4. Experimental Section
Infrared (IR) spectra were obtained using a PerkinElmer FT-IR spectrometer 1720X (Perkin Elmer, Waltham, MA, USA). High Resolution Mass Spectra (HRMS) were obtained using a JEOL JMS-700 (2) mass spectrometer (JEOL, Tokyo, Japan). NMR spectra were recorded at 27 °C on Agilent 300-, 400-MR-DD2 (Agilent Technologies, Santa Clara, CA, USA), Agilent 600-DD2 (Agilent Technologies, Santa Clara, CA, USA), Varian Marcury-300BB (Varian, Palo Alto, CA, USA), and Varian Unity-500 spectrometers in CDCl3 using tetramethylsilane (TMS) as the internal standard. Liquid column chromatography was conducted on silica gel (Nacalai, silica gel 60, mesh 70–230 or 230–400). Analytical thin layer chromatography (TLC) was performed on pre-coated Merck glass plates (silica gel 60 F254) and compounds were detected by dipping the plates in an ethanol solution of phosphomolybdic acid, followed by heating. Microwave-aided reactions were carried out using a Biotage Initiator® reactor (PartnerTech Atvidaberg AB for Biotage Sweden AB, Uppsala, Sweden). Flash chromatography was carried out using Biotage Isolera One® purification system (PartnerTech Atvidaberg AB for Biotage Sweden AB, Uppsala, Sweden). Dry CH2Cl2, dry tetrahydrofuran (THF), NaBH4, trifluoroacetic acid (TFA), pyridine, and NBS were purchased from Wako Pure Chemical Industries (Wako Pure Chemical Industries, Tokyo, Japan). meta-Chloroperbenzoic Acid (mCPBA) and hexamethyldisilazane (HMDS) were purchased from nacalai tesque, Inc. (nacalai tesque, Inc., Kyoto, Japan). Cyclohexanone, camphorsulfonic acid (CSA), HCl in Et2O, trifluoroacetone, BF3·Et2O, and n-BuLi were purchased from Sigma-Aldrich Co. LLC. (St. Louis, MO, USA). Tf2O, CsOAc, and Dess-Martin periodinane (DMP) were purchased from TCI (Tokyo Chemical Industry Co. Ltd., Tokyo, Japan). (−)-Shikimic acid was purchased from Carbosynth, Ltd. (UK). (−)-Quinic acid was purchased from Merck (Merck & Co., Inc., Darmstadt, Germany). α-Glucosidase (Yeast, lot. 26010), β-glucosidase (Sweet Almond, lot. 81241), α-mannosidase (Jack Bean, lot. 055K7047), and deoxymannojirimycin were purchased from Sigma-Aldrich Co. LLC. (St. Louis, MO, USA). 1-Deoxynojirimycin was isolated from leaves of the plant Morus alba L.
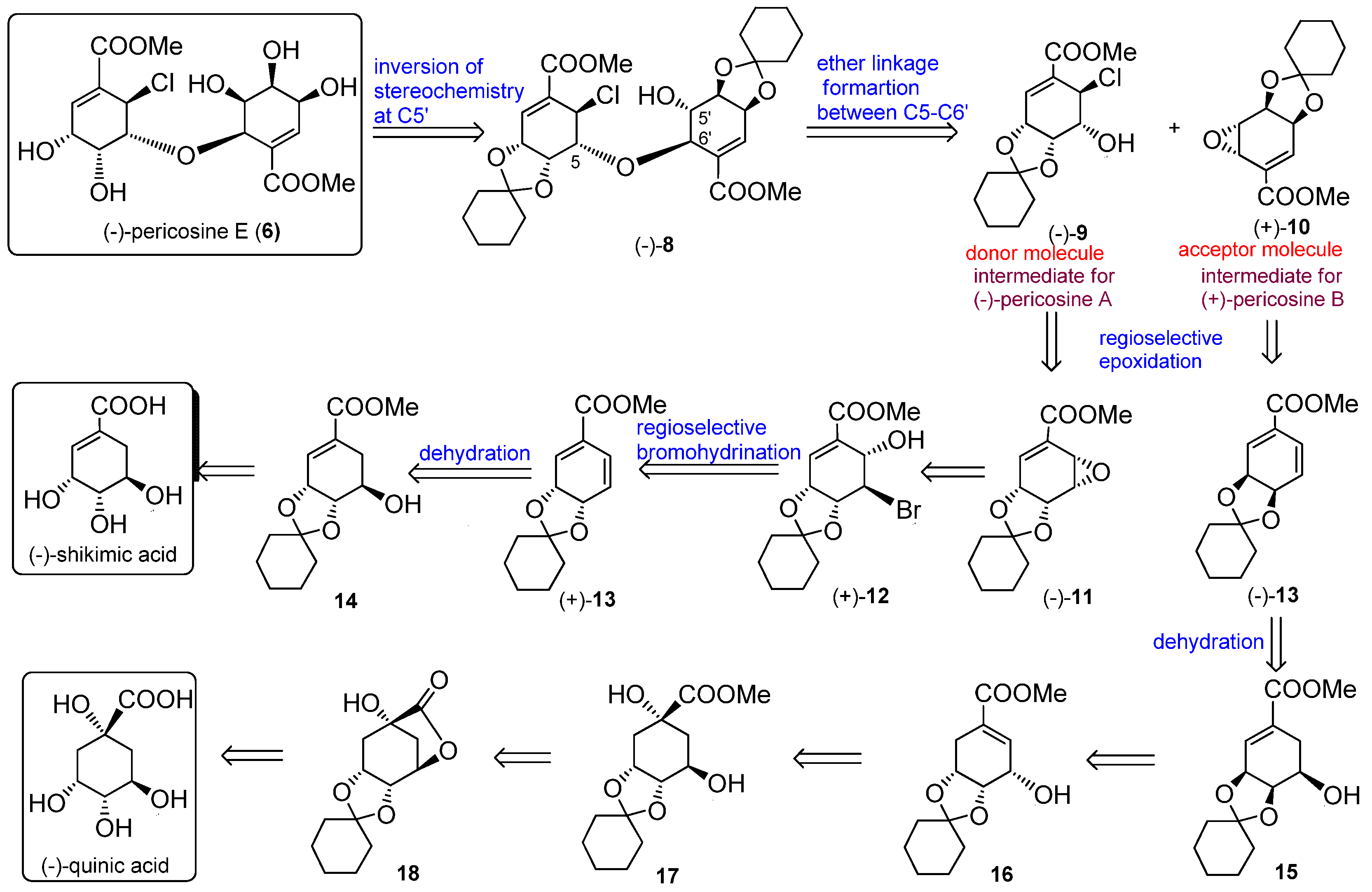
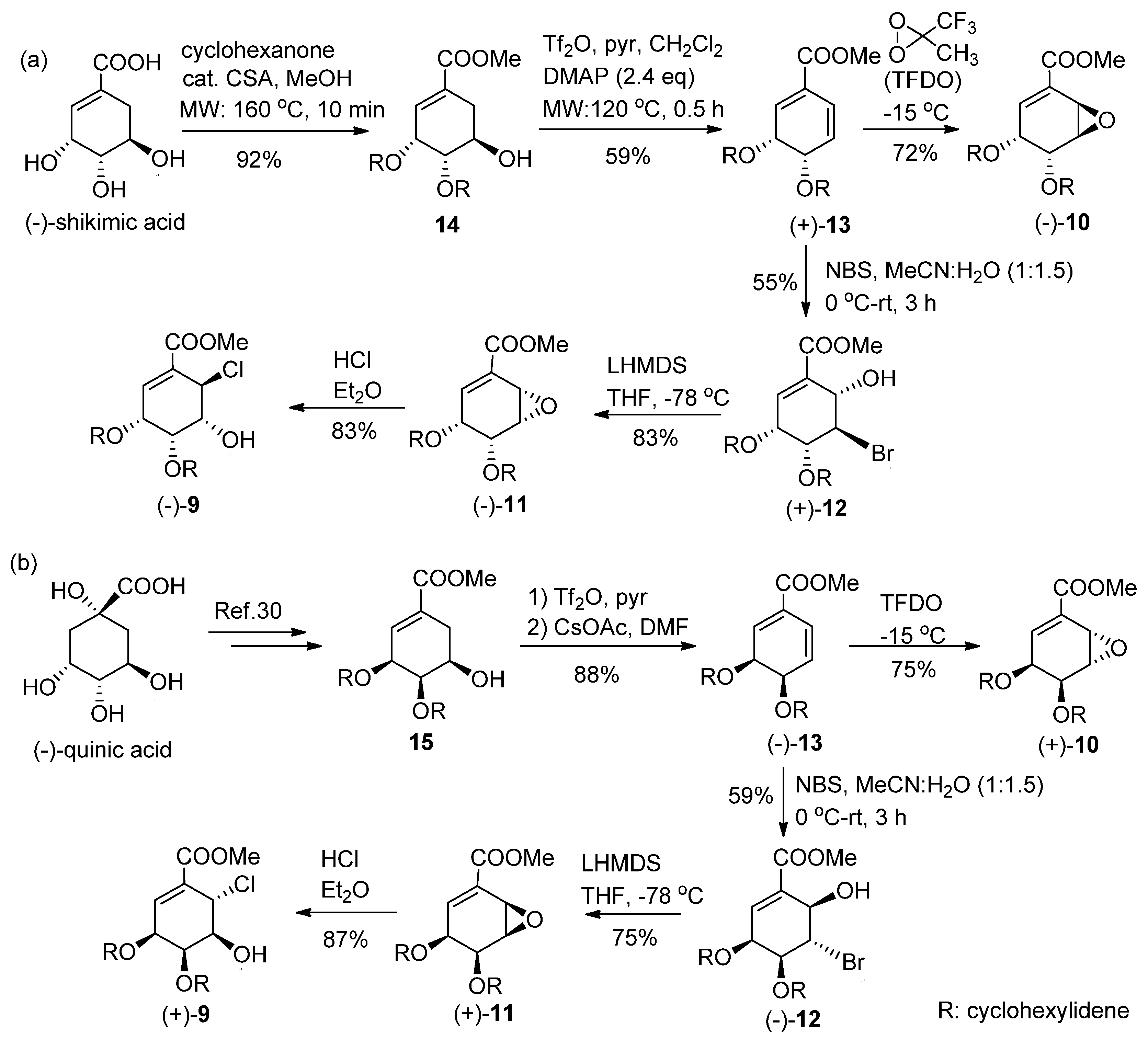
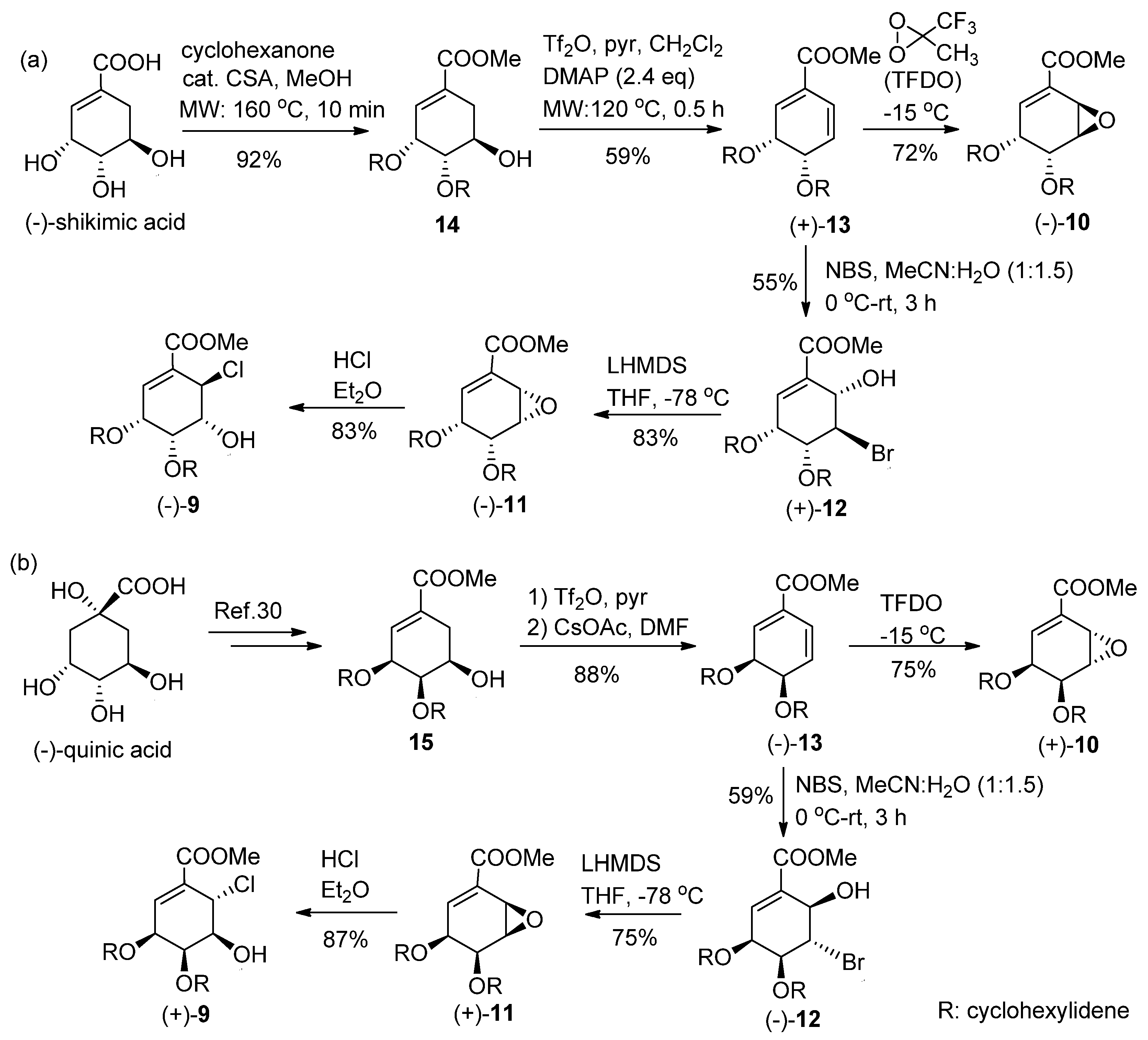
4.1. Bromohydrination of (+)-13
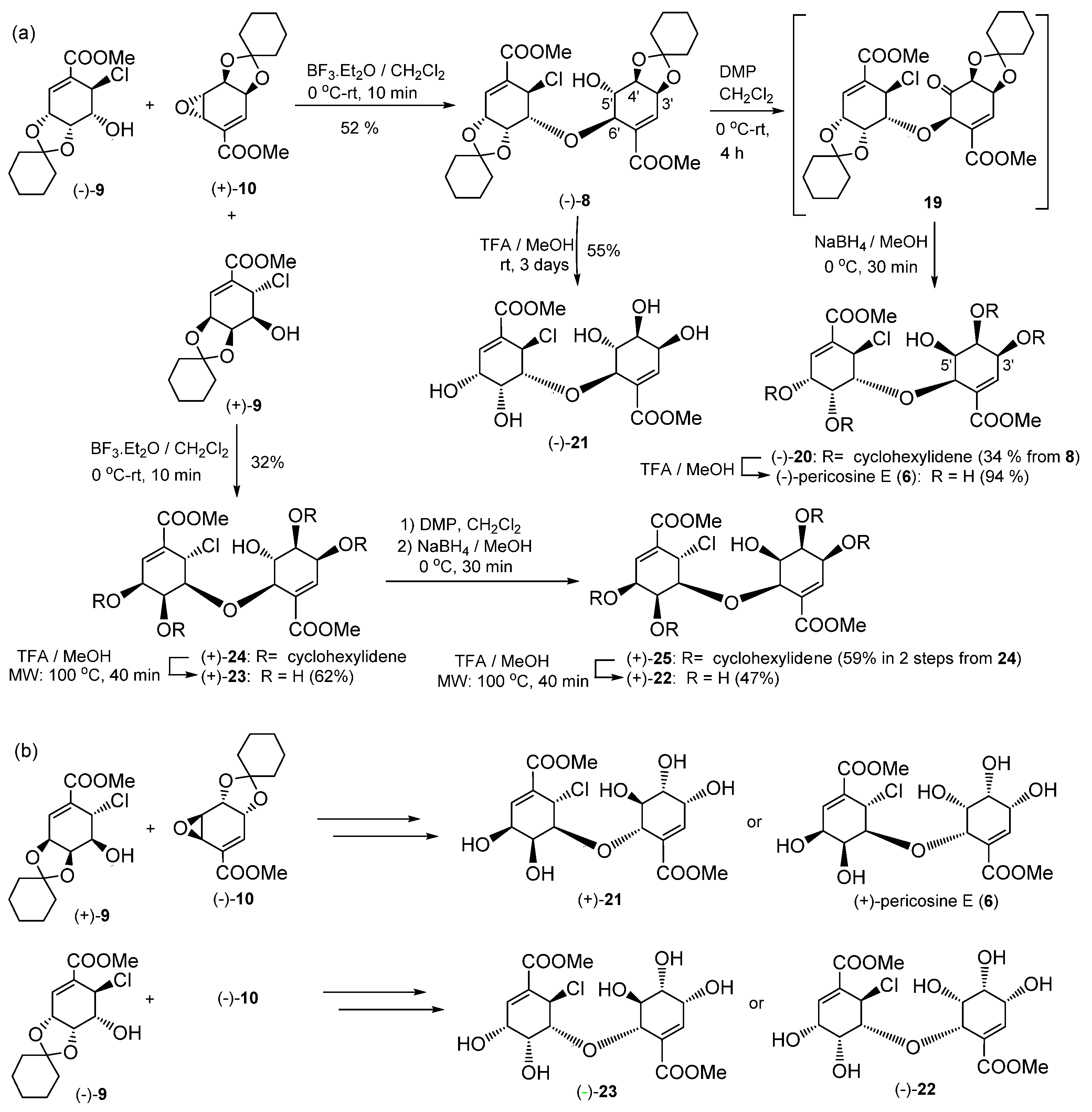
NBS (48 mg, 1.2 mmol) at 0 °C was added with stirring to a solution of diene (+)-13 (58 mg, 0.23 mmol) in acetonitrile-H2O (1:1.5, 12 mL). After stirring for 3 h at room temperature (RT), the reaction mixture was treated with aqueous (aq.) Na2S2O3 (10 mL) and saturated (sat.) aq. NaHCO3 (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure to afford a crude residue. Purification by silica gel column chromatography (CH2Cl2) afforded (+)-12 (44 mg, 55%).
(+)-12: Colorless oil; [α] +23.1 (c 0.06, CHCl3); IR (liquid film) νmax 3524 (OH), 1715 (C=O), 1660 (C=C) cm−1; 1H-NMR (CDCl3, 400 MHz, ppm) δ 1.25–1.70 (10H, m), 3.46 (1H, d, J = 10.7 Hz, 6-OH), 3.84 (3H, s, COOMe), 4.59 (1H, dd, J = 4.0, 2.8 Hz, H-5), 4.67 (1H, dddd, J = 4.9, 4.1, 1.9, 1.1 Hz, H-4), 4.74 (1H, br dd, J = 10.7, 2.7 Hz, H-6), 4.84 (1H, dd, J = 4.5, 3.3 Hz, H-3), 6.88 (1H, dd, J = 3.3, 1.0 Hz, H-2); 13C-NMR (CDCl3, 100 MHz, ppm) δ 23.6, 23.8, 24.7, 35.8, 37.7, 45.1, 52.4, 66.8, 71.3, 75.4, 112.8, 129.8, 135.6, 165.9; HRMS m/z calcd. for C14H19O579Br (M)+, 346.0416; found, 346.0415, m/z calcd. for C14H19O581Br (M)+, 348.0396; found, 348.0391.
4.2. Methyl (3R,4R,5S,6S)-3,4-O-cyclohexilidene-3,4-dihydroxy-5,6-epoxy-1-cyclohex-ene-1-carboxy-late (−)-11
To a solution of 1,1,1,3,3,3-hexamethyldisilazane (0.19 mL, 0.81 mmol) in dry THF (3 mL), 1.6 M n-BuLi in hexane (0.56 mL, 0.81 mmol) was added at −78 °C to give LHMDS. After 30 min the prepared LHMDS solution was added dropwise to a solution of (+)-12 (0.28 g, 0.81 mmol) in THF (5 mL) at −78 °C through a steel cannula under argon atmosphere. After stirring the reaction mixture for 1 h at −78 °C, followed by warming to RT over 1 h, the reaction mixture was treated with sat. aq. NH4Cl (20 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over MgSO4 and filtered, and the solvent was removed under reduced pressure to afford a crude residue. Purification by silica gel column chromatography (Hexane–EtOAc, 3:1) afforded (−)-11 (0.18 g, 83%) as crystals. (−)-11: White crystals (CH2Cl2); mp 81–84 °C; [α] −5.4 (c 0.98, CHCl3); IR (KBr) νmax 1731 (C=O), 1650 (C=C) cm−1; 1H-NMR (CDCl3, 400 MHz, ppm) δ 1.35–1.80 (10H, m), 3.74 (1H, ddd, J = 4.1, 2.8, 2.0 Hz, H-5), 3.83 (3H, s, COOMe), 4.03 (1H, dd, J = 4.1, 2.1 Hz, H-6), 4.48 (1H, dd, J = 6.6, 2.8 Hz, H-4), 4.73 (1H, ddd, J = 6.6, 5.5, 2.0 Hz, H-3), 7.14 (1H, dd, J = 5.5, 2.0 Hz, H-2); 13C-NMR (CDCl3, 100 MHz, ppm) δ 23.9, 24.1, 25.1, 34.5, 36.8, 49.0, 52.4, 56.2, 69.7, 72.1, 109.4, 132.5, 137.5, 165.4; HRMS m/z calcd. for C14H18O5 (M)+, 266.1155; found. 266.1150.
4.3. Methyl (3R,4R,5R,6R)-6-chloro-3,4-O-cyclohexylidene-3,4,5-trihydroxy-1-cyclo hexene-1-carbo-xylate (−)-9
To a solution of (−)-11 (55.9 mg, 0.21 mmol) in dry Et2O (1 mL), 1 M HCl in Et2O (0.31 mL, 0.31 mmol) was added at 0 °C. After stirring for 1 h, the solvent was removed under vacuum to afford a crude residue that was purified by preparative TLC (Hexane–EtOAc, 4:1) to afford (−)-9 (57.5 mg, 90%) as colorless crystals. (−)-9: Colorless crystals (CHCl3) mp 125–128 °C; [α] −165.0 (c 0.3, CHCl3); IR (KBr) νmax 3360 (OH), 1725 (C=O), 1649 (C=C) cm−1; 1H-NMR (CDCl3, 400 MHz, ppm) δ 1.20–1.80 (10H, m), 2.66 (1H, d, J = 2.4 Hz, 5-OH), 3.83 (3H, s, COOMe), 4.30 (1H, ddd, J = 3.9, 3.8, 2.3 Hz, H-5), 4.70 (1H, ddd, J = 7.5, 3.9, 0.4 Hz, H-4), 4.77 (1H, dd, J = 7.5, 3.0 Hz, H-3), 5.04 (1H, d, J = 3.9 Hz, H-6), 7.18 (1H, d, J = 3.2 Hz, H-2); 13C-NMR (CDCl3, 100 MHz, ppm) δ 23.5, 23.9, 25.0, 33.4, 36.1, 50.9, 52.4, 67.2, 69.6, 71.4, 110.8, 130.2, 137.8, 164.9; HRMS m/z calcd. for C14H19O535Cl (M)+ 302.0921, found 302.0925, m/z calcd. for C14H19O537Cl (M)+, 304.0891; found, 304.0903.
4.4. Synthesis of Anti-Epoxide (−)-10
To a solution of (+)-13 (60.0 mg, 0.24 mmol) and NaHCO3 (0.20 g, 2.4 mmol) in trifluoroacetone-H2O (1:1, 2 mL) at −15 °C, Oxone® was added every 15 min (four portions, each portion 0.073 g, 0.12 mmol) with stirring. After 3 h, tert-butyl methyl ether (TBME) (10 mL) was added and the reaction mixture was filtered through Celite. The filtrate was treated with sat. aq. NaHCO3 (10 mL) and extracted with TBME (3 × 10 mL). The combined organic layers were dried over MgSO4, filtered, and the solvent was removed under reduced pressure to afford a crude residue, which was purified by column chromatography (Hexane–EtOAc, 5:1) to afford (−)-10 (46.0 mg, 72%) as a colorless oil. (−)-10: Colorless oil; [α] −20.4 (c 0.29, CHCl3); IR (liquid film) νmax 1730 (C=O), 1647 (C=C) cm−1; 1H-NMR (CDCl3, 500 MHz, ppm) δ 1.35–1.70 (10H, m), 3.69 (1H, br dd, J = 3.7, 2.1 Hz, H-5), 3.84 (3H, s, COOMe), 3.99 (1H, ddd, J = 3.7, 1.6, 0.7 Hz, H-6), 4.58 (1H, dd, J = 6.9, 2.3 Hz, H-3), 4.80 (1H, br d, J = 6.9 Hz, H-4), 6.83 (1H, m, H-2); 13C-NMR (CDCl3, 125 MHz, ppm) δ 23.7, 23.9, 24.9, 35.3, 37.5, 46.1, 49.3, 52.3, 70.0, 70.8, 111.7, 127.2, 140.3, 165.5; HRMS m/z calcd. for C14H18O5 (M)+, 266.1156; found, 266.1158.
4.5. Bromohydrination of (−)-12
Diene (−)-13 (1.5 g, 5.9 mmol) was converted to (−)-12 (1.2 g, 57% yield) using the same procedure as for (+)-12.
4.6. Synthesis of (+)-11
Bromohydrin (−)-12 (556 mg, 4.3 mmol) was converted to (+)-11 (319 mg, 75% yield) using the same procedure described above.
4.7. Synthesis of (+)-9
Epoxide (+)-11 (386 mg, 1.4 mmol) was converted to chlorohydrin (+)-9 (395 mg, 90% yield) using the same procedure described above.
4.8. Synthesis of Anti-Epoxide (+)-10
Diene (−)-13 (1.5 g, 5.9 mmol) was oxidized to (+)-10 (1.2 g, 75% yield) using a procedure similar to that for (−)-10. (+)-10: Colorless oil; [α] +24.7 (c 0.68, CHCl3); IR (liquid film) νmax 1722 (C=O), 1654 (C=C) cm−1; 1H-NMR (CDCl3, 600 MHz, ppm) δ 1.34–1.70 (10H, m), 3.68 (1H, dd, J = 3.6, 2.4 Hz, H-5), 3.84 (3H, s, COOMe), 3.99 (1H, ddd, J = 3.8, 1.7, 0.6 Hz, H-6), 4.57 (1H, dd, J = 6.8, 2.4 Hz, H-3), 4.80 (1H, br d, J = 6.8 Hz, H-4), 6.83 (1H, m, H-2); 13C-NMR (CDCl3, 150 MHz, ppm) δ 23.7, 23.9, 24.8, 35.2, 37.4, 46.1, 49.3, 52.3, 70.0, 70.8, 111.6, 127.1, 140.3, 165.5; HRMS m/z calcd. for C14H18O5 (M)+, 266.1156; found, 266.1161.
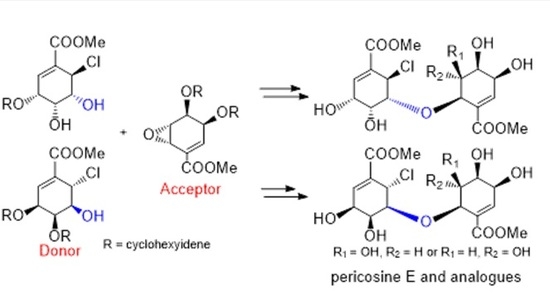
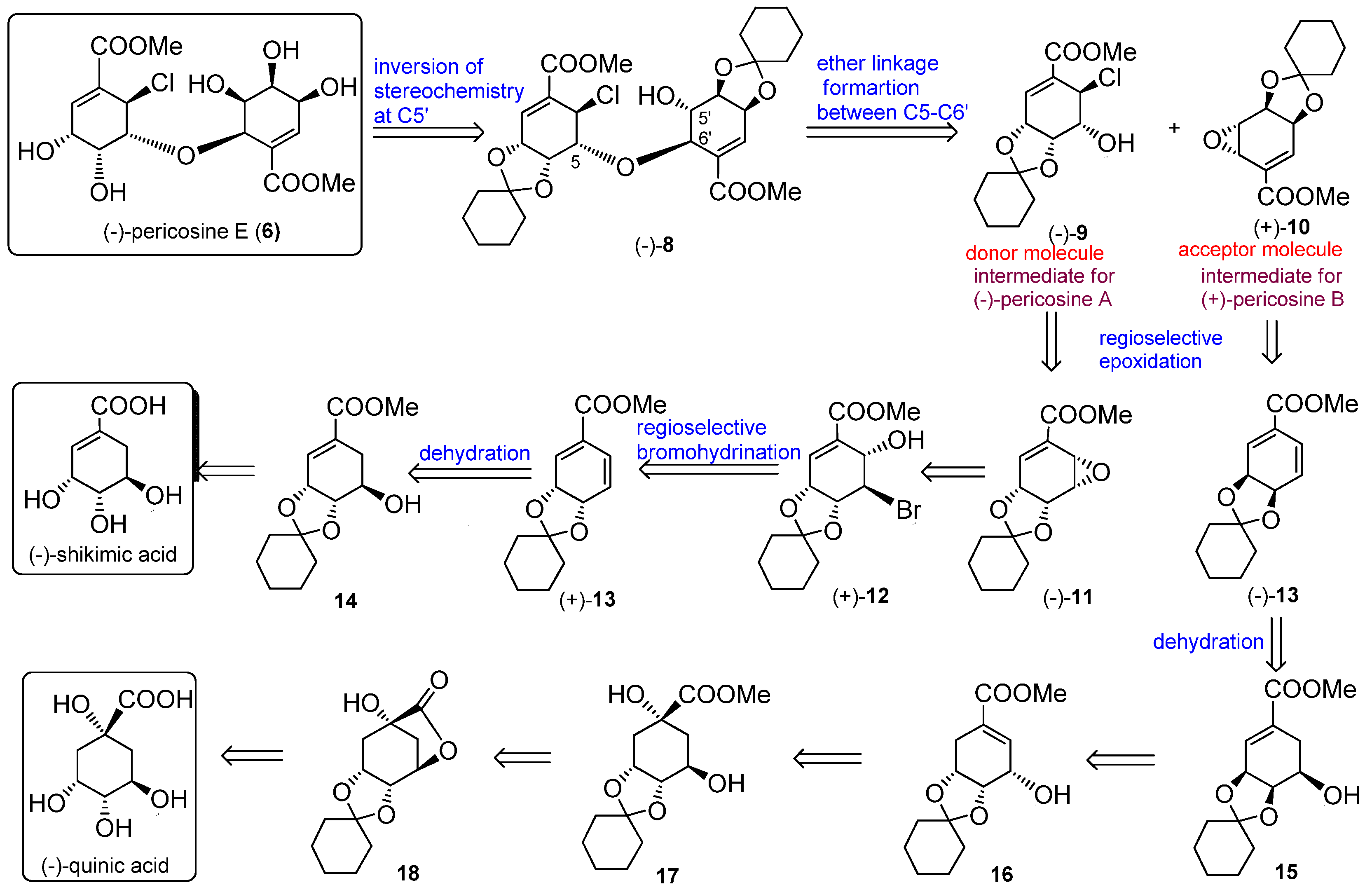
4.9. Synthesis of (−)-8 from (−)-9 and (+)-10
To a solution of chlorohydrin (–)-9 (73.0 mg, 0.24 mmol) and epoxide (+)-10 (53.5 mg, 0.20 mmol) in CH2Cl2 (0.6 mL), BF3·Et2O (5 μL, 0.019 mmol) was added at 0 °C. After stirring for 10 min at RT, the reaction mixture was treated with Et3N (5 μL, 0.035 mmol) and concentrated under vacuum to afford a crude residue, which was purified by silica gel column chromatography (Hexane–EtOAc, 3:1) to afford (–)-8 (58.9 mg, 52%) as a white amorphous solid. (−)-8: White amorphous solid; [α] −68.3 (c 0.21, CHCl3); IR (liquid film) νmax 3431 (OH), 1729 (C=O), 1657 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 1.30–1.80 (20H, m), 3.68 (3H, s, COOMe-8′), 3.81 (3H, s, COOMe-8), 4.01 (1H, ddd, J = 7.3, 6.1, 3.6 Hz, H-5′), 4.21 (1H, dd, J = 7.3, 6.2 Hz, H-4′), 4.37 (1H, ddd, J = 6.2, 1.4, 1,2 Hz, H-6′), 4.49 (1H, dd, J = 4.7, 3.9 Hz, H-5), 4.69 (1H, ddd, J = 6.2, 3.8, 1.2 Hz, H-3′), 4.74 (1H, d, J = 3.8 Hz, 5′-OH), 4.84 (1H, dd, J = 7.0, 3.9 Hz, H-4), 4.88 (1H, ddd, J = 7.0, 2.9, 0.6 Hz, H-3), 5.15 (1H, d, J = 4.4 Hz, H-6), 6.54 (1H, dd, J = 3.8, 1.4 Hz, H-2′), 6.95 (1H, d, J = 2.9 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 24.4, 24.4, 24.7, 24.7, 25.7, 25.8, 34.9, 36.3, 36.6, 38.8, 51.6, 52.2, 52.4, 71.0, 71.6, 72.6, 73.7, 76.3, 78.9, 79.5, 111.4, 111.6, 131.5, 133.2, 134.8, 138.8, 165.8, 167.2; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2073.
4.10. Dess-Martin Oxidation of (−)-8 Followed by NaBH4 Reduction
To a solution of alcohol (−)-8 (0.22 g, 0.38 mmol) in CH2Cl2 (8 mL), DMP (0.20 g, 0.46 mmol) was added at 0 °C, with stirring. The reaction mixture was warmed to RT and stirred for 4 h. The reaction mixture was treated with aqueous Na2S2O3 (20 mL) and sat. aq. NaHCO3 (10 mL), then extracted with TBME (3 × 20 mL). The combined organic layers were washed with brine (30 mL) and water (30 mL), dried over MgSO4, filtered, and concentrated under vacuum to afford an inseparable mixture (0.20 g) of enone 19 and undefined compound 19u, whose carbon skeleton is same as 19 suggested by 2D-NMR analysis of the mixture. Crude 19 and 19u: Oil; HRMS m/z calcd. for C28H35O1035Cl (M)+ of 19, 566.1919; found, 566.1923; 1H-NMR (CDCl3, 600 MHz, ppm) δ 19: 1.20–2.00 (20H, m), 3.77 (3H, s, COOMe), 3.85 (3H, s, COOMe), 4.30 (1H, dd, J = 6.2, 3.2 Hz, H-5), 4.58 (1H, dd, J = 6.5, 0.9 Hz, H-4′), 4.70 (1H, ddd, J = 5.9, 3.0, 1.2 Hz, H-3), 4.77 (1H, dd, J = 5.9, 3.2 Hz, H-4), 4.96 (1H, ddd, J = 6.5, 4.1, 0.9 Hz, H-3′), 5.05 (1H, br d, J = 6.2 Hz, H-6), 5.12 (1H, m, H-6′), 6.75 (1H, br d, J = 3.0 Hz, H-2), 6.83 (1H, dd, J = 4.1, 1.8 Hz, H-2′); 19a: 1.20–2.00 (20H, m), 3.79 (3H, s, COOMe), 3.83 (3H, s, COOMe), 4.15 (1H, ddd, J = 5.9, 1.5, 0.9 Hz, H-4′), 4.21 (1H, br s, H-6′), 4.45 (1H, t, J = 4.4 Hz, H-5), 4.69 (1H, ddd, J = 5.9, 3.5, 0.6 Hz, H-3′), 4.78 (1H, dd, J = 7.6, 2.7 Hz, H-3), 4.90 (1H, dd, J = 7.6, 4.4 Hz, H-4), 5.14 (1H, d, J = 4.4 Hz, H-6), 6.78 (1H, dd, J = 3.5, 0.9 Hz, H-2′), 7.03 (1H, d, J = 2.7 Hz, H-2); 13C-NMR (CDCl3, 150 MHz, ppm) δ 23.52 (CH2), 23.54 (CH2), 23.66 (CH2), 23.75 (CH2), 23.78 (CH2), 23.83 (CH2), 23.88 (CH2), 23.92 (CH2), 24.86 (CH2), 24.91 (CH2), 25.0 (CH2), 25.1 (CH2), 29.7 (CH2), 34.7 (CH2), 35.4 (CH2), 35.6 (CH2), 36.0 (CH2), 36.5 (CH2), 37.0 (CH2), 37.3 (CH2), 49.2 (CH, 19u), 52.22 (CH3), 52.26 (CH3), 52.34 (CH3), 52.4 (CH3), 53.1 (CH, 19), 70.4 (CH, 19u), 71.3 (CH, 19), 71.8 (CH, 19), 72.6 (CH, 19u), 73.2 (CH, 19), 74.8 (CH, 19), 76.2 (CH, 19), 77.2 (CH, 19), 77.7 (CH, 19a), 78.8 (CH, 19), 79.1 (CH, 19u), 79.8 (CH, 19u), 93.7 (Cq, 19), 111.3 (Cq), 111.4 (Cq), 112.3 (Cq), 113.7 (Cq), 129.1 (Cq, 19u), 129.7 (Cq, 19u), 130.8 (Cq, 19), 133.6 (CH, 19), 134.0 (Cq, 19), 136.9 (CH, 19), 137.7 (CH, 19u), 139.6 (CH, 19u), 164.9 (Cq, 19u), 165.2 (Cq, 19), 165.5 (Cq, 19), 166.9 (Cq, 19u), 201.2 (Cq, 19).
To a solution of NaBH4 (13.2 mg, 0.35 mmol) in MeOH (2.5 mL), a solution of crude 19 and 19u (202 mg) in MeOH (10 mL) was added dropwise at 0 °C. After stirring for 30 min, the reaction mixture was treated with sat. aq. NH4Cl (30 mL) and extracted with CH2Cl2 (3 × 30 mL). The organic layer was washed with brine (30 mL), dried over MgSO4, filtered, and concentrated under vacuum to afford a crude residue. Purification by silica gel column chromatography (Hexane–EtOAc, 3:1) afforded (−)-20 (74.0 mg, 34% from 7) as a white amorphous solid. (−)-20: White amorphous solid; [α] −67.5 (c 0.30, CHCl3); IR (liquid film) νmax 3421 (OH), 1719 (C=O), 1656 (C=C) cm−1; 1H-NMR (CDCl3, 600 MHz, ppm) δ 1.20–2.00 (20H, m), 3.71 (1H, m, H-5′), 3.77 (3H, s, COOMe), 3.86 (3H, s, COOMe), 4.34 (1H, dd, J = 5.0, 4.7 Hz, H-5), 4.43–4.45 (1H, m, H-4′), 4.55 (1H, br d, J = 6.5 Hz, H-6′), 4.60 (1H, ddd, J = 5.6, 3.5, 0.6 Hz, H-3′), 4.77 (1H, dd, J = 7.6, 2.7 Hz, H-3), 4.88 (1H, dd, J = 7.6, 4.7 Hz, H-4), 5.05 (1H, br s, OH), 5.07 (1H, d, J = 5.0 Hz, H-6), 6.78 (1H, dd, J = 3.2, 0.9 Hz, H-2′), 6.69 (1H, d, J = 2.7 Hz, H-2); 13C-NMR (CDCl3, 150 MHz, ppm) δ 23.50, 23.55, 23.76, 23.83, 24.9, 25.1, 33.1, 35.0, 36.0, 37.5, 50.5, 52.2, 52.4, 68.6, 70.6, 72.1, 72.3, 74.8, 74.9, 78.9, 111.1, 112.0, 129.2, 129.8, 138.6, 139.5, 165.1, 166.7; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2076.
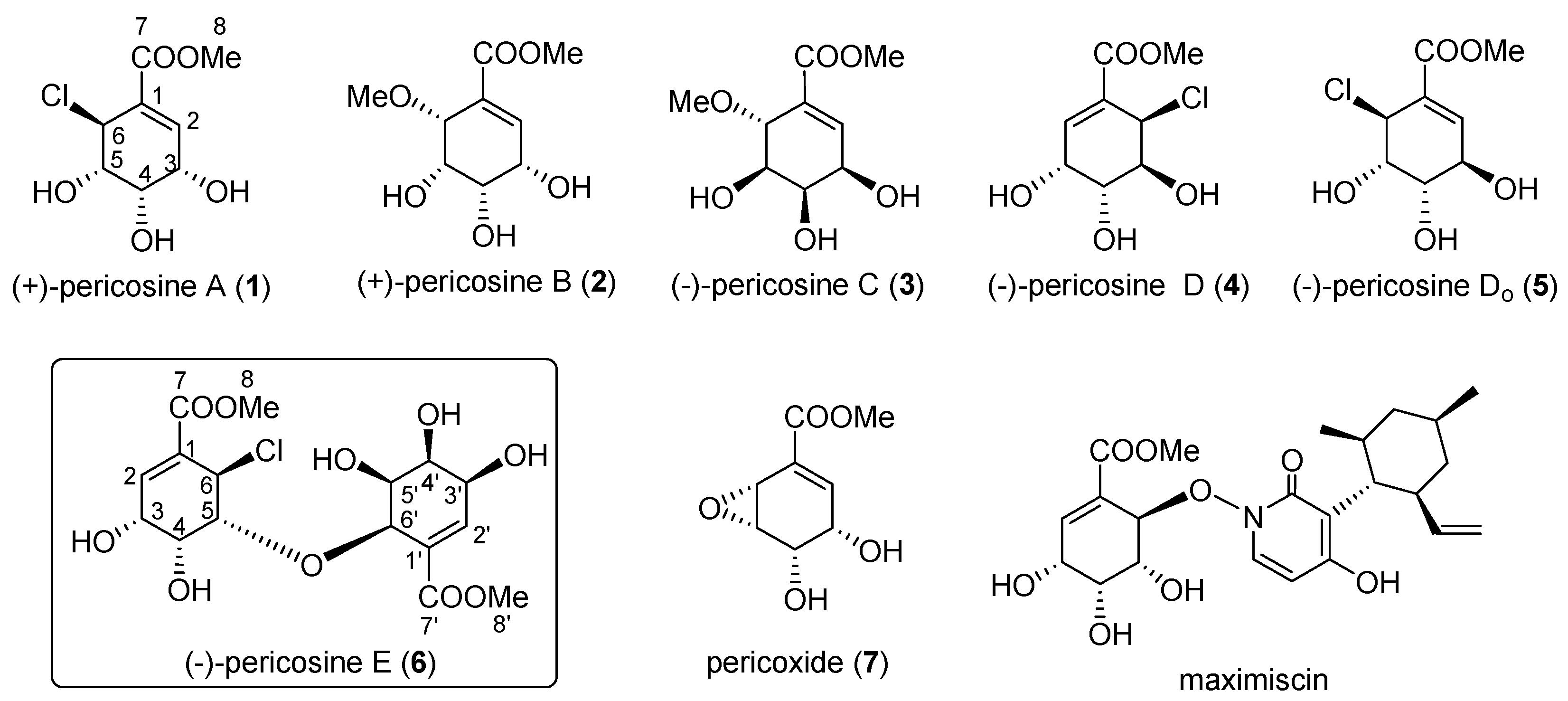
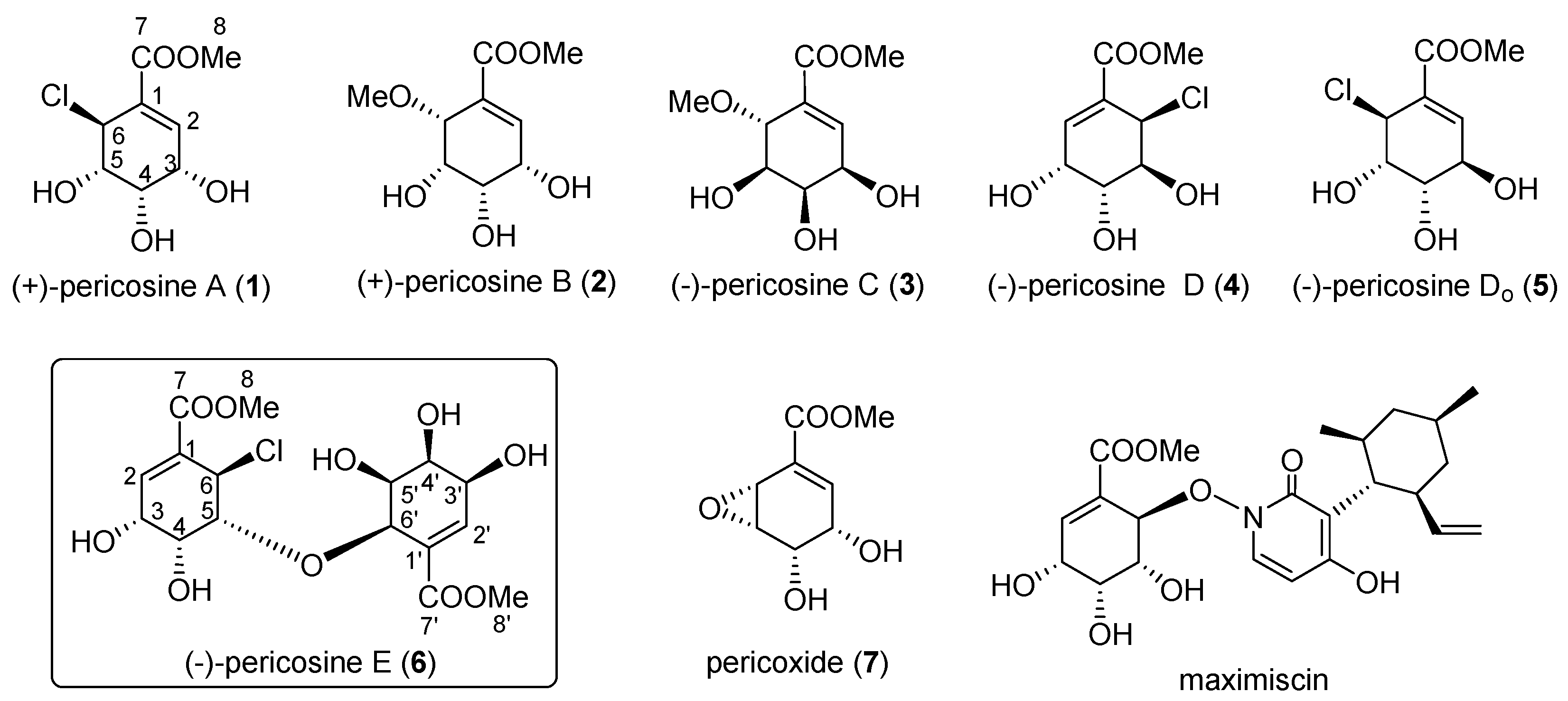
4.11. Synthesis of (−)-Pericosine E (6)
To a solution of alcohol (−)-20 (13.3 mg, 0.023 mmol) in MeOH (0.2 mL), TFA (1.8 mL) was added dropwise dropwise at 0 °C. After stirring for 5 h at RT, the reaction mixture was concentrated under vacuum to afford white crystals. The product was purified by preparative TLC (MeOH-CH2Cl2, 1:9) to afford (−)-6 (9.0 mg, 94%). (−)-6: white crystal; [α] −68.3 (c 0.06, EtOH); IR (liquid film) νmax 3431 (OH), 1729 (C=O), 1657 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 3.76 (1H, dd, J = 4.1, 2.1 Hz, H-5′), 3.790 (3H, s, COOMe), 3.793 (3H, s, COOMe), 4.06 (1H, br s, H-4′), 4.18 (1H, br d, J = 11.1 Hz, OH), 4.20–4.22 (1H, m, H-4′), 4.21 (1H, br s, H-3), 4.23–4.25 (1H, m, H-3′), 4.34–4.36 (1H, m, H-5), 4.53 (1H, d, J = 4.1 Hz, H-6′), 5.23 (1H, d, J = 2.9 Hz, H-6), 5.33 (1H, br s, OH), 5.61 (1H, br dd, J = 8.8, 0.5 Hz, OH), 6.74 (1H, dd, J = 2.4, 1.4 Hz, H-2′), 7.01 (1H, d, J = 4.4 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 52.45 (CH3), 52.49 (CH3), 53.1 (CH2), 65.5 (CH2), 66.8 (CH2), 69.3 (CH2), 70.5 (CH2), 72.4 (CH2), 77.1 (CH2), 85.6 (CH2), 129.3 (C), 130.4 (C), 143.2 (CH2), 143.5 (CH2), 166.1 (Cq), 166.9 (Cq); HRFABMS m/z calcd. for C16H22O1035Cl (M + H)+, 409.0901; found, 409.0908.
Spectroscopic data of natural
6 [
17]: Oil; [α]
D −31.5 (
c 0.43, EtOH) (racemate as plate from MeOH; mp 213–215 °C; [α]
D 0); IR (liquid film) ν
max 3326 (OH), 1721 (C=O), 1636 (C=C) cm
−1;
1H-NMR (acetone-
d6, 500 MHz, ppm) δ 3.76 (1H, br s, H-5′), 3.79 (3H, s, COOMe), 3.79 (3H, s, COOMe), 4.07 (1H, br s, H-4′), 4.22 (1H, m, H-2), 4.23 (1H, br s, 4′-OH), 4.25 (1H, br s, H-3), 4.26 (1H, br s, H-3′), 4.36 (1H, m, H-5), 4.53 (1H, d,
J = 4.1 Hz, H-6′), 5.24 (1H, d,
J = 3.0 Hz, H-6), 5.37 (1H, br s, 4-OH), 5.64 (1H, br s, 5′-OH), 6.74 (1H, t,
J = 1.8 Hz, H-2′), 7.01 (1H, d,
J = 3.9 Hz, H-2);
13C-NMR (acetone-
d6, 125 MHz, ppm) δ 52.44 (CH
3-8), 52.48 (CH
3-8′), 53.06 (CH
2-6), 65.57 (CH
2-3), 66.75 (CH
2-4), 69.22 (CH
2-3′), 70.43 (CH
2-5′), 72.42 (CH
2-4′), 77.07 (CH
2-6′), 85.52 (CH
2-6), 129.23 (Cq-1), 129.91 (Cq-1′), 143.17 (CH
2-2), 143.50 (CH
2-2′), 166.09 (CH
3-8), 166.87 (CH
3-8′); HRMS
m/
z calcd. for C
16H
22O
1035Cl (M + H)
+, 409.0900; found, 409.0904.
4.12. Synthesis of (−)-21
To a solution of alcohol (−)-8 (21.6 mg, 0.038 mmol) in MeOH (0.2 mL), TFA (1.8 mL) was added dropwise at 0 °C. After stirring for 3 days at RT, the reaction mixture was concentrated under vacuum to afford white crystals. The product was purified by preparative TLC (MeOH-CH2Cl2, 1:9) to give (–)-21 (8.6 mg, 55%). (−)-21: white crystal; [α] −47.1 (c 0.09, EtOH); IR (liquid film) νmax 3344 (OH), 1723 (C=O), 1657 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 3.72 (3H, s, COOMe), 3.80 (3H, s, COOMe), 4.00 (1H, br dddd, J = 5.3, 4.1, 1.2, 0.8 Hz, H-4′), 4.11 (1H, dd, J = 5.3, 2.4 Hz, H-4), 4.15 (1H, br t, J = 5.0 Hz, H-3), 4.24 (1H, br dd, J = 2.7, 2.6 Hz, H-5), 4.25 (1H, br d, 3.2 Hz, H-6′), 64.46 (1H, ddd, J = 4.1, 2.7, 0.9 Hz, H-3′), 4.50 (1H, dd, J = 5.3, 3.3 Hz, H-5′), 5.23 (1H, d, J = 3.0 Hz, H-6), 6.79 (1H, ddd, J = 2.7, 1.2, 0.6 Hz, H-2′), 7.01 (1H, d, J = 4.7 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 52.24 (CH3), 52.52 (CH3), 52.95 (CH2), 65.67 (CH2), 65.76 (CH2), 66.6 (CH2), 69.06 (CH2), 70.11 (CH2), 76.69 (CH2), 83.54 (CH2), 129.47 (C), 129.54 (C), 142.5 (CH2), 142.7 (CH2), 166.0 (Cq), 166.9 (Cq); HRMS m/z calcd. for C16H21O1035Cl (M)+, 408.0823; found, 408.0821.
4.13. Synthesis of (+)-24
To a solution of (+)-10 (0.22 g, 0.83 mmol) and (+)-9 (0.26 g, 0.86 mmol) in CH2Cl2 (3.0 mL), BF3·Et2O (3 μL, 0.011 mmol) was added at 0 °C, then the reaction mixture was stirred at RT. After 10 min, Et3N (20 μL, 0.175 mmol) was added and the mixture was stirred for another 3 h. Solvent removal under reduced pressure gave a crude residue, which was purified by silica gel column chromatography (Hexane–EtOAc, 3:1) to afford (+)-24 as an amorphous solid (0.15 g, 32%). (+)-24: Rf 0.14 (Hexane–EtOAc, 3:1); [α] +131.1 (c 0.135, CHCl3); IR (liquid film) νmax 3471 (OH), 1724 (C=O), 1659 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 1.26–1.70 (20H, m), 3.78 (3H, s, COOMe), 3.80 (3H, s, COOMe), 4.03 (1H, dd, J = 9.6, 4.7 Hz, H-5′), 4.23 (1H, dd, J = 5.9, 5.6 Hz, H-4′), 4.32 (1H, dd, J = 4.7, 3.8 Hz, H-5), 4.35 (1H, d, J = 4.1 Hz, H-6′), 4.57 (1H, d, J = 4.4 Hz, OH), 4.67 (1H, ddd, J = 6.1, 3.8, 0.9 Hz, H-3′), 4.69 (1H, dd, J = 6.7, 3.8 Hz, H-4), 4.78 (1H, dd, J = 6.5, 3.0 Hz, H-3), 5.16 (1H, d, J = 4.7 Hz, H-6), 6.63 (1H, d, J = 3.9 Hz, H-2′), 6.93 (1H, d, J = 2.9 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 24.6 (2C), 24.8 (2C), 25.8, 25.9, 35.6, 36.0, 37.0, 38.4, 52.2, 52.4, 52.7, 70.7, 71.1, 71.2, 72.6, 76.2, 77.3, 79.5, 111.2, 111.3, 131.4, 133.2, 135.0, 138.9, 165.9, 167.3; HRMS m/z calcd. for C28H37O10 35Cl (M)+, 568.2075; found, 568.2074.
4.14. Synthesis of (+)-25
To a solution of (+)-24 (170 mg, 0.30 mmol) in CH2Cl2 (5 mL), DMP (190 mg, 0.39 mmol) was added at 0 °C, and the mixture was stirred for 4 h at RT. The reaction was quenched by the addition of sat. aq. Na2S2SO4 and sat. aq. NaHCO3 (10 mL) and extracted with TBME (3 × 20 mL). The combined organic layers were washed with brine (30 mL), dried over MgSO4, filtered, and the solvent was removed under reduced pressure to give a crude residue (180 mg). Without purification, the residue was taken up in methanol (5 mL) and the methanol solution (1 mL) of NaBH4 (11 mg, 0.29 mmol) was added in four portions at 0 °C. After 30 min, the reaction mixture was quenched by the addition of sat aq. NH4Cl (30 mL), extracted with CH2Cl2 (3 × 30 mL). The organic layer was washed with brine, dried over MgSO4, filtered, and the solvent was removed under reduced pressure to give a crude residue, which was purified by silica gel column chromatography (Hexane–EtOAc, 3:1) to afford (+)-25 (100 mg, 59% in two steps) as an oil. (+)-25: Rf 0.14 (Hexane–EtOAc, 3:1); [α] +60.2 (c 0.665, CHCl3); IR (liquid film) νmax 3460 (OH), 1722 (C=O), 1653 (C=C) cm−1; 1H-NMR (acetone-d6, 400 MHz, ppm) δ 1.20–1.70 (20H, m), 3.79 (3H, s, COOMe), 3.81 (3H, s, COOMe), 3.78–3.81 (1H, m, H-5′), 4.37 (1H, dd, J = 5.3, 3.9 Hz, H-4′), 4.48 (1H, br dd, J = 6.4, 3.5 Hz, H-4), 4.58 (1H, d, J = 4.9 Hz, H-6′), 4.75 (1H, d, J = 3.7 Hz, H-3′), 4.76 (1H, d, J = 3.6 Hz, H-5), 4.81 (1H, ddd, J = 6.8, 2.6, 0.8 Hz, H-3), 5.21 (1H, d, J = 5.3 Hz, H-6), 6.76 (1H, dd, J = 3.9, 0.8 Hz, H-2′), 6.88 (1H, d, J = 2.7 Hz, H-2); 13C-NMR (acetone-d6, 100 MHz, ppm) δ 24.5, 24.6, 24.6, 24.8, 25.8, 25.8, 35.1, 35.6, 36.7, 37.7, 52.3, 52.6, 53.3, 69.0, 71.4, 72.7, 72.7, 74.6, 75.1, 79.7, 111.1, 111.8, 131.1, 131.1, 138.2, 140.0, 166.4, 166.8; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2077.
4.15. Microwave-Aided Deprotection toward (+)-22
To a methanol solution (0.2 mL) of 25 (23.7 mg, 0.042 mmol) in a microwave vial, TFA (1.8 mL) was added at 0 °C. The vial was sealed and irradiated in the MW reactor at 100 °C for 30 min. After cooling, the reaction mixture was condensed under reduced pressure to give a crude residue, which was purified by silica gel column chromatography (MeOH–CH2Cl2, 1:9) to afford (+)-22 (8.0 mg, 47%) as an oil. (+)-22: Rf 0.3 (MeOH-CH2Cl2, 1:9); [α] +5.7 (c 0.12, EtOH); IR (liquid film) νmax 3404 (OH), 1713 (C=O), 1651 (C=C) cm−1; 1H-NMR (acetone-d6, 400 MHz, ppm) δ 3.64-3.82 (2H, m, OH, H-5), 3.78 (3H, s, COOMe), 3.80 (3H, s, COOMe), 3.84-3.98 (2H, m, OH), 3.92 (1H, br d, J = 7.3 Hz, OH), 3.99 (1H, s, H-4′), 4.02–4.10 (2H, m, H-4, OH), 4.10–4.24 (2H, m, H-3, OH), 4.27 (1H, br s, H-3′), 4.48 (1H, dd, J = 3.1, 2.3 Hz, H-5), 4.57 (1H, d, J = 4.3 Hz, H-6′), 5.38 (1H, d, J = 3.3 Hz, H-6), 6.80 (1H, br d, J = 1.0 Hz, H-2′), 7.04 (1H, d, J = 4.7 Hz, H-2); 13C-NMR (acetone-d6, 100 MHz, ppm) δ 51.49 51.52, 52.9, 64.9, 65.3, 68.2, 68.4, 71.2, 73.1, 82.2, 129.0, 129.1, 141.6, 142.5, 165.0, 166.4; HRMS m/z calcd. for C16H22O1035Cl (M + H)+, 409.0901; found, 409.0896.
4.16. Microwave Aided Deprotection toward (+)-23
Using the same procedure as for (+)-22, (+)-24 (22.6 mg, 0.040 mmol) was converted to (+)-23 (10.0 mg, 62%). (+)-23: white crystals; Rf 0.11 (MeOH-CH2Cl2, 1:9); [α] +75.4 (c 0.340, EtOH); IR (liquid film) νmax 3392 (OH), 1714 (C=O), 1652 (C=C) cm−1; 1H-NMR (acetone-d6, 300 MHz, ppm) δ 3.75 (1H, br s, H-4′), 3.79 (6H, s, COOMe ×2), 4.02 (1H, br s, H-5′), 4.10 (1H, br s, H-4), 4.20 (1H, br s, H-3), 4.37 (2H, br m, H-3′, H-6′), 4.43 (1H, br s, H-5), 5.24 (1H, br d, J = 3.2 Hz, H-6), 6.80 (1H, br s, H-2′), 6.99 (1H, br d, J = 2.9 Hz, H-2); 13C-NMR (acetone-d6, 75 MHz, ppm) δ 52.5, 52.5, 54.5, 66.3, 66.4, 66.6, 70.5, 71.9, 77.1, 83.7, 129.7, 130.7, 141.3, 142.8, 166.1, 167.7; HRMS m/z calcd. for C16H21O1035Cl (M)+, 408.0823; found, 408.0819.
4.17. Synthesis of (+)-8
Using the procedure described for (−)-8, (+)-8 (0.092 g, 39%) was prepared from (+)-9 (0.15 g, 0.5 mmol) and (−)-10 (0.11 g, 0.4 mmol). (+)-8: White amorphous solid; [α] +72.7 (c 0.42, CHCl3); IR (liquid film) νmax 3431 (OH), 1729 (C=O), 1653 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 1.28–1.80 (20H, m), 3.68 (3H, s, COOMe-8′), 3.81 (3H, s, COOMe-8), 4.03 (1H, ddd, J = 7.7, 6.2, 3.5 Hz, H-5′), 4.21 (1H, dd, J = 7.7, 6.2 Hz, H-4′), 4.37 (1H, ddd, J = 6.2, 1.5, 1,2 Hz, H-6′), 4.49 (1H, dd, J = 4.4, 4.1 Hz, H-5), 4.68(1H, ddd, J = 6.1, 4.1, 1.2 Hz, H-3′), 4.73(1H, d, J = 3.5 Hz, OH), 4.84 (1H, dd, J = 7.0, 3.9 Hz, H-4), 4.89 (1H, ddd, J = 7.0, 3.0, 0.6 Hz, H-3), 5.15 (1H, d, J = 4.7 Hz, H-6), 6.54 (1H, dd, J = 4.1, 1.4 Hz, H-2′), 6.95 (1H, d, J = 2.9 Hz, H-2): 13C-NMR (acetone-d6, 150 MHz, ppm) δ 24.4, 24.4, 24.7, 24.7, 25.7, 25.8, 34.9, 36.3, 36.6, 38.8, 51.6, 52.2, 52.4, 71.0, 71.6, 72.6, 73.7, 76.3, 78.9, 79.5, 111.4, 111.6, 131.5, 133.2, 134.8, 138.8, 165.8, 167.2; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2074.
4.18. Synthesis of (+)-20
Using the same procedure described for (−)-20, (+)-20 (17.0 mg, 22%) was prepared from (+)-8 (78.8 mg, 0.14 mmol). (+)-20: White amorphous solid; [α] +62.1 (c 0.355, CHCl3); IR (liquid film) νmax 3423 (OH), 1722 (C=O), 1656 (C=C) cm−1; 1H-NMR (CDCl3, 300 MHz, ppm) δ 1.20–2.00 (20H, m), 3.70–3.80 (1H, m, H-5′), 3.77 (3H, s, COOMe), 3.86 (3H, s, COOMe), 4.34 (1H, t, J = 4.6 Hz, H-5), 4.42–4.46 (1H, m, H-4′), 4.55 (1H, br d, J = 6.9 Hz, H-6′), 4.61 (1H, dd, J = 5.5, 3.3 Hz, H-3′), 4.77 (1H, dd, J = 7.5, 2.3 Hz, H-3), 4.88 (1H, dd, J = 7.5, 4.3 Hz, H-4), 5.06 (1H, br s, OH), 5.07 (1H, d, J = 4.8 Hz, H-6), 6.78 (1H, dd, J = 3.4, 0.9 Hz, H-2′), 6.98 (1H, d, J = 2.3 Hz, H-2); 13C-NMR (CDCl3, 75 MHz, ppm) δ 23.5, 23.5, 23.8, 23.8, 24.9, 25.0, 33.1, 35.0, 36.0, 37.5, 50.5, 52.2, 52.5, 68.6, 70.6, 72.1, 72.3, 74.8, 74.8, 78.9, 111.1, 112.0, 129.2, 129.8, 138.6, 139.5, 165.1, 166.8; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2079.
4.19. Synthesis of (+)-6
Using the same procedure described for (−)-6, (+)-6 (11.3 mg, 90%) was prepared from (+)-20 (17.4 mg, 0.031 mmol). (+)-6: white crystal; [α] +73.3 (c 0.085, EtOH); IR (KBr) νmax 3435 (OH), 1713 (C=O), 1643 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 3.74–3.78 (1H, m, H-5′), 3.788 (3H, s, COOMe), 3.789 (3H, s, COOMe), 4.07 (1H, br s, H-4′), 4.20–4.30 (4H, m, OH, H-4′, H-3, H-3′), 4.36 (1H, br dd, J = 1.4, 1.2 Hz, H-5), 4.53 (1H, d, J = 4.1 Hz, H-6′), 5.24 (1H, d, J = 2.9 Hz, H-6), 5.40 (1H, br s, OH), 5.66 (1H, br d, J = 7.0 Hz, OH), 6.74 (1H, s, H-2′), 7.01 (1H, d, J = 4.2 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 52.46 (CH3), 52.50 (CH3), 53.1 (CH2), 65.6 (CH2), 66.7 (CH2), 69.2 (CH2), 70.4 (CH2), 72.4 (CH2), 77.1 (CH2), 85.5 (CH2), 129.2 (C), 129.9 (C), 143.2 (CH2), 143.5 (CH2), 166.1 (Cq), 166.9 (Cq); HRMS m/z calcd. for C16H22O1035Cl (M)+, 408.0823; found, 408.0819.
4.20. Synthesis of (+)-21
Using the same procedure described for (−)-21, (+)-21 (0. 9 mg, 63%) was prepared from (+)-8 (20.0 mg, 0.035 mmol). (+)-21: oil; [α] −40.5 (c 0.035, EtOH); IR (liquid film) νmax 3389 (OH), 1721 (C=O), 1653 (C=C) cm−1; 1H-NMR (acetone-d6, 300 MHz, ppm) δ 3.72 (3H, s, COOMe), 3.80 (3H, s, COOMe), 4.00 (1H, br s, H-4′), 4.09–4.20 (2H, m, H-3, H-4), 4.20–4.30 (1H, m, H-5, H-6′), 4.46 (1H, br s, H-3′), 4.50 (1H, br s, H-5′), 5.23 (1H, d, J = 2.6 Hz, H-6), 6.79 (1H, s, H-2′), 7.01 (1H, br d, J = 3.5 Hz, H-2); 13C-NMR (acetone-d6, 75 MHz, ppm) δ 52.3 (CH3), 52.5 (CH3), 52.9 (CH2), 65.7 (CH2), 65.8 (CH2), 66.7 (CH2), 69.1 (CH2), 70.1 (CH2), 76.7 (CH2), 83.5 (CH2), 129.5 (C), 129.5 (C), 142.5 (CH2), 142.7 (CH2), 166.0 (Cq), 166.9 (Cq); HRMS m/z calcd. for C16H21O1035Cl (M)+, 408.0823; found, 408.0819.
4.21. Synthesis of (−)-24
Using the same procedure described for (+)-24, (−)-24 (0.25 g, 35%) was prepared from (−)-9 (0.45 g, 1.5 mmol) and (−)-10 (0.33 g, 1.2 mmol). (−)-24: amorphous solid; Rf 0.14 (Hexane–EtOAc, 3:1); [α] −126.47 (c 0.825, CHCl3); IR (liquid film) νmax 3470 (OH), 1722 (C=O), 1658 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 1.26–1.70 (20H, m), 3.78 (3H, s, COOMe), 3.80 (3H, s, COOMe), 4.03 (1H, br ddd, J = 5.0, 4.7, 4.4 Hz, H-5′), 4.22 (1H, dd, J = 6.1, 5.3 Hz, H-4′), 4.32 (1H, dd, J = 4.7, 3.8 Hz, H-5), 4.35 (1H, dd, J = 4.4, 0.9 Hz, H-6′), 4.55 (1H, d, J = 4.7 Hz, OH), 4.67 (1H, ddd, J = 6.1, 3.8, 1.1 Hz, H-3′), 4.69 (1H, dd, J = 6.4, 3.8 Hz, H-4), 4.78 (1H, ddd, J = 6.7, 3.0, 0.6 Hz, H-3), 5.16 (1H, d, J = 4.7 Hz, H-6), 6.63 (1H, d, J = 3.8 Hz, H-2′), 6.93 (1H, d, J = 2.9 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 24.6 (2C), 24.8 (2C), 25.8, 25.9, 35.6, 36.0, 36.9, 38.4, 52.2, 52.4, 52.7, 70.8, 71.1, 71.2, 72.6, 76.2, 77.3, 79.5, 111.2, 111.3, 131.4, 133.2, 135.0, 138.9, 165.9, 167.3; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2079.
4.22. Synthesis of (−)-25
Using the same procedure described for (–)-21, (+)-21 (0.066 g, 39%) was prepared from (−)-24 (0.17 g, 0.30 mmol). (−)-25: oil; Rf 0.14 (Hexane–EtOAc, 3:1); [α] −43.3 (c 0.085, CH2Cl2); IR (liquid film) νmax 3523 (OH), 1717 (C=O), 1653 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 1.20–1.70 (20H, m), 3.79 (3H, s, COOMe), 3.81 (3H, s, COOMe), 3.78–3.81 (1H, m, H-5′), 4.36 (1H, dd, J = 5.3, 3.9 Hz, H-4′), 4.48 (1H, dd, J = 6.5, 3.5 Hz, H-4), 4.58 (1H, d, J = 5.0 Hz, H-6′), 4.75 (1H, d, J = 3.9 Hz, H-3′), 4.76 (1H, dd, J = 3.8, 0.9 Hz, H-5), 4.81 (1H, ddd, J = 6.7, 2.7, 0.9 Hz, H-3), 5.21 (1H, d, J = 5.3 Hz, H-6), 6.76 (1H, dd, J = 3.8, 0.9 Hz, H-2′), 6.88 (1H, d, J = 2.7 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 24.5, 24.6, 24.6, 24.9, 25.8, 25.9, 35.1, 35.7, 36.7, 37.7, 52.3, 52.6, 53.3, 69.0, 71.4, 72.7, 72.8, 74.6, 75.2, 79.7, 111.1, 111.8, 131.1, 131.2, 138.2, 140.0, 166.4, 166.8; HRMS m/z calcd. for C28H37O1035Cl (M)+, 568.2075; found, 568.2076.
4.23. Synthesis of (−)-22
Using the same procedure described above for (−)-22, (+)-22 (5.9 mg, 33%) was prepared from (−)-25 (25 mg, 0.043 mmol). (−)-22: oil; Rf 0.3 (MeOH-CH2Cl2, 1:9); [α] −5.0 (c 0.225, EtOH); IR (liquid film) νmax 3391 (OH), 1716 (C=O), 1651 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 3.68 (1H, br d, J = 10.0 Hz, OH), 3.76–3.82 (1H, m, H-5), 3.78 (3H, s, COOMe), 3.80 (3H, s, COOMe), 3.86 (1H, br d, J = 10.3 Hz, OH), 3.92 (1H, br d, J = 7.3 Hz, OH), 3.99 (1H, s, H-4′), 4.01–4.08 (2H, m, H-4, OH), 4.12–4.18 (2H, m, H-3, OH), 4.26 (1H, s, H-3′), 4.47 (1H, dd, J = 2.9, 2.4 Hz, H-5), 4.57 (1H, d, J = 4.1 Hz, H-6′), 5.38 (1H, d, J = 3.3 Hz, H-6), 6.80 (1H, dd, J = 2.3, 1.2 Hz, H-2′), 7.04 (1H, d, J = 5.0 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 52.40, 52.43, 53.87, 65.89, 66.20, 69.09, 69.33, 72.18, 74.01, 83.18, 129.93, 130.07, 142.56, 143.34, 165.95, 167.33; HRFABMS m/z calcd. for C16H22O1035Cl (M + H)+, 409.0901; found, 409.0900.
4.24. Synthesis of (−)-23
Using the same procedure described above for (+)-23, (−)-23 (7.6 mg, 50%) was prepared from (−)-24 (21.3 mg, 0.038 mmol). (−)-23: white crystals; Rf 0.11 (MeOH-CH2Cl2, 1:9); [α] −75.3 (c 0.095, EtOH); IR (liquid film) νmax 3349 (OH), 1716 (C=O), 1593 (C=C) cm−1; 1H-NMR (acetone-d6, 600 MHz, ppm) δ 3.75 (1H, dd, J = 6.7, 4.1 Hz, H-4′), 3.79 (6H, s, COOMe), 4.02 (1H, d, J = 6.8, 4.1 Hz, H-5′), 4.10 (1H, br s, H-4), 4.21 (1H, br s, H-3), 4.36 (1H, d, J = 4.1 Hz, H-6′), 4.37 (1H, dd, J = 4.1, 3.8 Hz, H-3′), 4.43 (1H, dd, J = 3.6, 2.0 Hz, H-5), 5.24 (1H, d, J = 3.5 Hz, H-6), 6.79 (1H, d, J = 3.5 Hz, H-2′), 6.99 (1H, d, J = 4.4 Hz, H-2); 13C-NMR (acetone-d6, 150 MHz, ppm) δ 52.4, 52.5, 54.5, 66.3, 66.4, 66.6, 70.6, 71.9, 77.2, 83.7, 129.8, 130.8, 141.3, 1412.7, 166.1, 167.1; HRMS m/z calcd. for C16H21O1035Cl (M)+, 408.0823; found, 408.0821.
4.25. Glycosidase Assays of Synthesized Compounds
Assay of α-Glucosidase inhibition: The assay reaction mixture consisted of 0.1 M acetate buffer (pH 5.0, 45 μL),
p-nitrophenyl α-
d-glucopyranoside solution (25 μL, 250 mM), and α-glucosidase solution (25 μL, a stock solution of 1.0 mg/mL in 50 mM Tris-HCl-buffer at pH 7.8 diluted 200-fold with 10 mM phosphate buffer at pH 7.0 just prior to the assay), with the test samples
6,
21–
23, or DNJ (25 μL solution, concentration range 0.1–20 mg/mL). After incubation for 20 min at 37 °C, the reaction was interrupted by the addition of 0.5 M sodium carbonate (100 μL). The amount of
p-nitrophenol liberated was measured colorimetrically at 400 nm (optical density at 400 nm: ODtest). The inhibition rates (%) were calculated using the formula 100 − 100 × (ODtest − ODblank)/(control ODtest − control ODblank) and the IC
50 values were obtained from the inhibition curves. Assays for β-glucosidase and α-mannosidase were carried out as outlined above using
p-nitrophenyl β-
d-glucopyranoside and α-
d-mannopyranoside as the substrates. The IC
50 values are shown in
Table 1.
Assays on β-glucosidase and α-mannnosidase inhibition of synthesized carbadisaccharides 6 and 21–23 were carried out in a similar fashion.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}