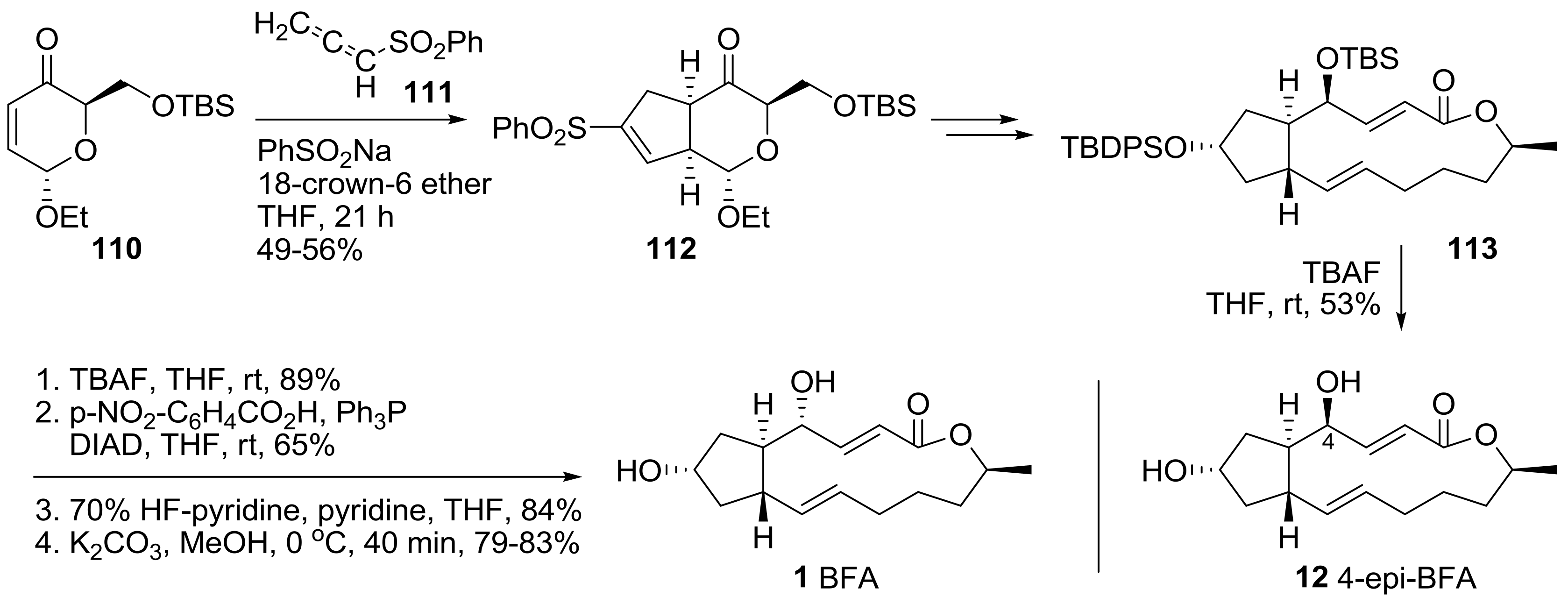
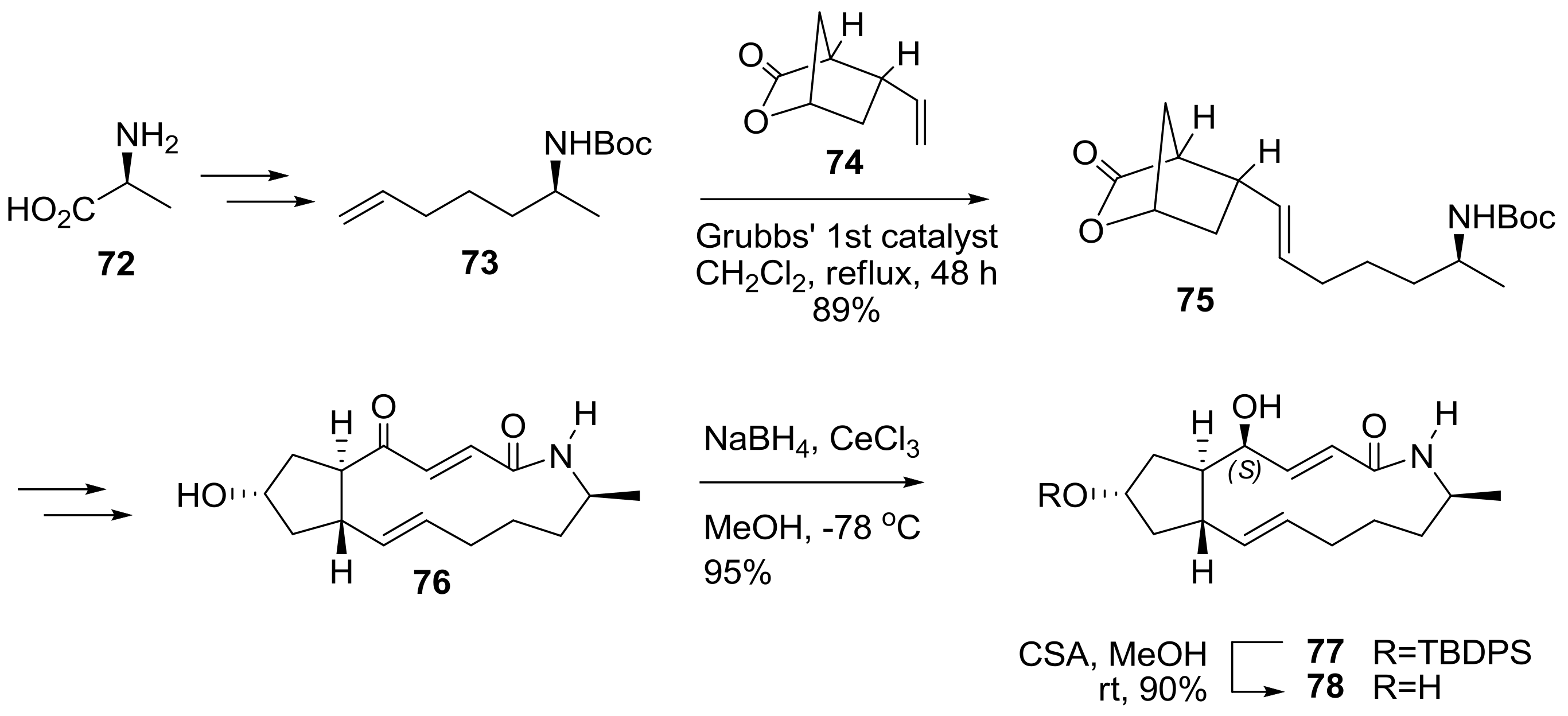
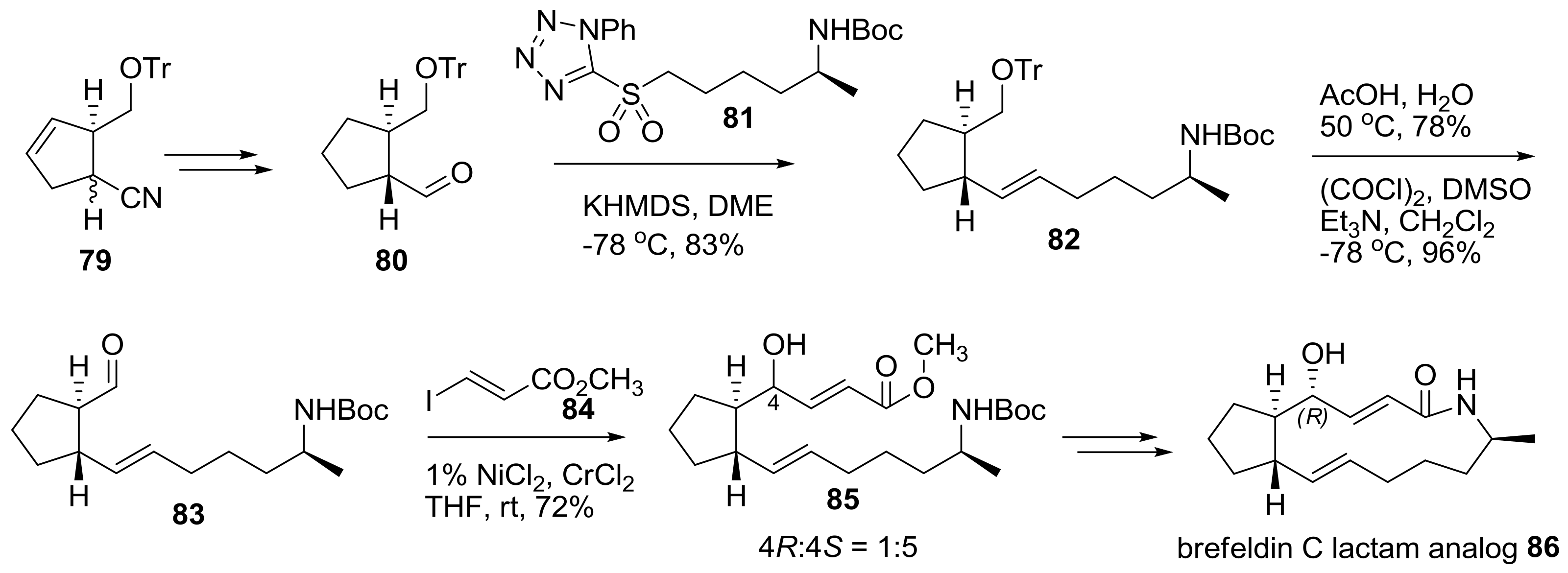
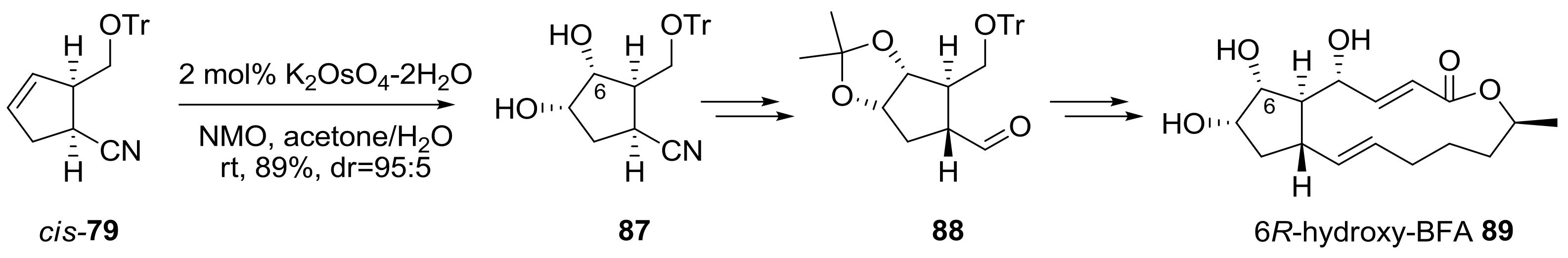
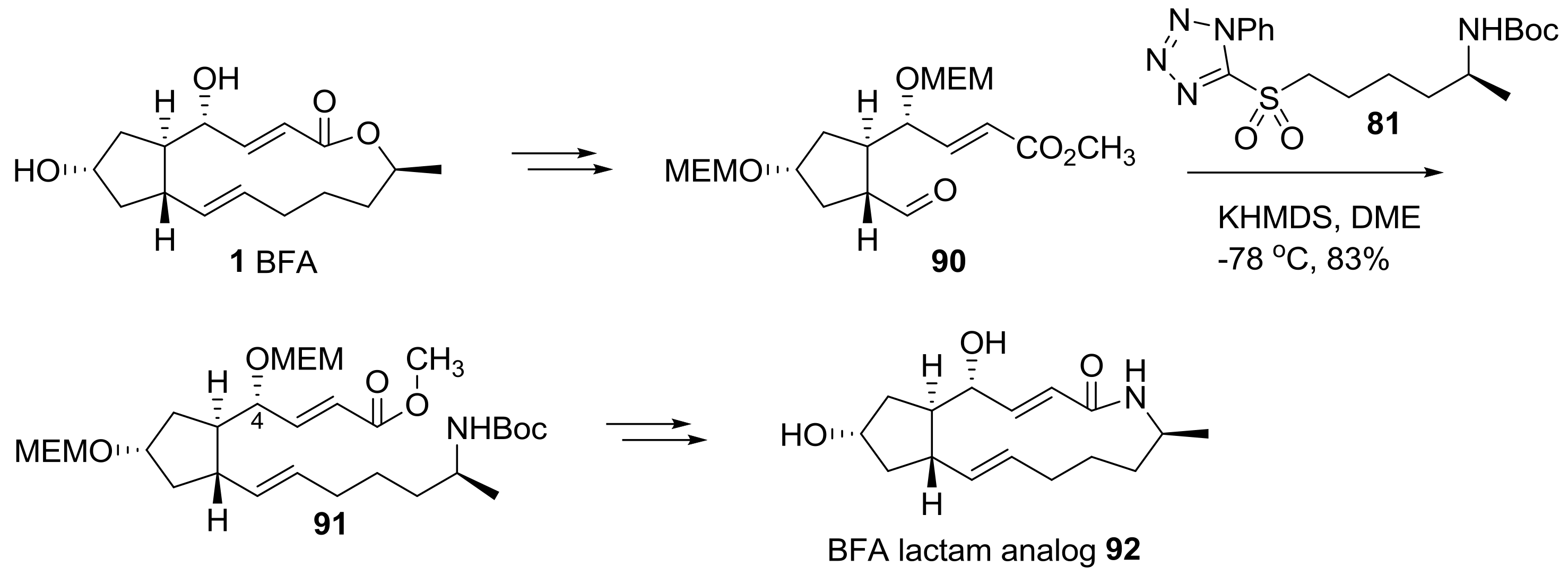
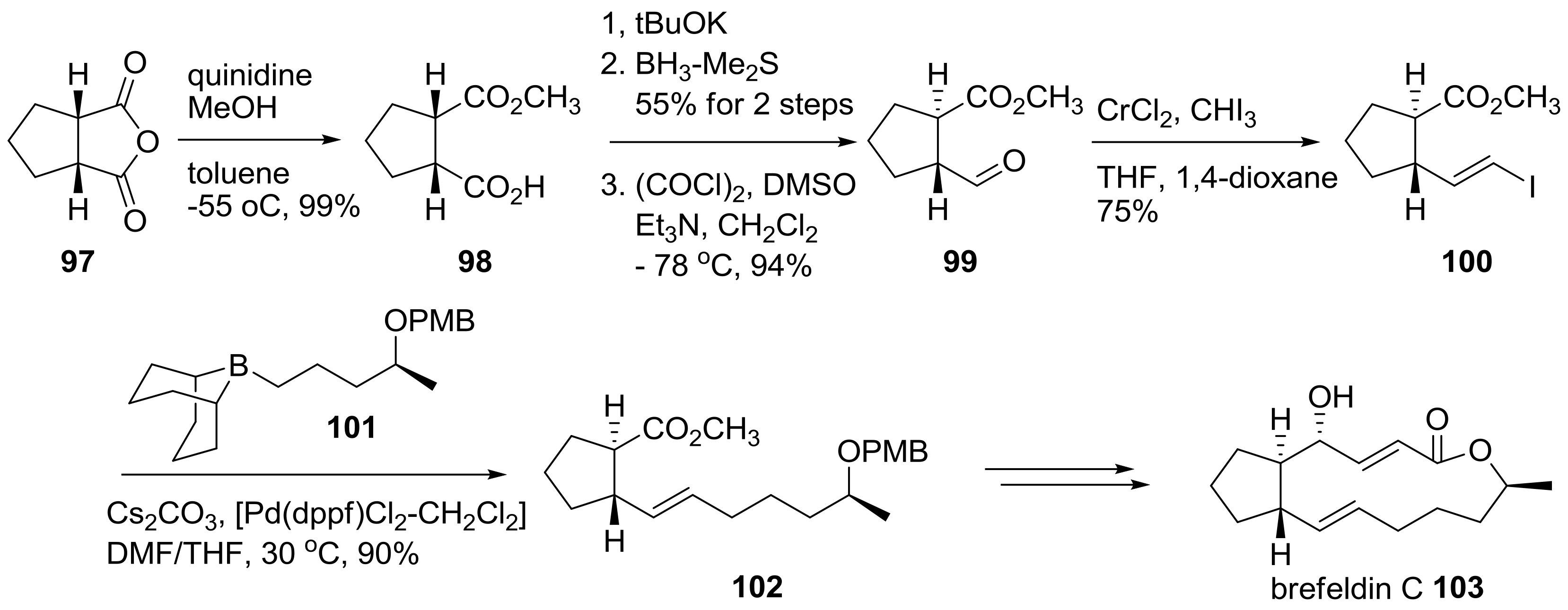
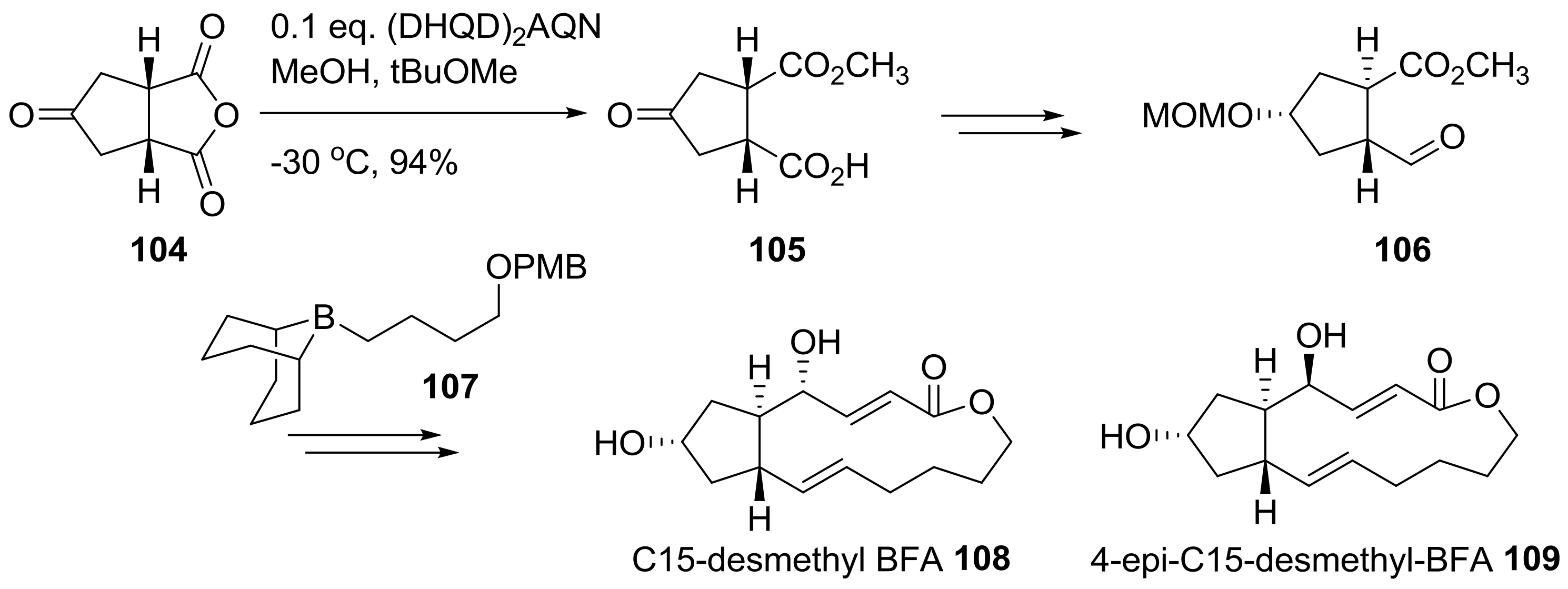
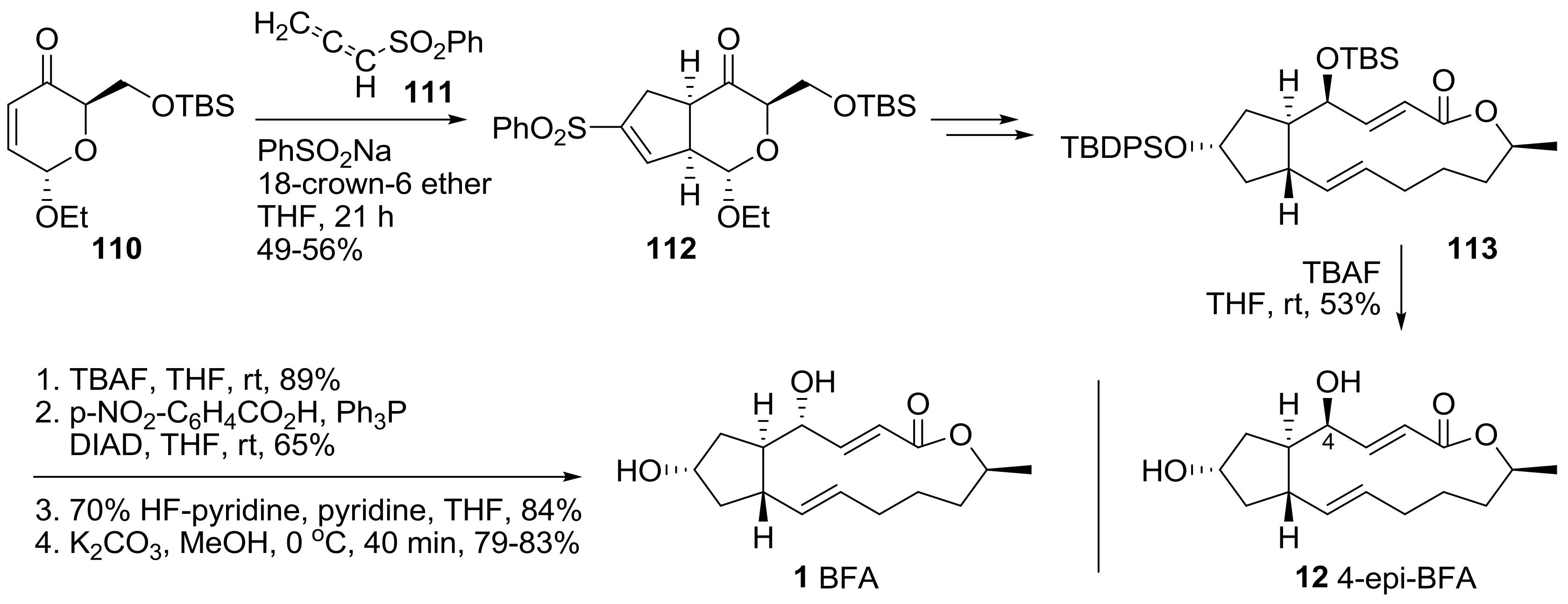
2.2. Modification of BFA Based on Total Synthesis
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Singleton, V.L.; Bohonos, N.; Ullstrup, A.J. Decumbin, a New Compound from a Species of Penicillium. Nature 1958, 181, 1072. [Google Scholar] [CrossRef] [PubMed]
- Betina, V. Effects of the Macrolide Antibiotic Cyanein on HeLa Cells Growth and Metabolism. Neoplasma 1969, 16, 23–32. [Google Scholar] [PubMed]
- Haerri, E.; Loeffler, W.; Sigg, H.P.; Stähelin, H.; Tamm, C.H. Isolation of New Metabolic Products from Penicillium Brefeldianum. Helv. Chim. Acta 1963, 46, 1235–1243. [Google Scholar]
- Tamura, G.; Ando, K.; Suzuki, S.; Takatsuki, A.; Arima, K. Antiviral Activity of Brefeldin A and Verrugarin A. J. Antibiot. 1968, 21, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.E.; Wickstrom, M.; Hassan, S.; Oberg, K.; Granberg, D. The Cytotoxic Agents NSC-95397, Brefeldin A, Bortezomib and Sanguinarine Induce Apoptosis in Neuroendocrine Tumors in Vitro. Anticancer Res. 2010, 30, 149–156. [Google Scholar] [PubMed]
- Moon, J.L.; Kim, S.Y.; Shin, S.W.; Park, J. Regulation of Brefeldin A-Induced ER Stress and Apoptosis by Mitochondrial NADP+-Dependent Isocitrate Dehydrogenase. Biochem. Biophys. Res. Commun. 2012, 417, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid Redistribution of Golgi Proteins into the ER in Cells Treated with Brefeldin A: Evidence for Membrane Cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Dinter, A.; Berger, E.G. Golgi-Disturbing Agents. Histochem. Cell Biol. 1998, 109, 571–590. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, Y.; Zheng, Y.; Chen, W.; Zhu, Q. Synthesis and Cytotoxic Evaluation of Acylated Brefeldin a Derivatives as Potential Anticancer Agents. Chem. Biol. Drug Des. 2013, 82, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Trisuwan, K.; Rukachaisirikul, V.; Sukpondma, Y.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Lactone Derivatives from the Marine-Derived Fungus Penicillium sp. PSU-F44. Chem. Pharm. Bull. 2009, 57, 1100–1102. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Qin, L.; Ding, W.; Liu, Y.; Ma, Z. New Analogues of Brefeldin A from Sediment-Derived Fungus Penicillium sp. DT-F29. Nat. Prod. Res. 2016, 30, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Ishikawa, T.; Kneusel, R.E.; Matern, U.; Lottspeich, F.; Wieland, F.T. Brefeldin A Binds to Glutathione S-Transferase and is Secreted as Glutathione and Cysteine Conjugates by Chinese Hamster Ovary Cells. J. Biol. Chem. 1992, 267, 7726–7732. [Google Scholar] [PubMed]
- Phillips, L.R.; Supko, J.G.; Malspeis, L. Analysis of Brefeldin A in Plasma by Gas Chromatography with Electron Capture Detection. Anal. Biochem. 1993, 211, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.R.; Wolfe, T.L.; Malspeis, L.; Supko, J.G. Analysis of Brefeldin A and the Prodrug Breflate in Plasma by Gas Chromatography with Mass Selective Detection. J. Pharm. Biomed. Anal. 1998, 16, 1301–1309. [Google Scholar] [CrossRef]
- Fuchs, M.; Fürstner, A. trans-Hydrogenation: Application to a Concise and Scalable Synthesis of Brefeldin A. Angew. Chem. Int. Ed. Engl. 2015, 54, 3978–3982. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Yelleni, M.K.R. Application of Ru(II)-Catalyzed Enyne Cyclization in the Synthesis of Brefeldin A. J. Org. Chem. 2016, 81, 10912–10921. [Google Scholar] [CrossRef] [PubMed]
- Heasley, B. Recent Developments in the Stereocontrolled Synthesis of Highly Substituted Cyclopentane Core Structures: From Drug Discovery Research to Natural Product Synthesis. Curr. Org. Chem. 2014, 18, 641–686. [Google Scholar] [CrossRef]
- Zhu, J.; Hori, H.; Nojiri, H.; Tsukuda, T.; Taira, Z. Synthesis and Activity of Brefeldin A Analogs as Inducers of Cancer Cell Differentiation and Apoptosis. Bioorg. Med. Chem. Lett. 1997, 7, 139–144. [Google Scholar] [CrossRef]
- Proksa, B.; Uhrin, D.; Adamcova, J.; Fuska, J. Oxidation of Brefeldin A. Pharmazie 1992, 47, 582–584. [Google Scholar] [PubMed]
- Zhu, J.; Nagasawa, H.; Nagura, F.; Mohamad, S.B.; Uto, Y.; Ohkura, K.; Hori, H. Elucidation of Strict Structural Requirements of Brefeldin A as an Inducer of Differentiation and Apoptosis. Bioorg. Med. Chem. 2000, 8, 455–463. [Google Scholar] [CrossRef]
- Argade, A.B.; Devraj, R.; Vroman, J.A.; Haugwitz, R.D.; Hollingshead, M.; Cushman, M. Design and Synthesis of Brefeldin A Sulfide Derivatives as Prodrug Candidates with Enhanced Aqueous Solubilities. J. Med. Chem. 1998, 41, 3337–3346. [Google Scholar] [CrossRef] [PubMed]
- Argade, A.B.; Haugwitz, R.D.; Devraj, R.; Kozlowski, J.; Fanwick, P.E.; Cushman, M. Highly Efficient Diastereoselective Michael Addition of various Thiols to (+)-Brefeldin A. J. Org. Chem. 1998, 63, 273–278. [Google Scholar] [CrossRef]
- Fox, B.M.; Vroman, J.A.; Fanwick, P.E.; Cushman, M. Preparation and Evaluation of Sulfide Derivatives of the Antibiotic Brefeldin a as Potential Prodrug Candidates with Enhanced Aqueous Solubilities. J. Med. Chem. 2001, 44, 3915–3924. [Google Scholar] [CrossRef] [PubMed]
- Anadu, N.O.; Davisson, V.J.; Cushman, M. Synthesis and Anticancer Activity of Brefeldin A Ester Derivatives. J. Med. Chem. 2006, 49, 3897–3905. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Xu, F.; Gao, X.; Han, T.; Li, J.; Pan, H.; Zang, L.; Li, D.; Li, Z.; Uchita, T. Nitric Oxide-Releasing Derivatives of Brefeldin A as Potent and Highly Selective Anticancer Agents. Eur. J. Med. Chem. 2017, 136, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Förster, S.; Persch, E.; Tverskoy, O.; Rominger, F.; Helmchen, G.; Klein, C.; Gönen, B.; Brügger, B. Syntheses and Biological Properties of Brefeldin Analogues. Eur. J. Org. Chem. 2011, 2011, 878–891. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, X.; Yang, Y.; Hu, Q.; Huang, J. Enantioselective Total Synthesis of (+)-Brefeldin A and 7-epi-Brefeldin A. J. Org. Chem. 2004, 69, 3857–3865. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Yang, Y.; Hu, Q.; Huang, J.; Gao, J.; Wu, Y. Synthesis of 15-Demethyl Brefeldin A and its 7-epi Isomer. Chin. J. Chem. 2007, 25, 802–807. [Google Scholar] [CrossRef]
- Gao, J.; Huang, Y.; Wu, Y. Enantioselective Total Synthesis of 13-O-Brefeldin A. Tetrahedron 2008, 64, 11105–11109. [Google Scholar] [CrossRef]
- Paek, S. Synthesis of Tetrasubstituted Alkenes via Metathesis. Molecules 2012, 17, 3348–3358. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.; Seo, S.; Min, K.; Shin, D.M.; Chung, Y.K.; Suh, Y. Synthesis of Lactam Analog of 4-epi-Brefeldin A. Heterocycles 2007, 71, 1059–1066. [Google Scholar]
- Förster, S.; Helmchen, G. Stereoselective Synthesis of a Lactam Analogue of Brefeldin C. Synlett 2008, 2008, 831–836. [Google Scholar]
- Langhans, M.; Förster, S.; Helmchen, G.; Robinson, D.G. Differential Effects of the Brefeldin A Analogue (6R)-Hydroxy-BFA in Tobacco and Arabidopsis. J. Exp. Bot. 2011, 62, 2949–2957. [Google Scholar] [CrossRef] [PubMed]
- Seehafer, K.; Rominger, F.; Helmchen, G.; Langhans, M.; Robinson, D.G.; Özata, B.; Brügger, B.; Strating, J.R.; van Kuppeveld, F.J.; Klein, C.D. Synthesis and Biological Properties of Novel Brefeldin A Analogues. J. Med. Chem. 2013, 56, 5872–5884. [Google Scholar] [CrossRef] [PubMed]
- Archambaud, S.; Aphecetche-Julienne, K.; Guingant, A. A New Total Synthesis of (+)-Brefeldin C. Synlett 2005, 2005, 139–143. [Google Scholar] [CrossRef]
- Archambaud, S.; Legrand, F.; Aphecetche-Julienne, K.; Collet, S.; Guingant, A.; Evain, M. Total Synthesis of (+)-Brefeldin C, (+)-nor-Me Brefeldin A and (+)-4-epi-nor-Me Brefeldin A. Eur. J. Org. Chem. 2010, 2010, 1364–1380. [Google Scholar] [CrossRef]
- Xiong, Z.; Hale, K.J. Total Synthesis of the Antitumor Macrolides, (+)-Brefeldin A and 4-Epi-Brefeldin A from d-Glucose: Use of the Padwa Anionic Allenylsulfone [3+2]-Cycloadditive Elimination to Construct Trans-Configured Chiral Cyclopentane Systems. Org. Lett. 2016, 18, 4254–4257. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Corpina, R.A.; Goldberg, J. Crystal Structure of ARF1·Sec7 Complexed with Brefeldin A and Its Implications for the Guanine Nucleotide Exchange Mechanism. Mol. Cell 2003, 12, 1403–1411. [Google Scholar] [CrossRef]
- Cherfils, J.; Menetrey, J.; Mathieu, M.; Le Bras, G.; Robineau, S.; Beraud-Dufour, S.; Antonny, B.; Chardin, P. Structure of the Sec7 domain of the Arf exchange factor ARNO. Nature 1998, 392, 101–105. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
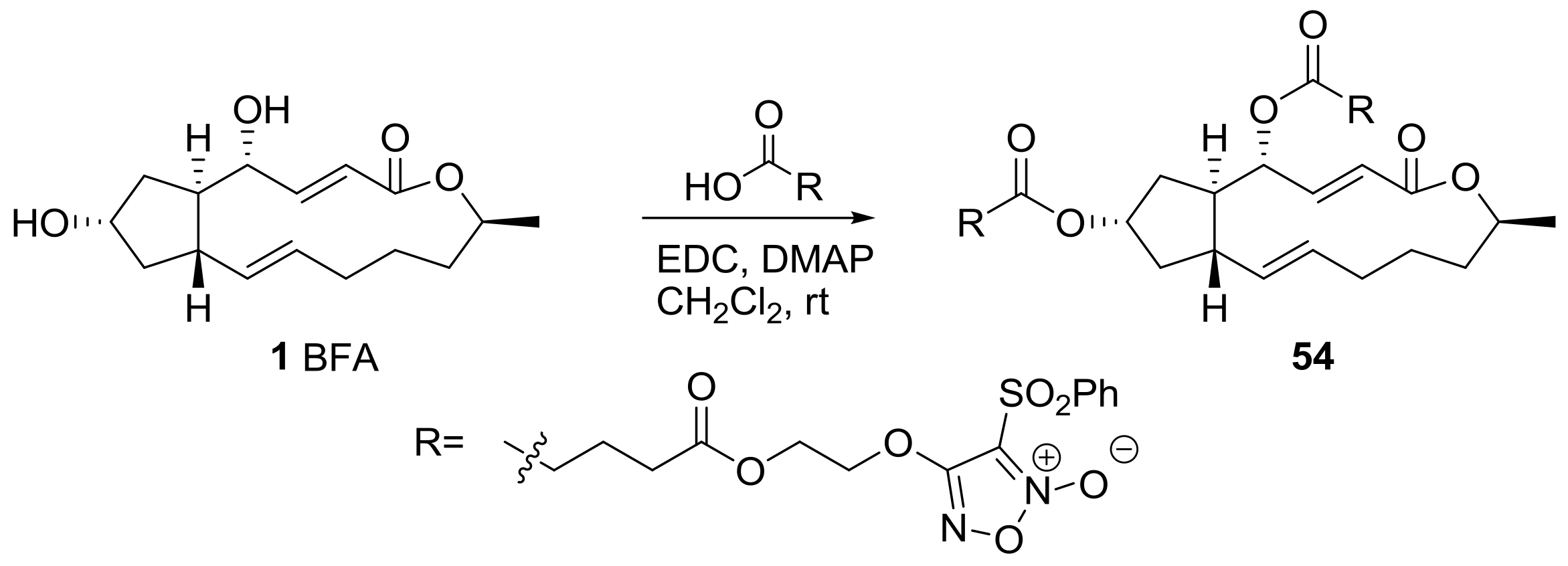{kind=link}
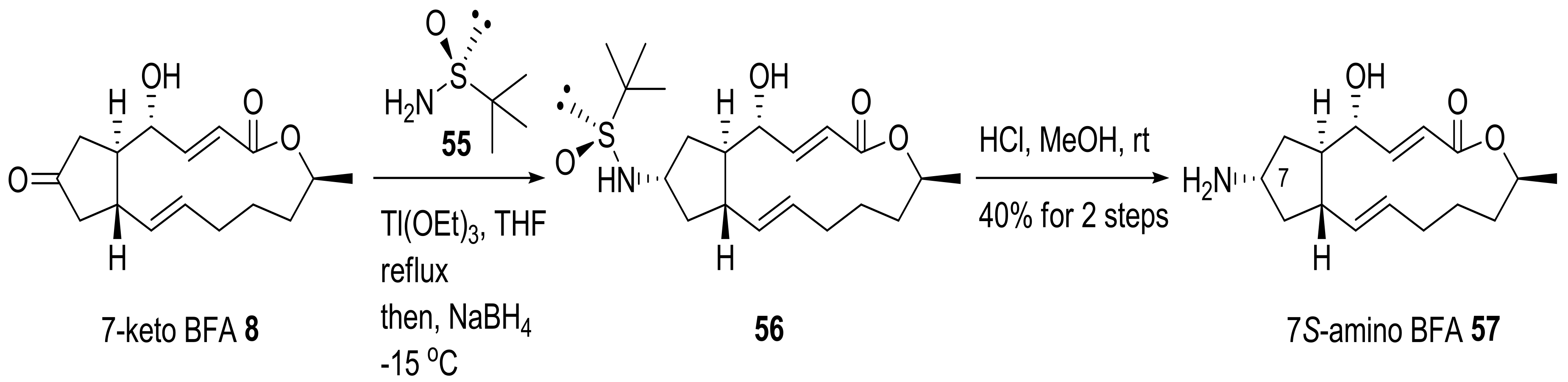{kind=link}
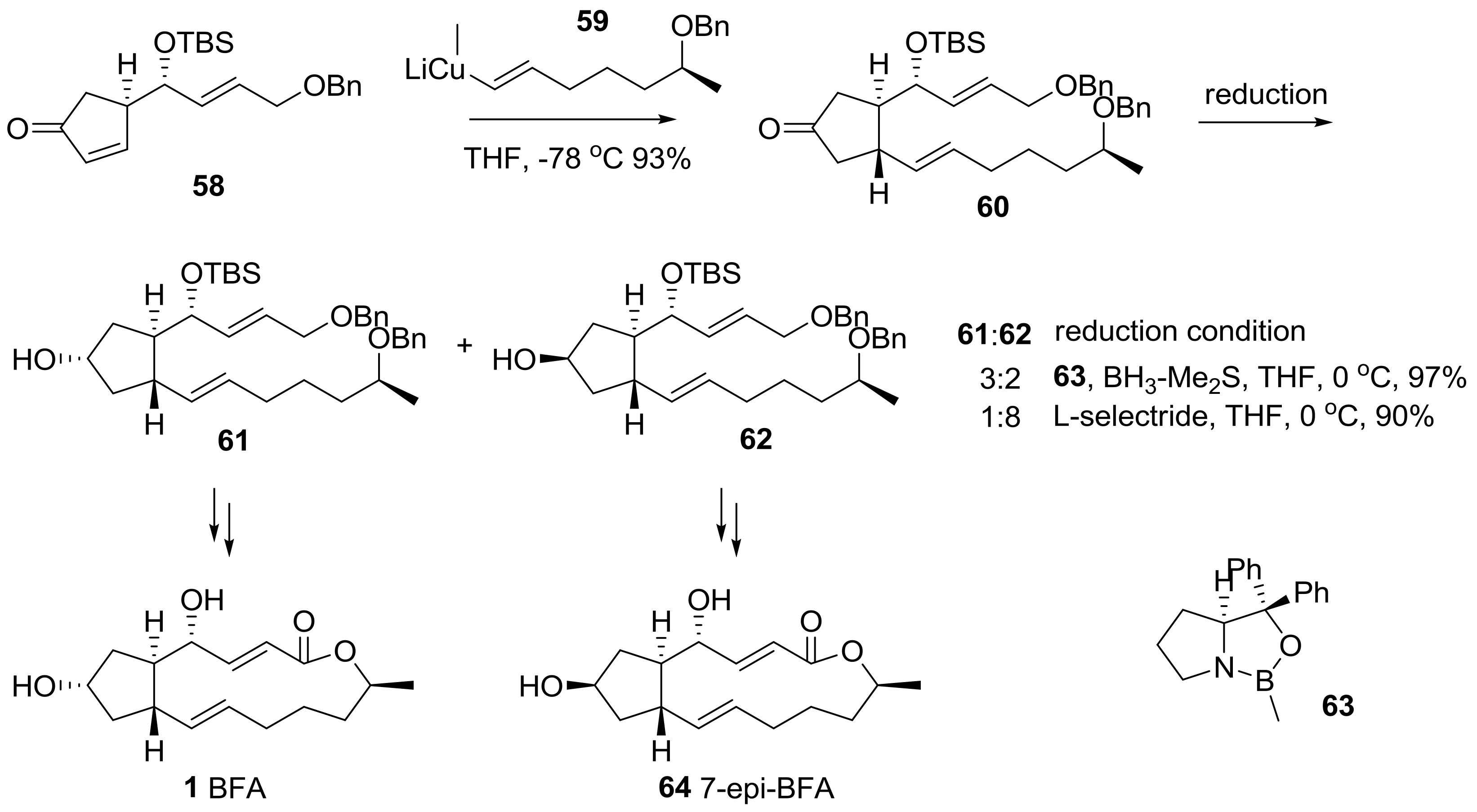{kind=link}
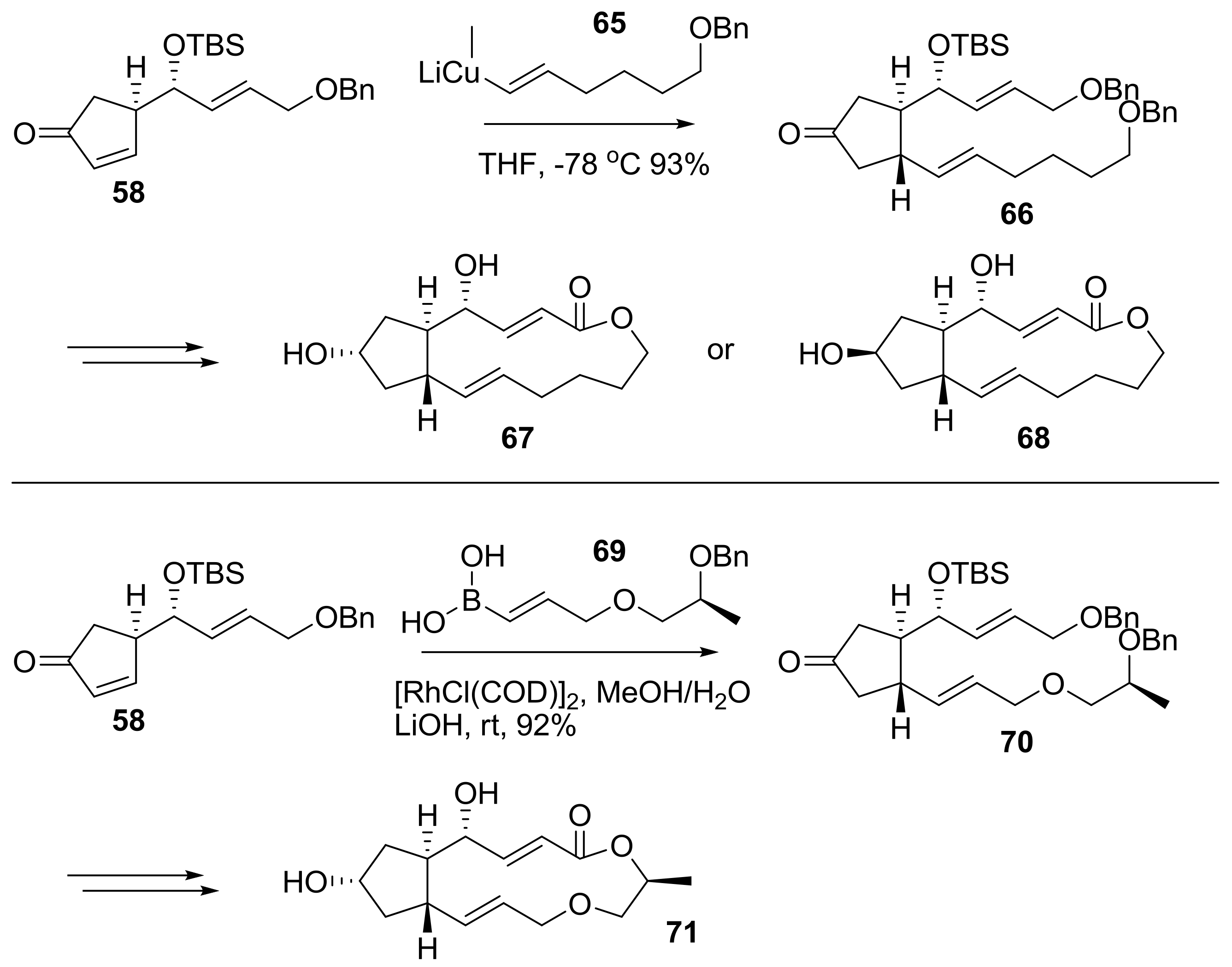{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC 1 (µM) | IC50 2 (µM) | Compound | EC (µM) | IC50 (µM) |
|---|---|---|---|---|---|
| 1 BFA | 0.11 | 0.2 | 9 | ND | - |
| 2 | 0.27 | 0.1 | 10 | 4.1 | 20 |
| 3 | 0.27 | 2.0 | 11 | ND | - |
| 4 | 0.16 | 0.2 | 12 | 70 | 60 |
| 5 | ND 3 | - | 13 | ND | - |
| 6 | ND | - | 14 | ND | - |
| 7 | ND | 3.0 | 15 | ND | - |
| 8 | ND | - |
| Compound | MGM 1 of GI50 (µM) | Compound | MGM of GI50 (µM) |
|---|---|---|---|
| 16 | 4.1 ± 0.076 | 25 | 0.35 |
| 17 | 3.2 ± 0 | 26 | 0.037 ± 0.007 |
| 18 | 0.68 ± 0.062 | 27 | 0.20 ± 0.07 |
| 19 | 20 ± 1.8 | 28 | 0.11 ± 0.07 |
| 20 | 2.5 | 29 | 0.055 ± 0.028 |
| 21 | 1.8 ± 0.18 | 30 | 0.096 ± 0.077 |
| 22 | 3.00 ± 0.31 | 31 | 0.84 ± 0.61 |
| 23 | 36.3 | 32 | 1.47 ± 0.88 |
| 24 | 38 | 33 | 0.028 ± 0.017 |
| 1 BFA | 0.040 ± 0.019 | 34 | 0.048 ± 0.023 |
| Compound | MGM 1 of GI50 (µM) | Compound | MGM of GI50 (µM) |
|---|---|---|---|
| 1 BFA | 0.04 ± 0.019 | 45 | 0.47 ± 0.001 |
| 42 | 13.00 | 46 | 5.10 |
| 43 | 97.72 | 50 | 29.51 |
| 44 | 0.11 ± 0.003 | 51 | 0.16 ± 0.002 |
| Compound | GI50 (µM) 1 | TGI (µM) 2 | Compound | GI50 (µM) | TGI (µM) |
|---|---|---|---|---|---|
| 1 BFA | 0.021 | 3.48 | 96h | 0.081 | 5.05 |
| 96a | 0.321 | 5.49 | 96i | 0.030 | 2.04 |
| 96b | 0.045 | 5.82 | 96j | 0.034 | 2.68 |
| 96c | 0.032 | 4.91 | 96k | 0.152 | 5.70 |
| 96d | 0.593 | 13.35 | 96l | 0.21 | 5.97 |
| 96e | 2.570 | 38.00 | 96m | 0.074 | 2.28 |
| 96f | 0.178 | 7.69 | 96n | 0.261 | 7.61 |
| 96g | 0.090 | 5.78 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paek, S.-M. Recent Synthesis and Discovery of Brefeldin A Analogs. Mar. Drugs 2018, 16, 133. https://doi.org/10.3390/md16040133
Paek S-M. Recent Synthesis and Discovery of Brefeldin A Analogs. Marine Drugs. 2018; 16(4):133. https://doi.org/10.3390/md16040133
Chicago/Turabian StylePaek, Seung-Mann. 2018. "Recent Synthesis and Discovery of Brefeldin A Analogs" Marine Drugs 16, no. 4: 133. https://doi.org/10.3390/md16040133
APA StylePaek, S.-M. (2018). Recent Synthesis and Discovery of Brefeldin A Analogs. Marine Drugs, 16(4), 133. https://doi.org/10.3390/md16040133