
Identification of Tight-Binding Plasmepsin II and Falcipain 2 Inhibitors in Aqueous Extracts of Marine Invertebrates by the Combination of Enzymatic and Interaction-Based Assays

 , ,
, , 
Abstract
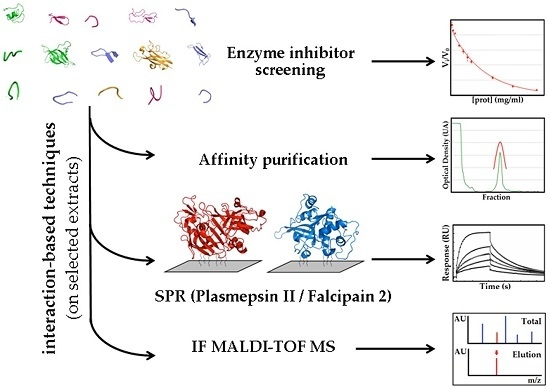
:
1. Introduction
2. Results
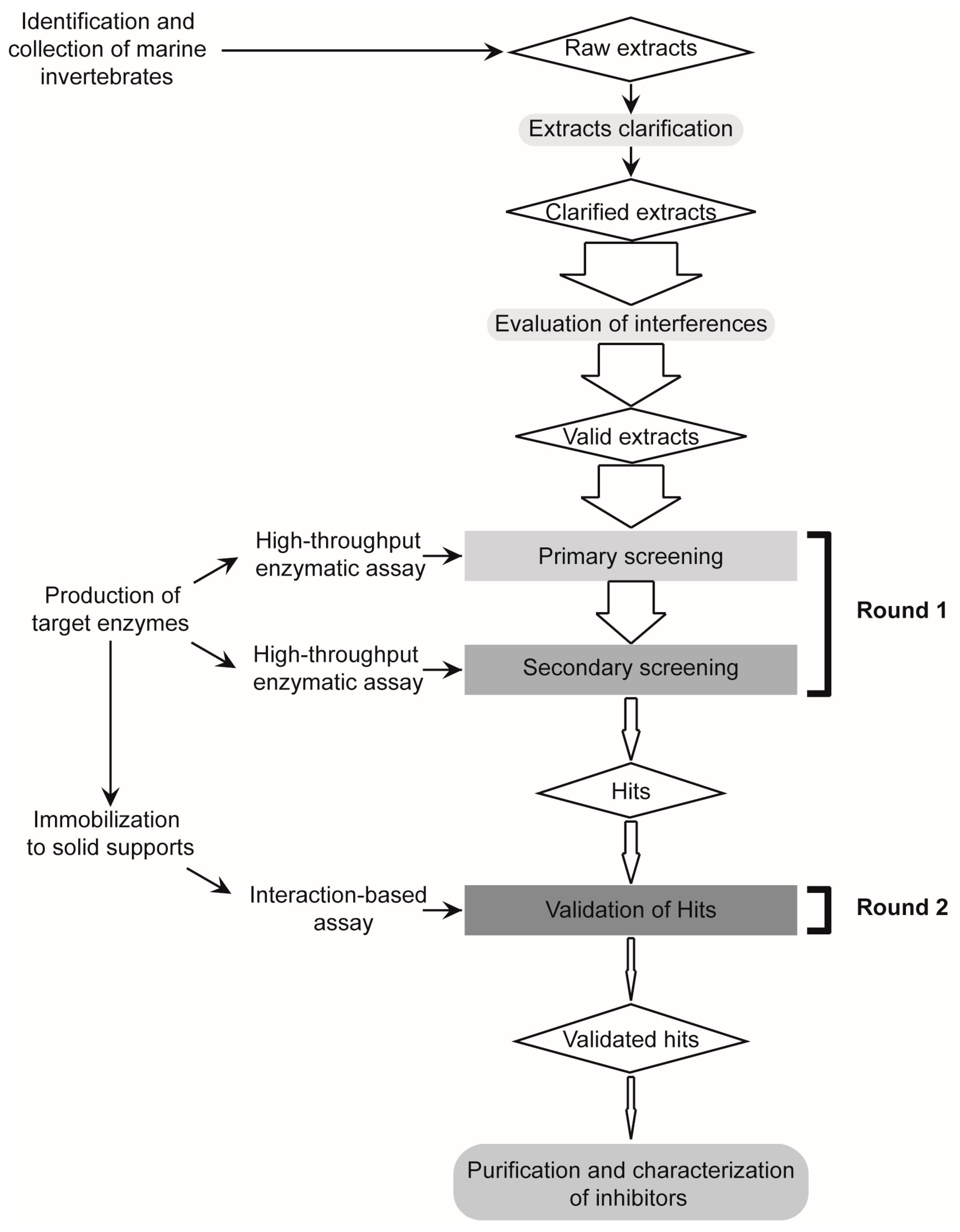
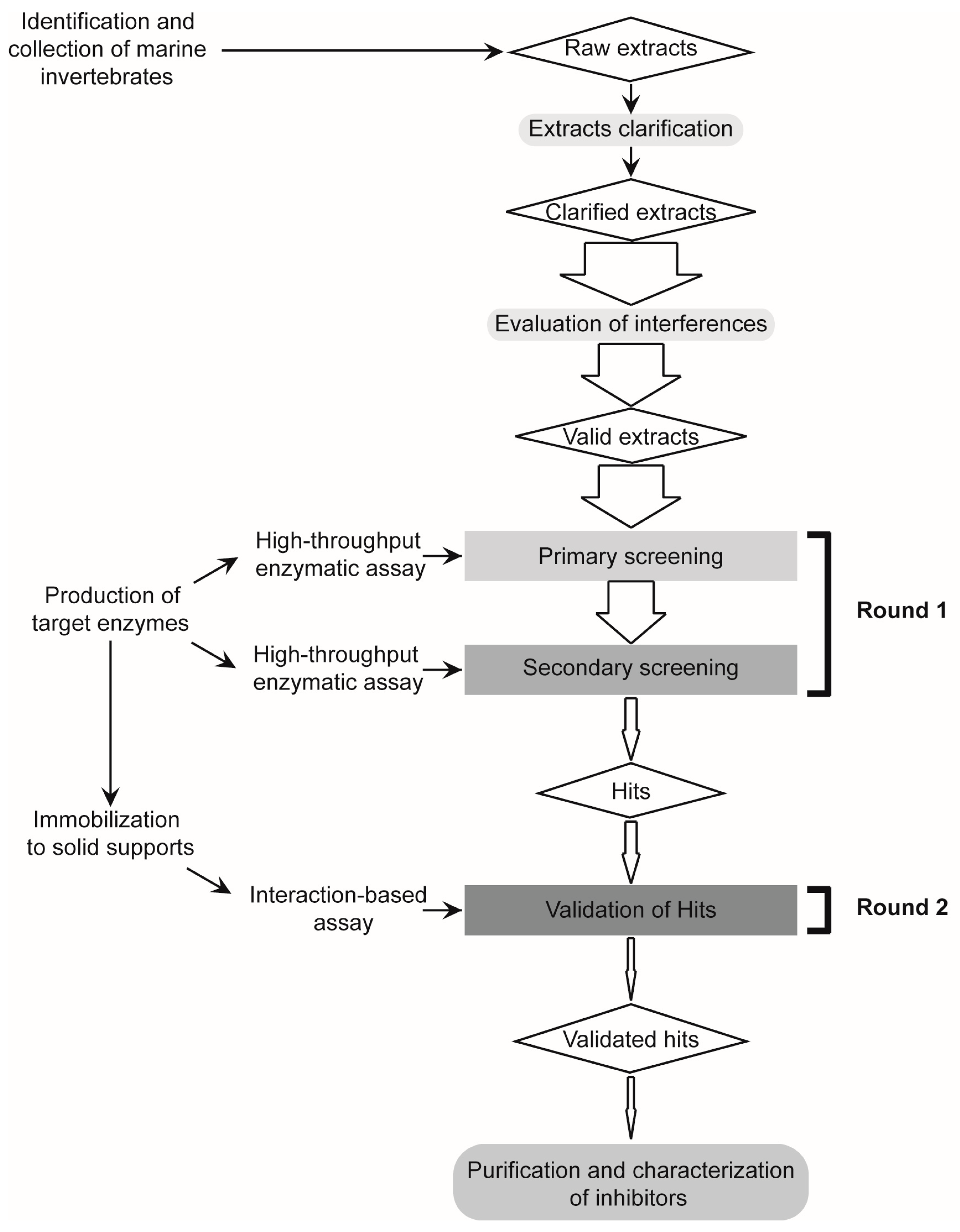
2.1. Design and Validation of a Two-Round Screening Strategy Using Five Reference Extracts of Marine Organisms
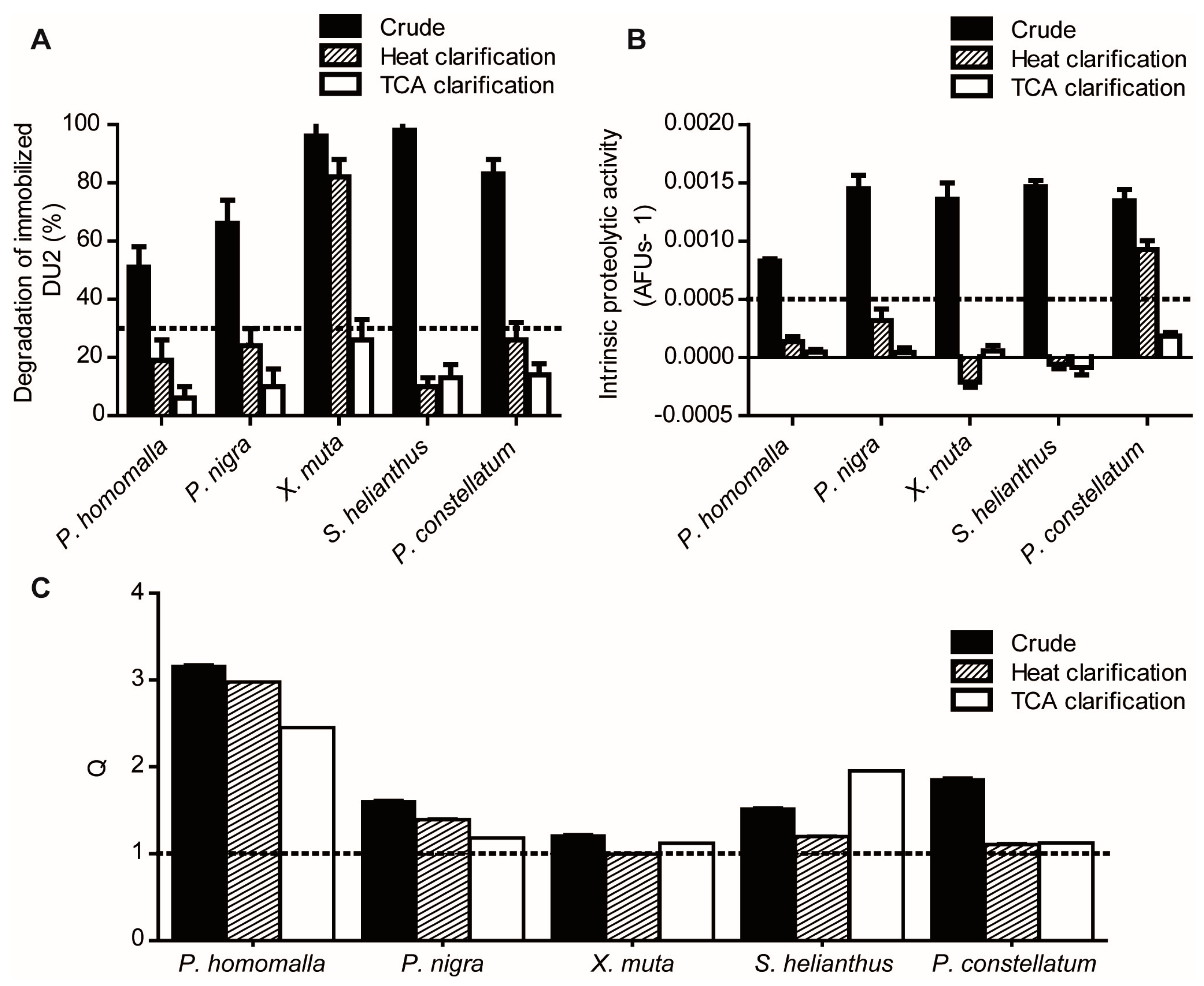
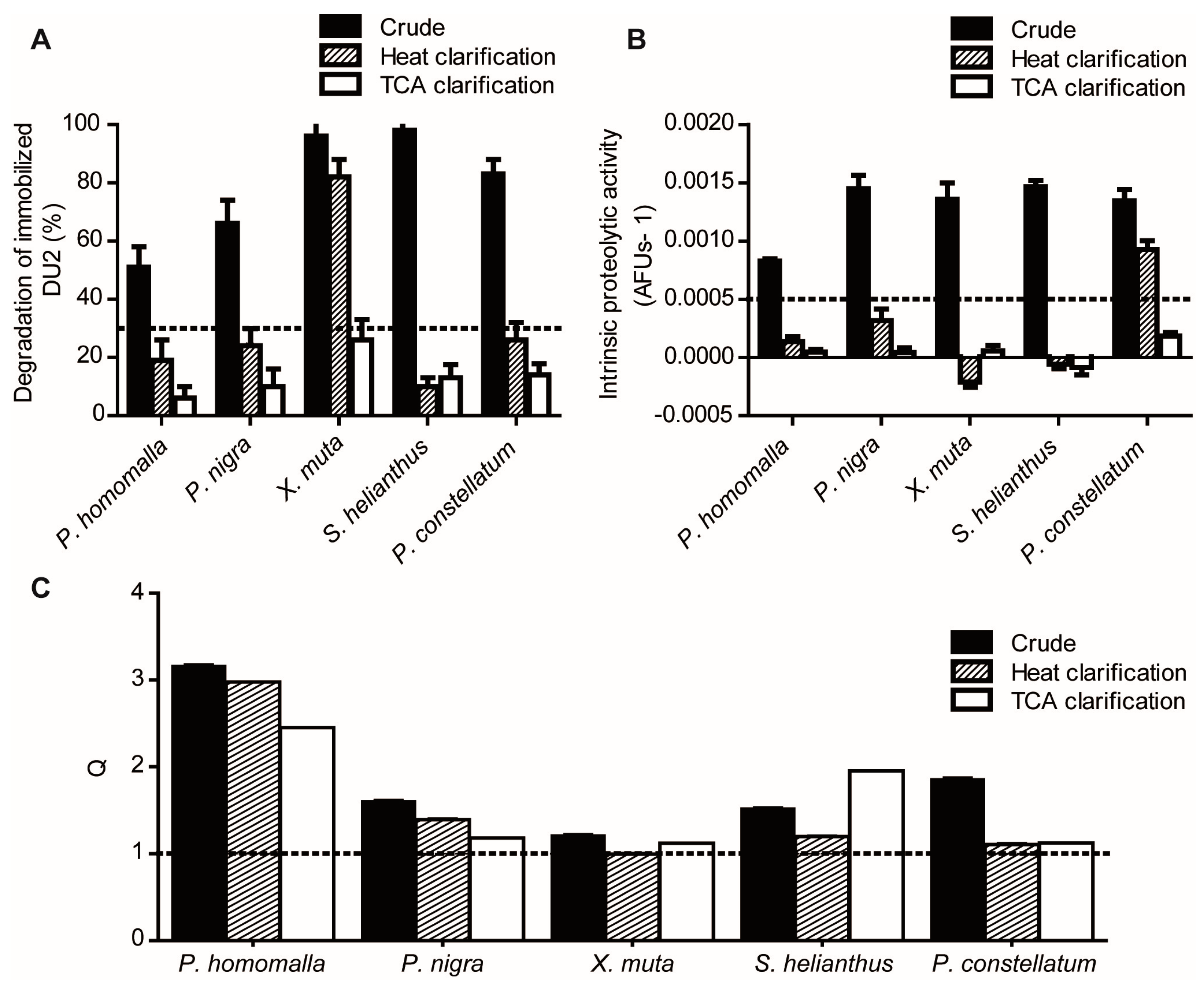
2.1.1. Reduction of Interference Caused by Raw Extracts on Enzymatic Assays
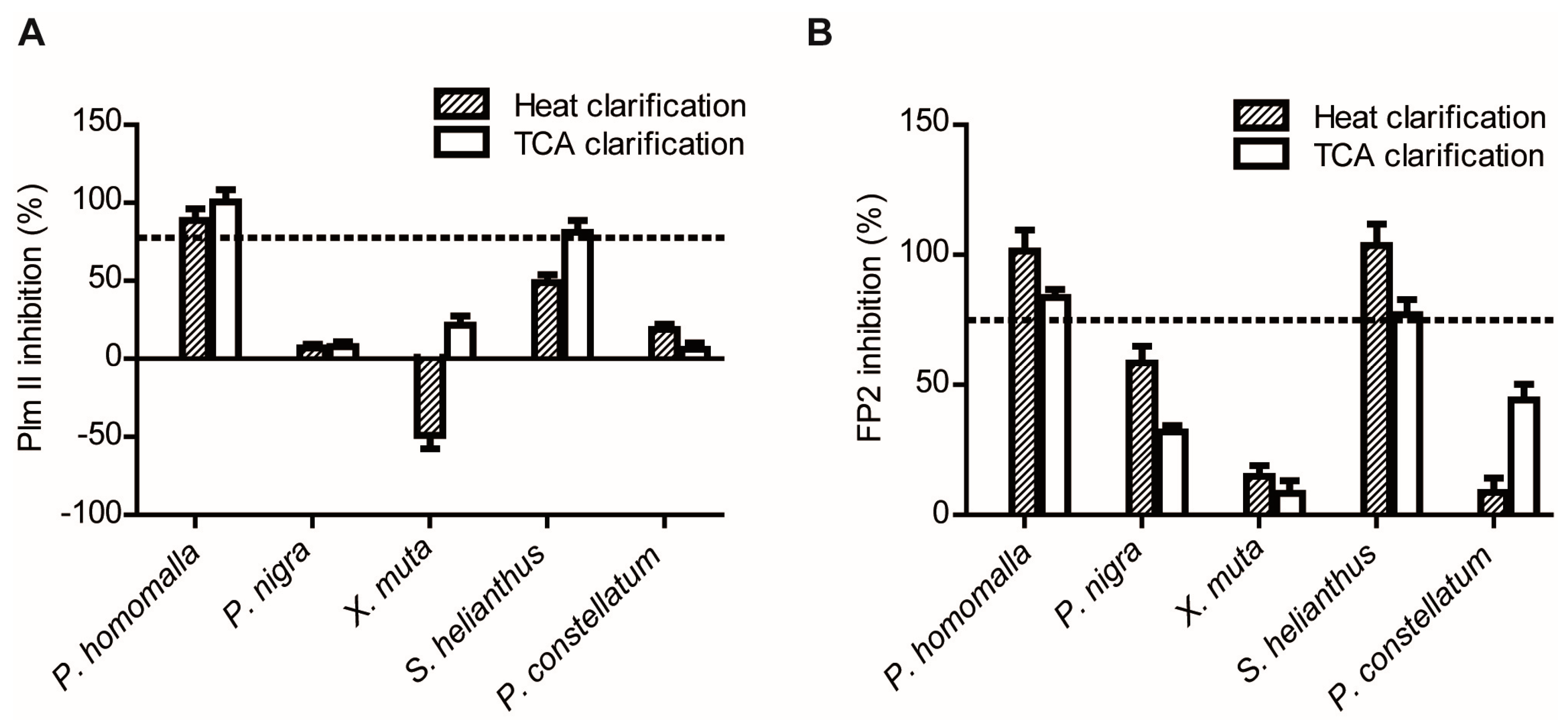
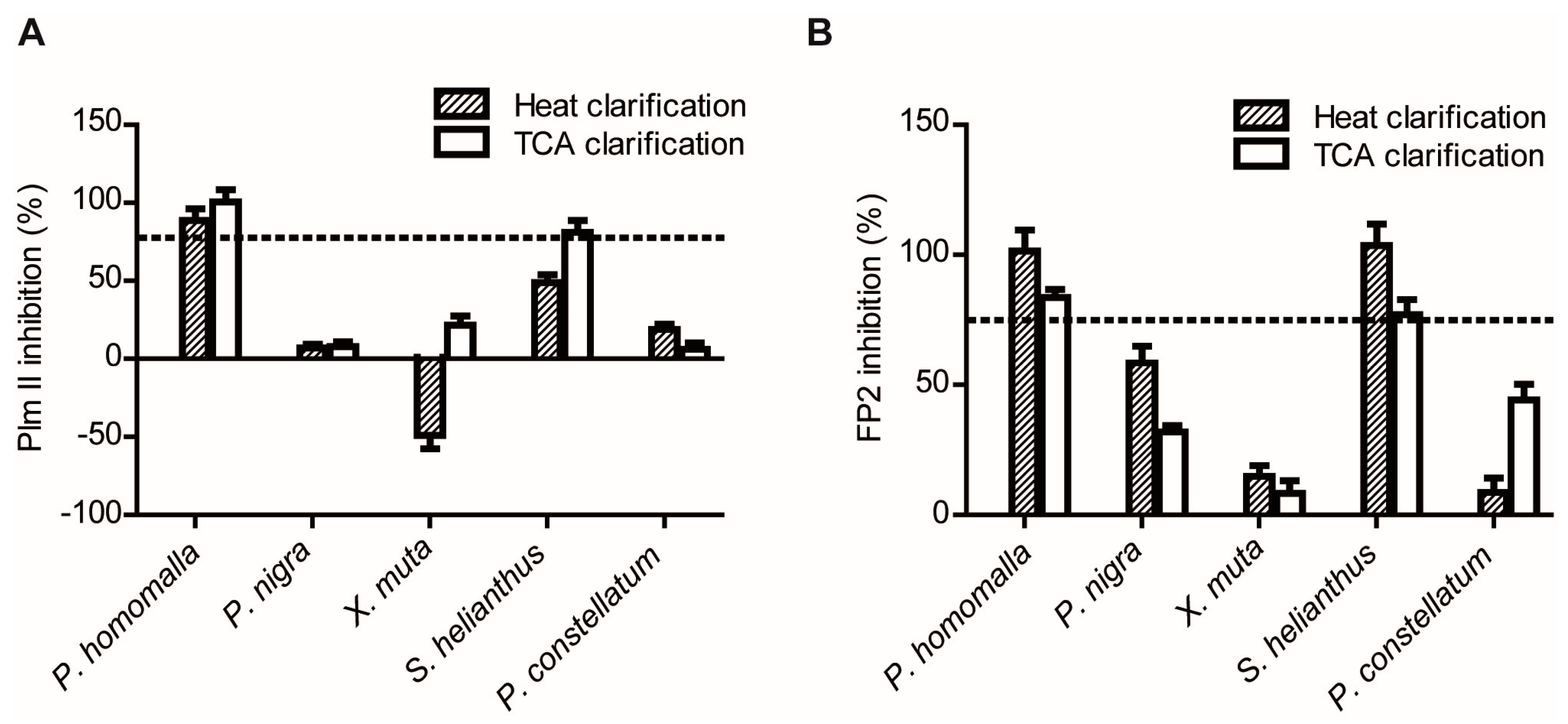
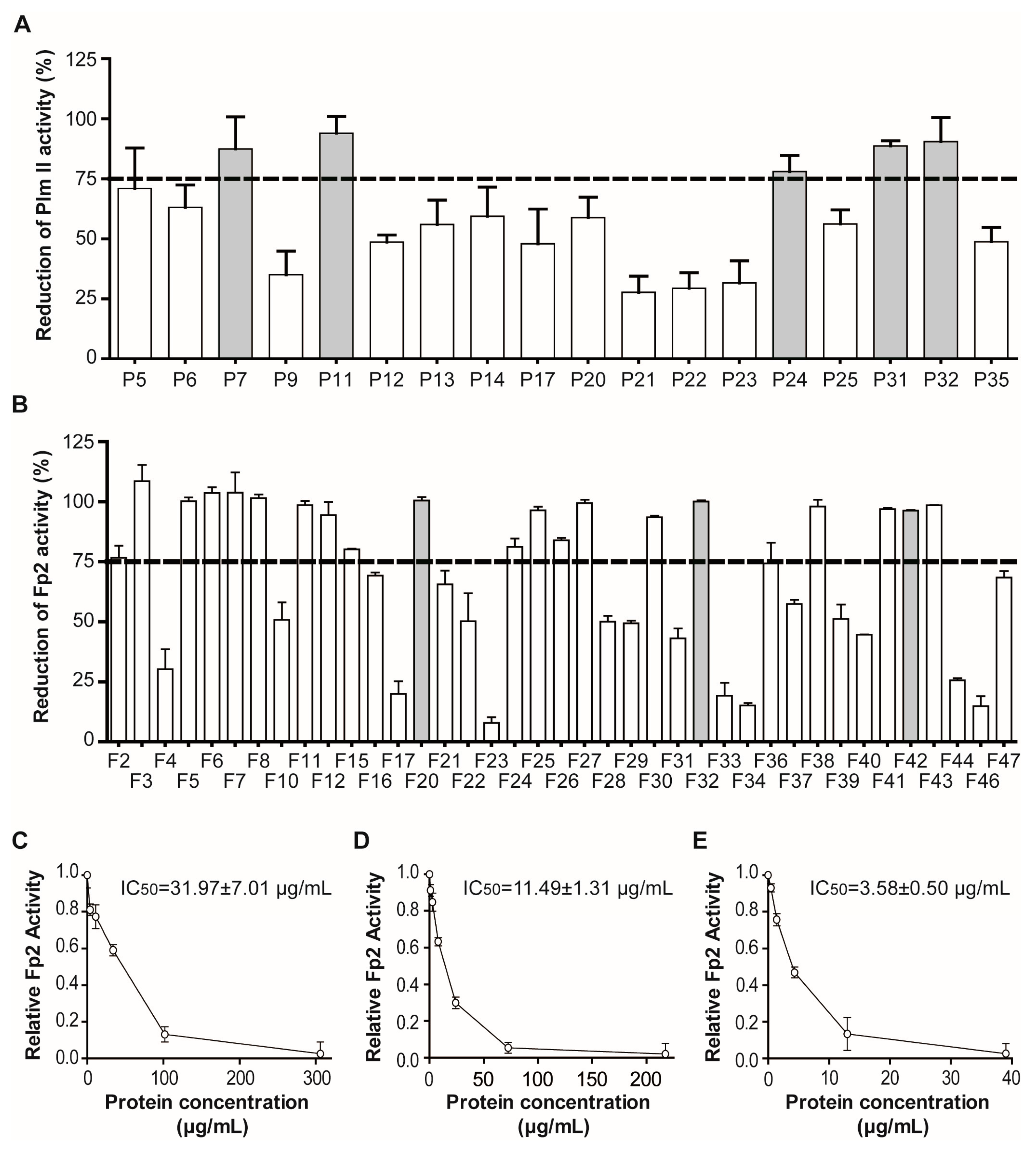
2.1.2. Screening for Tight-Binding Inhibitors Using Specific Activity Assays and Selection of Positive Hit Threshold
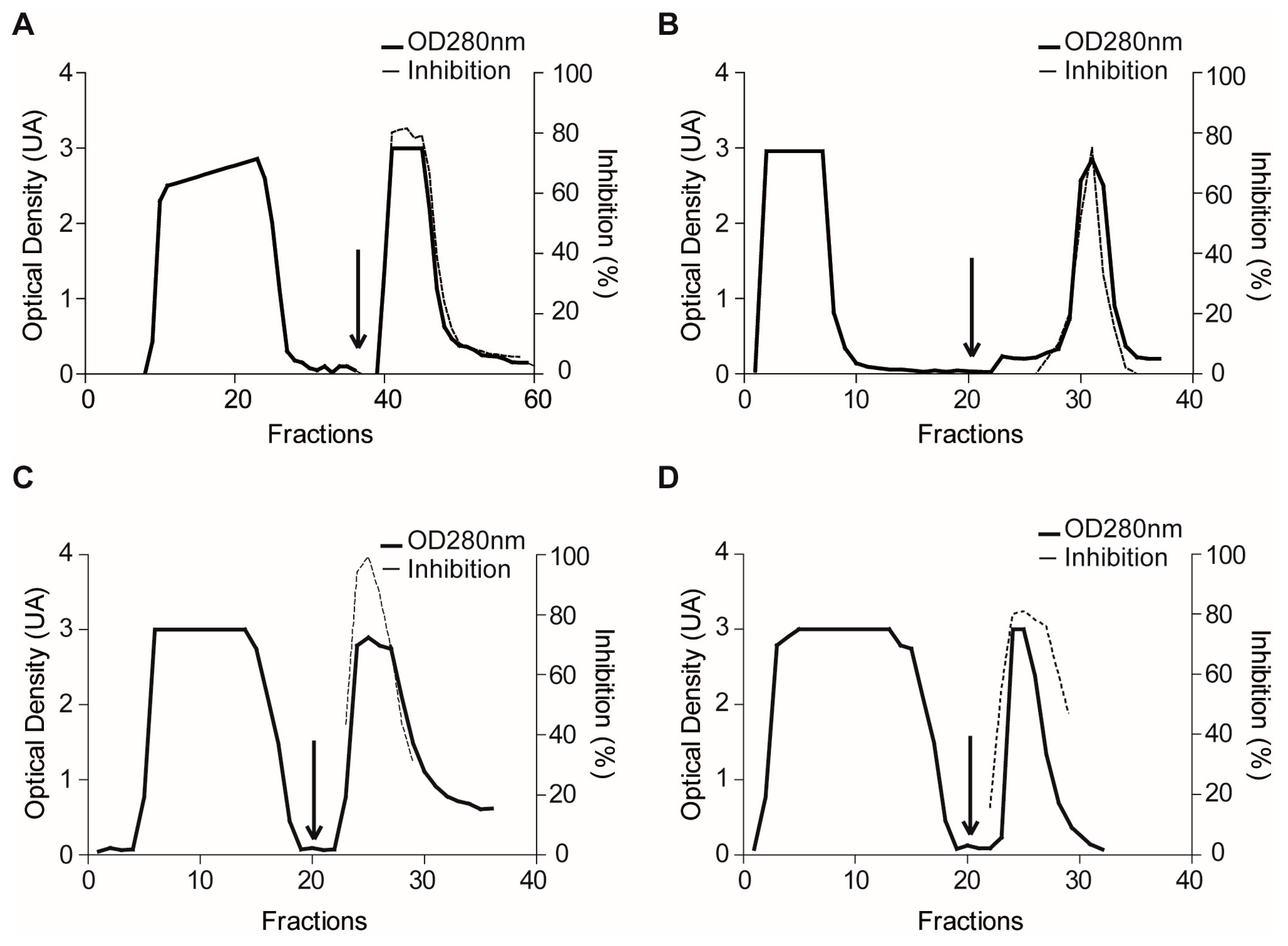
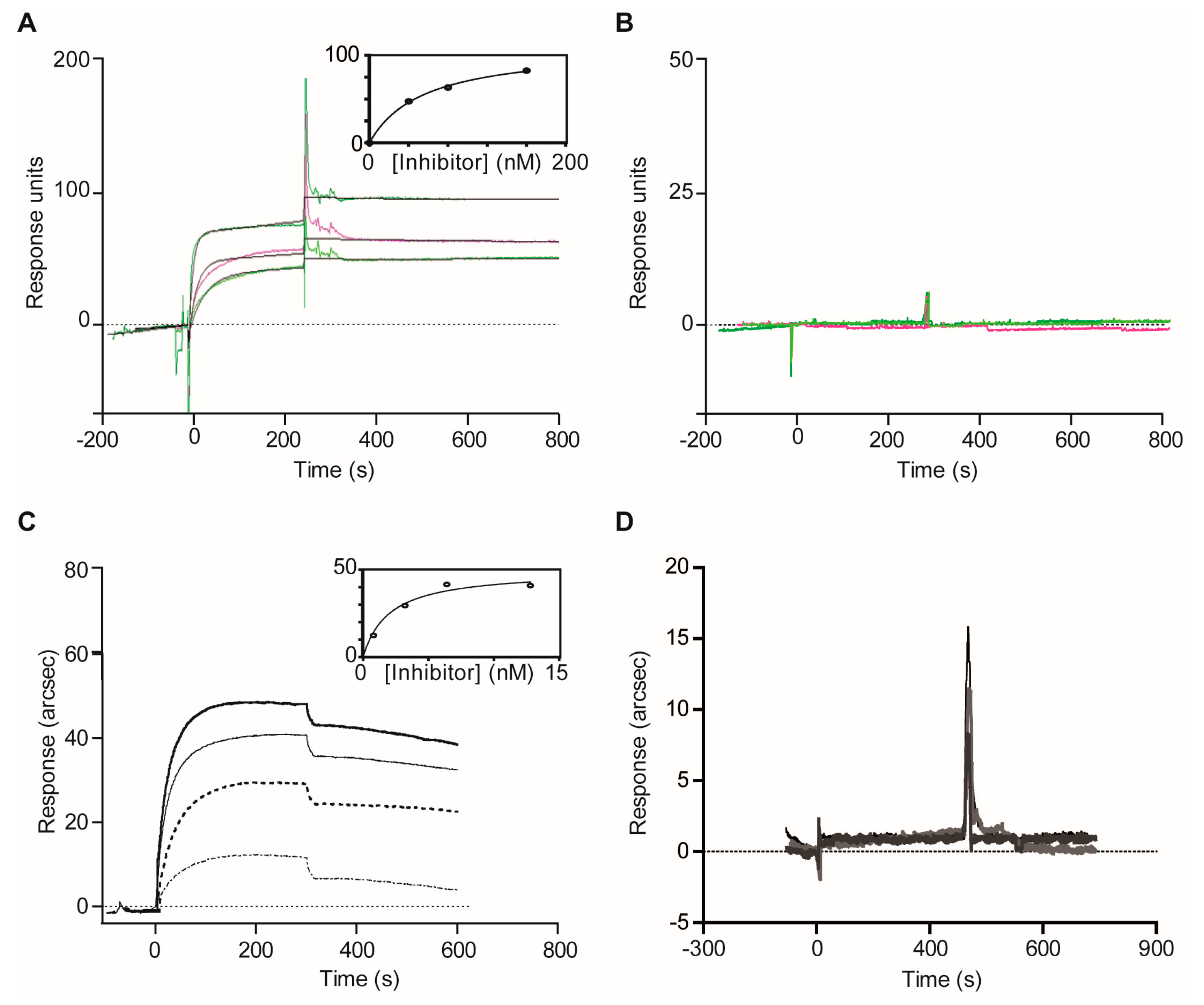
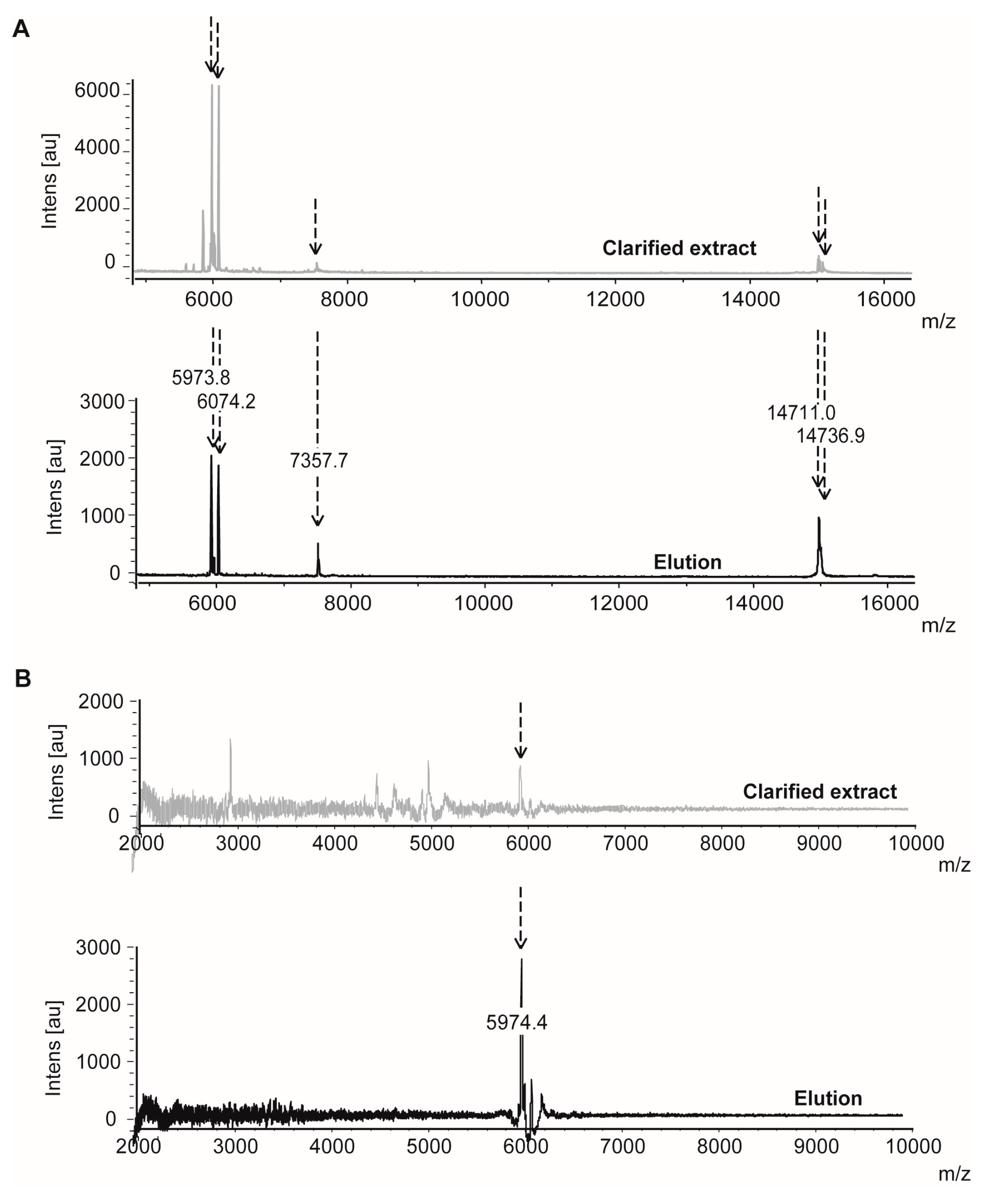
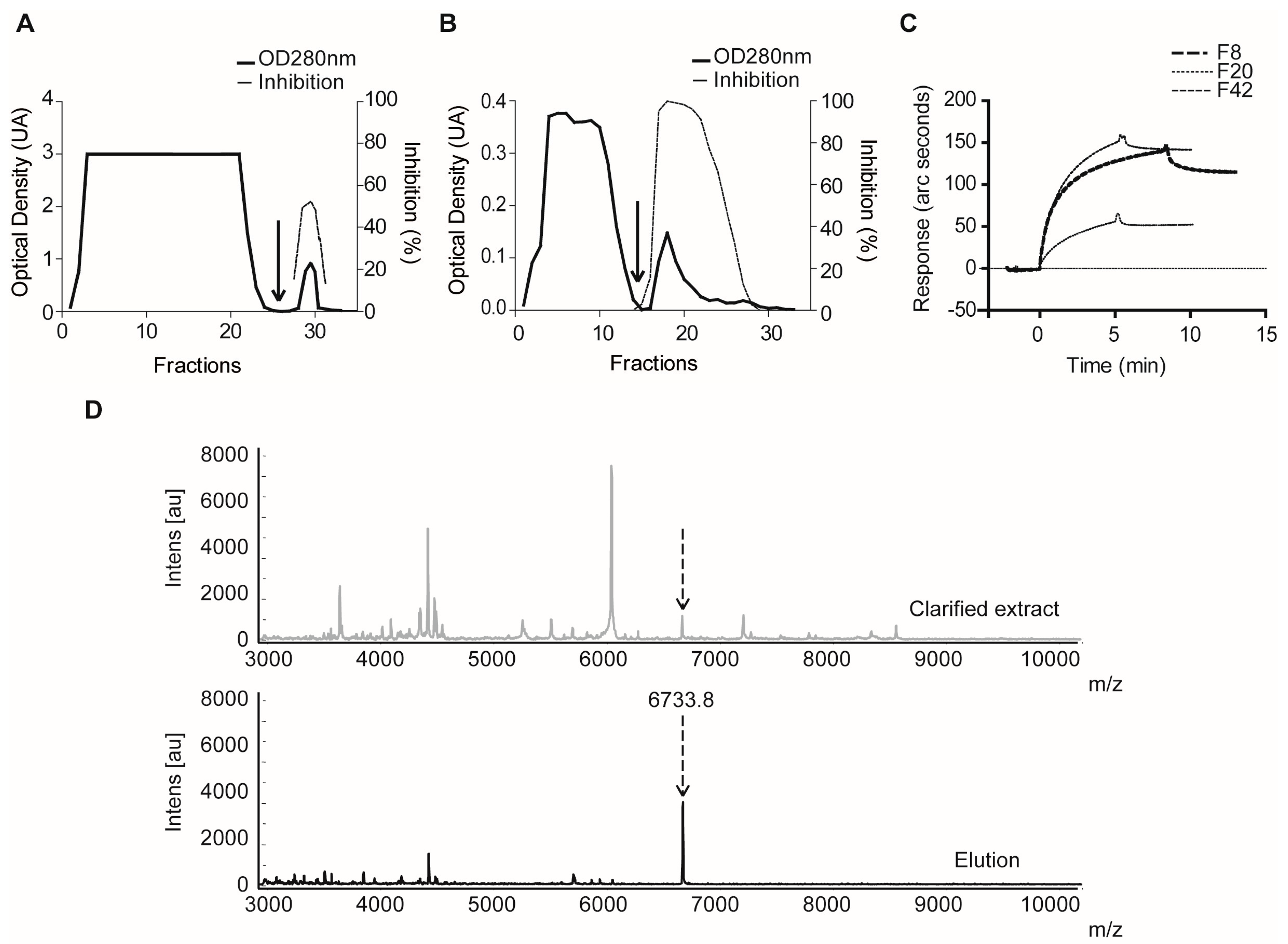
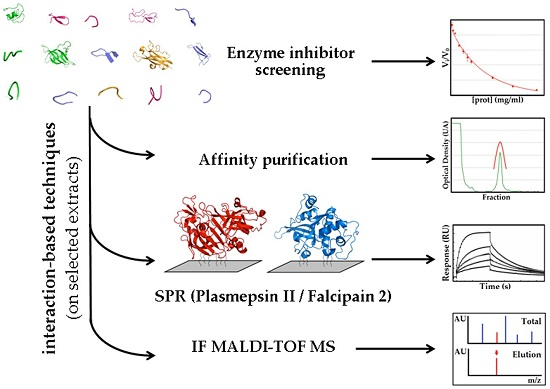
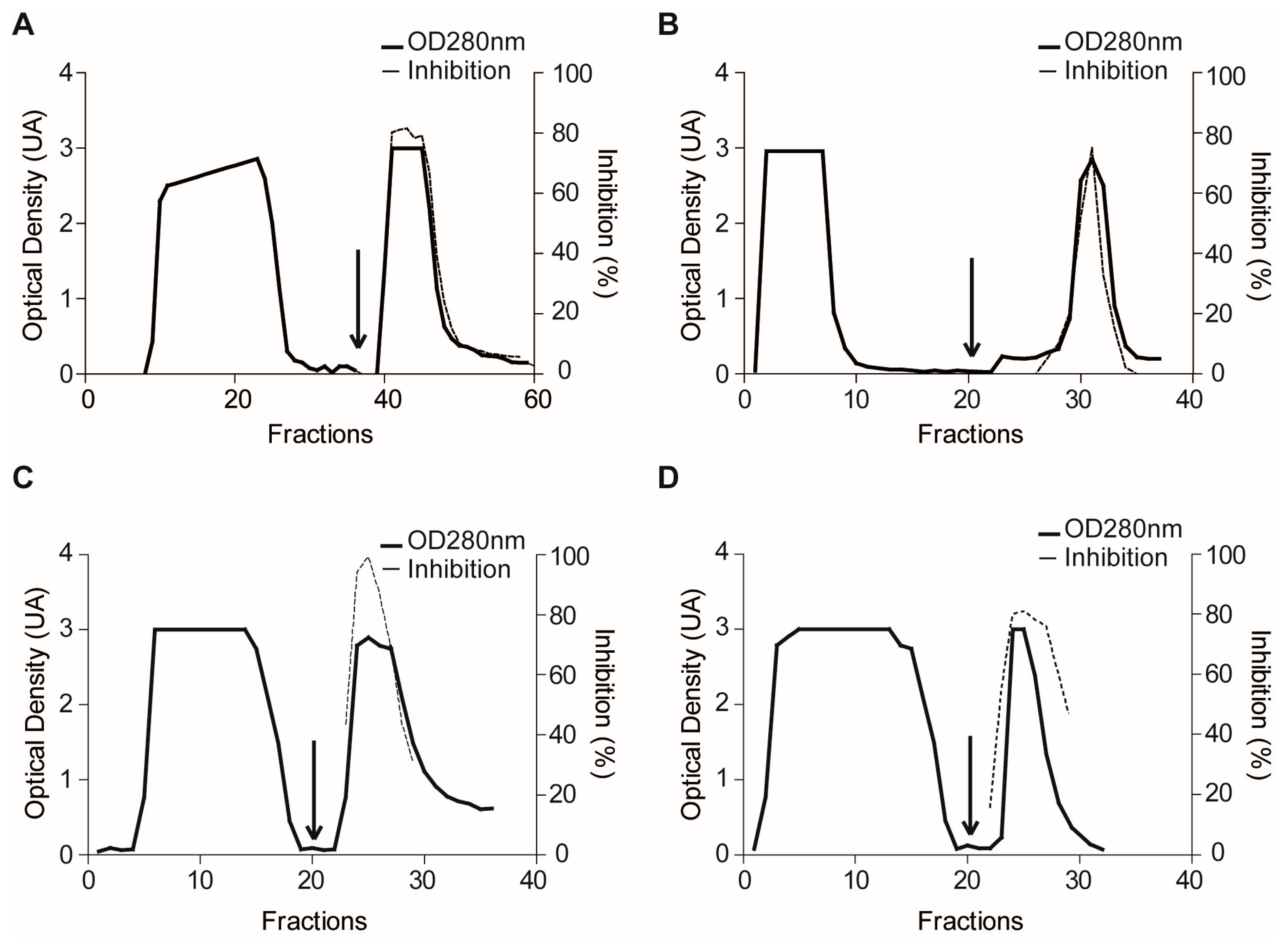
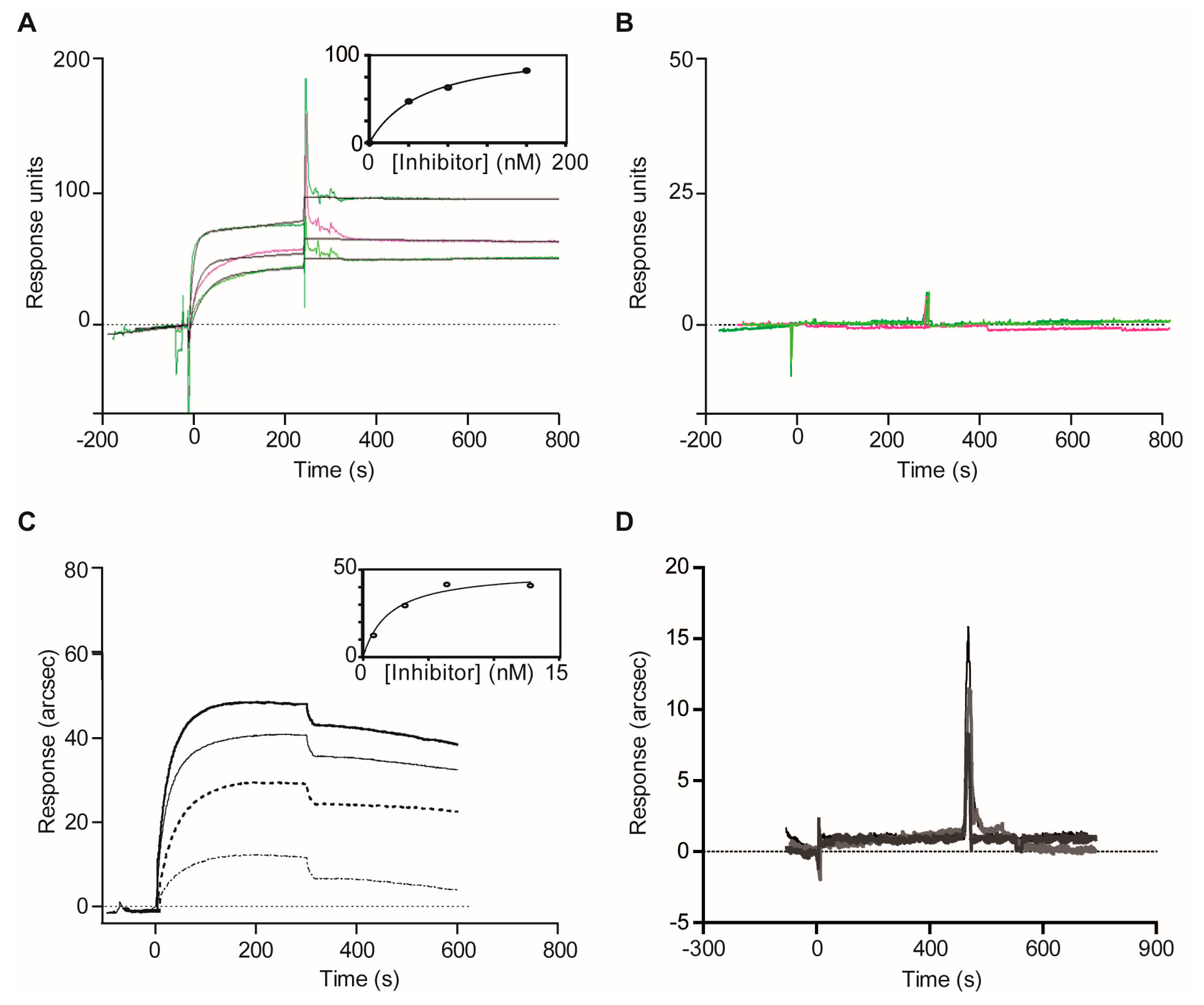
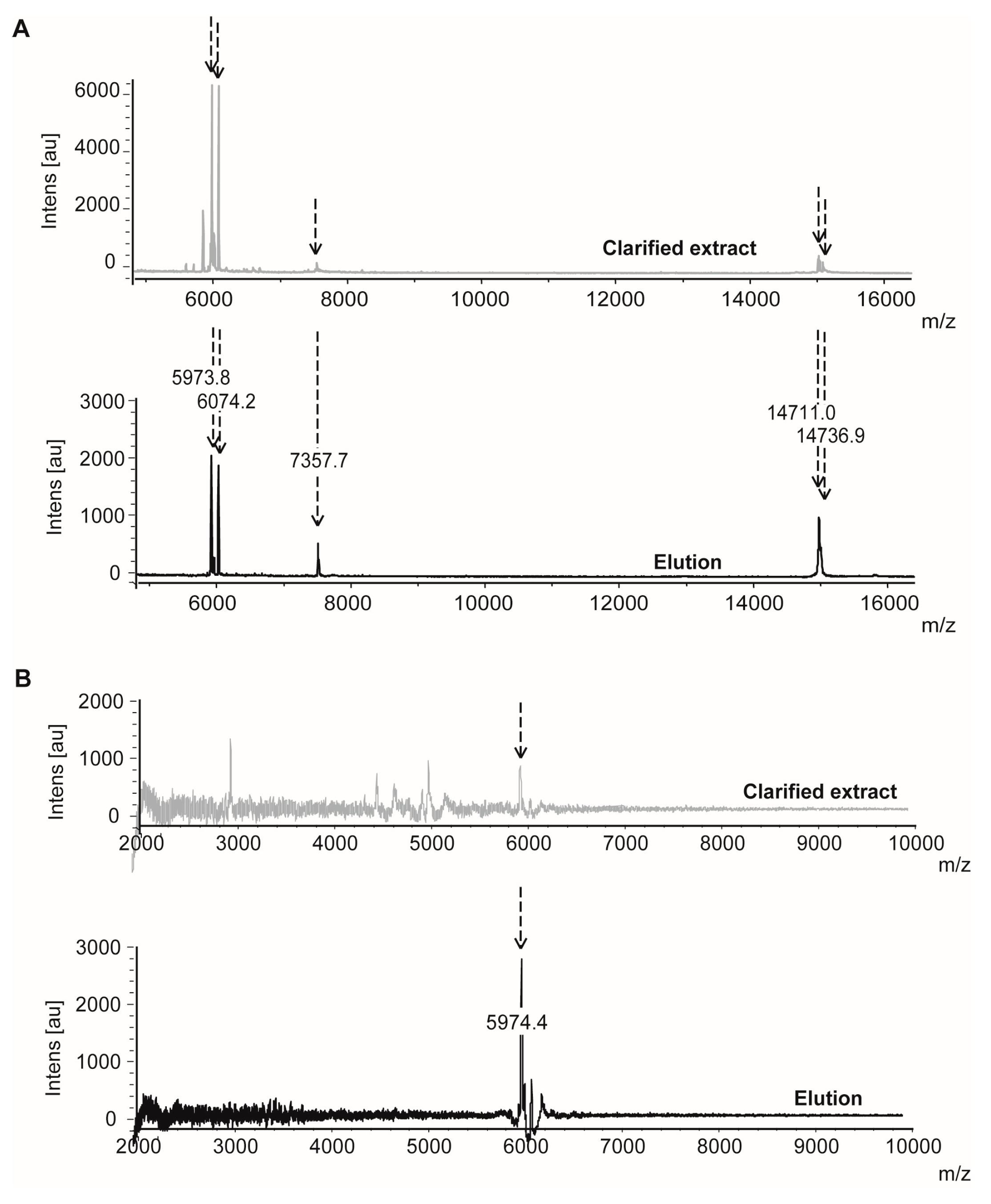
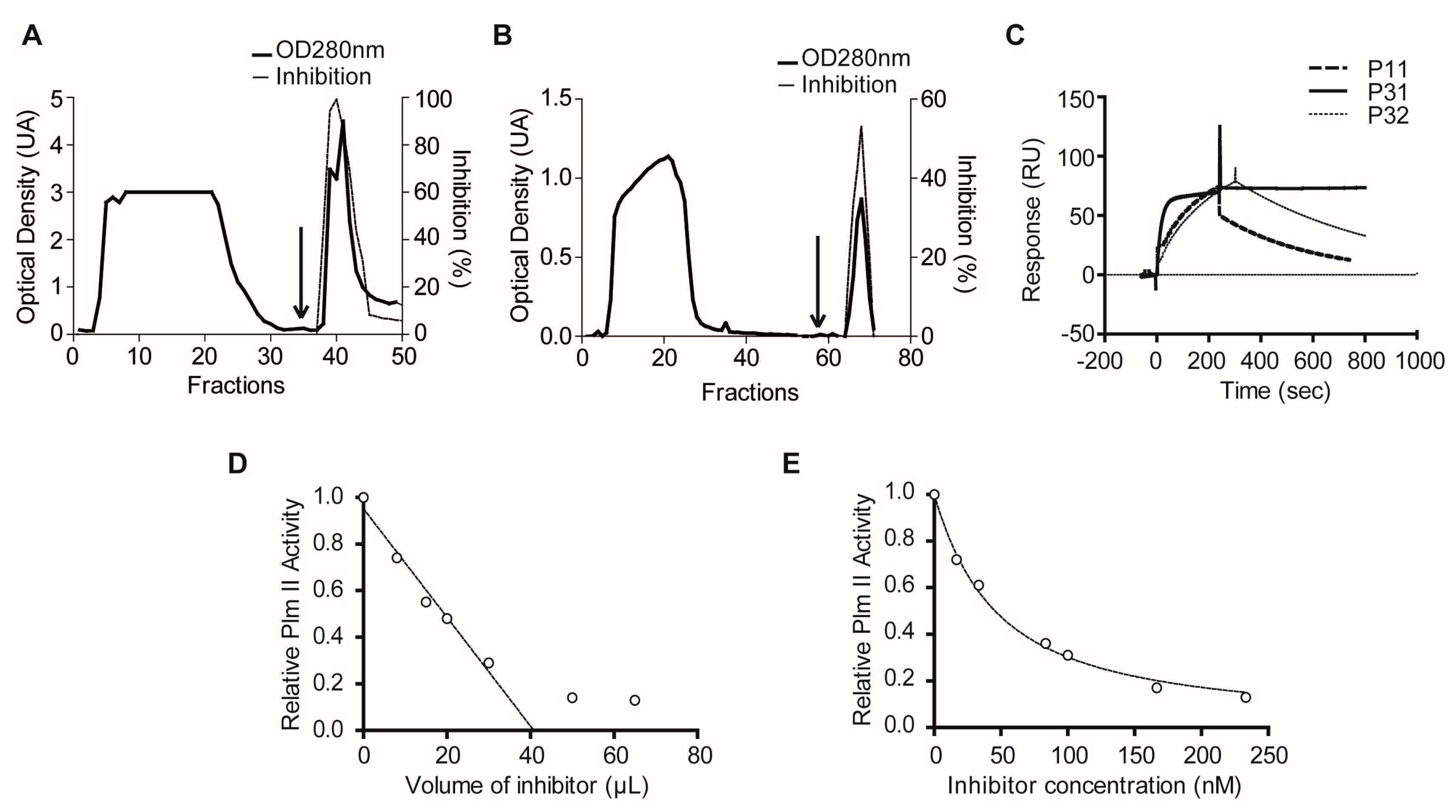
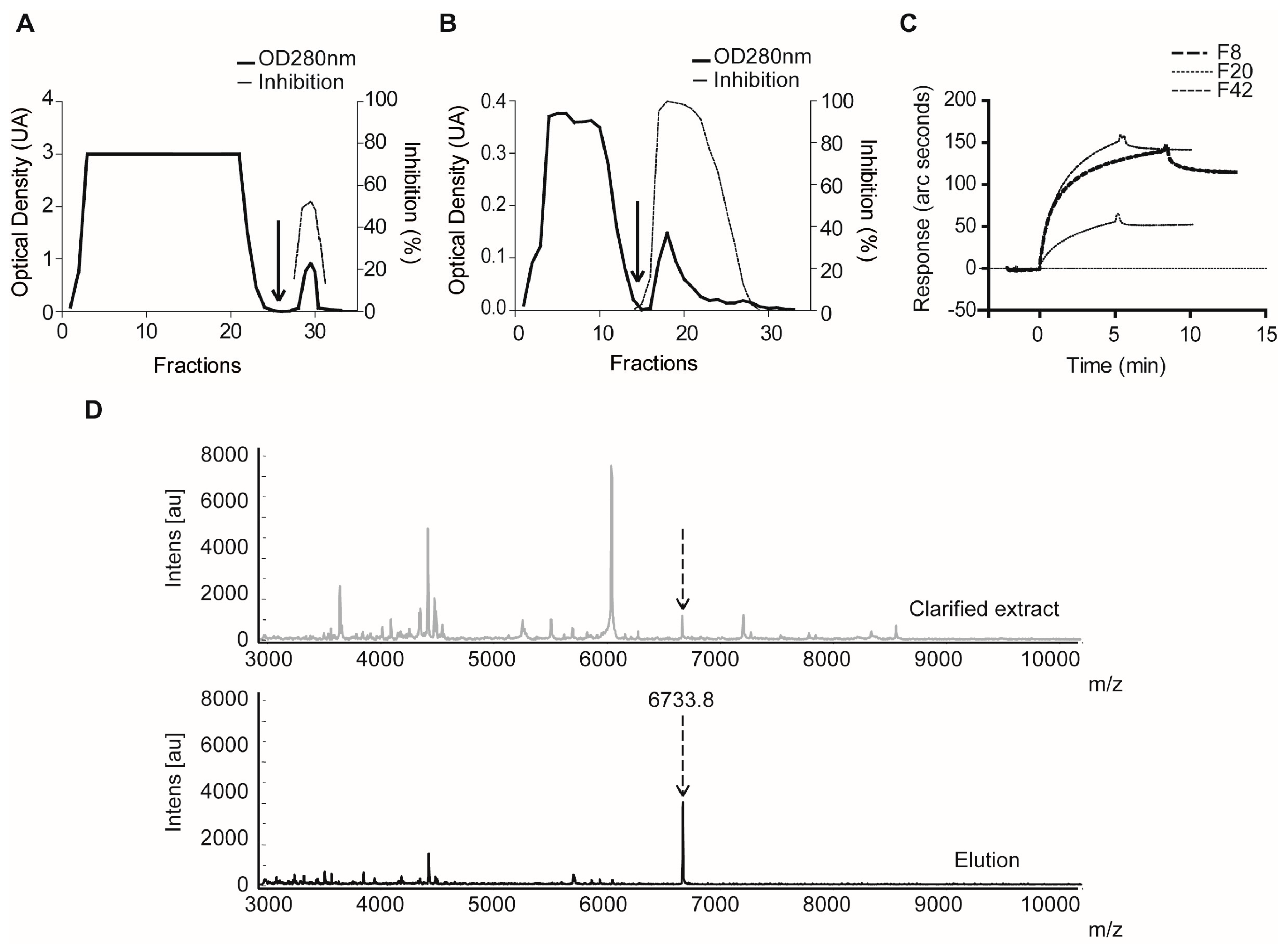
2.1.3. Confirmatory Interaction Assays Using Affinity Chromatography, SPR and IF MALDI-TOF MS
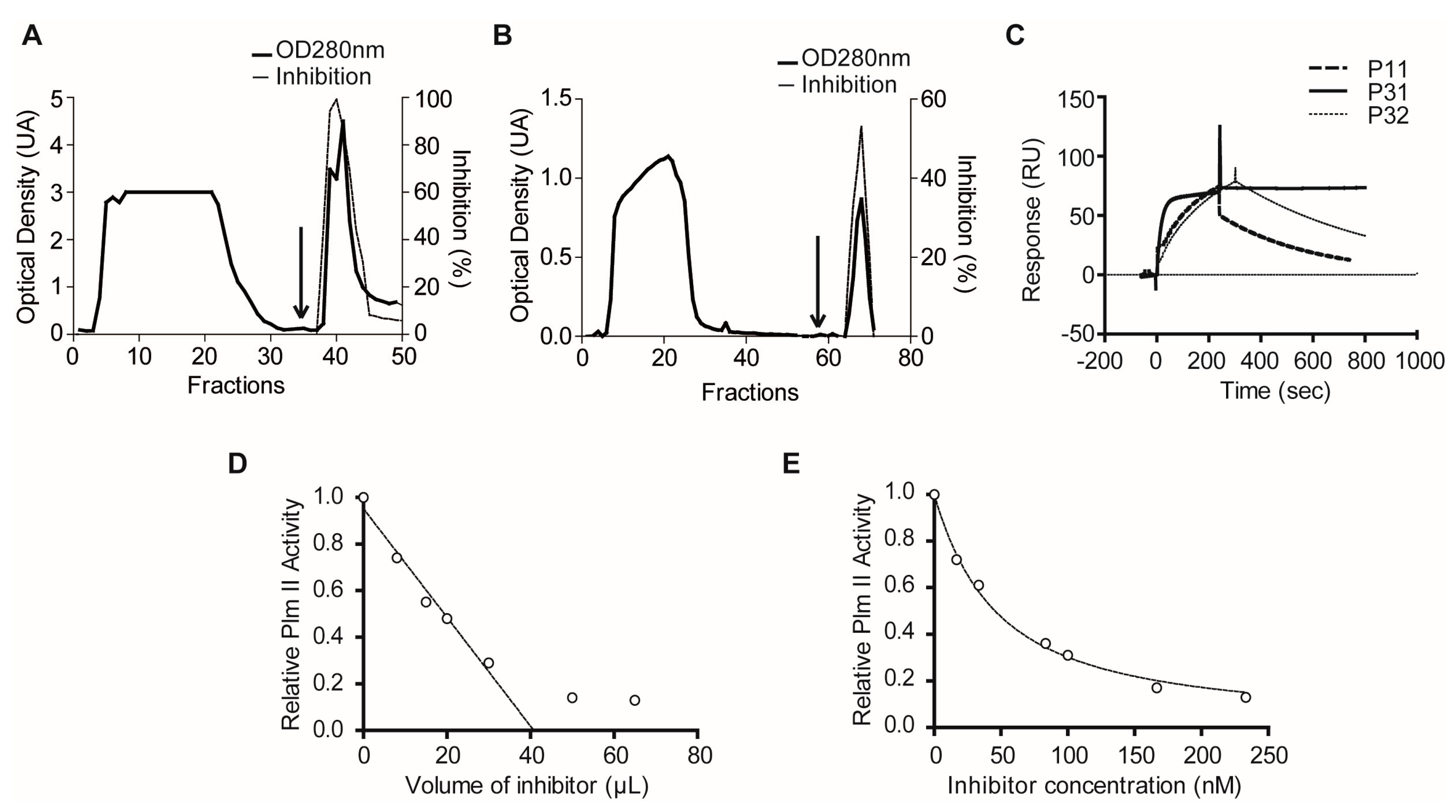
2.2. Massive Screening of Aqueous Extracts of Cuban Marine Invertebrates
3. Discussion
4. Materials and Methods
4.1. Collection of Marine Organisms. Preparation and Clarification of Aqueous Extracts
4.2. Expression, Purification and Refolding of Plm II and FP
4.3. Enzymatic Assays
4.3.1. Plasmepsin II
4.3.2. Falcipain 2
4.4. Estimation of the Active Concentration of Inhibitors by Titration with Enzymes
4.5. Kinetic Estimation of Ki Values
4.6. Generation of the Immobilized Enzyme Support
4.7. Validation Experiments by Affinity Chromatography
4.8. Validation Experiments with Optical Biosensor
4.9. Validation Experiments by Intensity Fading MALDI-TOF MS
4.10. Purification of Plexaura homomalla Plasmepsin Inhibitor (PhPI) by PlmII Affinity- and Size-Exclusion Chromatography
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AFU | arbitrary fluorescence units |
| AMC | 7-amino-4-methyl coumarin |
| AU | absorbance units |
| CMD | carboxymethyl dextran |
| DU2 | PlmII-related peptide for immunoenzymatic assays |
| E. coli | Escherichia coli |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EI | enzyme-inhibitor complex |
| E-64 | trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane |
| FP2 | falcipain 2 |
| IC50 | half-maximal inhibitory concentration |
| IF | intensity fading |
| MALDI-TOF | matrix assisted laser desorption/ionization time-of-flight |
| MS | mass spectrometry |
| MW | molecular weight |
| NaAc | sodium acetate buffer |
| NHS | N-hydroxysuccinimide |
| OD | Optical density |
| O.N. | overnight |
| PhPI | Plexaura homomalla plasmepsin inhibitor |
| Plm II | plasmepsin II |
| RT | room temperature |
| SPR | surface plasmon resonance |
| TCA | trichloroacetic acid |
| TFA | trifluoracetic acid |
| Z-FR-AMC | benzyloxycarbonyl-Phenyl-Arginyl-7-amino-4-methyl coumarin |
References
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug. Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, D.; Turk, V. Protease signalling: The cutting edge. EMBO J. 2012, 31, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Tolle, D.P.; Barrett, A.J. Evolutionary families of peptidase inhibitors. Biochem. J. 2004, 378, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, T.N.; Parveen, R.; Fatima, S. Characterization, biomedical and agricultural applications of protease inhibitors: A review. Int. J. Biol. Macromol. 2016, 91, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Krugliak, M.; Zhang, J.; Ginsburg, H. Intraerythrocytic plasmodium falciparum utilizes only a fraction of the amino acids derived from the digestion of host cell cytosol for the biosynthesis of its proteins. Mol. Biochem. Parasitol. 2002, 119, 249–256. [Google Scholar] [CrossRef]
- Goldberg, D.E. Complex nature of malaria parasite hemoglobin degradation [corrected]. Proc. Natl. Acad. Sci. USA 2013, 110, 5283–5284. [Google Scholar] [CrossRef] [PubMed]
- Chugh, M.; Sundararaman, V.; Kumar, S.; Reddy, V.S.; Siddiqui, W.A.; Stuart, K.D.; Malhotra, P. Protein complex directs hemoglobin-to-hemozoin formation in plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2013, 110, 5392–5397. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.A.; Gulnik, S.V.; Afonina, E.; Yu, B.; Ellman, J.A.; Haque, T.S.; Silva, A.M. Novel uncomplexed and complexed structures of plasmepsin ii, an aspartic protease from plasmodium falciparum. J. Mol. Biol. 2003, 327, 173–181. [Google Scholar] [CrossRef]
- Luker, K.E.; Francis, S.E.; Gluzman, I.Y.; Goldberg, D.E. Kinetic analysis of plasmepsins I and II aspartic proteases of the plasmodium falciparum digestive vacuole. Mol. Biochem. Parasitol. 1996, 79, 71–78. [Google Scholar] [CrossRef]
- Silva, A.M.; Lee, A.Y.; Gulnik, S.V.; Maier, P.; Collins, J.; Bhat, T.N.; Collins, P.J.; Cachau, R.E.; Luker, K.E.; Gluzman, I.Y.; et al. Structure and inhibition of plasmepsin II, a hemoglobin-degrading enzyme from plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1996, 93, 10034–10039. [Google Scholar] [CrossRef] [PubMed]
- Shenai, B.R.; Sijwali, P.S.; Singh, A.; Rosenthal, P.J. Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of plasmodium falciparum. J. Biol. Chem. 2000, 275, 29000–29010. [Google Scholar] [CrossRef] [PubMed]
- Drew, M.E.; Banerjee, R.; Uffman, E.W.; Gilbertson, S.; Rosenthal, P.J.; Goldberg, D.E. Plasmodium food vacuole plasmepsins are activated by falcipains. J. Biol. Chem. 2008, 283, 12870–12876. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E. Hemoglobin degradation. Curr. Top. Microbiol. Immunol. 2005, 295, 275–291. [Google Scholar] [PubMed]
- Hanspal, M.; Dua, M.; Takakuwa, Y.; Chishti, A.H.; Mizuno, A. Plasmodium falciparum cysteine protease falcipain-2 cleaves erythrocyte membrane skeletal proteins at late stages of parasite development. Blood 2002, 100, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Boss, C.; Richard-Bildstein, S.; Weller, T.; Fischli, W.; Meyer, S.; Binkert, C. Inhibitors of the plasmodium falciparum parasite aspartic protease plasmepsin II as potential antimalarial agents. Curr. Med. Chem. 2003, 10, 883–907. [Google Scholar] [CrossRef] [PubMed]
- Corminboeuf, O.; Dunet, G.; Hafsi, M.; Grimont, J.; Grisostomi, C.; Meyer, S.; Binkert, C.; Bur, D.; Jones, A.; Prade, L.; et al. Inhibitors of plasmepsin II-potential antimalarial agents. Bioorg. Med. Chem. Lett. 2006, 16, 6194–6199. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.E.; Lee, G.K.; Semenov, A.; Rosenthal, P.J. Antimalarial effects in mice of orally administered peptidyl cysteine protease inhibitors. Bioorg. Med. Chem. 1999, 7, 633–638. [Google Scholar] [CrossRef]
- Micale, N.; Kozikowski, A.P.; Ettari, R.; Grasso, S.; Zappala, M.; Jeong, J.J.; Kumar, A.; Hanspal, M.; Chishti, A.H. Novel peptidomimetic cysteine protease inhibitors as potential antimalarial agents. J. Med. Chem. 2006, 49, 3064–3067. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Bova, F.; Zappala, M.; Grasso, S.; Micale, N. Falcipain-2 inhibitors. Med. Res. Rev. 2010, 30, 136–167. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, J.A.; Bonilla, T.D.; Yowell, C.A.; Fujioka, H.; Dame, J.B. Critical roles for the digestive vacuole plasmepsins of plasmodium falciparum in vacuolar function. Mol. Microbiol. 2007, 65, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Sijwali, P.S.; Koo, J.; Singh, N.; Rosenthal, P.J. Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of plasmodium falciparum. Mol. Biochem. Parasitol. 2006, 150, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.W.; Spaccapelo, R.; Schwarzer, E.; Sajid, M.; Annoura, T.; Deroost, K.; Ravelli, R.B.; Aime, E.; Capuccini, B.; Mommaas-Kienhuis, A.M.; et al. Replication of plasmodium in reticulocytes can occur without hemozoin formation, resulting in chloroquine resistance. J. Exp. Med. 2015, 212, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Tyas, L.; Phylip, L.H.; Kay, J.; Dunn, B.M.; Berry, C. High level expression and characterisation of plasmepsin II, an aspartic proteinase from plasmodium falciparum. FEBS Lett. 1994, 352, 155–158. [Google Scholar] [CrossRef]
- Sijwali, P.S.; Brinen, L.S.; Rosenthal, P.J. Systematic optimization of expression and refolding of the plasmodium falciparum cysteine protease falcipain-2. Protein Expr. Purif. 2001, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Prade, L.; Jones, A.F.; Boss, C.; Richard-Bildstein, S.; Meyer, S.; Binkert, C.; Bur, D. X-ray structure of plasmepsin II complexed with a potent achiral inhibitor. J. Biol. Chem. 2005, 280, 23837–23843. [Google Scholar] [CrossRef] [PubMed]
- Hogg, T.; Nagarajan, K.; Herzberg, S.; Chen, L.; Shen, X.; Jiang, H.; Wecke, M.; Blohmke, C.; Hilgenfeld, R.; Schmidt, C.L. Structural and functional characterization of falcipain-2, a hemoglobinase from the malarial parasite plasmodium falciparum. J. Biol. Chem. 2006, 281, 25425–25437. [Google Scholar] [CrossRef] [PubMed]
- Dan, N.; Bhakat, S. New paradigm of an old target: An update on structural biology and current progress in drug design towards plasmepsin II. Eur. J. Med. Chem. 2015, 95, 324–348. [Google Scholar] [CrossRef] [PubMed]
- Mane, U.R.; Gupta, R.C.; Nadkarni, S.S.; Giridhar, R.R.; Naik, P.P.; Yadav, M.R. Falcipain inhibitors as potential therapeutics for resistant strains of malaria: A patent review. Expert Opin. Ther. Pat. 2013, 23, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Flotow, H.; Leong, C.Y.; Buss, A.D. Development of a plasmepsin ii fluorescence polarization assay suitable for high throughput antimalarial drug discovery. J. Biomol. Screen. 2002, 7, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Salas, E.; Ramirez, A.; Otero-Bilbao, A.; Vazquez, R.; Reyes, O.; Mendiola, J.; Duarte, C.A.; Otero-Gonzalez, A.; Gutierrez, O.A.; Chavez, M.A. A heterogeneous enzymatic assay for quantification of plasmepsin II activity and the evaluation of its inhibitors. J. Pharm. Biomed. Anal. 2004, 34, 833–840. [Google Scholar] [CrossRef]
- Machon, U.; Buchold, C.; Stempka, M.; Schirmeister, T.; Gelhaus, C.; Leippe, M.; Gut, J.; Rosenthal, P.J.; Kisker, C.; Leyh, M.; et al. On-bead screening of a combinatorial fumaric acid derived peptide library yields antiplasmodial cysteine protease inhibitors with unusual peptide sequences. J. Med. Chem. 2009, 52, 5662–5672. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, J.; Chen, L.; Liu, X.; Chen, T.; Zhu, J.; Lu, W.; Shen, X.; Li, J.; Hilgenfeld, R.; et al. Identification of novel falcipain-2 inhibitors as potential antimalarial agents through structure-based virtual screening. J. Med. Chem. 2009, 52, 4936–4940. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Puga, J.; Serodio, J.; Gomes, N.C.; Calado, R. Trends in the discovery of new marine natural products from invertebrates over the last two decades—Where and what are we bioprospecting? PLoS ONE 2012, 7, e30580. [Google Scholar] [CrossRef] [PubMed]
- Suleria, H.A.; Osborne, S.; Masci, P.; Gobe, G. Marine-based nutraceuticals: An innovative trend in the food and supplement industries. Mar. Drugs 2015, 13, 6336–6351. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Fusetani, N. Enzyme inhibitors from marine invertebrates. J. Nat. Prod. 2007, 70, 689–710. [Google Scholar] [CrossRef] [PubMed]
- Frazao, B.; Vasconcelos, V.; Antunes, A. Sea anemone (cnidaria, anthozoa, actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.B. Proteases and protease inhibitors: A balance of activities in host-pathogen interaction. Immunobiology 2006, 211, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Delfin, J.; Martinez, I.; Antuch, W.; Morera, V.; Gonzalez, Y.; Rodriguez, R.; Marquez, M.; Saroyan, A.; Larionova, N.; Diaz, J.; et al. Purification, characterization and immobilization of proteinase inhibitors from stichodactyla helianthus. Toxicon 1996, 34, 1367–1376. [Google Scholar] [CrossRef]
- Lenarcic, B.; Ritonja, A.; Strukelj, B.; Turk, B.; Turk, V. Equistatin, a new inhibitor of cysteine proteinases from actinia equina, is structurally related to thyroglobulin type-1 domain. J. Biol. Chem. 1997, 272, 13899–13903. [Google Scholar] [PubMed]
- Pascual, I.; Gil-Parrado, S.; Cisneros, M.; Joseph-Bravo, P.; Diaz, J.; Possani, L.D.; Charli, J.L.; Chavez, M. Purification of a specific inhibitor of pyroglutamyl aminopeptidase ii from the marine annelide hermodice carunculata. In vivo effects in rodent brain. Int. J. Biochem. Cell Biol. 2004, 36, 138–152. [Google Scholar] [CrossRef]
- Gonzalez, Y.; Tanaka, A.S.; Hirata, I.Y.; del Rivero, M.A.; Oliva, M.L.; Araujo, M.S.; Chavez, M.A. Purification and partial characterization of human neutrophil elastase inhibitors from the marine snail cenchritis muricatus (mollusca). Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 146, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Alonso-del-Rivero, M.; Trejo, S.A.; Reytor, M.L.; Rodriguez-de-la-Vega, M.; Delfin, J.; Diaz, J.; Gonzalez-Gonzalez, Y.; Canals, F.; Chavez, M.A.; Aviles, F.X. Tri-domain bifunctional inhibitor of metallocarboxypeptidases a and serine proteases isolated from marine annelid sabellastarte magnifica. J. Biol. Chem. 2012, 287, 15427–15438. [Google Scholar] [CrossRef] [PubMed]
- Covaleda, G.; del Rivero, M.A.; Chavez, M.A.; Aviles, F.X.; Reverter, D. Crystal structure of novel metallocarboxypeptidase inhibitor from marine mollusk nerita versicolor in complex with human carboxypeptidase a4. J. Biol. Biol. Chem. 2012, 287, 9250–9258. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.R.; Guerra, Y.; Otero, A.; Garcia, B.; Berry, C.; Mendiola, J.; Hernandez-Zanui, A.; Chavez Mde, L. Generation of an affinity matrix useful in the purification of natural inhibitors of plasmepsin II, an antimalarial-drug target. Biotechnol. Appl. Biochem. 2009, 52, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Reytor, M.E.; González, Y.; Pascual, I.; Hernández, A.; Chávez, M.Á.; Alonso del Rivero, M. Screening of protease inhibitory activity in extracts of five ascidian species from cuban coasts. Biotecnol. Apl. 2011, 28, 77–82. [Google Scholar]
- Martis, E.A.; Radhakrishnan, R.; Badve, R.R. High-throughput screening: The hits and leads of drug discovery—An overview. J. Appl. Pharm. Sci. 2011, 1, 2–10. [Google Scholar]
- Duong-Thi, M.D.; Meiby, E.; Bergstrom, M.; Fex, T.; Isaksson, R.; Ohlson, S. Weak affinity chromatography as a new approach for fragment screening in drug discovery. Anal. Biochem. 2011, 414, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Borch, J.; Roepstorff, P. Screening for enzyme inhibitors by surface plasmon resonance combined with mass spectrometry. Anal. Chem. 2004, 76, 5243–5248. [Google Scholar] [CrossRef] [PubMed]
- Yanes, O.; Villanueva, J.; Querol, E.; Aviles, F. Functional screening of serine protease inhibitors in the medical leech hirudo medicinalis monitored by intensity fading maldi-tof ms. Mol. Cell. Proteom. 2005, 4, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Yanes, O.; Villanueva, J.; Querol, E.; Aviles, F. Detection of non-covalent protein interactions by ‘intensity fading’ maldi-tof mass spectrometry: Applications to proteases and protease inhibitors. Nat. Protoc. 2007, 2, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Salas-Sarduy, E.; Cabrera-Munoz, A.; Cauerhff, A.; Gonzalez-Gonzalez, Y.; Trejo, S.A.; Chidichimo, A.; Chavez-Planes, M.D.; Cazzulo, J.J. Antiparasitic effect of a fraction enriched in tight-binding protease inhibitors isolated from the caribbean coral plexaura homomalla. Exp. Parasitol. 2013, 135, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, Y.; Araujo, M.S.; Oliva, M.L.; Sampaio, C.A.; Chavez, M.A. Purification and preliminary characterization of a plasma kallikrein inhibitor isolated from sea hares aplysia dactylomela rang, 1828. Toxicon 2004, 43, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Bandaranayake, W.M. The nature and role of pigments of marine invertebrates. Nat. Prod. Rep. 2006, 23, 223–255. [Google Scholar] [CrossRef] [PubMed]
- Bieth, J.G. Theoretical and practical aspects of proteinase inhibition kinetics. Methods Enzymol. 1995, 248, 59–84. [Google Scholar] [PubMed]
- Copeland, R. Enzymes: A practical Introduction to Structure, Mechanism, and Data Analysis; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 2005, 46, 1–265. [Google Scholar] [PubMed]
- Gutierrez, O.A.; Chavez, M.; Lissi, E. A theoretical approach to some analytical properties of heterogeneous enzymatic assays. Anal. Chem. 2004, 76, 2664–2668. [Google Scholar] [CrossRef] [PubMed]
- Backman, D.; Monod, M.; Danielson, U.H. Biosensor-based screening and characterization of HIV-1 inhibitor interactions with sap 1, sap 2, and sap 3 from candida albicans. J. Biomol. Screen. 2006, 11, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Shuman, C.F.; Hamalainen, M.D.; Danielson, U.H. Kinetic and thermodynamic characterization of HIV-1 protease inhibitors. J. Mol. Recognit. 2004, 17, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Malo, N.; Hanley, J.A.; Cerquozzi, S.; Pelletier, J.; Nadon, R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006, 24, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Macht, M. Mass spectrometric top-down analysis of proteins. Bioanalysis 2009, 1, 1131–1148. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. Merops: The peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.O.; Garcia-Gomes, A.S.; Catanho, M.; Sodré, C.L.; Santos, A.L.; Branquinha, M.H.; d’Avila-Levy, C.M. Aspartic peptidases of human pathogenic trypanosomatids: Perspectives and trends for chemotherapy. Curr. Med. Chem. 2013, 20, 3116–3133. [Google Scholar] [CrossRef] [PubMed]
- Zucca, M.; Savoia, D. Current developments in the therapy of protozoan infections. Open Med. Chem. J. 2011, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Gary, W. Pharmaceutical Biotechnology: Concepts and Applications; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Sarduy, E.S.; Muñoz, A.C.; Trejo, S.A.; Chavez Planes, M.D. High-level expression of falcipain-2 in escherichia coli by codon optimization and auto-induction. Protein Expr. Purif. 2012, 83, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Otero, C.; Ballesteros, A.; Guisán, J.M. Immobilization/stabilization of lipase from candida rugosa. Appl. Biochem. Biotechnol. 1988, 19, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Yanes, O.; Querol, E.; Serrano, L.; Aviles, F.X. Identification of protein ligands in complex biological samples using intensity-fading maldi-tof mass spectrometry. Anal. Chem. 2003, 75, 3385–3395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Phylum | Identifier | |
|---|---|---|---|
| Falcipain 2 | Plasmepsin II | ||
| Hermodice carunculata | Annelida | F30 (+) | — |
| Sabellastarte magnifica (b.c.) | Annelida | F22 | — |
| Sabellastarte magnifica (b.) | Annelida | F23 | — |
| Bugula plumosa | Bryozoa | F39 | P25 |
| Copidozoum sp. | Bryozoa | — | P24 (+) |
| Zoobotrium sp. | Bryozoa | — | P30 |
| Zoobotrium sp. (TCA clarified) | Bryozoa | — | P33 |
| Ascidia sydneiensis | Chordata | F31 | — |
| Ascidia sydneiensis (TCA clarified) | Chordata | F44 | P28 |
| Diplosoma listerianum | Chordata | F33 | — |
| Ecteinascidia turbinata | Chordata | F28 | — |
| Microcosmus sp. | Chordata | — | P15 |
| Phallusia nigra | Chordata | F21 | — |
| Phallusia nigra (TCA clarified) | Chordata | F45 | — |
| Acetabularia sp. | Chlorophyta | — | P17 |
| Bryopsis pennata | Chlorophyta | — | P21 |
| Halimeda opuntia | Chlorophyta | F34 | — |
| Penicillus capitatus | Chlorophyta | — | P9 |
| Bartholomea annulata | Cnidaria | — | P11 (++) |
| Bunodosoma granulifera | Cnidaria | F11 (+) | P8 |
| Bunodosoma granulifera (TCA clarified) | Cnidaria | F32 (++) | — |
| Cassiopeia xamachana | Cnidaria | — | P29 |
| Condylactis gigantea | Cnidaria | F3 (+) | — |
| Condylactis gigantea (TCA clarified) | Cnidaria | F29 | — |
| Eunicea calyculata | Cnidaria | — | P13 |
| Lenubrea danae (p.c.) | Cnidaria | F16 | — |
| Lenubrea danae (w.b.) | Cnidaria | F17 | — |
| Lenubrea danae (w.b.) (TCA clarified) | Cnidaria | F36 (+) | — |
| Linuche unguiculata | Cnidaria | F15 (+) | — |
| Palythoa caribbaeorum | Cnidaria | F2 (+) | P5 |
| Palythoa caribbaeorum (TCA clarified) | Cnidaria | F12 (+) | — |
| Physalia physalis | Cnidaria | F47 | — |
| Plexaura homomalla | Cnidaria | F8 (++) | P31 (++) |
| Stichodactyla helianthus | Cnidaria | F6 (++) | P3 |
| Stichodactyla helianthus (TCA clarified) | Cnidaria | F38 (+) | P35 |
| Zhoanthus pulchelus | Cnidaria | F7 (+) | P22 |
| Zhoanthus sp. | Cnidaria | — | P16 |
| Echinaster echinophorus | Echinodermata | F35 | P4 |
| Echinometra lucunter | Echinodermata | — | P23 |
| Eucidaris tribuloides | Echinodermata | F24 (+) | — |
| Holothuria mexicana | Echinodermata | F42 (++) | P6 |
| Holothuria sp. | Echinodermata | F37 | — |
| Holothuria sp. | Echinodermata | — | P18 |
| Holothuria sp. | Echinodermata | — | P19 |
| Holothuria sp. | Echinodermata | — | P20 |
| Isostochopus badionotus | Echinodermata | F26 (+) | — |
| Luidia senegala | Echinodermata | — | P26 |
| Lytechinus variegatus (w.b.) | Echinodermata | F40 | — |
| Oreaster reticulates | Echinodermata | — | P27 |
| Tripneustes ventricosus (s.) | Echinodermata | F4 | — |
| Tripneustes ventricosus (g.) | Echinodermata | F5 (+) | — |
| Tripneustes ventricosus (w.b.) | Echinodermata | F10 | P2 |
| Cenchritis muricatus | Mollusca | F43 (+) | P7 (+) |
| Nerita peloronta | Mollusca | F20 (++) | — |
| Nerita versicolor | Mollusca | F41 (+) | — |
| Purpura patula | Mollusca | — | P12 |
| Ectyoplasia ferox | Porifera | — | P32 (++) |
| Lissodendorix isodyctialis | Porifera | F25 (+) | P1 |
| Xetospongia muta | Porifera | F46 | P10 * |
| Xetospongia muta (TCA clarified) | Porifera | — | P34 |
| Galaxaura sp. | Rhodophyta | F27 (+) | — |
| Laurencia sp. | Rhodophyta | — | P14 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salas-Sarduy, E.; Guerra, Y.; Covaleda Cortés, G.; Avilés, F.X.; Chávez Planes, M.A. Identification of Tight-Binding Plasmepsin II and Falcipain 2 Inhibitors in Aqueous Extracts of Marine Invertebrates by the Combination of Enzymatic and Interaction-Based Assays. Mar. Drugs 2017, 15, 123. https://doi.org/10.3390/md15040123
Salas-Sarduy E, Guerra Y, Covaleda Cortés G, Avilés FX, Chávez Planes MA. Identification of Tight-Binding Plasmepsin II and Falcipain 2 Inhibitors in Aqueous Extracts of Marine Invertebrates by the Combination of Enzymatic and Interaction-Based Assays. Marine Drugs. 2017; 15(4):123. https://doi.org/10.3390/md15040123
Chicago/Turabian StyleSalas-Sarduy, Emir, Yasel Guerra, Giovanni Covaleda Cortés, Francesc Xavier Avilés, and María A. Chávez Planes. 2017. "Identification of Tight-Binding Plasmepsin II and Falcipain 2 Inhibitors in Aqueous Extracts of Marine Invertebrates by the Combination of Enzymatic and Interaction-Based Assays" Marine Drugs 15, no. 4: 123. https://doi.org/10.3390/md15040123
APA StyleSalas-Sarduy, E., Guerra, Y., Covaleda Cortés, G., Avilés, F. X., & Chávez Planes, M. A. (2017). Identification of Tight-Binding Plasmepsin II and Falcipain 2 Inhibitors in Aqueous Extracts of Marine Invertebrates by the Combination of Enzymatic and Interaction-Based Assays. Marine Drugs, 15(4), 123. https://doi.org/10.3390/md15040123