Diverse and Abundant Secondary Metabolism Biosynthetic Gene Clusters in the Genomes of Marine Sponge Derived Streptomyces spp. Isolates

 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Antimicrobial Activities
2.2. Genome Sequencing
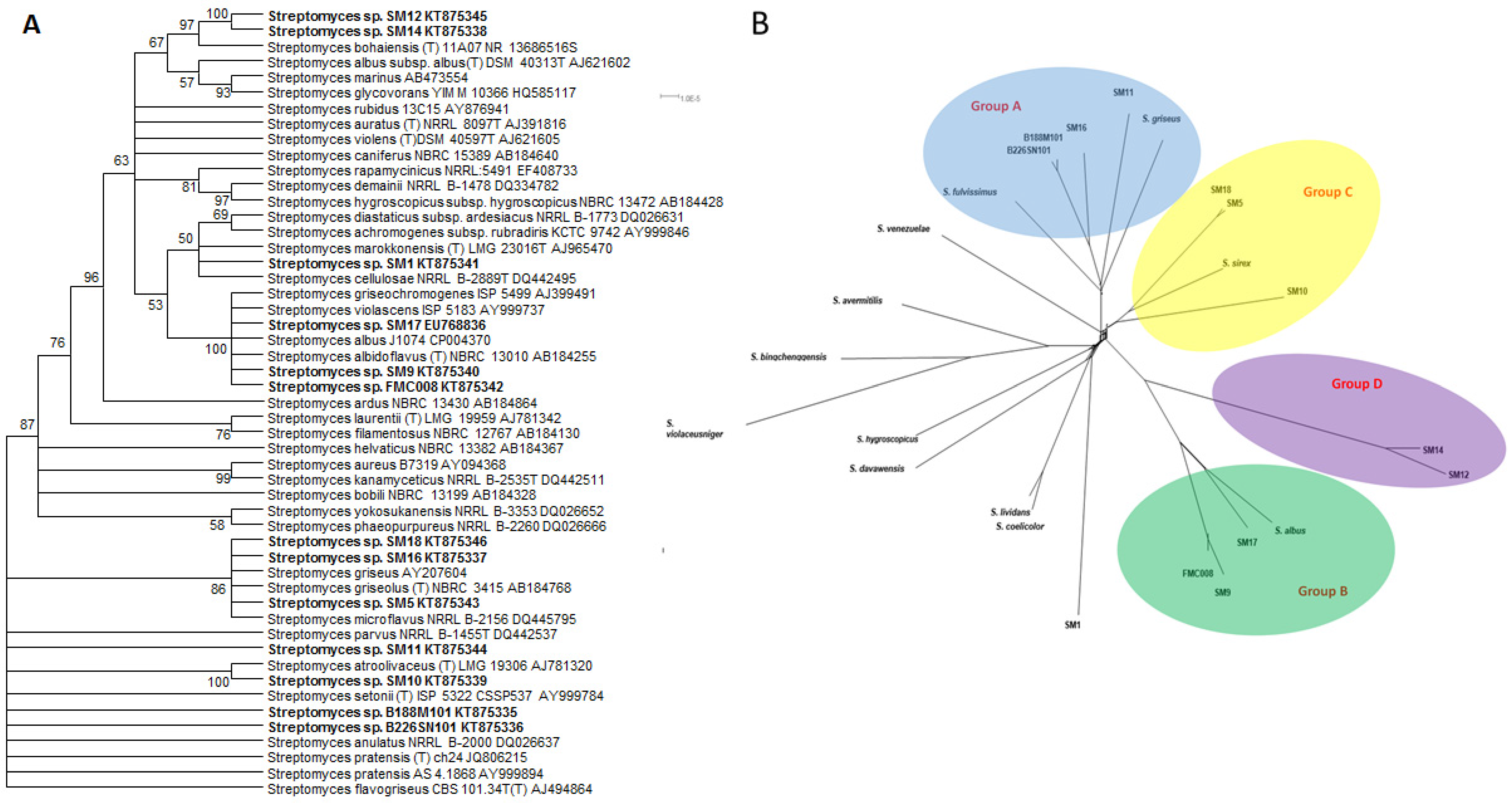
2.3. Taxonomy and Phylogeny
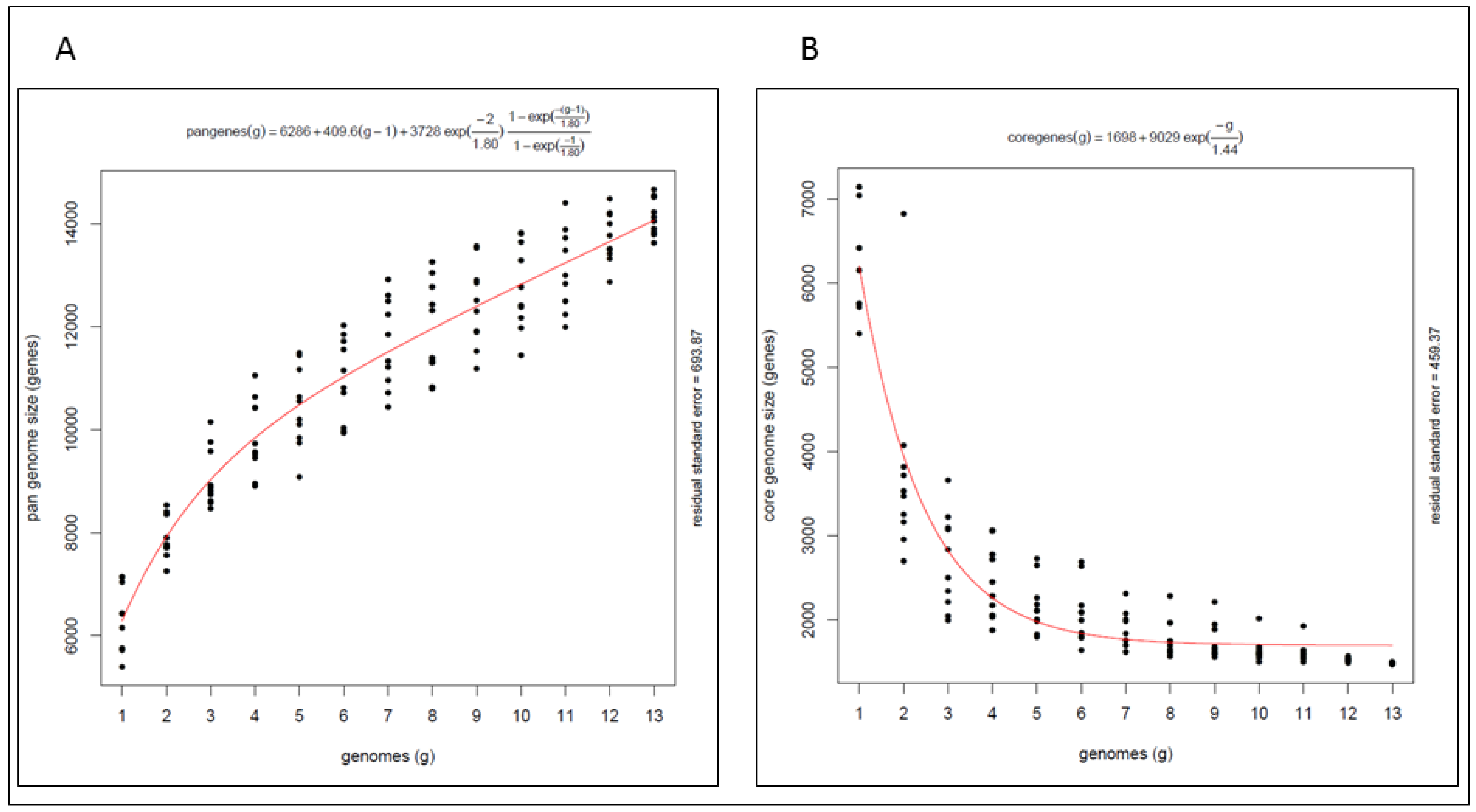
2.4. Pan Genome and Core Genome
2.5. Secondary Metabolism Biosynthetic Gene Clusters
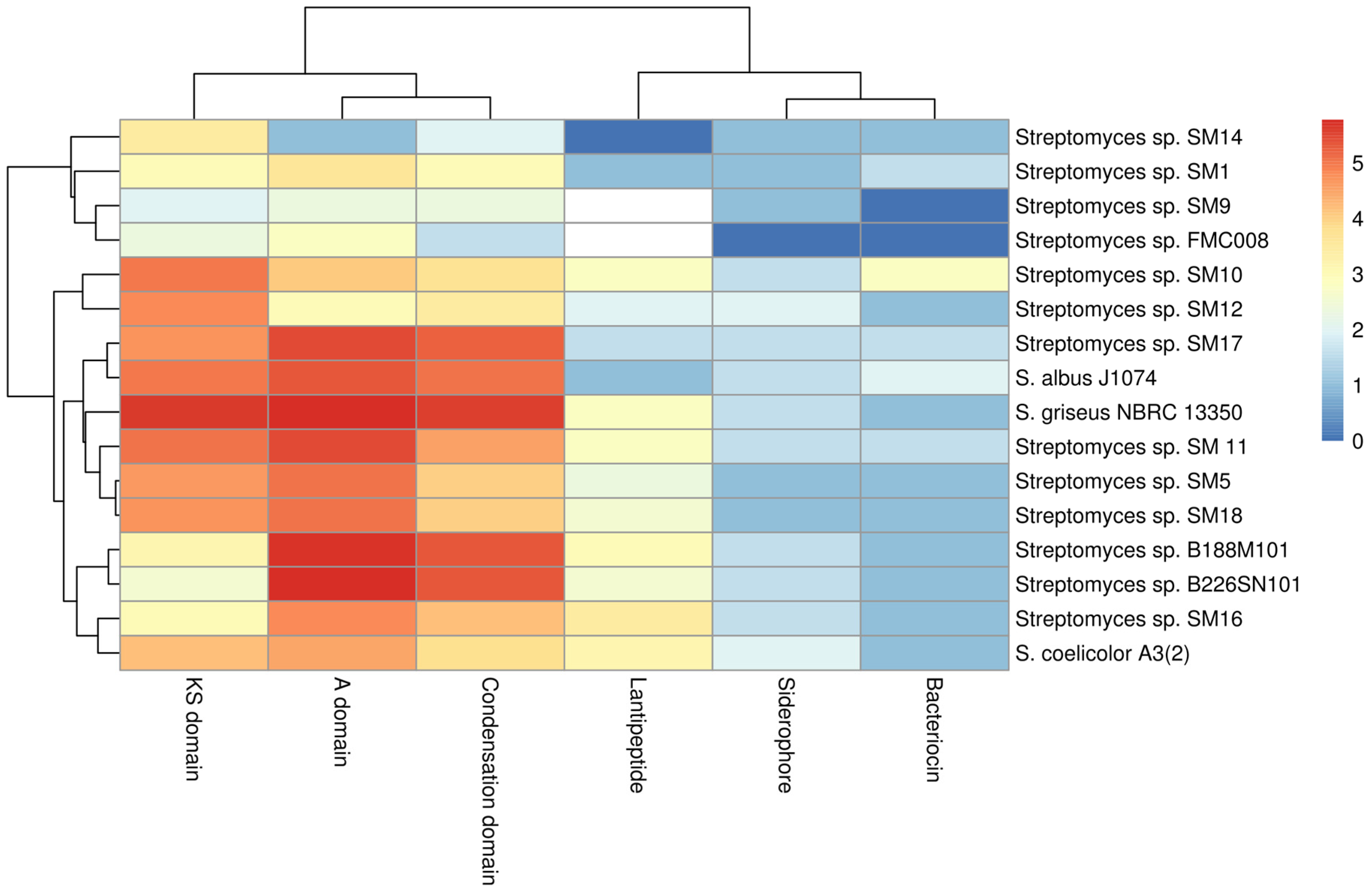
2.6. Protein Family (Pfam) Domain Analysis
2.6.1. KS Domains of PKS Gene Clusters
2.6.2. Nrps Gene Clusters
2.6.3. Siderophores, Bacteriocins and Lantibiotics
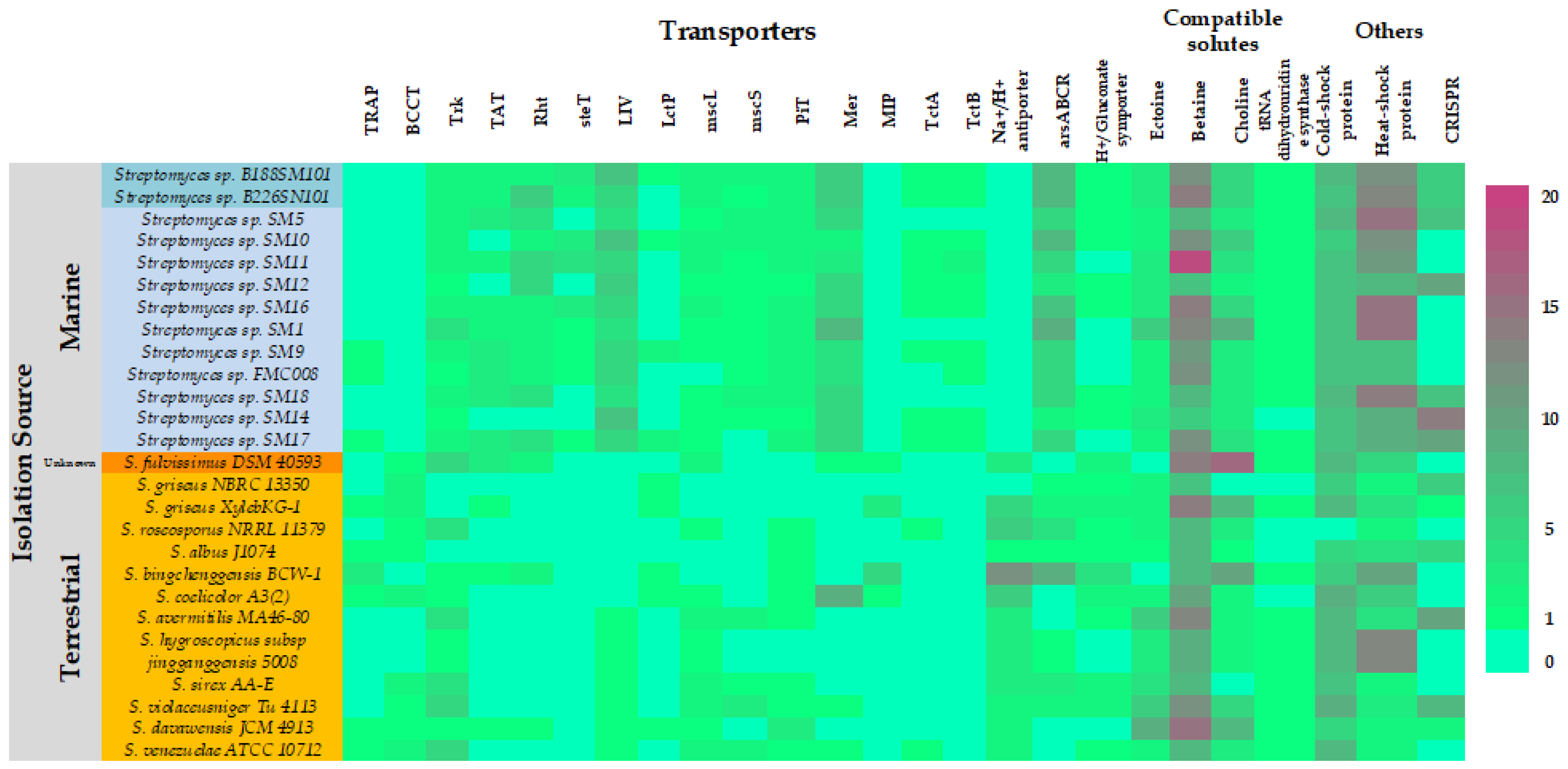
2.7. Marine Adaptations
3. Materials and Methods
3.1. Sponge Sampling
3.2. Culture Isolation
3.3. DNA Extraction
3.4. 16S rRNA Gene Sequencing
3.5. 16S rRNA Based Phylogenetic Analysis
3.6. Bioactivity Screening
3.7. Whole Genome Sequencing
3.8. Bioinformatic Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Subramani, R.; Aalbersberg, W. Culturable rare actinomycetes: Diversity, isolation and marine natural product discovery. Appl. Microbiol. Biotechnol. 2013, 97, 9291–9321. [Google Scholar] [CrossRef] [PubMed]
- Berdy, J. Bioactive microbial metabolites. J. Antibiot. (Tokyo) 2005, 58, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 2015, 14, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Antibiotics: A New Hope. Chem. Biol. 2012, 19, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.W.; Chang, F.Y.; Kim, J.H.; Obiajulu, S.C.; Brady, S.F. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl. Environ. Microbiol. 2010, 767, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Ekkers, D.M.; Creitoiu, M.S.; Kielak, A.M.; Elsas, J.D. The great sreen anomaly—A new frontier in product discovery through functional metagenomics. Appl. Microbiol. Bitechnol. 2012, 93, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Seyedsayamdost, M.R. High-throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc. Natl. Acad. Sci. USA 2014, 111, 7266–7271. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Seyedsayamdost, M.R. Synergy and Target Promiscuity Drive Structural Divergence in Bacterial Alkylquinolone Biosynthesis. Cell Chem. Biol. 2017, 24, 1437–1444.e3. [Google Scholar] [CrossRef] [PubMed]
- Smanski, M.J.; Zhou, H.; Claesen, J.; Shen, B.; Fischbach, M.A.; Voigt, C.A. Synthetic biology to access and expand nature’s chemical diversity. Nat. Rev. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.J.; Romano, S.; Dobson, A.D.; O’Gara, F. The Sound of Silence: Activating Silent Biosynthetic Gene Clusters in Marine Microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar] [CrossRef] [PubMed]
- Rosen, P.C.; Seyedsayamdost, M.R. Though Much Is Taken, Much Abides: Finding New Antibiotics Using Old Ones. Biochemistry 2017, 56, 4925–4926. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Nazari, B.; Moon, K.; Bushin, L.B.; Seyedsayamdost, M.R. Discovery of a Cryptic Antifungal Compound from Streptomyces albus J1074 Using High-Throughput Elicitor Screens. J. Am. Chem. Soc. 2017, 139, 9203–9212. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escribano, J.; Alt, S.; Bibb, M. Next Generation Sequencing of Actinobacteria for the Discovery of Novel Natural Products. Mar. Drugs 2016, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Moore, B.S.; Fenical, W. The marine actinomycete genus Salinispora: A model organism for secondary metabolite discovery. Nat. Prod. Rep. 2015, 32, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Alt, S.; Wilkinson, B. Biosynthesis of the Novel Macrolide Antibiotic Anthracimycin. ACS Chem. Biol. 2015, 10, 2468–2479. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Ahn, C.H.; Shin, Y.; Won, T.H.; Ko, K.; Lee, S.K.; Oh, K.B.; Shin, J.; Nam, S.I.; Oh, D.C. New benzoxazine secondary metabolites from an arctic actinomycete. Mar. Drugs 2014, 12, 2526–5238. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Yu, Y.; Li, H.R.; Dong, N.; Zhang, X.H. Phylogenetic diversity and biological activity of actinobacteria isolated from the Chukchi shelf marine sediments in the Arctic Ocean. Mar. Drugs 2014, 12, 1281–1297. [Google Scholar] [CrossRef] [PubMed]
- Kamjam, M.; Sivalingam, P.; Deng, Z.; Hong, K. Deep Sea Actinomycetes and Their Secondary Metabolites. Front. Microbiol. 2017, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Li, S.; Chen, Y.; Tian, X.; Zhang, H.; Zhang, G.; Zhang, W.; Yang, X.; Zhang, S.; Ju, J.; et al. Lobophorins E and F, new spirotetronate antibiotics from a South China sea-derived Streptomyces sp. SCSIO 01127. J. Antibiot. 2011, 64, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, Q.; Liu, X.; Chen, Y.; Zhang, Y.; Sun, A.; Zhang, W.; Zhang, J.; Ju, J. Cyclic hexapeptides from the deep south China sea-derived Streptomyces scopuliridis SCSIO ZJ46 active against pathogenic gram-positive bacteria. J. Nat. Prod. 2014, 77, 1937–1941. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, H.B.; Li, J.; Song, Y.X.; Jiang, R.W.; Liu, J.; Zhang, S.; Hua, Y.; Ju, J. New antiinfective cycloheptadepsipeptide congeners and absolute stereochemistry from the deep sea-derived Streptomyces drozdowiczii SCSIO 10141. Tetrahedron 2014, 70, 7795–7801. [Google Scholar] [CrossRef]
- Song, Y.; Huang, H.; Chen, Y.; Ding, J.; Zhang, Y.; Sun, A.; Zhang, W.; Ju, J. Cytotoxic and antibacterial Marfuraquinocins from the deep South China sea-derived Streptomyces niveus SCSIO 3406. J. Nat. Prod. 2013, 76, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Baker, P.; Piper, C.; Cotter, P.D.; Walsh, M.; Mooij, M.J.; Bourke, M.B.; Rea, M.C.; O’Conner, P.M.; Ross, R.P.; et al. Isolation and analysis of bacteria with antimicrobial activities from the marine sponge Haliclona simulans collected from Irish waters. Mar. Biotechnol. 2009, 11, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Flemer, B.; Jackson, S.A.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D.W. Evidence of a putative deep sea specific microbiome in marine sponges. PLoS ONE 2014, 9, e91092. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.A.; Flemer, B.; McCann, A.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D. Archaea appear to dominate the microbiome of Inflatella pellicula deep-sea sponges. PLoS ONE 2013, 8, e84438. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.E.; Jun, S.-R.; Wu, G.A.; Kim, S.-H. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc. Natl. Acad. Sci. USA 2009, 106, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Muller, R.; Wohlleben, W.; et al. antiSMASH 3.0―A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Borchert, E.; Jackson, S.A.; O’Gara, F.; Dobson, A.D.W. Diversity of natural product biosynthetic genes in the microbiome of the deep sea sponges Inflatella pellicula, Poecillastra compressa and Stelletta normani. Front. Microbiol. 2016, 7, 1027. [Google Scholar] [CrossRef] [PubMed]
- Sandy, M.; Butler, A. Microbial iron acquisition: marine and terrestrial siderophores. Chem. Rev. 2009, 109, 4580–4595. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.K.; Braucher, O. Observations of dissolved iron concentrations in the world ocean: Implications and constraints for ocean biogeochemical models. Biogeosci. Discuss. 2007, 4, 1241–1277. [Google Scholar] [CrossRef]
- Krasko, A.; Schröder, H.C.; Bate, R.L.; Grebenjuk, V.A.; Steffen, R.; Müller, I.M.; Müller, W.E.G. Iron induces proliferation and morphogenesis in primmorphs from the marine sponge Suberites domuncula. DNA Cell Biol. 2004, 21, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Mayzel, B.; Aizenberg, J.; Ilan, M. The elemental composition of Demospongiae from the Red Sea, Gulf of Aqaba. PLoS ONE 2014, 9, e99918. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, Z.; Yang, T.; Chen, M.; Li, J.; Chen, F.; Yang, J.; Li, W.; Zhang, B.; Zhang, Z.; et al. Comparative Genomics Analysis of Streptomyces Species Reveals Their Adaptation to the Marine Environment and Their Diversity at the Genomic Level. Front. Microbiol. 2016, 7, 998. [Google Scholar] [CrossRef] [PubMed]
- Karen, R.; Mayzel, B.; Lavy, A.; Polishchuk, I.; Levy, D.; Fakra, S.C.; Pokroy, B.; Ilan, M. Sponge-associated bacteria mineralize arsenic and barium on intracellular vesicles. Nat. Commun. 2017, 8, 14393. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; Wiley: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002. [Google Scholar] [CrossRef]
- Nakhleh, L.; Jin, G.; Zhao, F.; Mellor-Crummey, J. Reconstructing Phylogenetic Networks Using Maximum Parsimony. In Proceedings of the 2005 IEEE Computational Systems Bioinformatics Conference, Stanford, CA, USA, 8–11 August 2005. [Google Scholar]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Bibi, F. Diversity of antagonistic bacteria isolated from medicinal plant Peganum harmala L. Saudi J. Biol. Sci. 2017, 24, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package or scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Strain | Gram Negative Bacteria | Gram Positive Bacteria | Yeasts | ||||
|---|---|---|---|---|---|---|---|
| E. coli NCIMB 12210 | P. aeruginosa PAO1 | Bacillus pp. | Staphylococcus spp. | L. monocyte genes F2365 | Candida spp. | A. fumigatus ATCC 46645 | |
| SM1 * | + | − | + 1,2 | + a | + | + | n.d. |
| SM5 * | + | − | + 1,2 | + a | − | − | n.d. |
| SM9 * | + | − | − | − | − | − | n.d. |
| SM10 * | + | n.d. | − | − | − | − | n.d. |
| SM11 * | − | − | + 2 | + a,b | − | − | n.d. |
| SM12 * | − | + | − | + a | − | − | n.d. |
| SM14 * | − | − | + 1,2 | + a | − | − | n.d. |
| SM16 * | − | + | + 2 | + c | − | − | n.d. |
| SM17 * | + | − | − | + b | − | + | n.d. |
| SM18 * | − | − | + 2 | + b | − | − | n.d. |
| FMC008 * | − | + | + 2 | + d | − | − | n.d. |
| B226SN101 | − | − | − 3 | − | n.d. | + | + |
| B188M101 | − | − | − 3 | − | n.d. | + | + |
| Isolate ID | No. of Contigs | Total Length (Mb) |
|---|---|---|
| B188SM101 | 609 | 8.23 |
| B226SN101 | 580 | 8.39 |
| SM5 | 469 | 7.62 |
| SM10 | 195 | 7.48 |
| SM11 | 311 | 8 |
| SM12 | 910 | 6.5 |
| SM16 | 388 | 8.44 |
| SM1 | 1057 | 8.08 |
| SM9 | 1592 | 6.47 |
| FMC008 | 1369 | 6.5 |
| SM18 | 403 | 7.6 |
| SM14 | 639 | 6.41 |
| SM17 | 674 | 7.106 |
| Isolate ID | PKS * | NRPS | PKS/NRPS Hybrid | Bacteriocin | Lantipeptide | Siderophore | Terpene | Butyrolactone | Ectoine | Other | TOTAL |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B188M101 | 4 | 19 | 4 | 2 | 4 | 2 | 5 | 2 | 2 | 9 | 53 |
| B226SN101 | 2 | 20 | 4 | 2 | 3 | 2 | 5 | 2 | 2 | 9 | 51 |
| SM5 | 5 | 9 | 5 | 2 | 1 | 1 | 6 | 1 | 1 | 7 | 38 |
| SM10 | 16 | 3 | 3 | 5 | 3 | 2 | 4 | 2 | 1 | 5 | 44 |
| SM11 | 10 | 11 | 5 | 2 | 3 | 2 | 6 | 2 | 2 | 11 | 54 |
| SM12 | 18 | 7 | - | 1 | 3 | 4 | 1 | 1 | 1 | 4 | 40 |
| SM16 | 2 | 5 | 6 | 1 | 5 | 2 | 7 | 2 | 2 | 7 | 39 |
| SM1 | 4 | 8 | 1 | 2 | 2 | 2 | 4 | 1 | - | 5 | 28 |
| SM9 | 3 | 2 | - | 1 | - | 2 | 6 | - | 1 | 1 | 16 |
| FMC008 | 1 | 3 | - | 1 | - | 1 | 5 | - | 1 | 3 | 15 |
| SM18 | 9 | 9 | 6 | 2 | 2 | 1 | 6 | 1 | 1 | 4 | 41 |
| SM14 | 8 | 2 | - | - | 1 | 2 | - | 1 | - | 3 | 17 |
| SM17 | 14 | 18 | 2 | 3 | 2 | 3 | 5 | - | 1 | 1 | 49 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, S.A.; Crossman, L.; Almeida, E.L.; Margassery, L.M.; Kennedy, J.; Dobson, A.D.W. Diverse and Abundant Secondary Metabolism Biosynthetic Gene Clusters in the Genomes of Marine Sponge Derived Streptomyces spp. Isolates. Mar. Drugs 2018, 16, 67. https://doi.org/10.3390/md16020067
Jackson SA, Crossman L, Almeida EL, Margassery LM, Kennedy J, Dobson ADW. Diverse and Abundant Secondary Metabolism Biosynthetic Gene Clusters in the Genomes of Marine Sponge Derived Streptomyces spp. Isolates. Marine Drugs. 2018; 16(2):67. https://doi.org/10.3390/md16020067
Chicago/Turabian StyleJackson, Stephen A., Lisa Crossman, Eduardo L. Almeida, Lekha Menon Margassery, Jonathan Kennedy, and Alan D.W. Dobson. 2018. "Diverse and Abundant Secondary Metabolism Biosynthetic Gene Clusters in the Genomes of Marine Sponge Derived Streptomyces spp. Isolates" Marine Drugs 16, no. 2: 67. https://doi.org/10.3390/md16020067
APA StyleJackson, S. A., Crossman, L., Almeida, E. L., Margassery, L. M., Kennedy, J., & Dobson, A. D. W. (2018). Diverse and Abundant Secondary Metabolism Biosynthetic Gene Clusters in the Genomes of Marine Sponge Derived Streptomyces spp. Isolates. Marine Drugs, 16(2), 67. https://doi.org/10.3390/md16020067