Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs


Abstract
:1. Introduction
2. Results
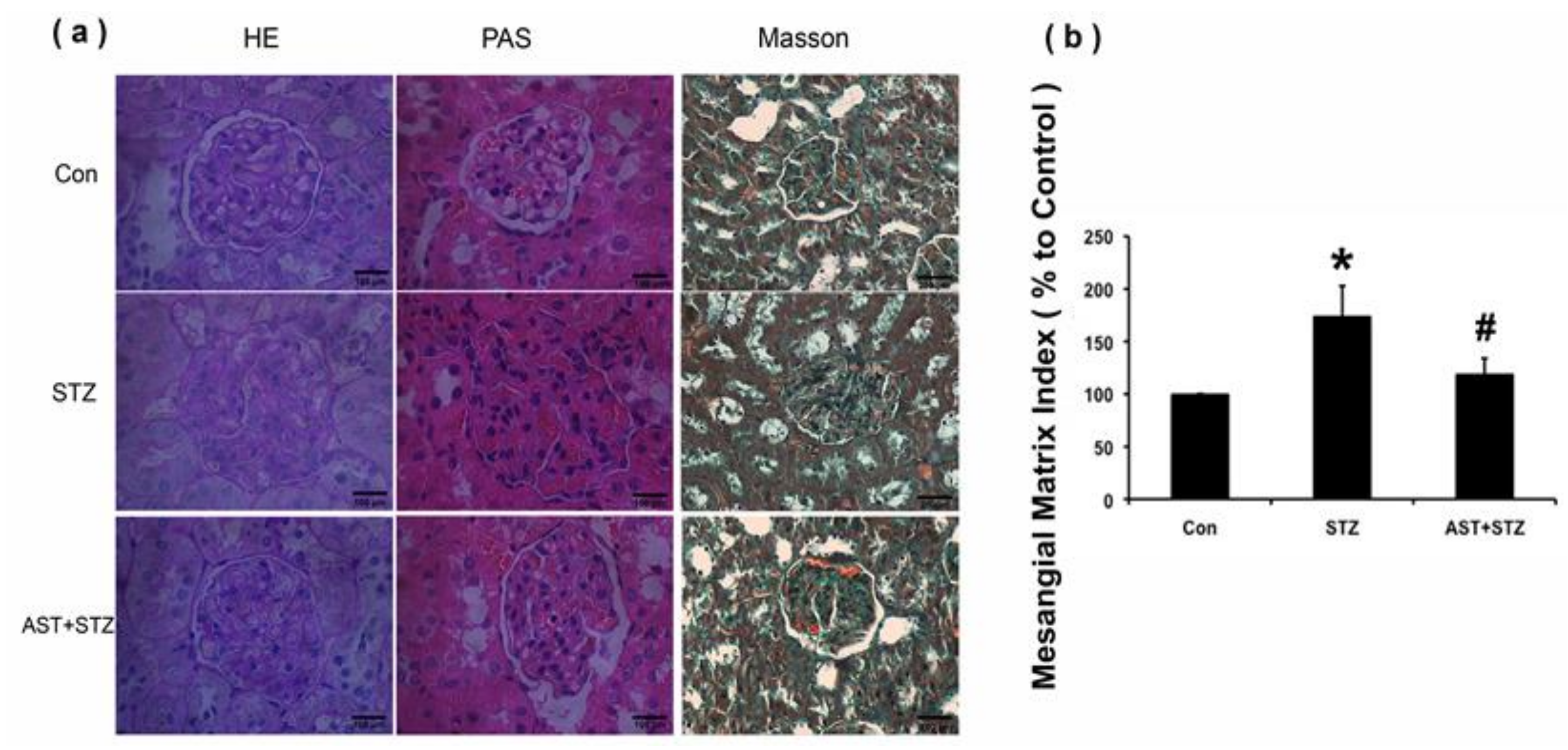
2.1. Effect of AST on Metabolic Parameters, Renal Morphology and ECM Accumulation in Streptozotocin (STZ)-Induced Diabetic Rats
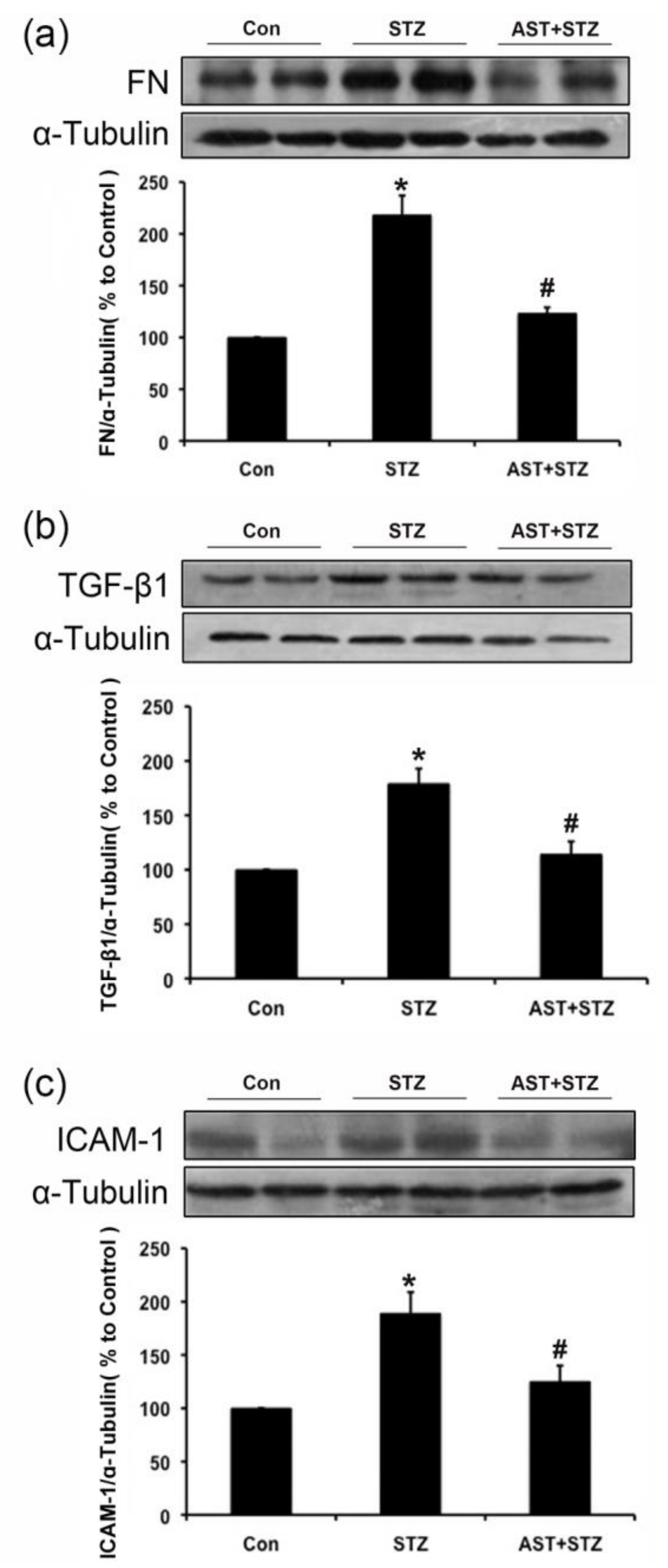
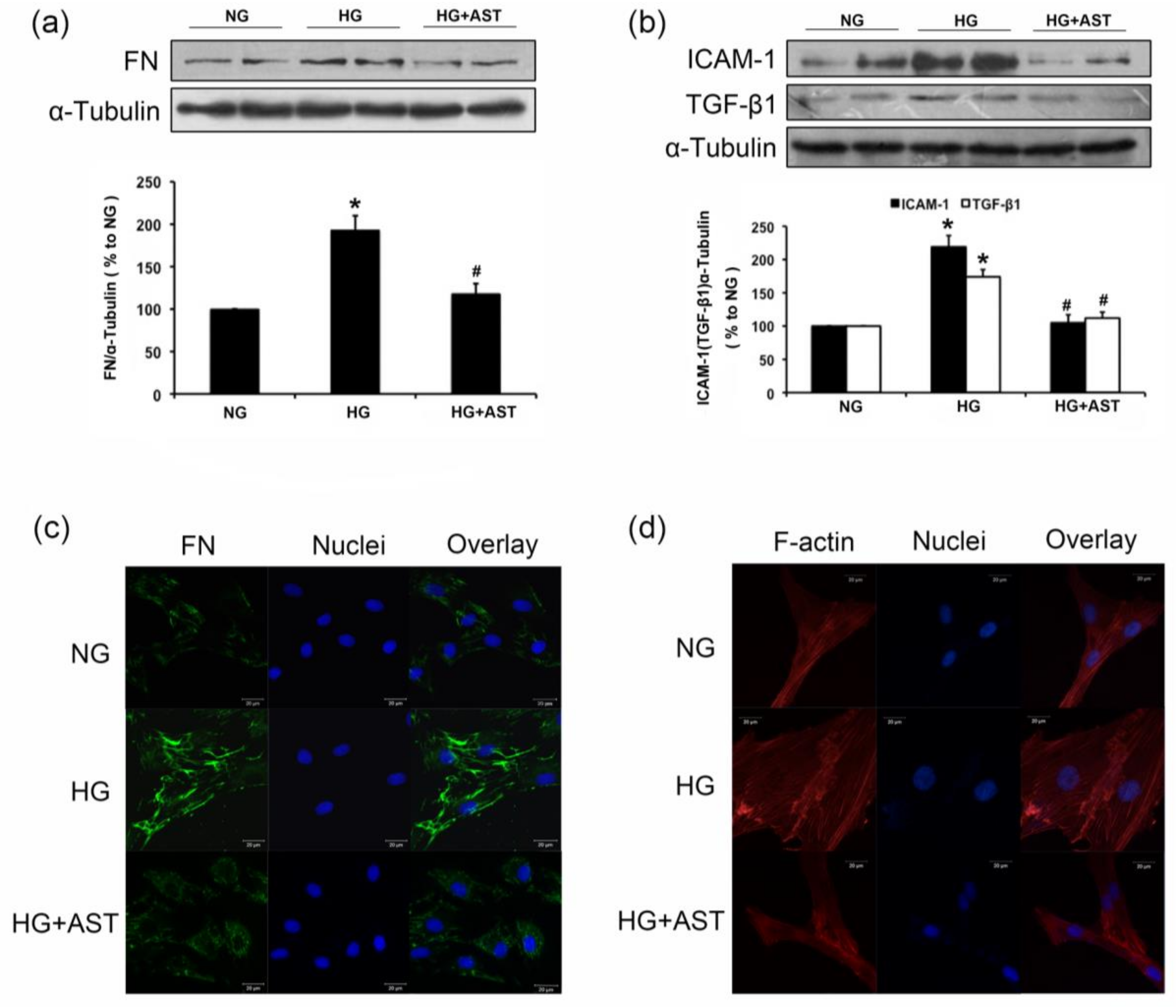
2.2. Effect of AST on the Protein Expression of FN, TGF-β1 and ICAM-1 in the Kidneys of Diabetic Rats
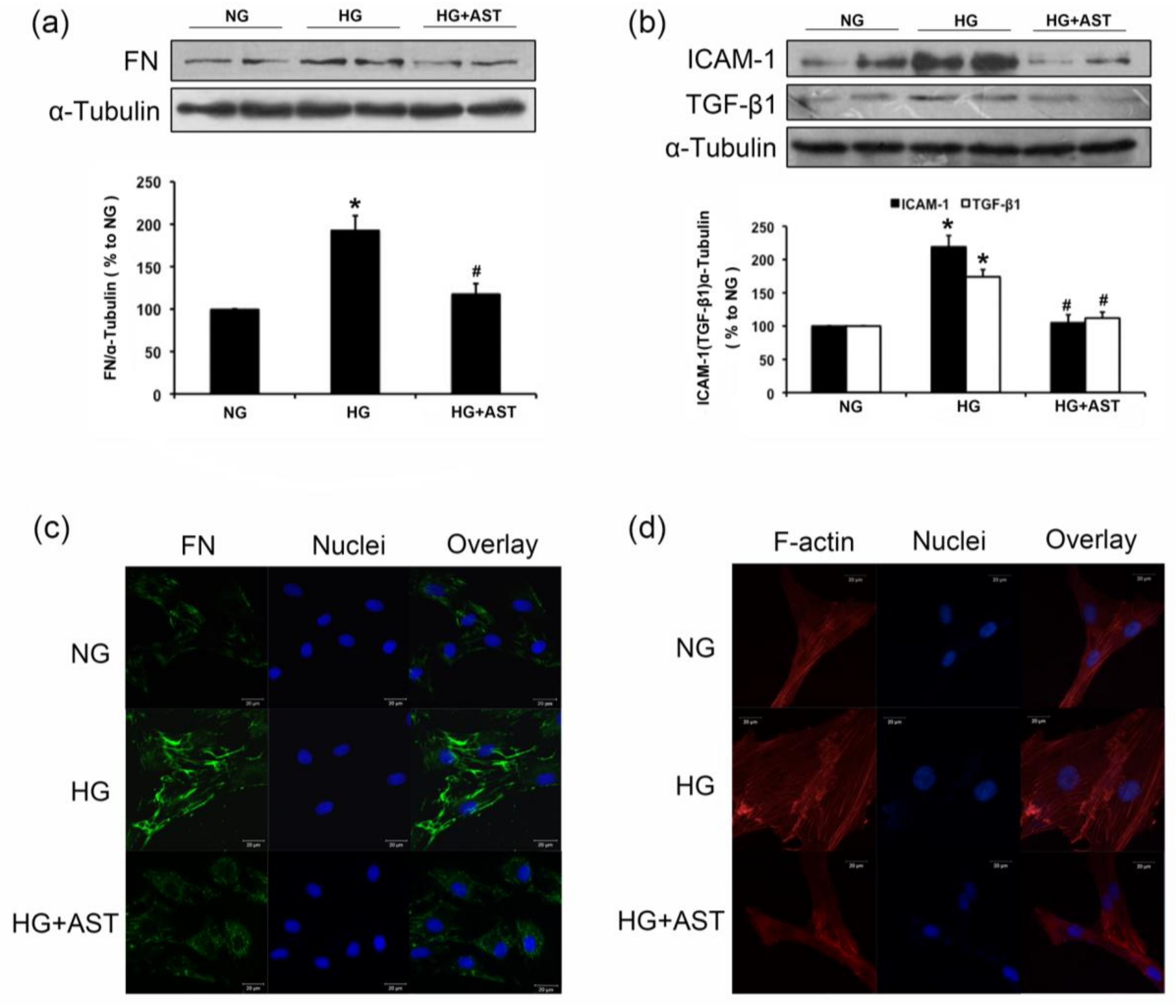
2.3. AST Treatment Prevented the Increased F-Action Formation and Protein Expression of FN, TGF-β1 and ICAM-1 in GMCs Challenged with HG
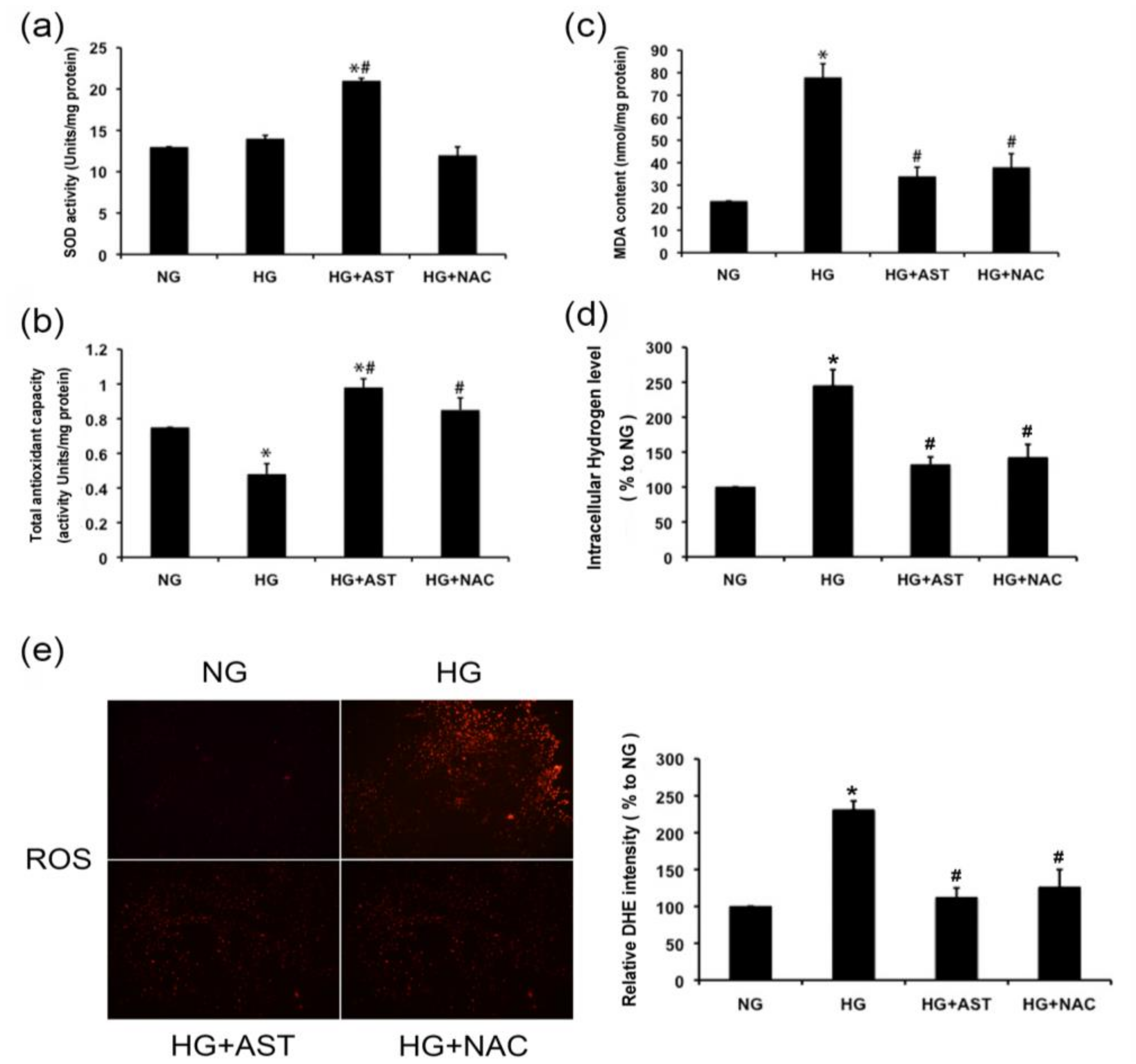
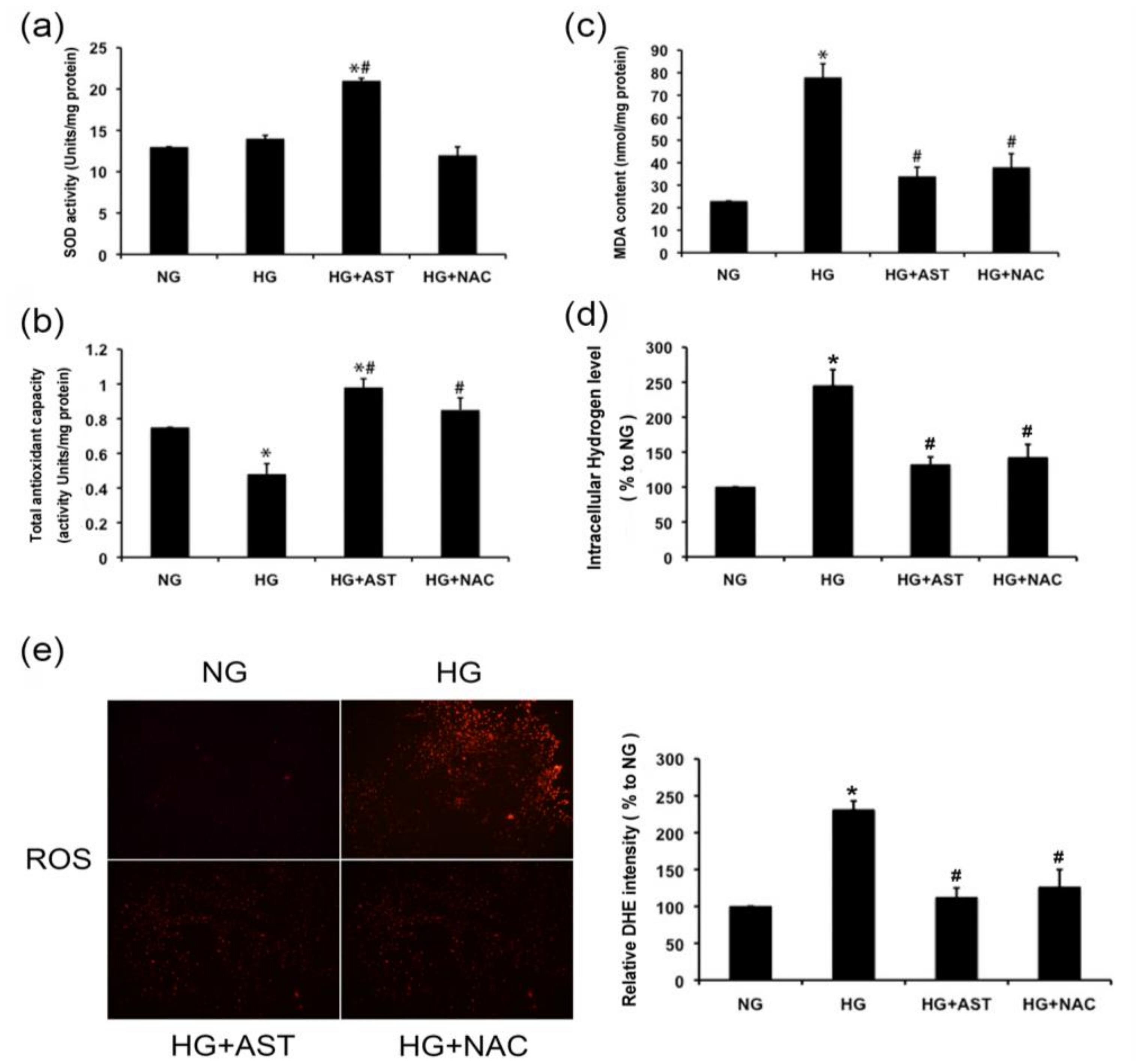
2.4. AST Boosted the Activity of SOD and Total Antioxidative Capacity, and Reduced the Malondialdehyde (MDA) Content and H2O2 Level in GMCs Treated by HG
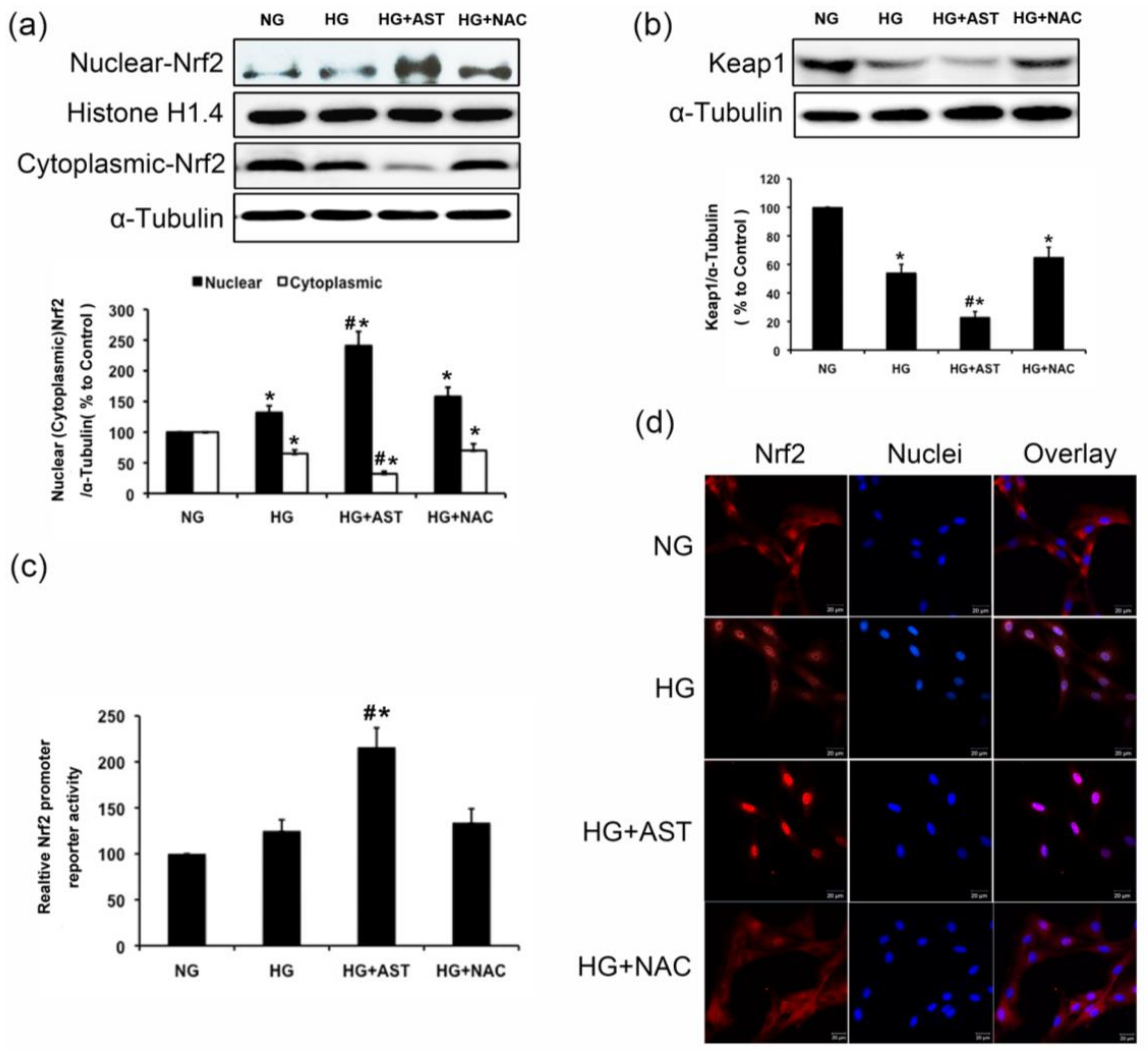
2.5. Nrf2/ARE Signaling Was Enhanced by AST Treatment in HG-Induced GMCs
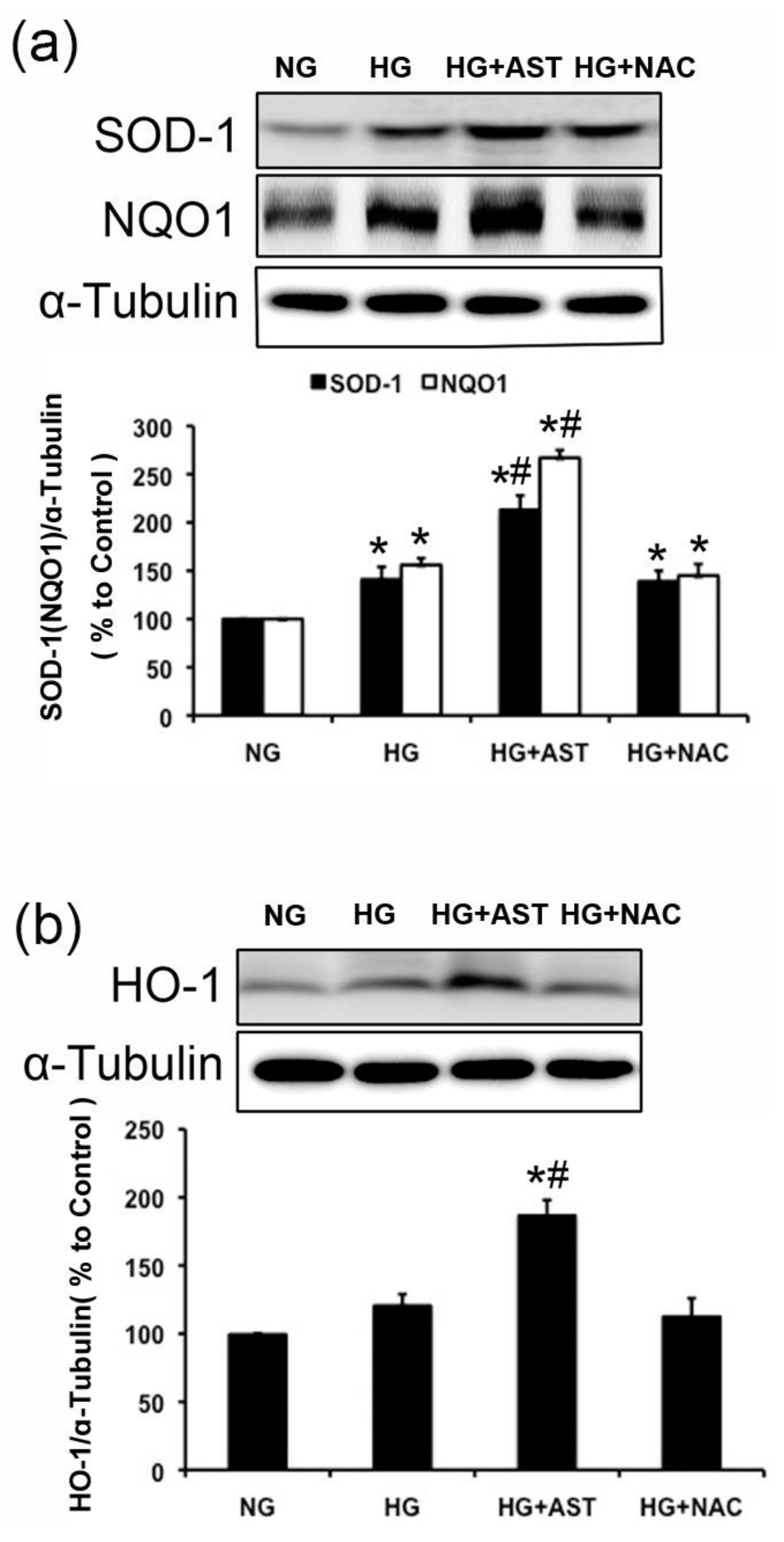
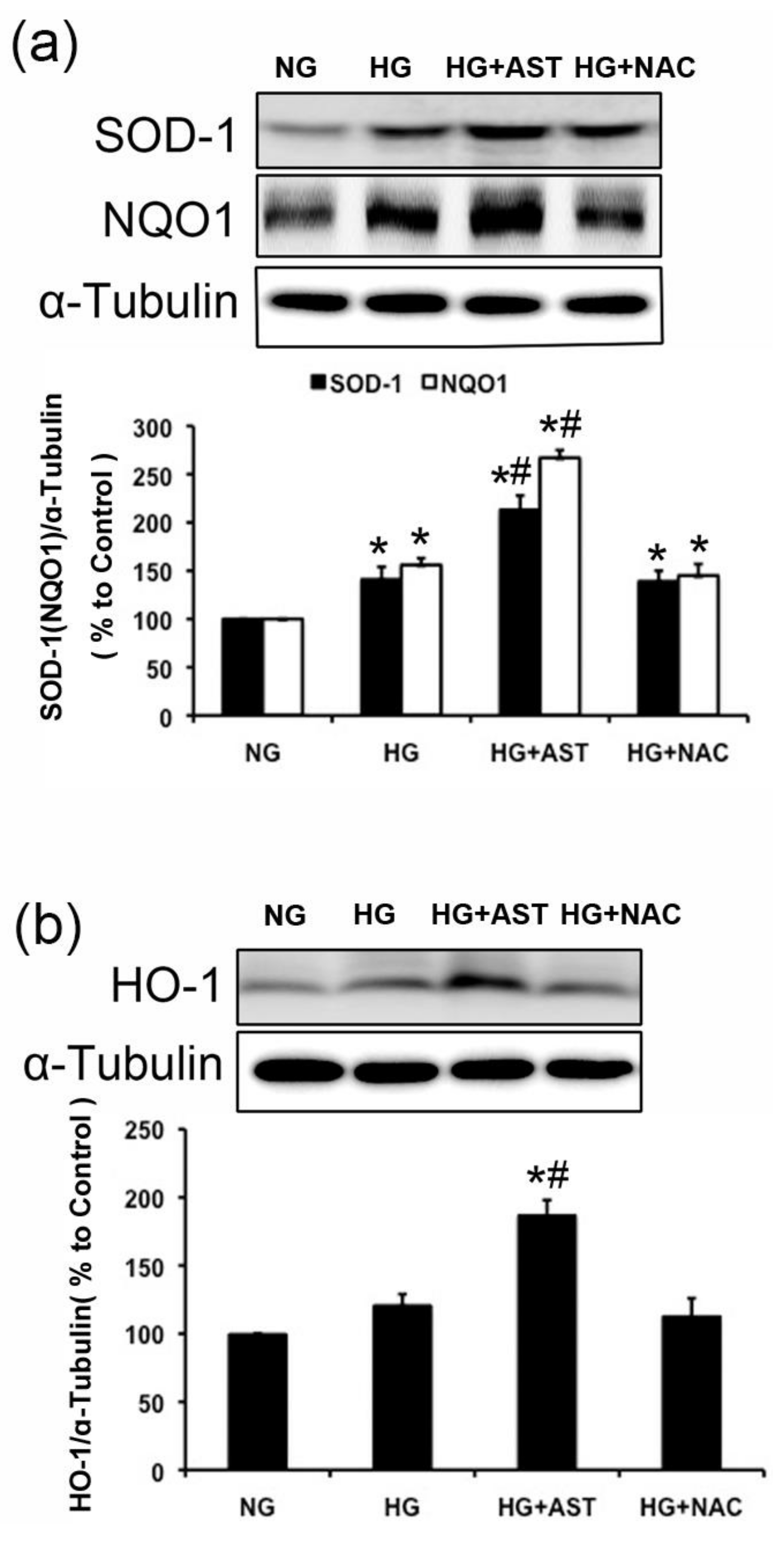
2.6. The Protein Expression of SOD1, HO-1 and NQO1 Were Further Enhanced by AST Treatment in GMCs Challenged with HG
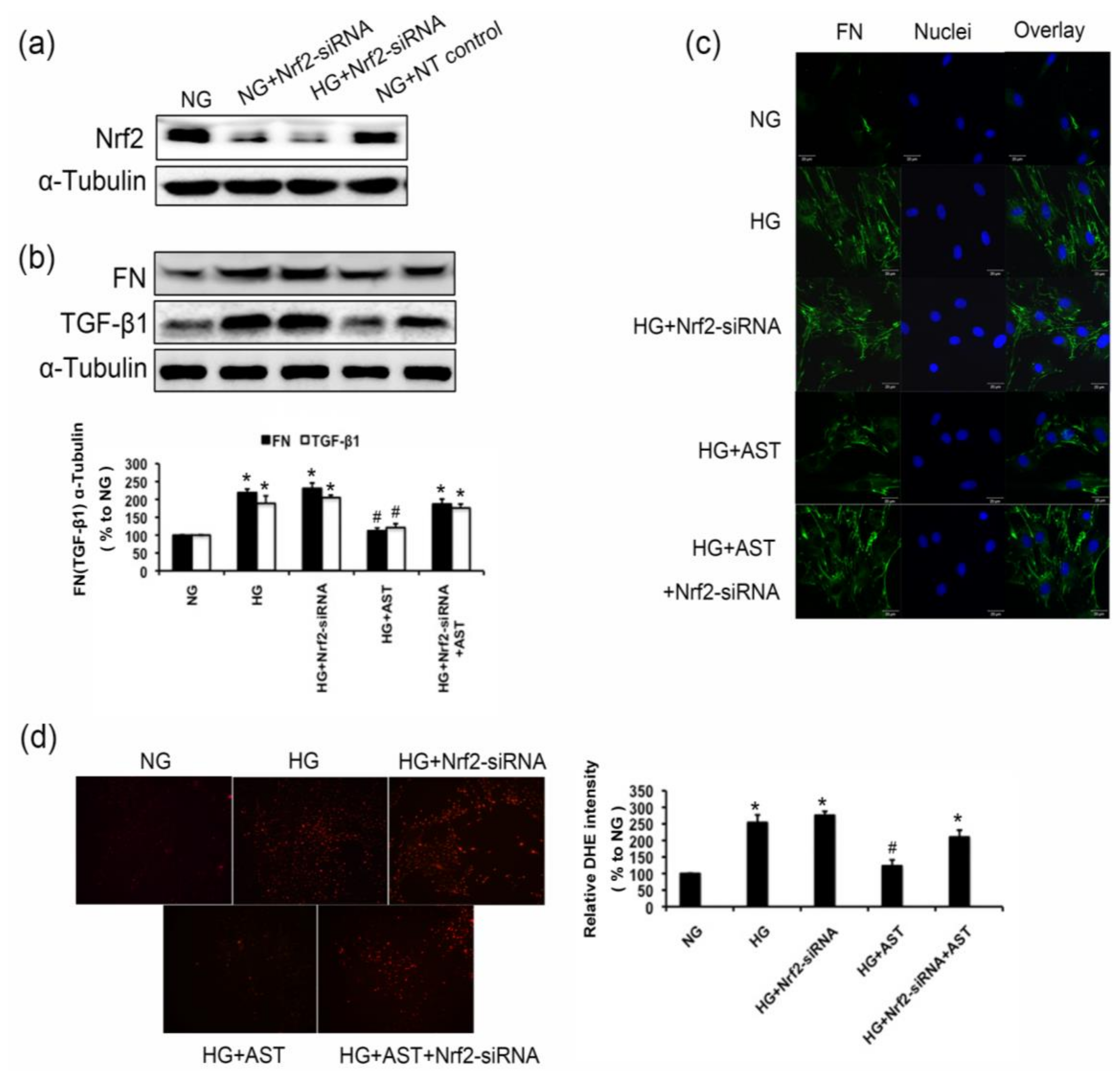
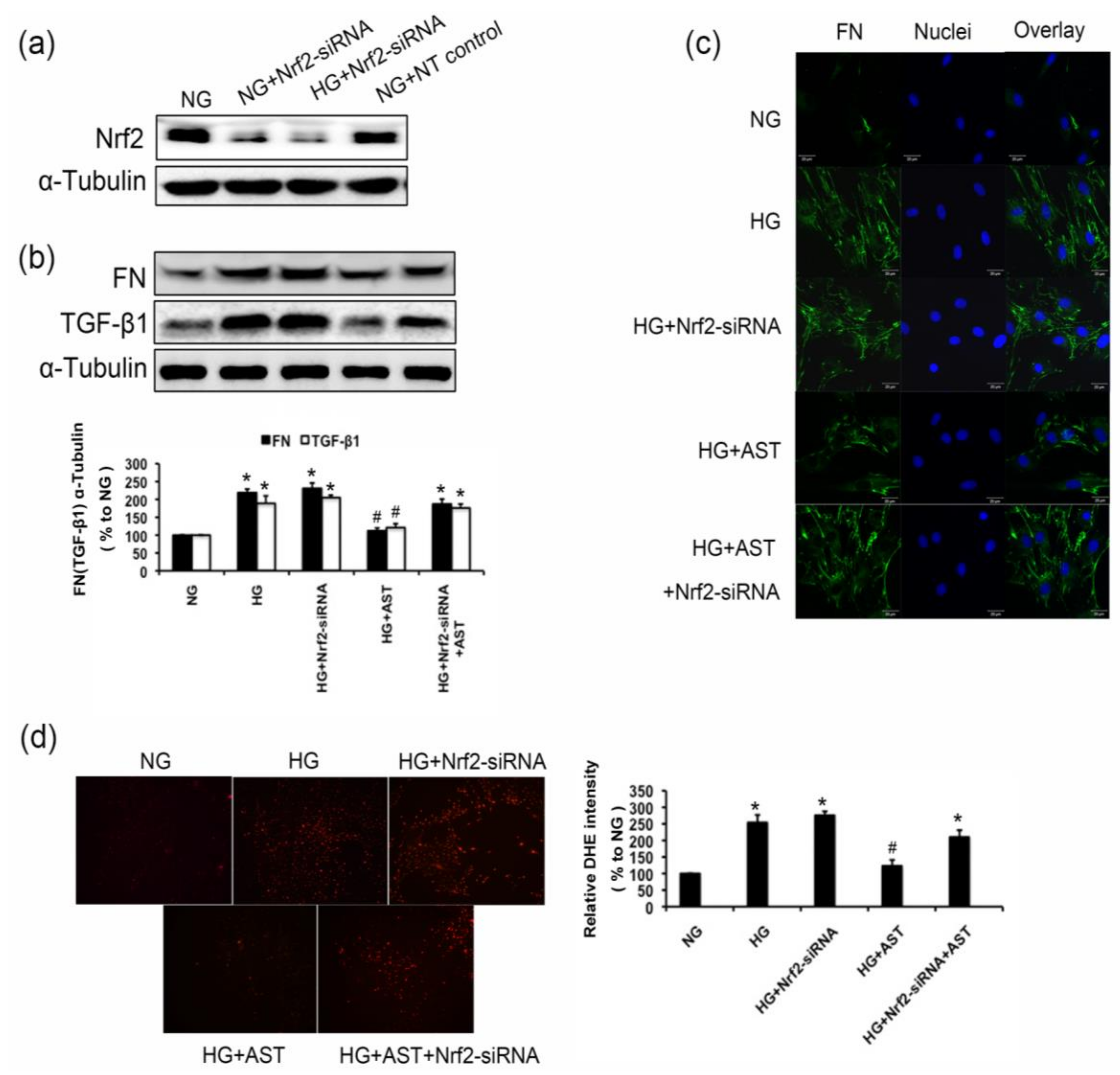
2.7. Nrf2 Deficiency Abrogated the Inhibition of FN and TGF-β1 Expression of AST, and Decreased the Antioxidant Capacity of AST in GMCs Treated by HG
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Transfection
4.3. Animal Experiment
4.4. Biochemical and Morphological Studies
4.5. Western Blot Assay
4.6. Dual Luciferase Reporter Assay
4.7. Detection of Intracellular Reactive Oxygen Species (ROS)
4.8. Determination of Hydrogen Peroxide Generation
4.9. Determination of Intracellular Superoxide Dismutase (SOD) and Lipid Peroxidation
4.10. Confocal Laser Scanning Fluorescence Microscopy (LSCM)
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kanwar, Y.S.; Sun, L.; Xie, P.; Liu, F.Y.; Chen, S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 2011, 6, 395–423. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Diabetic nephropathy—Emerging epigenetic mechanisms. Nat. Rev. Nephrol. 2014, 10, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Schlondorff, D.; Banas, B. The mesangial cell revisited: No cell is an island. J. Am. Soc. Nephrol. 2009, 20, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Daiber, A.; Dopheide, J.F.; Munzel, T.; Espinola-Klein, C. Peripheral artery disease, redox signaling, oxidative stress—Basic and clinical aspects. Redox Biol. 2017, 12, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, N.; Haruna, Y.; Kondeti, V.K.; Kanwar, Y.S. Oxidative stress in diabetic nephropathy. Curr. Med. Chem. 2010, 17, 4256–4269. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 3), S241–S245. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Li, J.; Chen, Z.; Huang, J.; Chen, Q.; Cai, W.; Liu, P.; Huang, H. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating CKIP-1 to resist HG-induced up-regulation of FN and ICAM-1 in GMCs and diabetic mice kidneys. Free Radic. Biol. Med. 2017, 106, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem. 2006, 281, 12132–12142. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; He, S.; Liu, S.; Kong, Z.; Wang, J.; Zhang, Y. Effects of different exercise durations on Keap1-Nrf2-ARE pathway activation in mouse skeletal muscle. Free Radic. Res. 2015, 49, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Nazir, S.; Wang, H.; Kohli, S.; Ranjan, S.; Mertens, P.R.; Nawroth, P.P.; et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 2016, 6, 34228. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chang, X.; Chen, L.; Huang, K.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine ameliorates experimental diabetes-induced renal inflammation and fibronectin by inhibiting the activation of RhoA/ROCK signaling. Mol. Cell. Endocrinol. 2013, 381, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Sila, A.; Ghlissi, Z.; Kamoun, Z.; Makni, M.; Nasri, M.; Bougatef, A.; Sahnoun, Z. Astaxanthin from shrimp by-products ameliorates nephropathy in diabetic rats. Eur. J. Nutr. 2015, 54, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Murillo, A.G.; Fernandez, M.L. Potential of Dietary Non-Provitamin A Carotenoids in the Prevention and Treatment of Diabetic Microvascular Complications. Adv. Nutr. 2016, 7, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Shimidzu, N.; Goto, M.; Miki, W. Carotenoids as singlet oxygen quenchers in marine organisms. Fish. Sci. 1996, 62, 134–137. [Google Scholar] [CrossRef]
- Naito, Y.; Uchiyama, K.; Aoi, W.; Hasegawa, G.; Nakamura, N.; Yoshida, N.; Maoka, T.; Takahashi, J.; Yoshikawa, T. Prevention of diabetic nephropathy by treatment with astaxanthin in diabetic db/db mice. Biofactors 2004, 20, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Manabe, E.; Handa, O.; Naito, Y.; Mizushima, K.; Akagiri, S.; Adachi, S.; Takagi, T.; Kokura, S.; Maoka, T.; Yoshikawa, T. Astaxanthin protects mesangial cells from hyperglycemia-induced oxidative signaling. J. Cell. Biochem. 2008, 103, 1925–1937. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-beta1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2016, 102, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, Y.A.; Yokozawa, T. Protection against oxidative stress, inflammation, and apoptosis of high-glucose-exposed proximal tubular epithelial cells by astaxanthin. J. Agric. Food Chem. 2009, 57, 8793–8797. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Xu, F.H.; Roh, S.S.; Song, Y.O.; Uebaba, K.; Noh, J.S.; Yokozawa, T. Astaxanthin and Corni Fructus protect against diabetes-induced oxidative stress, inflammation, and advanced glycation end product in livers of streptozotocin-induced diabetic rats. J. Med. Food 2015, 18, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic nephropathy and extracellular matrix. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2012, 60, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Peng, J.; Chang, X.; Huang, K.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Activation of RhoA/ROCK regulates NF-kappaB signaling pathway in experimental diabetic nephropathy. Mol. Cell. Endocrinol. 2013, 369, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Bunting, K.; Rao, S.; Hardy, K.; Woltring, D.; Denyer, G.S.; Wang, J.; Gerondakis, S.; Shannon, M.F. Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappaB transcription factor c-Rel. J. Immunol. 2007, 178, 7097–7109. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Shikata, K.; Matsuda, M.; Ogawa, D.; Usui, H.; Kido, Y.; Nagase, R.; Wada, J.; Shikata, Y.; Makino, H. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes 2003, 52, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shaw, S.; Amiri, F.; Eaton, D.C.; Marrero, M.B. Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes 2003, 51, 3505–3509. [Google Scholar] [CrossRef]
- Miranda-Diaz, A.G.; Pazarin-Villasenor, L.; Yanowsky-Escatell, F.G.; Andrade-Sierra, J. Oxidative Stress in Diabetic Nephropathy with Early Chronic Kidney Disease. J. Diabetes Res. 2016, 2016, 7047238. [Google Scholar] [CrossRef] [PubMed]
- Lal, M.A.; Brismar, H.; Ekl‚àö‚àÇf, A.C.; Aperia, A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002, 61, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Huang, J.; Xie, X.; Wang, S.; Chen, C.; Shen, X.; Liu, P.; Huang, H. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-beta1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2013, 65, 528–40. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C. S.; Meyer, C. J.; Li, X.; Cai, L.; Cui, T. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; Rojo de la Vega, M.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Taira, J.; Sonamoto, M.; Uehara, M. Dual Biological Functions of a Cytoprotective Effect and Apoptosis Induction by Bioavailable Marine Carotenoid Fucoxanthinol through Modulation of the Nrf2 Activation in RAW264.7 Macrophage Cells. Mar. Drugs 2017, 15, 305. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, K.; Wiernsperger, N.; Lagarde, M.; El Bawab, S. Bimodal effect of advanced glycation end products on mesangial cell proliferation is mediated by neutral ceramidase regulation and endogenous sphingolipids. J. Biol. Chem. 2004, 279, 34343–34352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Liu, P.; Wu, X.; Liu, W.; Shen, X.; Lan, T.; Xu, S.; Peng, J.; Xie, X.; Huang, H. Berberine attenuates lipopolysaccharide-induced extracelluar matrix accumulation and inflammation in rat mesangial cells: Involvement of NF-kappaB signaling pathway. Mol. Cell. Endocrinol. 2011, 331, 34–40. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control (n = 8) | STZ (n = 8) | STZ + AST (n = 8) |
|---|---|---|---|
| Body weight (g) | 484.67 ± 8.28 | 218.23 ± 13.98 * | 319.23 ± 21.51 *,# |
| Kidney weight (g) | 2.37 ± 0.09 | 3.31 ± 0.59 * | 2.73 ± 0.33 * |
| KW/BW (%) | 0.47 ± 0.04 | 1.19 ± 0.11 * | 0.75 ± 0.18 *,# |
| Blood glucose (mM) | 5.09 ± 0.37 | 23.4 ± 2.61 * | 21.7 ± 1.81 * |
| BUN (mM) | 5.49 ± 1.11 | 15.07 ± 3.10 * | 9.23 ± 1.52 # |
| Cr (μM) | 26.51 ± 3.29 | 44.23 ± 7.21 * | 27.82 ± 8.55 # |
| UP (mg/24 h) | 13.34 ± 2.78 | 97.87 ± 27.23 * | 66.23 ± 19.34 *,# |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Tao, J.; Xie, X. Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Mar. Drugs 2018, 16, 117. https://doi.org/10.3390/md16040117
Chen Q, Tao J, Xie X. Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Marine Drugs. 2018; 16(4):117. https://doi.org/10.3390/md16040117
Chicago/Turabian StyleChen, Qing, Jun Tao, and Xi Xie. 2018. "Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs" Marine Drugs 16, no. 4: 117. https://doi.org/10.3390/md16040117
APA StyleChen, Q., Tao, J., & Xie, X. (2018). Astaxanthin Promotes Nrf2/ARE Signaling to Inhibit HG-Induced Renal Fibrosis in GMCs. Marine Drugs, 16(4), 117. https://doi.org/10.3390/md16040117