Inhibitory Growth of Oral Squamous Cell Carcinoma Cancer via Bacterial Prodigiosin


 , ,
, , 
Abstract

1. Introduction
2. Results and Discussion
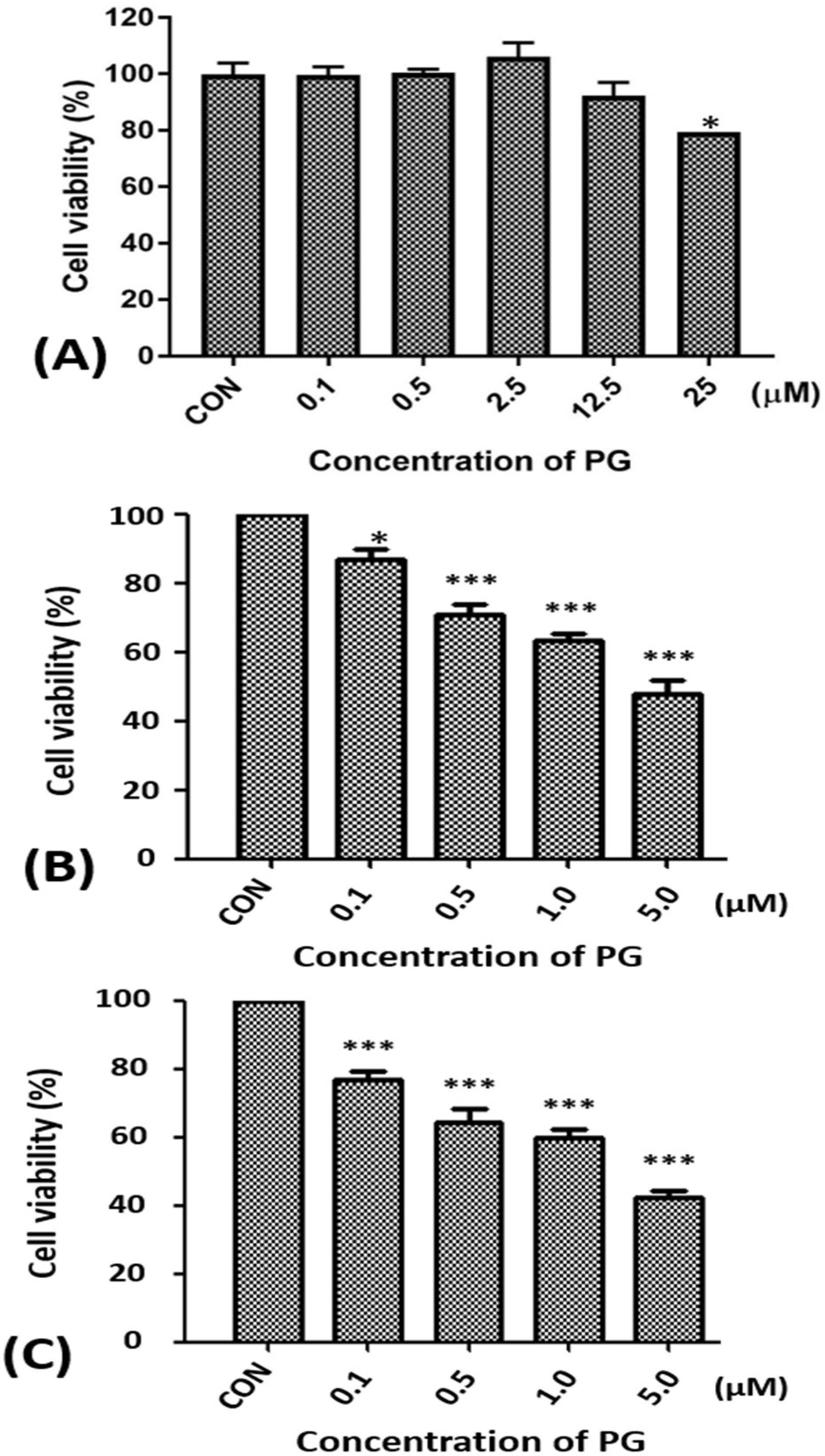
2.1. Effect of Prodigiosin on the Cell Viability of Oral Cancer Cells
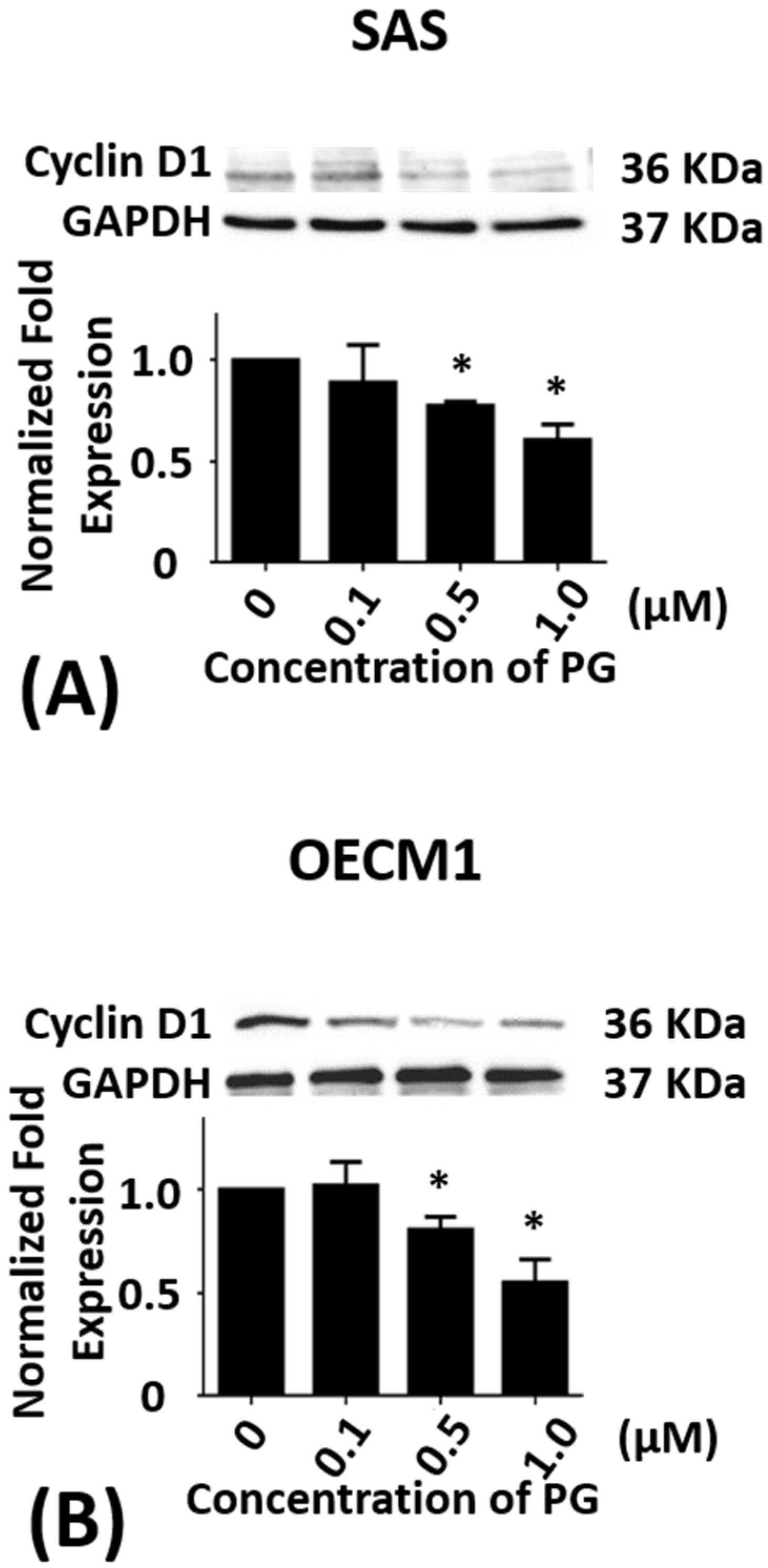
2.2. Effect of Prodigiosin on the Cell Cycle of Oral Cancer Cells
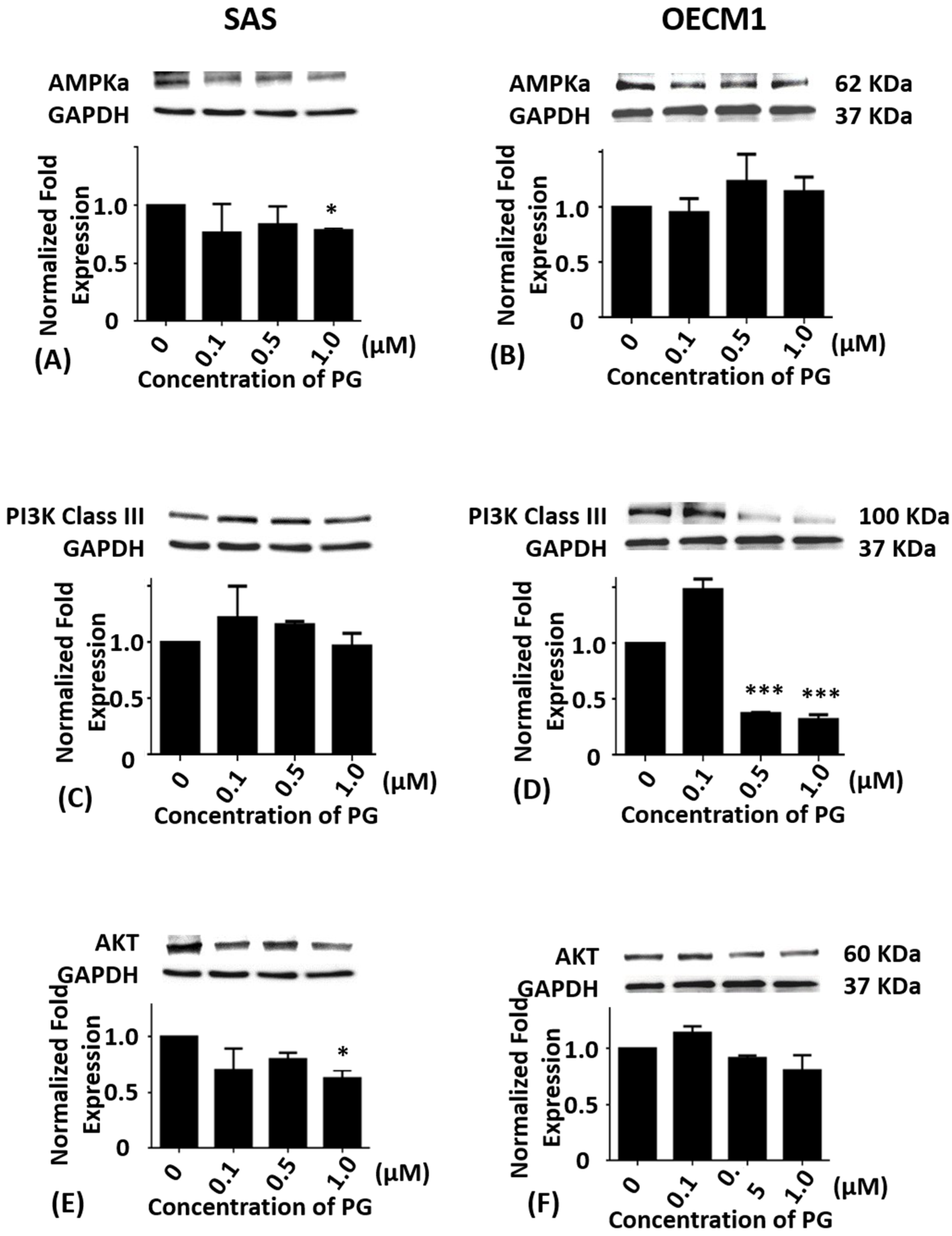
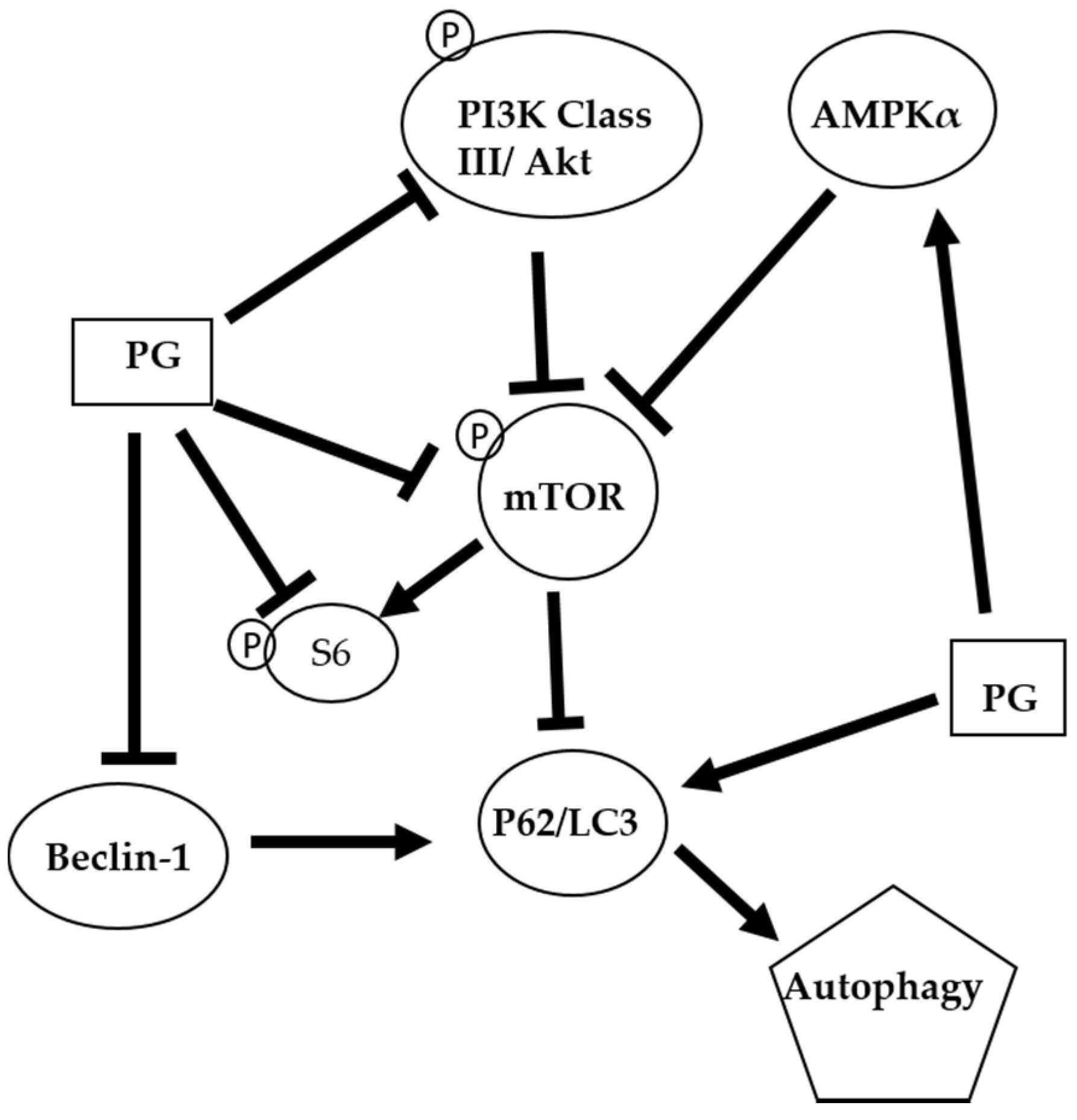
2.3. Effects of Prodigiosin on AMPKα, PI3K Class III and Akt Protein Levels in Oral Cancer Cells
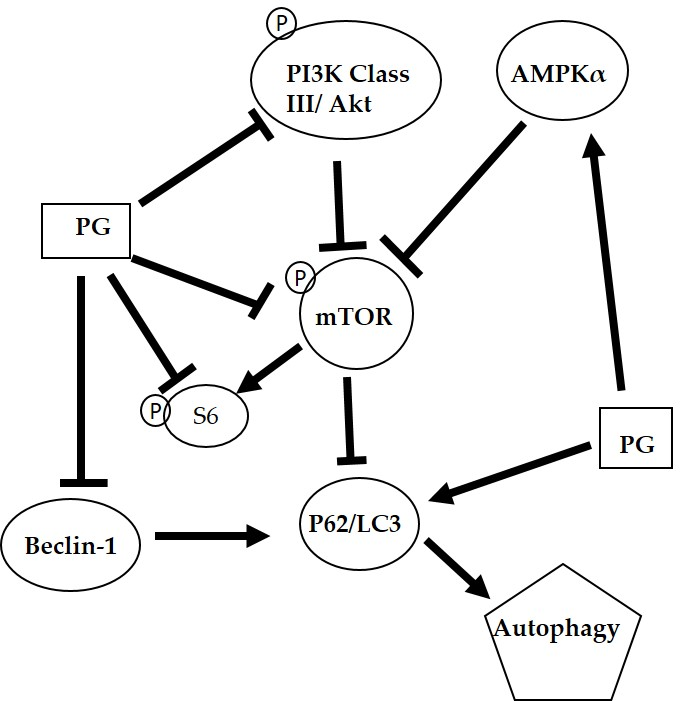
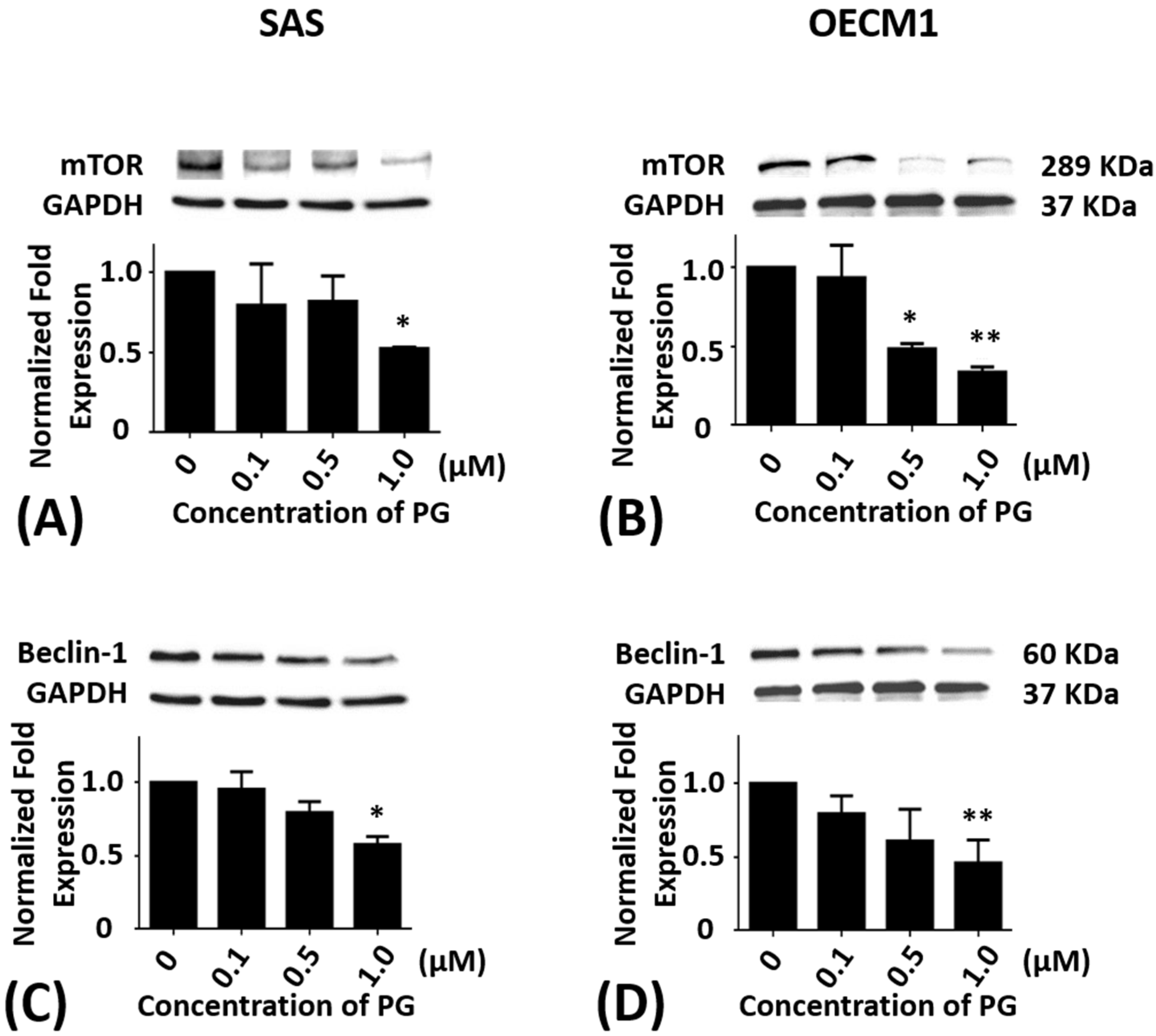
2.4. Effects of Prodigiosin on mTOR and Beclin-1 Protein Levels in Oral Cancer Cells
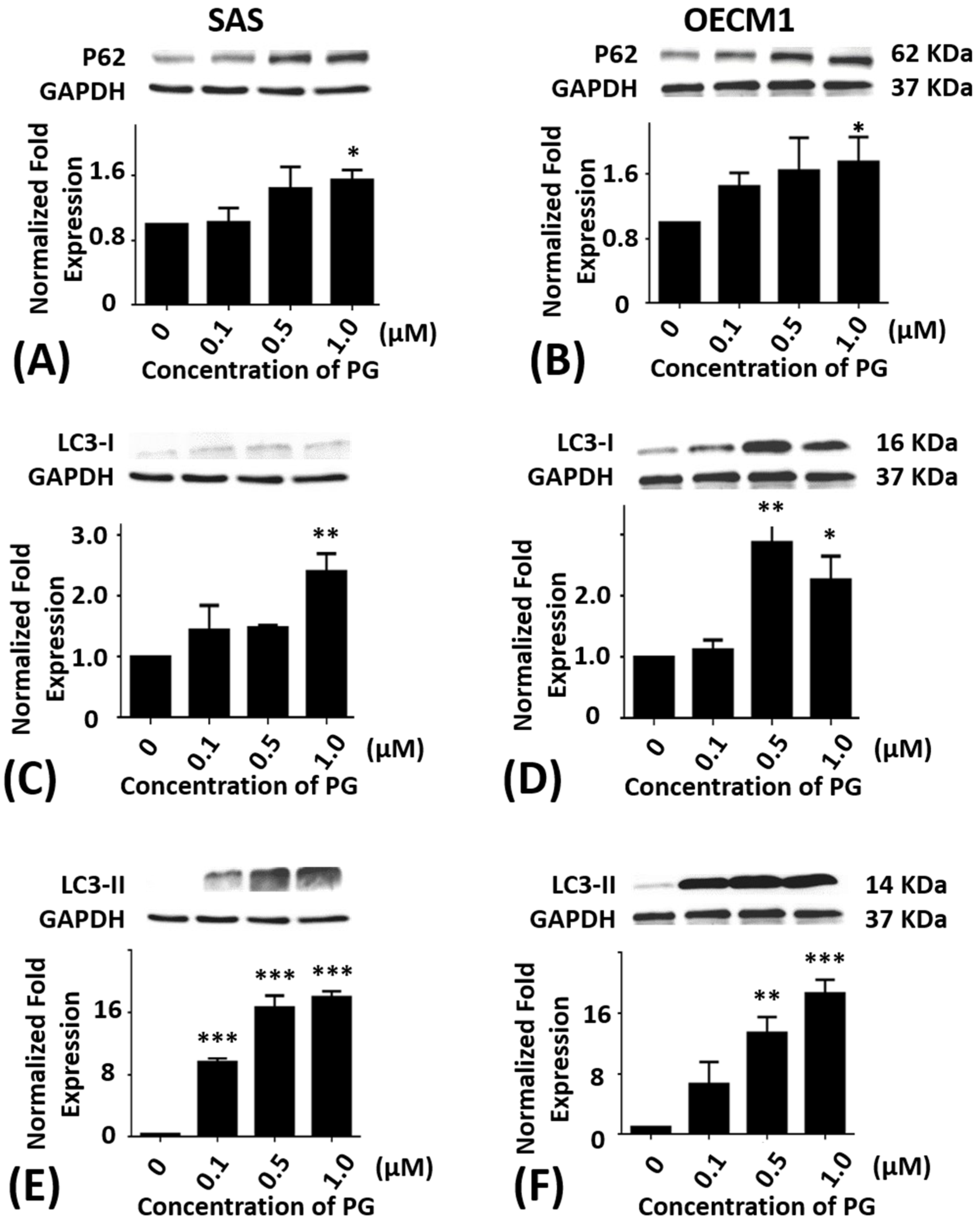
2.5. Effects of Prodigiosin on P62, LC3-I and LC3-II Protein Levels in Oral Cancer Cells
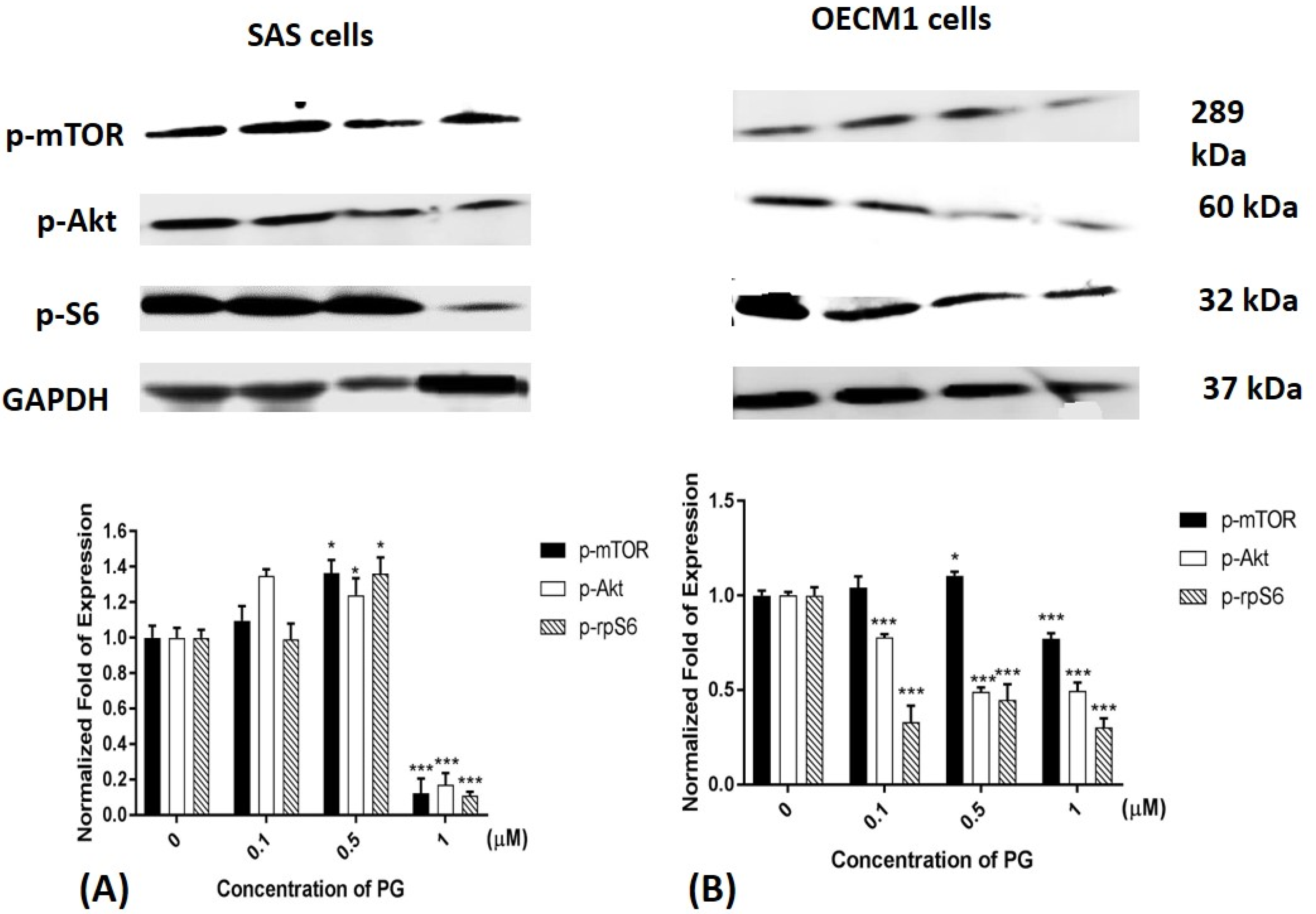
2.6. Phosphorylated Protein Levels of mTOR, Akt, and Ribosomal Protein S6 after PG Treatment
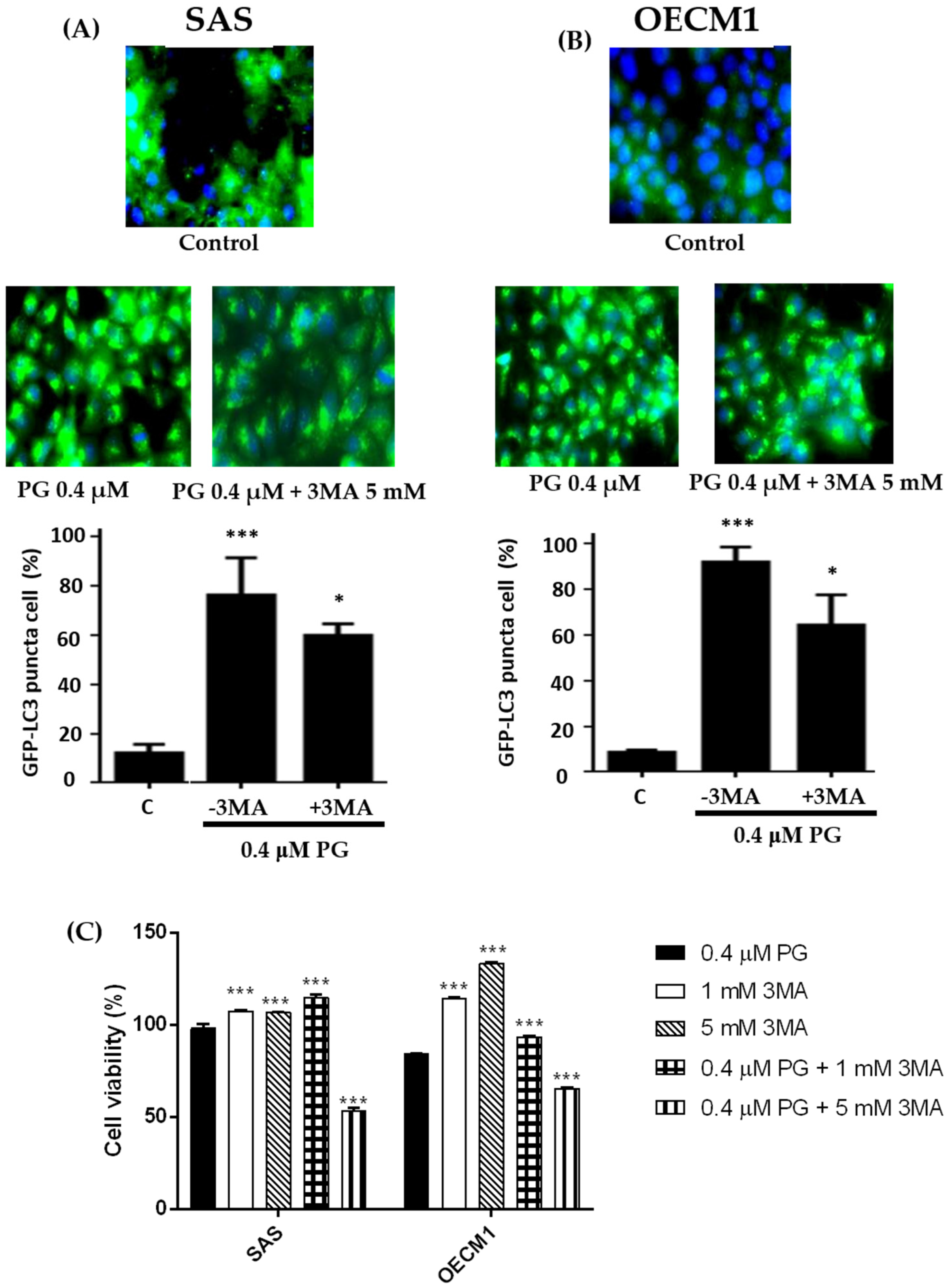
2.7. Effect of Prodigiosin on Autophagosome Formation in Oral Cancer Cells
3. Materials and Methods
3.1. Preparations of Prodigiosin
3.2. Cell Cultures
3.3. Cytotoxicity Assay
3.4. Cell Cycle Analysis
3.5. Western Blotting
3.6. Autophagosome Formation Analysis
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marques, L.A.; Eluf-Neto, J.; Figueiredo, R.A.; Gois-Filho, J.F.; Kowalski, L.P.; Carvalho, M.B.; Abrahao, M.; Wunsch-Filho, V. Oral health, hygiene practices and oral cancer. Rev. Saude Publica 2008, 42, 471–479. [Google Scholar] [CrossRef]
- Sciubba, J.J. Oral cancer. The importance of early diagnosis and treatment. Am. J. Clin. Dermatol. 2001, 2, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.M.; Moore, E.J.; Koch, C.A.; Kasperbauer, J.L.; Olsen, K.D. Oral cavity and oropharynx squamous cell carcinoma with metastasis to the parotid lymph nodes. Oral Oncol. 2011, 47, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Singletary, K.; Milner, J. Diet, autophagy, and cancer: A review. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1596–1610. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Quijano, C.; Chen, E.; Liu, H.; Cao, L.; Fergusson, M.M.; Rovira, I.I.; Gutkind, S.; Daniels, M.P.; Komatsu, M.; et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging 2009, 1, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Cai, J.; Lin, J.Z.; Luo, W.S.; Luo, H.S.; Jiang, Y.Y.; Zhang, Y. Autophagy inhibition promotes gambogic acid-induced suppression of growth and apoptosis in glioblastoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 6211–6216. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of the akt/mtor/p70s6k and erk1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007, 3, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Y.; Kim, H.; Jeong, S.H.; Lim, D.S.; Ryu, M.H. Sulfasalazine induces autophagic cell death in oral cancer cells via akt and erk pathways. Asian Pac. J. Cancer Prev. 2014, 15, 6939–6944. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.A.; Berhow, M.A.; Singletary, K.W. Inhibition of akt signaling and enhanced erk1/2 activity are involved in induction of macroautophagy by triterpenoid b-group soyasaponins in colon cancer cells. Carcinogenesis 2006, 27, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Lv, Y.C.; Zhang, M.; Xie, W.; Tan, Y.L.; Gong, D.; Cheng, H.P.; Liu, D.; Li, L.; Liu, X.Y.; et al. Apelin-13 impedes foam cell formation by activating class iii pi3k/beclin-1-mediated autophagic pathway. Biochem. Biophys. Res. Commun. 2015, 466, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of akt/pkb by the rictor-mtor complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Degtyarev, M.; De Maziere, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; van Dijk, S.; Wu, H.; Gray, D.C.; et al. Akt inhibition promotes autophagy and sensitizes pten-null tumors to lysosomotropic agents. J. Cell Biol. 2008, 183, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Pattanaik, A.K.; Das, J.; Bhutia, S.K.; Behera, B.; Singh, P.; Maiti, T.K. Role of pi3k/akt/mtor and mek/erk pathway in concanavalin a induced autophagy in hela cells. Chem. Biol. Interact. 2014, 210, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Ma, L.; Chen, G.; Huang, Y.; Sun, X.; Jiang, C.; Liu, H. Autophagy inhibitor 3-methyladenine enhances the sensitivity of nasopharyngeal carcinoma cells to chemotherapy and radiotherapy. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2016, 41, 9–18. [Google Scholar] [PubMed]
- Chang, C.C.; Chen, W.C.; Ho, T.F.; Wu, H.S.; Wei, Y.H. Development of natural anti-tumor drugs by microorganisms. J. Biosci. Bioeng. 2011, 111, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Marchal, E.; Smithen, D.A.; Uddin, M.I.; Robertson, A.W.; Jakeman, D.L.; Mollard, V.; Goodman, C.D.; MacDougall, K.S.; McFarland, S.A.; McFadden, G.I.; et al. Synthesis and antimalarial activity of prodigiosenes. Org. Biomol. Chem. 2014, 12, 4132–4142. [Google Scholar] [CrossRef] [PubMed]
- Lapenda, J.C.; Silva, P.A.; Vicalvi, M.C.; Sena, K.X.; Nascimento, S.C. Antimicrobial activity of prodigiosin isolated from serratia marcescens ufpeda 398. World J. Microbiol. Biotechnol. 2015, 31, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, A.; Hosokawa, K.; Soliev, A.B.; Osaka, I.; Arakawa, R.; Enomoto, K. Cytotoxic prodigiosin family pigments from pseudoalteromonas sp. 1020r isolated from the pacific coast of Japan. Biosci. Biotechnol. Biochem. 2012, 76, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Kimyon, O.; Das, T.; Ibugo, A.I.; Kutty, S.K.; Ho, K.K.; Tebben, J.; Kumar, N.; Manefield, M. Serratia secondary metabolite prodigiosin inhibits pseudomonas aeruginosa biofilm development by producing reactive oxygen species that damage biological molecules. Front. Microbiol. 2016, 7, 972–987. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, G.; Li, J.; Huang, H.; Zhang, X.; Zhang, H.; Ju, J. Cytotoxic and antibacterial angucycline- and prodigiosin-analogues from the deep-sea derived streptomyces sp. Scsio 11594. Mar. Drugs 2015, 13, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Kancharla, P.; Lu, W.; Salem, S.M.; Kelly, J.X.; Reynolds, K.A. Stereospecific synthesis of 23-hydroxyundecylprodiginines and analogues and conversion to antimalarial premarineosins via a rieske oxygenase catalyzed bicyclization. J. Org. Chem. 2014, 79, 11674–11689. [Google Scholar] [CrossRef] [PubMed]
- Perez-Tomas, R.; Vinas, M. New insights on the antitumoral properties of prodiginines. Curr. Med. Chem. 2010, 17, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Sam, S.; Sam, M.R.; Esmaeillou, M.; Safaralizadeh, R. Effective targeting survivin, caspase-3 and microrna-16–1 expression by methyl-3-pentyl-6-methoxyprodigiosene triggers apoptosis in colorectal cancer stem-like cells. Pathol. Oncol. Res. 2016, 22, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.J.; Ou, J.H.; Wang, M.L.; Jialielihan, N.; Liu, Y.H. Elevated survivin mediated multidrug resistance and reduced apoptosis in breast cancer stem cells. J. BUON 2015, 20, 1287–1294. [Google Scholar] [PubMed]
- Papireddy, K.; Smilkstein, M.; Kelly, J.X.; Shweta; Salem, S.M.; Alhamadsheh, M.; Haynes, S.W.; Challis, G.L.; Reynolds, K.A. Antimalarial activity of natural and synthetic prodiginines. J. Med. Chem. 2011, 54, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Wang, Y.H.; Chern, C.M.; Liou, K.T.; Hou, Y.C.; Peng, Y.T.; Shen, Y.C. Prodigiosin inhibits gp91(phox) and inos expression to protect mice against the oxidative/nitrosative brain injury induced by hypoxia-ischemia. Toxicol. Appl. Pharmacol. 2011, 257, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Dalili, D.; Fouladdel, S.; Rastkari, N.; Samadi, N.; Ahmadkhaniha, R.; Ardavan, A.; Azizi, E. Prodigiosin, the red pigment of serratia marcescens, shows cytotoxic effects and apoptosis induction in ht-29 and t47d cancer cell lines. Nat. Prod. Res. 2012, 26, 2078–2083. [Google Scholar] [PubMed]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Krishna, P.S.; Vani, K.; Prasad, M.R.; Samatha, B.; Bindu, N.S.; Charya, M.A.; Reddy Shetty, P. In silico molecular docking analysis of prodigiosin and cycloprodigiosin as cox-2 inhibitors. Springerplus 2013, 2, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Espona-Fiedler, M.; Soto-Cerrato, V.; Quesada, R.; Perez-Tomas, R.; Guallar, V. Molecular interactions of prodiginines with the bh3 domain of anti-apoptotic bcl-2 family members. PLoS ONE 2013, 8, e57562. [Google Scholar] [CrossRef] [PubMed]
- Elahian, F.; Moghimi, B.; Dinmohammadi, F.; Ghamghami, M.; Hamidi, M.; Mirzaei, S.A. The anticancer agent prodigiosin is not a multidrug resistance protein substrate. DNA Cell Biol. 2013, 32, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.Y.; Shen, Y.C.; Lu, C.H.; Yang, S.Y.; Ho, T.F.; Peng, Y.T.; Chang, C.C. Prodigiosin activates endoplasmic reticulum stress cell death pathway in human breast carcinoma cell lines. Toxicol. Appl. Pharmacol. 2012, 265, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Huang, C.Y.; Li, K.T.; Li, R.N.; Liaw, C.C.; Wu, S.H.; Liu, J.R.; Sheu, J.H.; Chang, H.W. Sinuleptolide inhibits proliferation of oral cancer ca9–22 cells involving apoptosis, oxidative stress, and DNA damage. Arch. Oral Biol. 2016, 66, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Sun, C.; Huang, S.; Zhou, Q. Carvacrol suppresses proliferation and invasion in human oral squamous cell carcinoma. Onco Targets Ther. 2016, 9, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Lee, J.H.; Park, H.R.; Choi, Y.W. Acetylshikonin inhibits growth of oral squamous cell carcinoma by inducing apoptosis. Arch. Oral Biol. 2016, 70, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Peng, C.Y.; Liao, Y.W.; Lu, M.Y.; Tsai, M.L.; Yeh, J.C.; Yu, C.H.; Yu, C.C. Sulforaphane targets cancer stemness and tumor initiating properties in oral squamous cell carcinomas via mir-200c induction. J. Formos. Med. Assoc. 2017, 116, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Su, N.W.; Wu, S.H.; Chi, C.W.; Liu, C.J.; Tsai, T.H.; Chen, Y.J. Metronomic cordycepin therapy prolongs survival of oral cancer-bearing mice and inhibits epithelial-mesenchymal transition. Molecules 2017, 22, 629. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.H.; Kim, I.R.; Kim, H.J.; Park, B.S.; Yu, S.B. Alpha-mangostin induces apoptosis and cell cycle arrest in oral squamous cell carcinoma cell. Evid. Based Complement. Altern. Med. 2016, 2016, e5352412. [Google Scholar] [CrossRef] [PubMed]
- Tjioe, K.C.; Tostes Oliveira, D.; Gavard, J. Luteolin impacts on the DNA damage pathway in oral squamous cell carcinoma. Nutr. Cancer 2016, 68, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Utaipan, T.; Athipornchai, A.; Suksamrarn, A.; Jirachotikoon, C.; Yuan, X.; Lertcanawanichakul, M.; Chunglok, W. Carbazole alkaloids from murraya koenigii trigger apoptosis and autophagic flux inhibition in human oral squamous cell carcinoma cells. J. Nat. Med. 2017, 71, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Espona-Fiedler, M.; Soto-Cerrato, V.; Hosseini, A.; Lizcano, J.M.; Guallar, V.; Quesada, R.; Gao, T.; Perez-Tomas, R. Identification of dual mtorc1 and mtorc2 inhibitors in melanoma cells: Prodigiosin vs. Obatoclax. Biochem. Pharmacol. 2012, 83, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Smithen, D.A.; Forrester, A.M.; Corkery, D.P.; Dellaire, G.; Colpitts, J.; McFarland, S.A.; Berman, J.N.; Thompson, A. Investigations regarding the utility of prodigiosenes to treat leukemia. Org. Biomol. Chem. 2013, 11, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. Ampk and mtor regulate autophagy through direct phosphorylation of ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The ampk signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The beclin 1-vps34 complex—At the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, C.G.; Yim, J.H.; Lee, H.K.; Pyo, S. Ramalin-mediated apoptosis is enhanced by autophagy inhibition in human breast cancer cells. Phytother. Res. 2016, 30, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Vegliante, R.; Desideri, E.; Di Leo, L.; Ciriolo, M.R. Dehydroepiandrosterone triggers autophagic cell death in human hepatoma cell line hepg2 via jnk-mediated p62/sqstm1 expression. Carcinogenesis 2016, 37, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.C.; Xin, Z.Y.; Deborah, B.; Zhang, J.S.; Yuan, D.Y.; Xu, K.; Liu, X.B.; Jiang, H.Q.; Fan, Q.C.; Zhang, B.; et al. Inhibition of autophagy augments apoptosis in human oral squamous cell carcinoma under nutrient depletion. J. Oral Pathol. Med. 2015, 44, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.P.; Lima, C.F.; Pedro, D.F.; Wilson, J.M.; Kristiansen, K.; Pereira-Wilson, C. Ursolic acid induces cell death and modulates autophagy through jnk pathway in apoptosis-resistant colorectal cancer cells. J. Nutr. Biochem. 2013, 24, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Auberger, P. Ampk- and p62/sqstm1-dependent autophagy mediate resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy 2010, 6, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via jnk-mediated p62/sqstm1 expression and ampk activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Utaipan, T.; Athipornchai, A.; Suksamrarn, A.; Chunsrivirot, S.; Chunglok, W. Isomahanine induces endoplasmic reticulum stress and simultaneously triggers p38 mapk-mediated apoptosis and autophagy in multidrug-resistant human oral squamous cell carcinoma cells. Oncol. Rep. 2017, 37, 1243–1252. [Google Scholar] [PubMed]
- Shrotriya, S.; Tyagi, A.; Deep, G.; Orlicky, D.J.; Wisell, J.; Wang, X.J.; Sclafani, R.A.; Agarwal, R.; Agarwal, C. Grape seed extract and resveratrol prevent 4-nitroquinoline 1-oxide induced oral tumorigenesis in mice by modulating ampk activation and associated biological responses. Mol. Carcinog. 2015, 54, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015, 16, e138. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Ishii, K.A.; Saito, Y.; Miura, M.; Otagiri, A.; Kawakami, Y.; Shimano, H.; Hara, H.; Takekoshi, K. Inhibition of autophagy enhances sunitinib-induced cytotoxicity in rat pheochromocytoma pc12 cells. J. Pharmacol. Sci. 2013, 121, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Inhibition of pi3k/akt/mtor signaling by natural products. Anticancer Agents Med. Chem. 2013, 13, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mtor through atp competition. Sci. Rep. 2016, 6, e21772. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.N.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mtor signaling, activates amp-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.-N.; Codogno, P.; Shen, H.-M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class i and iii phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Karp, C.; Strohecker, A.M.; Guo, Y.; Mathew, R. Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 2010, 22, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Hassankhani, R.; Sam, M.R.; Esmaeilou, M.; Ahangar, P. Prodigiosin isolated from cell wall of serratia marcescens alters expression of apoptosis-related genes and increases apoptosis in colorectal cancer cells. Med. Oncol. 2015, 32, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Cosway, B.; Lovat, P. The role of autophagy in squamous cell carcinoma of the head and neck. Oral Oncol. 2016, 54, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Chen, F.F.; Lung, J.; Lo, C.H.; Lee, F.H.; Lu, Y.C.; Hung, C.H. Prognostic significance of p62/sqstm1 subcellular localization and lc3b in oral squamous cell carcinoma. Br. J. Cancer 2014, 111, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shen, Y.; Liu, J.; Wei, D. Antimetastatic effect of prodigiosin through inhibition of tumor invasion. Biochem. Pharmacol. 2005, 69, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Hong, B.; Allen, J.E.; Zhang, S.; Lulla, A.R.; Dicker, D.T.; El-Deiry, W.S. Small-molecule prodigiosin restores p53 tumor suppressor activity in chemoresistant colorectal cancer stem cells via c-jun-mediated δnp73 inhibition and p73 activation. Cancer Res. 2016, 76, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, B.; Zhou, L.; Yu, S.; Su, Z.; Song, J.; Sun, Q.; Sha, O.; Wang, X.; Jiang, W.; et al. Prodigiosin inhibits wnt/β-catenin signaling and exerts anticancer activity in breast cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 13150–13155. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell | Dosage (μM) | Sub G1 (%) | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|---|---|
| 12 h | 0 | 2.2 ± 0.8 | 40.3 ± 3.3 | 25.2 ± 4.4 | 32.4 ± 2.9 |
| 0.1 | 1.7 ± 0.8 | 45.3 ± 4.1 * | 23.2 ± 0.2 | 29.9 ± 0.9 | |
| 0.5 | 1.0 ± 0.2 | 51.2 ± 1.9 * | 21.1 ± 2.8 | 26.6 ± 0.7 * | |
| 1.0 | 1.4 ± 0.7 | 51.4 ± 1.2 * | 20.0 ± 2.7 | 27.2 ± 0.2 * | |
| 24 h | 0 | 0.9 ± 0.3 | 42.1 ± 2.7 | 20.4 ± 2.7 | 36.6 ± 2.1 |
| 0.1 | 1.4 ± 0.2 | 39.7 ± 2.2 | 25.0 ± 2.0 | 33.9 ± 3.6 | |
| 0.5 | 1.7 ± 0.3 * | 51.7 ± 3.2 * | 20.9 ± 0.8 | 25.7 ± 2.5 * | |
| 1.0 | 2.5 ± 0.7 * | 54.0 ± 3.7 * | 17.0 ± 0.7 | 26.3 ± 3.2 * |
| Cell | Dosage (μM) | Sub G1 (%) | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|---|---|
| 12 h | 0 | 0.5 ± 0.1 | 50.9 ± 1.7 | 16.6 ± 1.0 | 32.1 ± 0.4 |
| 0.1 | 0.5 ± 0.2 | 52.3 ± 0.5 | 17.0 ± 0.5 | 30.2 ± 2.2 | |
| 0.5 | 0.4 ± 0.1 | 63.2 ± 0.6 ** | 12.2 ± 0.2 * | 24.1 ± 1.3 * | |
| 1.0 | 0.4 ± 0.1 | 63.3 ± 0.4 ** | 10.5 ± 0.2 * | 25.7 ± 0.8 * | |
| 24 h | 0 | 1.2 ± 0.2 | 47.9 ± 2.3 | 14.0 ± 1.6 | 36.9 ± 3.1 |
| 0.1 | 1.1 ± 0.1 | 50.9 ± 3.8 | 21.9 ± 2.9 * | 26.1 ± 1.6 * | |
| 0.5 | 1.4 ± 0.1 | 60.2 ± 2.5 * | 19.8 ± 3.1 * | 18.7 ± 2.3 ** | |
| 1.0 | 1.2 ± 0.1 | 61.8 ± 0.4 * | 18.4 ± 2.6 * | 18.7 ± 3.3 ** |
| Antibody | MW(kDa) | Dilution | Sources |
|---|---|---|---|
| mTOR | 289 | 1:1000 | Cell Signalling |
| p-mTOR (Ser2448) | 289 | 1:200 | Santa Cruz |
| PI3K class III | 100 | 1:1000 | Cell Signalling |
| AMPKα | 62 | 1:1000 | Cell Signalling |
| P62 | 62 | 1:1000 | Cell Signalling |
| Akt | 60 | 1:1000 | Cell Signalling |
| P-Akt (Ser473) | 60 | 1:200 | Santa Cruz |
| Beclin-1 | 60 | 1:1000 | Cell Signalling |
| Cyclin D1 | 34 | 1:1000 | Cell Signalling |
| p-Ribosomal protein S6 (Ser235/236) | 32 | 1:200 | Santa Cruz |
| LC3-I | 16 | 1:1000 | Cell Signalling |
| LC3-II | 14 | 1:1000 | Cell Signalling |
| GADPH | 37 | 1:10,000 | Cell Signalling |
| anti-Rabbit (IgG) | - | 1:5000 | GeneTex |
| anti-Mouse (IgG) | - | 1:10,000 | GE |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.-F.; Lin, C.-S.; Chen, Y.-H.; Sung, P.-J.; Lin, S.-R.; Tong, Y.-W.; Weng, C.-F. Inhibitory Growth of Oral Squamous Cell Carcinoma Cancer via Bacterial Prodigiosin. Mar. Drugs 2017, 15, 224. https://doi.org/10.3390/md15070224
Cheng M-F, Lin C-S, Chen Y-H, Sung P-J, Lin S-R, Tong Y-W, Weng C-F. Inhibitory Growth of Oral Squamous Cell Carcinoma Cancer via Bacterial Prodigiosin. Marine Drugs. 2017; 15(7):224. https://doi.org/10.3390/md15070224
Chicago/Turabian StyleCheng, Ming-Fang, Chun-Shu Lin, Yu-Hsin Chen, Ping-Jyun Sung, Shian-Ren Lin, Yi-Wen Tong, and Ching-Feng Weng. 2017. "Inhibitory Growth of Oral Squamous Cell Carcinoma Cancer via Bacterial Prodigiosin" Marine Drugs 15, no. 7: 224. https://doi.org/10.3390/md15070224
APA StyleCheng, M.-F., Lin, C.-S., Chen, Y.-H., Sung, P.-J., Lin, S.-R., Tong, Y.-W., & Weng, C.-F. (2017). Inhibitory Growth of Oral Squamous Cell Carcinoma Cancer via Bacterial Prodigiosin. Marine Drugs, 15(7), 224. https://doi.org/10.3390/md15070224